
Mono- and Dinuclear Aluminium Complexes Derived from Biguanide and Carbothiamide Ligands


1
Institute of Inorganic and Analytical Chemistry, Friedrich Schiller University Jena, Humboldtstraße 8, 07743 Jena, Germany
2
Jena Center for Soft Matter (JCSM), Friedrich Schiller University Jena, Philosophenweg 7, 07743 Jena, Germany
*
Author to whom correspondence should be addressed.
Inorganics 2021, 9(7), 52; https://doi.org/10.3390/inorganics9070052
Submission received: 16 June 2021
/
Revised: 1 July 2021
/
Accepted: 1 July 2021
/
Published: 7 July 2021
(This article belongs to the Special Issue Organoaluminum Compounds)
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Dianionic N,N-chelating ligands play a crucial role in coordination chemistry, but reports on related complexes remain limited to certain types of ligands. In here, the reactions of two diprotic ligands, i.e., a biguanide and a carbothiamide, with trimethylaluminium, are reported, which give rise to mono- and dinuclear aluminium(III) complexes. In addition, single deprotonation of the diprotic biguanide using potassium bis(trimethylsilyl)amide gives rise to a one-dimensional coordination polymer. All complexes have been fully characterized, and their solid-state structures were determined by single crystal X-ray diffraction analysis.
1. Introduction
Diprotic N,N-chelating ligands are widely utilized in their dianionic form in the coordination chemistry of transition metals, main-group and rare-earth elements. In main-group chemistry, they have been particularly beneficial, and, thanks to the electronic and steric capabilities of diamide ligands, molecules that were formerly considered unstable such as boryllithium [1] and silylenes [2] have been isolated for the first time. While significant achievements could be realized using diamide ligands, their applicability is not universal which is why alternative dianionic N,N-chelates, Figure 1, have been used during the last decade to stabilize low-valent and electron-precise compounds of the Group 13 and 14 elements [3,4,5,6,7,8,9,10,11,12,13,14]. Furthermore, certain monoanionic ligands such as the well-established β-diketiminates, also called “NacNac”, can be transferred to their dianionic relatives [15,16]. This behavior is beneficial in terms of metal-ligand cooperativity [17,18], and the addition of hydrogen or protic substrates to gallium(III) β-diketiminate complexes generates an additional protic side within the ligand [19,20]. During our recent studies on dianionic bis(guanidine)s [21], we were able to isolate monoprotic biguanides as well as diprotic carbothiamides depending on the experimental conditions [22], and we wondered if diprotic biguanides are available as well when using a primary instead of a secondary amine. In here, we report the synthesis of the unprecedented biguanide 1 along with its reactivity towards trimethylaluminium and potassium bis(trimethylsilyl)amide. For comparison, the reactivity of the carbothiamide 2 with trimethylaluminium has been investigated as well.
2. Results and Discussion
Based on the synthetic protocol previously established for monoprotic biguanide ligands, [22] biguanide 1 could be obtained from the ethylene-bridged bis(thiourea), cyclohexylamine, and lead(II) oxide in a one-pot procedure, Scheme 1. The crystalline yield of 1 amounts to 16%, although 1 is formed in 40% based on the crude 1H NMR besides the carbothiamide 2 and the unsymmetric thiourea carrying one 2,6-diisopropylphenyl (Dipp) and one cyclohexyl substituent. Notably, such side products have also been observed in the synthesis of monoprotic biguanides [22].
The 1H NMR spectrum of 1 features four doublets and two septets for the methyl and methine resonances of the Dipp groups indicating hindered rotation about the Caryl–N bonds. The methylene resonances of the imidazoline ring appear as triplets at 3.28 and 3.98 ppm, and the NH protons resonate as a broadened singlet (3.93 ppm) and as a doublet (9.33 ppm). In order to distinguish which of the conceivable isomers, Scheme 1, prevails in solution (CDCl3), multidimensional NMR experiments have been conducted. The 1H,1H-COSY spectrum, Figure S3, shows distinct coupling between the broad NH singlet at 3.93 ppm and the triplet at 3.28 ppm, accounting for the CH2 group of the five-membered ring. The NH doublet at 9.33 ppm shows coupling with a broad singlet resonance at 2.96 ppm which is associated with the methine proton of the cyclohexyl ring. These observations are further supported by the 1H,13C-HMBC spectrum, Figure S5, and indicate that 1 is the predominant tautomer in solution at room temperature. This observation agrees well with the molecular structure in the solid state, which has been established by single-crystal X-ray diffraction, Figure 2. The amine protons reside on N1 and N5, and the C1–N3 and C4–N4 bond lengths (1.280(2) and 1.278(2) Å) are reminiscent of C=N double bonds. Hydrogen bonding, which is common for biguanides and used in crystal engineering [23], is also observed within 1, and an intramolecular hydrogen bond, i.e., N5H1∙∙∙N3, induces a pseudo-bicyclic system.
We next explored the reactivity of 1 towards potassium bis(trimethylsilyl)amide (KHMDS) and trimethylaluminium, Scheme 2. Deprotonation of 1 using 1.2 equivalents of KHMDS affords the potassium complex 3 in 30% crystalline yield. Single crystals, suitable for an X-ray diffraction analysis, allowed establishing the molecular structure in the solid state, Figure 3.
Complex 3 forms a polymeric one-dimensional network [24,25], in which the monoanionic ligands provide three donor sites. Hence, each tetracoordinated potassium ion binds to the nitrogen atom N1 and the phenyl ring of the related Dipp group of one ligand in a κ1 and η6 mode, in κ1 fashion to the N4 nitrogen atom of the next ligand and to one molecule of THF. The respective potassium-nitrogen bond lengths in the range of 2.695(2) to 2.735(2) Å, as well as the distance of the potassium ion to the centre of the C6 perimeter (2.857(2) Å), are in good agreement with previously reported potassium complexes [22,26]. Hence, deprotonation occurs exclusively at N1, while the amino function at N5 remains intact, which is most likely due to the stabilization of H5 by the intramolecular N5H1∙∙∙N3 hydrogen bridge. In C6D6 solution, 3 features well-resolved 1H NMR resonances, and the absence of one of the NH resonances (at 3.93 ppm) and the high-field shift to 10.31 ppm of the other one agree well with only a single deprotonation. The pattern of resonances of the Dipp groups, i.e., four methyl doublets and one methine septet, indicates a plane of symmetry within the molecule. Unfortunately, repeated attempts to obtain the double deprotonation product of 1 by using higher amounts of KHMDS remained without success.
The outcome of the reaction with trimethylaluminium is strongly dependent on the reaction conditions, but gives in both case dinuclear rather than mononuclear complexes. Reacting 1 with one equivalent of trimethylaluminium at room temperature affords a new species along with unreacted starting material in a 1:1 ratio. Hence, the reaction was repeated using two equivalents of trimethylaluminium which allowed isolating complex 4 in 55% crystalline yields. An X-ray diffraction analysis revealed its dinuclear nature, Figure 4a, which is reminiscent of a previously reported aluminium complex based on a monoprotic biguanide and resembles comparable Al–N and Al–C bond lengths [22]. i.e., the tetracoordinated aluminium centre is chelated by N3 and N4, forming a non-planar six-membered metallacycle. Again, the proton at N5 remains illustrating its robustness, and the donor-acceptor interaction between Al2 and N1 causes an elongation of the C1–N1 bond. The room temperature 1H NMR spectrum of 4 shows two overlapping singlet resonances of the bridging and terminal Al-CH3 groups, and the Dipp methyl and methine resonances appear as four doublets and two septets, respectively. Aiming to force deprotonation of the remaining NH function, the reaction with one equivalent of trimethylaluminium was repeated at 90 °C. The 1H NMR spectrum of the crude reaction mixture reveals the formation of one main species besides several side products. Complex 5 could be isolated from this mixture in 18% crystalline yield (the crude mixture contains about 50%) and fully characterized including an X-ray diffraction analysis. Complex 5 possesses a dinuclear structure in the solid state in which the ligand has been deconstructed by C–N bond cleavage. Two of the remaining monoanionic guanidine moieties are bridged via N1, N2, N4, and N5 forming an overall eight-membered dimetallacycle, while two protons reside on N3 and N6. The Al–N bond lengths are comparable and fall in between 1.9149(19) and 1.9306(19) Å, and the N–Al–N bite angles are more obtuse as compared to complex 4, in line with the larger ring size. The Dipp methyl and methine resonances appear as two doublets and one broad singlet in the 1H NMR spectrum of 5, which indicates conformational averaging on the NMR timescale at room temperature.
As double deprotonation of 1 did not work out, we considered the carbothiamide 2 as a suitable dianionic ligand and allowed it to react with two equivalents of trimethylaluminium either at room temperature or at 90 °C, Scheme 3. However, in both cases, only the mononuclear complex 6 could be isolated, although in different yields of 36% and 23%, respectively. Notably, the crude 1H NMR spectrum of the reaction performed at room temperature evidences a yield of about 70%. The 1H NMR spectrum of 6 features sharp, well-resolved resonances including one singlet at −0.53 ppm for the Al(CH3)2 and four doublets as well as two septets accounting for the iso-propyl groups of the Dipp residues, indicating hindered rotation about the Caryl-N bonds. However, only one of the NH functions has been deprotonated, as evidenced by a broad resonance at 3.48 ppm. This finding agrees well with the solid-state structure of 6, Figure 4c. The complex features comparable Al–N bond lengths within in the six-membered metallacycle that are in good agreement with those reported for thioacetamide heteroscorpionate ligands [27]. Finally, the residual proton could be located at N1.
3. Materials and Methods
3.1. General Considerations
The solvents and starting materials were purchased from ABCR, Sigma Aldrich, or VWR and used as delivered. 1,1′-(ethane-1,2-diyl)bis(3-(2,6-diisopropylphenyl)thiourea) and 2 were prepared as described elsewhere [22]. All preparations were performed under an inert atmosphere of dinitrogen by means of standard Schlenk-line techniques, while the samples for analytics were handled in a glovebox (GS-Systemtechnik and MBraun). Traces of oxygen and moisture were successively removed from the inert gas by passing it over a BASF R 3-11 (CuO/MgSiO3) catalyst, through concentrated sulfuric acid, over coarsely granulated silica gel, and finally P4O10. Toluene, n-pentane, and tetrahydrofuran were used as p.a. grade and distilled from Na/benzophenone prior to use. C6D6 was dried by distillation from potassium.
The NMR-spectra were recorded on Bruker Avance 300 and 400 spectrometers (T = 300 K) with δ (given in ppm) referenced to external tetramethylsilane (1H and 13C). 1H and 13C NMR spectra were calibrated by using the solvent residual peak (δ 1H (C6D5H) = 7.16, δ 1H (CHCl3) = 7.26), and the solvent peak (δ 13C (C6D6) = 128.06, δ 13C (CDCl3) = 77.16), respectively. The coupling constants J are given in Hertz (Hz). High-resolution mass spectra were measured by using a Waters LCT Micromass spectrometer. Infrared spectra were recorded on a Bruker ALPHA spectrometer equipped with a diamond ATR unit; the wavenumbers are given in cm−1. Elemental analysis was performed on a Vario micro cube (Elementar Analysensysteme GmbH); however, a few samples were consistently low on carbon content, while providing satisfactory H and N values.
3.2. Synthesis of the Protio-Ligand 1
A mixture of 9.75 g (19.5 mmol) 1,1′-(ethane-1,2-diyl)bis(3-(2,6-diisopropylphenyl)thiourea), 38.65 g (390.0 mmol) cyclohexylamine, and 8.73 g (39.0 mmol) PbO in 250 mL of toluene was stirred at 100 °C for 16 h. After cooling to room temperature, the solids were filtered off, washed with toluene (30 mL), and the combined filtrates were concentrated en vacuo. The residue was dissolved in boiling acetonitrile (50 mL), and the desired product 1 crystallized in the form of colourless blocks upon standing at r.t.
The results were as follows: 1.65 g, 3.1 mmol, 16%. 1H NMR (400 MHz, CDCl3): δ = 0.85 (br, 2H, (NCH(CH2)2(CH2)2(CH2)), 0.92 (br, 2H, (NCH(CH2)2(CH2)2(CH2)), 0.99 (br, 2H, (NCH(CH2)2(CH2)2(CH2)), 1.16 (d, 3JHH = 6.8 Hz, 6H, CHCH3), 1.18 (d, 3JHH = 6.8 Hz, 6H, CHCH3), 1.24 (d, 3JHH = 6.8 Hz, 6H, CHCH3), 1.26 (d, 3JHH = 6.8 Hz, 6H, CHCH3), 1.32 (br, 2H, (NCH(CH2)2(CH2)2(CH2)), 1.38 (br, 2H, (NCH(CH2)2(CH2)2(CH2)), 2.96 (br, 1H, CH2CH2CHNH), 3.09 (sept, 3JHH = 6.8 Hz, 2H, CHCH3), 3.20 (sept, 3JHH = 6.8 Hz, 2H, CHCH3), 3.28 (t, 3JHH = 7.8 Hz, 2H, NCH2CH2NHC), 3.93 (br, 1H, NCH2CH2NHC), 3.98 (t, 3JHH = 7.8 Hz, 2H, NCH2CH2NHC), 6.90 (t, 3JHH = 7.3 Hz, 1H, p-CHarom), 7.02 (d, 3JHH = 7.2 Hz, 2H, m-CHarom), 7.04 (t, 3JHH = 7.8 Hz, 1H, p-CHarom), 7.02 (d, 3JHH = 7.2 Hz, 2H, m-CHarom), 7.13 (d, 3JHH = 7.2 Hz, 2H, m-CHarom), 9.33 (d, 3JHH = 9.2 Hz, 1H, (HNcyclohexylamineCNDipp). 13C{1H} NMR (101 MHz, CDCl3): δ = 22.3 (CHCH3), 23.1 (CHCH3), 23.9 (CHCH3), 23.9 (CHCH3), 24.8 (NCH(CH2)2(CH2)2(CH2)), 25.7 (NCH(CH2)2(CH2)2(CH2)), 28.5 (CHCH3), 28.7 (CHCH3), 34.4 (NCH(CH2)2(CH2)2(CH2)), 38.9 (NCH2CH2NHC), 46.0 (NCH2CH2NHC), 49.8 (CNHC=N), 121.0 (p-CHarom), 122.3 (m-CHarom), 123.4 (m-CHarom), 123.4 (p-CHarom), 138.5 (o-Carom), 140.6 (o-Carom), 142.9 (HNcyclohexylamineCNDipp), 143.6 (i-Carom), 145.3 (i-Carom), 151.5 (NHCNDipp). IR [cm−1]: ν(NH) = 3378, ν(NH) = 3143, ν(CH3) = 2959, ν(CN) = 1541. HR-ESI-MS: calcd. for C34H51N5 [M + H]+ 530.4222; found 530.4180.
3.3. Synthesis of the Complexes 3–6
For 3: A mixture of 0.53 g (1.0 mmol) of 1 and 0.23 g (1.2 mmol) potassium bis(trimethylsilyl)amide in 20 mL of toluene and 1 mL of tetrahydrofuran was stirred at 90 °C for 16 h. Solids were filtered off, and the filtrate was concentrated to dryness en vacuo. The thus obtained residue was dissolved in a boiling toluene/pentane mixture (2:1, 7 mL), and 3 crystallized as colourless blocks upon standing at room temperature; 0.19 g, 0.3 mmol, 30%. 1H NMR (400 MHz, C6D6): δ = 0.99–1.02 (m, 3H, (NCH(CH2)2(CH2)2(CH2)), 1.13–1.44 (m, 7H, (NCH(CH2)2(CH2)2(CH2)), O(CH2)2(CH2)2), 1.18 (d, 3JHH = 6.8 Hz, 6H, CHCH3), 1.27 (d, 3JHH = 6.8 Hz, 6H, CHCH3), 1.30 (d, 3JHH = 6.8 Hz, 6H, CHCH3), 1.39 (d, 3JHH = 6.8 Hz, 6H, CHCH3), 1.57 (br, 2H, (NCH(CH2)2(CH2)2(CH2)), 1.88 (br, 2H, (NCH(CH2)2(CH2)2(CH2)), 2.64 (br, 2H, NCH2CH2N), (2.99 (br, 1H, CH2CH2CHNH), 3.36–3.54 (m, 4H, O(CH2)2(CH2)2), 3.52 (sept, 3JHH = 6.8 Hz, 2H, CHCH3), 3.89 (t, 3JHH = 7.8 Hz, 2H, NCH2CH2N), 6.95 (br, 1H, CHarom), 7.07–7.11 (m, 3H, CHarom), 7.19 (d, 3JHH = 7.2 Hz, 2H, m-CHarom). 13C{1H} NMR (101 MHz, C6D6): δ = 22.5 (CHCH3), 23.1 (CHCH3), 24.2 (CHCH3), 25.1 (NCH(CH2)2(CH2)2(CH2)), 25.8 (O(CH2)2(CH2)2), 25.9 (NCH(CH2)2(CH2)2(CH2)), 28.7 (CHCH3), 29.1 (CHCH3), 35.0 (NCH(CH2)2(CH2)2(CH2)), 40.0 (NCH2CH2N), 46.6 (NCH2CH2N), 50.2 (NCH(CH2)2(CH2)2(CH2)), 67.8 (O(CH2)2(CH2)2), 121.7 (CHarom), 122.8 (CHarom), 123.6 (CHarom), 137.6 (Carom), 138.8 (Carom), 141.3 (NcyclohexylamineCNDipp), 144.0 (Carom), 145.9 (N = CNDipp). IR [cm−1]: ν(NH) = 3428, ν(CH3) = 2958, ν(CH3) = 2927, ν(CH3) = 2864, ν(CN) = 1570. Anal. Calcd for C38H58KN5O: C, 71.31; H, 9.13; N, 10.94. Found: C, 69.41; H, 8.67; N, 10.44.
For 4: 0.53 g (1.0 mmol) of 1 were dissolved in 20 mL of toluene. Then, 1 mL of a trimethylaluminium solution (2 mmol, 2 M in toluene) was added at room temperature, and the solution was stirred for 16 h. The mixture was filtered, and the filtrate was concentrated to dryness en vacuo. The residue was dissolved in a boiling toluene/pentane mixture (2:1, 7 mL), and 4 crystallized as colourless blocks upon standing at room temperature; 0.36 g, 0.5 mmol, 55%. 1H NMR (400 MHz, C6D6): δ = −0.57 (s, 9H, Al(CH3)3), −0.56 (s, 6H, Al(CH3)2), 0.40–0.50 (m, 2H, (NCH(CH2)2(CH2)2(CH2)), 0.53–0.62 (m, 1H, (NCH(CH2)2(CH2)2(CH2)), 0.75–0.84 (m, 2H, (NCH(CH2)2(CH2)2(CH2)), 1.12 (d, 3JHH = 6.8 Hz, 6H, CHCH3), 1.13 (d, 3JHH = 6.8 Hz, 6H, CHCH3), 1.18–1.29 (m, 3H, (NCH(CH2)2(CH2)2(CH2)), 1.33 (d, 3JHH = 6.8 Hz, 6H, CHCH3), 1.40–1.44 (m, 2H, (NCH(CH2)2(CH2)2(CH2)), 1.60 (d, 3JHH = 6.8 Hz, 6H, CHCH3), 2.85 (br, 1H, CH2CH2CHNH), 3.04 (sept, 3JHH = 6.8 Hz, 2H, CHCH3), 3.23 (t, 3JHH = 7.3 Hz, 2H, NCH2CH2NHC), 3.29 (sept, 3JHH = 6.8 Hz, 2H, CHCH3), 3.72 (t, 3JHH = 7.3 Hz, 2H, NCH2CH2NHC), 4.02 (d, 3JHH = 9.2 Hz, 1H, (HNcyclohexylamineCNDipp), 6.99–7.09 (m, 3H, CHarom), 7.24 (br, 3H, CHarom). 13C{1H} NMR (101 MHz, C6D6): δ = −9.7 (Al(CH3)2), −6.1 (Al(CH3)3), 24.7 (NCH(CH2)2(CH2)2(CH2)), 24.8 (CHCH3), 24.9 (CHCH3), 25.1 (NCH(CH2)2(CH2)2(CH2)), 25.3 (CHCH3), 25.4 (CHCH3), 28.3 (CHCH3), 29.9 (CHCH3), 33.7 (NCH(CH2)2(CH2)2(CH2)), 47.9 (NCH2CH2NAl), 50.0 (NCH2CH2NAl), 55.5 (CNHC=N), 125.5 (m-CHarom), 125.8 (m-CHarom), 127.3 (p-CHarom), 129.0 (p-CHarom), 134.1 (i-Carom), 138.8 (i-Carom), 143.6 (o-Carom), 145.1 (o-Carom), 154.7 (NcyclohexylamineCNDipp), 160.5 (N=CNDipp). 27Al NMR (104 MHz, C6D6): δ = no signal. IR [cm−1]: ν(NH) = 3360, ν(CH3) = 2959, ν(CH3) = 2863, ν(CN) = 1606. Anal. Calcd for C39H65Al2N5: C, 71.20; H, 9.96; N, 10.64. Found: C, 70.90; H, 9.71; N, 10.62.
For 5: 0.53 g (1.0 mmol) of 1 were dissolved in 20 mL of toluene, and the solution was heated to 90 °C, before 0.5 mL of a trimethylaluminium solution (1 mmol, 2 M in toluene) were added. After stirring for 16 h at 90 °C, the mixture was filtered at room temperature, and the filtrate was concentrated to dryness en vacuo. The residue was dissolved in a boiling toluene/pentane mixture (2:1, 7 mL), and 5 crystallized as colourless blocks upon standing at room temperature; 0.11 g, 0.2 mmol, 18%. 1H NMR (400 MHz, C6D6): δ = −0.51 (br, 12H, Al(CH3)2), 1.19 (d, 3JHH = 6.8 Hz, 12H, CHCH3), 1.42 (d, 3JHH = 6.8 Hz, 12H, CHCH3), 2.33 (t, 3JHH = 7.8 Hz, 4H, NCH2CH2N), 3.50 (t, 3JHH = 7.8 Hz, 4H, NCH2CH2N), 3.55–3.70 (br, 5H, 2H, CHCH3, NCH2CH2NHC), 7.14 (br, 6H, CHarom). 13C{1H} NMR (101 MHz, C6D6): δ = -7.0 (Al(CH3)2), 24.3 (CHCH3), 26.0 (CHCH3), 28.0 (CHCH3), 42.8 (NCH2CH2N), 51.1 (NCH2CH2N), 124.7 (m-CHarom), 127.2 (p-CHarom), 141.5 (o-Carom), 146.0 (i-Carom), 168.9 (N=CNDipp). 27Al NMR (104 MHz, C6D6): δ = no signal. IR [cm−1]: ν(NH) = 3396, ν(CH3) = 2962, ν(CH3) = 2945, ν(CH3) = 2925, ν(CH3) = 2866, ν(CN) = 1593. Anal. Calcd for C34H56Al2N6 · 0.65 C7H8: C, 69.87; H, 9.31; N, 12.68. Found: C, 65.83; H, 9.01; N, 12.93.
For 6: 0.46 g (1.0 mmol) of 2 were dissolved in 20 mL of toluene, and 0.5 mL of a trimethylaluminium solution (1 mmol, 2 M in toluene) were added at room temperature. The mixture was stirred for 16 h, filtered, and the filtrate was concentrated to dryness en vacuo. The residue was dissolved in a boiling toluene/pentane mixture (2:1, 7 mL), and 6 crystallized as colourless blocks upon standing at room temperature; 0.22 g, 0.4 mmol, 36%. 1H NMR (400 MHz, C6D6): δ = −0.53 (br, 6H, Al(CH3)2), 0.98 (d, 3JHH = 6.8 Hz, 6H, CHCH3), 1.27 (d, 3JHH = 6.8 Hz, 6H, CHCH3), 1.46 (d, 3JHH = 6.8 Hz, 6H, CHCH3), 1.68 (d, 3JHH = 6.8 Hz, 6H, CHCH3), 1.92 (t, 3JHH = 8.4 Hz, 2H, NCH2CH2N), 3.36 (sept, 3JHH = 6.8 Hz, 2H, CHCH3), 3.48 (br, 1H, CH2CH2CNH), 3.62 (sept, 3JHH = 6.8 Hz, 2H, CHCH3), 4.23 (t, 3JHH = 8.4 Hz, 2H, NCH2CH2N), 7.00–7.14 (m, 3H, CHarom), 7.29 (br, 3H, CHarom). 13C{1H} NMR (101 MHz, C6D6): δ = −9.2 (Al(CH3)2), 24.3 (CHCH3), 25.1 (CHCH3), 25.3 (CHCH3), 26.7 (CHCH3), 28.4 (CHCH3), 29.3 (CHCH3), 38.4 (NCH2CH2N), 52.0 (NCH2CH2N), 124.7 (m-CHarom), 125.5 (m-CHarom), 127.2 (p-CHarom), 136.0 (o-Carom), 141.5 (o-Carom), 144.9 (i-Carom), 156.2 (HNCNDipp), 184.3 (S=CNDipp). 27Al NMR (104 MHz, C6D6): δ = no signal. IR [cm−1]: ν(NH) = 3420, ν(CH3) = 2966, ν(CH3) = 2927, ν(CH3) = 2867, ν(CN) = 1617. Anal. Calcd for C30H45AlN4S · 1.05 C7H8: C, 72.65; H, 8.72; N, 9.07. Found: C, 73.14; H, 8.64; N, 9.51.
3.4. Crystallographic Details
The single crystal X-ray diffraction data for 1 were recorded on a GV-50 diffractometer with a TitanS2 detector from Rigaku Oxford Diffraction (formerly Agilent Technologies) applying a Cu Kα radiation (λ = 1.54184 Å). Analytical absorption corrections were applied to the data [28]. The intensity data for the compounds 3, 4, 5, and 6 were collected on a Nonius KappaCCD diffractometer using graphite-monochromated Mo-Kα radiation. Data were corrected for Lorentz and polarization effects; absorption was taken into account on a semi-empirical basis using multiple-scans [29,30,31]. The structures were solved by intrinsic phases (SHELXT) [32] and refined by full-matrix least squares techniques against Fo2 (SHELXL-2014) [33]. The hydrogen atoms, bound to N1, N5 of compound 1, to N5 of 3 and 4, to N1, N4 of 5, as well as to N1 of 6, were located by difference Fourier synthesis and refined isotropically. All other hydrogen atoms were included at calculated positions with fixed thermal parameters. All non-hydrogen atoms were refined anisotropically [33]. Crystallographic data as well as structure solution and refinement details are summarized in Table S1 (in Supplementary Materials). Olex2 v1.2 was used for structure representations [34].
4. Conclusions
The diprotic biguanide 1 is readily available using a facile one-pot procedure. Single deprotonation of 1 is feasible, while attempts of a two-fold deprotonation using either potassium bis(trimethylsilyl)amide or trimethylaluminium remained without success. In the case of the potassium complex 3, a one-dimensional coordination polymer could be obtained, while in the case of aluminium, two dinuclear complexes, i.e., 4 and 5, each possessing one intact NH function, were isolated. While the complexes 3 and 4 possess a different element-dependent coordination behaviour, the tautomeric form of the monoanionic ligand is the same; that is, the proton resides at the exocyclic cyclohexyl-substituted nitrogen atom N5. Substituting the cyclohexyl-rest is an option to alter the acidity of the respective protons and hence the overall characteristics of the ligand. In the case of the carbothiamide 2, mono-deprotonation was also the exclusive event in the reaction with trimethylaluminium. While based on these results neither the diprotic biguanide 1 nor the carbothiamide 2 appear as a suitable dianionic ligand for alkylaluminium fragments, this might be different when substituting the metal or the additional ligands. Furthermore, the multiple binding sites possibly give rise to di- or polynuclear complexes, which is why the exploration of the coordination behaviour of both 1 and 2 towards elements of the s-, p-, d-, and f-block is worthy of pursuit. Finally, the presence of a NH function indicates the potential of related complexes to be used for metal-ligand cooperativity, which we will investigate in the future.
Supplementary Materials
The following are available online at https://www.mdpi.com/article/10.3390/inorganics9070052/s1: Table S1: Crystal data and refinement details for the X-ray structure determinations; Figures S1–S25: 1H, 13C, and 27Al NMR spectra of 1, 3, 4, 5, and 6; Figures S26–S30: ATR-IR spectra of 1, 3, 4, 5, and 6; CIF and CheckCIF files of 1, 3, 4, 5, and 6.
Author Contributions
Conceptualization, R.K.; validation, M.D., H.G., and R.K.; investigation, M.D. and H.G.; writing—original draft preparation, M.D.; writing—review and editing, R.K.; visualization, M.D.; supervision, R.K.; project administration, R.K.; funding acquisition, R.K. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by Deutsche Forschungsgemeinschaft, grant number KR4782/3-1, and the Friedrich Schiller University Jena.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
NMR and IR spectra are given in the supporting information, while crystallographic data (excluding structure factors) have been deposited with the Cambridge Crystallographic Data Centre as supplementary publication CCDC-20910018 for 1, CCDC-20910019 for 3, CCDC-20910020 for 4, CCDC-20910021 for 5, and CCDC-20910022 for 6. Copies of the data can be obtained free of charge on application to CCDC, 12 Union Road, Cambridge CB2 1EZ, UK (E-mail: [email protected]).
Acknowledgments
This work is dedicated to Hansgeorg Schnöckel on the occasion of his 80th birthday.
Conflicts of Interest
The authors declare no conflict of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript; or in the decision to publish the results.
References
- Segawa, Y.; Yamashita, M.; Nozaki, K. Boryllithium: Isolation, characterization, and reactivity as a boryl anion. Science 2006, 314, 113–115. [Google Scholar] [CrossRef]
- Denk, M.; Lennon, R.; Hayashi, R.; West, R.; Belyakov, A.V.; Verne, H.P.; Haaland, A.; Wagner, M.; Metzler, N. Synthesis and Structure of a Stable Silylene. J. Am. Chem. Soc. 1994, 116, 2691–2692. [Google Scholar] [CrossRef]
- Liew, S.K.; Al-Rafia, S.M.I.; Goettel, J.T.; Lummis, P.A.; McDonald, S.M.; Miedema, L.J.; Ferguson, M.J.; McDonald, R.; Rivard, E. Expanding the steric coverage offered by bis(amidosilyl) chelates: Isolation of low-coordinate N-heterocyclic germylene complexes. Inorg. Chem. 2012, 51, 5471–5480. [Google Scholar] [CrossRef] [PubMed]
- Oetzel, J.; Weyer, N.; Bruhn, C.; Leibold, M.; Gerke, B.; Pöttgen, R.; Maier, M.; Winter, R.F.; Holthausen, M.C.; Siemeling, U. Redox-Active N-Heterocyclic Germylenes and Stannylenes with a Ferrocene-1,1′-diyl Backbone. Chem. Eur. J. 2017, 23, 1187–1199. [Google Scholar] [CrossRef] [PubMed]
- Schwamm, R.J.; Coles, M.P.; Hill, M.S.; Mahon, M.F.; McMullin, C.L.; Rajabi, N.A.; Wilson, A.S.S. A Stable Calcium Alumanyl. Angew. Chem. Int. Ed. 2020, 59, 3928–3932. [Google Scholar] [CrossRef] [PubMed]
- Weyer, N.; Heinz, M.; Schweizer, J.I.; Bruhn, C.; Holthausen, M.C.; Siemeling, U. A Stable N-Heterocyclic Silylene with a 1,1′-Ferrocenediyl Backbone. Angew. Chem. Int. Ed. 2021, 60, 2624–2628. [Google Scholar] [CrossRef]
- Kristinsdóttir, L.; Oldroyd, N.L.; Grabiner, R.; Knights, A.W.; Heilmann, A.; Protchenko, A.V.; Niu, H.; Kolychev, E.L.; Campos, J.; Hicks, J.; et al. Synthetic, structural and reaction chemistry of N-heterocyclic germylene and stannylene compounds featuring N-boryl substituents. Dalton Trans. 2019, 48, 11951–11960. [Google Scholar] [CrossRef]
- Böserle, J.; Zhigulin, G.; Ketkov, S.; Jambor, R.; Růžička, A.; Dostál, L. Diverse reactivity of a boraguanidinato germylene toward organic pseudohalides. Dalton Trans. 2018, 47, 14880–14883. [Google Scholar] [CrossRef]
- Driess, M.; Yao, S.; Brym, M.; van Wüllen, C.; Lentz, D. A New Type of N-Heterocyclic Silylene with Ambivalent Reactivity. J. Am. Chem. Soc. 2006, 128, 9628–9629. [Google Scholar] [CrossRef]
- Driess, M.; Yao, S.; Brym, M.; van Wüllen, C. A Heterofulvene-Like Germylene with a Betain Reactivity. Angew. Chem. Int. Ed. 2006, 45, 4349–4352. [Google Scholar] [CrossRef]
- Fedushkin, I.L.; Skatova, A.A.; Chudakova, V.A.; Khvoinova, N.M.; Baurin, A.Y.; Dechert, S.; Hummert, M.; Schumann, H. Stable Germylenes Derived from 1,2-Bis(arylimino)acenaphthenes. Organometallics 2004, 23, 3714–3718. [Google Scholar] [CrossRef]
- Schwamm, R.J.; Anker, M.D.; Lein, M.; Coles, M.P. Reduction vs. Addition: The Reaction of an Aluminyl Anion with 1,3,5,7-Cyclooctatetraene. Angew. Chem. Int. Ed. 2019, 58, 1489–1493. [Google Scholar] [CrossRef]
- Schwamm, R.J.; Anker, M.D.; Lein, M.; Coles, M.P.; Fitchett, C.M. Indyllithium and the Indyl Anion InL-: Heavy Analogues of N-Heterocyclic Carbenes. Angew. Chem. Int. Ed. 2018, 57, 5885–5887. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Inoue, S.; Yao, S.; Driess, M. Reactivity of N-Heterocyclic Germylene Toward Ammonia and Water. Organometallics 2011, 30, 6490–6494. [Google Scholar] [CrossRef]
- Camp, C.; Arnold, J. On the non-innocence of “Nacnacs”: Ligand-based reactivity in β-diketiminate supported coordination compounds. Dalton Trans. 2016, 45, 14462–14498. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Bellows, S.M.; Holland, P.L. Tuning steric and electronic effects in transition-metal β-diketiminate complexes. Dalton Trans. 2015, 44, 16654–16670. [Google Scholar] [CrossRef] [Green Version]
- Greb, L.; Ebner, F.; Ginzburg, Y.; Sigmund, L.M. Element-Ligand Cooperativity with p-Block Elements. Eur. J. Inorg. Chem. 2020, 2020, 3030–3047. [Google Scholar] [CrossRef]
- Elsby, M.R.; Baker, R.T. Strategies and mechanisms of metal-ligand cooperativity in first-row transition metal complex catalysts. Chem. Soc. Rev. 2020, 49, 8933–8987. [Google Scholar] [CrossRef]
- Abdalla, J.A.B.; Riddlestone, I.M.; Tirfoin, R.; Aldridge, S. Cooperative bond activation and catalytic reduction of carbon dioxide at a group 13 metal center. Angew. Chem. Int. Ed. 2015, 54, 5098–5102. [Google Scholar] [CrossRef]
- Hitzfeld, P.S.; Kretschmer, R. Cooperative H-X Bond Activation by Electron-Precise Aluminium and Gallium Compounds Incorporating β-Diketiminate Ligands. Eur. J. Inorg. Chem. 2020, 2020, 1624–1630. [Google Scholar] [CrossRef]
- Vass, V.; Dehmel, M.; Lehni, F.; Kretschmer, R. A Facile One-Pot Synthesis of 1,2,3-Tri- and 1,1,2,3-Tetrasubstituted Bis(guanidines) from Bis(thioureas). Eur. J. Org. Chem. 2017, 2017, 5066–5073. [Google Scholar] [CrossRef]
- Dehmel, M.; Vass, V.; Prock, L.; Görls, H.; Kretschmer, R. Synthesis and Coordination Chemistry of 3,4-Ethylene-Bridged 1,1,2,5-Tetrasubstituted Biguanides. Inorg. Chem. 2020, 59, 2733–2746. [Google Scholar] [CrossRef]
- Şerb, M.-D.; Kalf, I.; Englert, U. Biguanide and squaric acid as pH-dependent building blocks in crystal engineering. CrystEngComm 2014, 16, 10631–10639. [Google Scholar] [CrossRef]
- Fromm, K.M. Coordination polymer networks with s-block metal ions. Coord. Chem. Rev. 2008, 252, 856–885. [Google Scholar] [CrossRef] [Green Version]
- Fromm, K.M.; Sagué, J.L.; Mirolo, L. Coordination Polymer Networks: An Alternative to Classical Polymers? Macromol. Symp. 2010, 291–292, 75–83. [Google Scholar] [CrossRef] [Green Version]
- Smith, J.D. Organometallic compounds of the heavier alkali metals. Adv. Organomet. Chem. 1998, 43, 267. [Google Scholar]
- Castro-Osma, J.A.; Alonso-Moreno, C.; Lara-Sánchez, A.; Otero, A.; Fernández-Baeza, J.; Sánchez-Barba, L.F.; Rodríguez, A.M. Catalytic behaviour in the ring-opening polymerisation of organoaluminiums supported by bulky heteroscorpionate ligands. Dalton Trans. 2015, 44, 12388–12400. [Google Scholar] [CrossRef]
- CrysAlisPro, version 171.38.42b; Agilent Technologies Inc.: Oxford, UK, 2015.
- COLLECT; Data Collection Software; Nonius B.V.: Delft, The Netherlands, 1998.
- Otwinowski, Z.; Minor, W. Processing of X-ray diffraction data collected in oscillation mode. In Methods in Enzymology: Macromolecular Crystallography Part A; Carter, C.W., Jr., Ed.; Academic Press: Cambridge, MA, USA, 1997; pp. 307–326. ISBN 0076-6879. [Google Scholar]
- SADABS 2.10; Bruker-AXS Inc.: Madison, WI, USA, 2002.
- Sheldrick, G.M. SHELXT—Integrated space-group and crystal-structure determination. Acta Cryst. A 2015, 71, 3–8. [Google Scholar] [CrossRef] [Green Version]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Cryst. C 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. OLEX2: A complete structure solution, refinement and analysis program. J. Appl. Crystallogr. 2009, 42, 339–341. [Google Scholar] [CrossRef]
Figure 1.
(a) Selected examples of dianionic N,N-chelating ligands. (b) The diprotic biguanide 1 and carbothiamide 2 used in this study.
Figure 1.
(a) Selected examples of dianionic N,N-chelating ligands. (b) The diprotic biguanide 1 and carbothiamide 2 used in this study.

Scheme 1.
Synthesis of the diprotic biguanide 1 and conceivable (but not observed) tautomers.

Figure 2.
Solid-state structure (hydrogen atoms except the NH are omitted for the sake of clarity) with selected bond lengths (Å) and angles (deg) of 1: C1–N1 1.364(2), C4–N4 1.278(2), C1–N3 1.280(2), N2–C4–N5 124.45(11), N2–C1–N3 114.63(10).
Figure 2.
Solid-state structure (hydrogen atoms except the NH are omitted for the sake of clarity) with selected bond lengths (Å) and angles (deg) of 1: C1–N1 1.364(2), C4–N4 1.278(2), C1–N3 1.280(2), N2–C4–N5 124.45(11), N2–C1–N3 114.63(10).

Scheme 2.
Synthesis of potassium and aluminium complexes originating from 1.

Figure 3.
(a) Solid-state structure (hydrogen atoms except the NH are omitted for the sake of clarity) with selected bond lengths (Å) and angles (deg) of 3: K1–N1 2.695(2), K1–N4′ 2.735(2), K1–O1 2.776(2), K1–Ph-Ring 2.857(2), N1–K1–N4′ 103.06(7), N4′–K1–O1 85.88(7); symmetry transformations used to generate equivalent atoms (marked with an ‘): −x + 3/2, y + 1/2, −z + 3/2. (b) 1D coordination polymer of 3.
Figure 3.
(a) Solid-state structure (hydrogen atoms except the NH are omitted for the sake of clarity) with selected bond lengths (Å) and angles (deg) of 3: K1–N1 2.695(2), K1–N4′ 2.735(2), K1–O1 2.776(2), K1–Ph-Ring 2.857(2), N1–K1–N4′ 103.06(7), N4′–K1–O1 85.88(7); symmetry transformations used to generate equivalent atoms (marked with an ‘): −x + 3/2, y + 1/2, −z + 3/2. (b) 1D coordination polymer of 3.

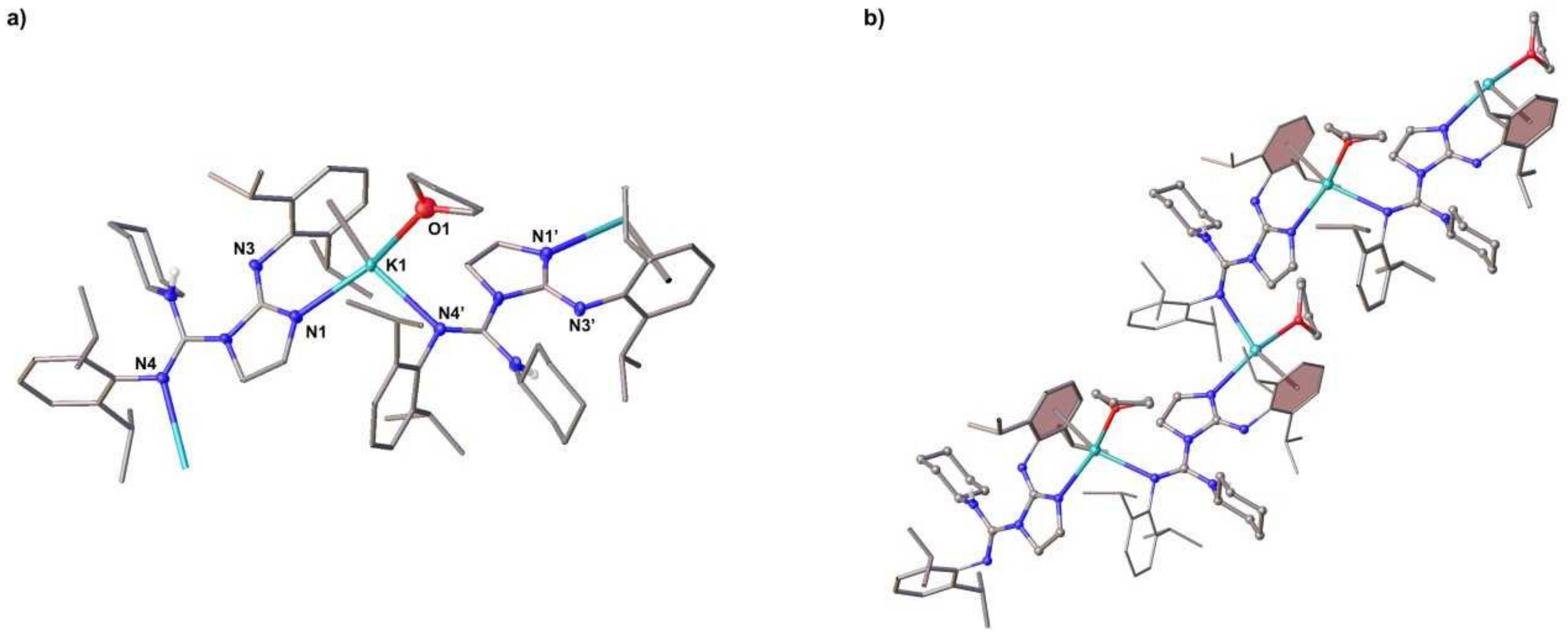
Figure 4.
(a) Solid-state structure (hydrogen atoms except the NH are omitted for the sake of clarity) with selected bond lengths (Å) and angles (deg) of: (a) 4: Al1–N4 1.9325(17), Al1–N3 1.9255(17), C1–N1 1.318(2), C1–N3 1.330(2), Al2–N1 2.0160(17), N4–Al1–N3 92.48(7), C35–Al1–C36 121.04(11); (b) 5: Al1–N1 1.9282(19), Al1–N5 1.9188(19), Al2–N2 1.9149(19), Al2–N4 1.9306(19), C13–N1 1.338(3), C13–N2 1.349(3), C16–N4 1.334(3), C16–N5 1.350(3), N1–Al1–N5 114.59(8), C31–Al1–C32 113.80(11), N2–Al2–N4 113.42(8), C33–Al2–C34 115.16(11); (c) 6: Al1–N4 1.9245(17), Al1–N3 1.9165(18), C1–N1 1.340(3), C1–N3 1.318(3), S1–C4 1.682(2), N3–Al1–N4 92.75(7), C29–Al1–C30 113.82(11).
Figure 4.
(a) Solid-state structure (hydrogen atoms except the NH are omitted for the sake of clarity) with selected bond lengths (Å) and angles (deg) of: (a) 4: Al1–N4 1.9325(17), Al1–N3 1.9255(17), C1–N1 1.318(2), C1–N3 1.330(2), Al2–N1 2.0160(17), N4–Al1–N3 92.48(7), C35–Al1–C36 121.04(11); (b) 5: Al1–N1 1.9282(19), Al1–N5 1.9188(19), Al2–N2 1.9149(19), Al2–N4 1.9306(19), C13–N1 1.338(3), C13–N2 1.349(3), C16–N4 1.334(3), C16–N5 1.350(3), N1–Al1–N5 114.59(8), C31–Al1–C32 113.80(11), N2–Al2–N4 113.42(8), C33–Al2–C34 115.16(11); (c) 6: Al1–N4 1.9245(17), Al1–N3 1.9165(18), C1–N1 1.340(3), C1–N3 1.318(3), S1–C4 1.682(2), N3–Al1–N4 92.75(7), C29–Al1–C30 113.82(11).

Scheme 3.
Reaction of the carbothiamide 2 with trimethylaluminium.

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Dehmel, M.; Görls, H.; Kretschmer, R. Mono- and Dinuclear Aluminium Complexes Derived from Biguanide and Carbothiamide Ligands. Inorganics 2021, 9, 52. https://doi.org/10.3390/inorganics9070052
AMA Style
Dehmel M, Görls H, Kretschmer R. Mono- and Dinuclear Aluminium Complexes Derived from Biguanide and Carbothiamide Ligands. Inorganics. 2021; 9(7):52. https://doi.org/10.3390/inorganics9070052
Chicago/Turabian StyleDehmel, Maximilian, Helmar Görls, and Robert Kretschmer. 2021. "Mono- and Dinuclear Aluminium Complexes Derived from Biguanide and Carbothiamide Ligands" Inorganics 9, no. 7: 52. https://doi.org/10.3390/inorganics9070052
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.