Evaluating Ligand Modifications of the Titanocene and Auranofin Moieties for the Development of More Potent Anticancer Drugs

 , , and
, , and
Abstract
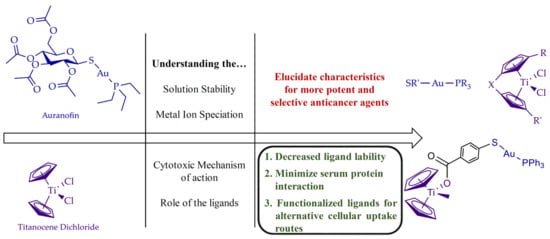
:
1. Introduction
2. Titanocene Dichloride and its Ligand Evaluation
2.1. Biologically Relevant Ti(IV) Coordination Chemistry
2.2. History of Ti(IV) in Anticancer Research
2.3. Ti(IV) in Anticancer Clinical Trials
2.4. Titanocene Dichloride Therapeutic Limitation due to Its Formulation and Speciation in the Body
2.5. Titanocenyl Modification and Encapsulation to Alter Method of Transport for Improved Cytotoxic Potency
3. Auranofin and its Ligand Evaluation
3.1. Biologically Relevant Au(I) Coordination Chemistry
3.2. Brief History of the Medical Use of Gold
3.3. Drug Repurposing of AF for Anticancer Application and an Evaluation of Its Mechanism of Action
3.4. Auranofin Therapeutic Limitation due to Its Speciation in the Body
3.5. Modifying the Phosphine and Thiosugar Ligands of Auranofin to Understand Their Mechanistic Contribution
3.6. AF in Anticancer Clinical Trials
4. Fusing the Auranofin and Titanocene Moieties to Create a Bimetallic Complex with Heightened Cytotoxic Potency
5. Conclusions and Future Directions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Institute for Health Metrics and Evaluation, Global Burden of Disease. Available online: https://vizhub.healthdata.org/cod/ (accessed on 29 November 2019).
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2019. CA Cancer J. Clin. 2019, 69, 7–34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Approved Drug Products with Therapeutic Equivalence Evaluations (Orange Book). Available online: https://www.fda.gov/drugs/resources-information-approved-drugs/approved-drug-products-therapeutic-equivalence-evaluations-orange-book (accessed on 1 November 2019).
- Rosenberg, B.; Vancamp, L.; Krigas, T. Inhibition of cell division in Escherichia coli by electrolysis products from a platinum electrode. Nature 1965, 205, 698–699. [Google Scholar] [CrossRef] [PubMed]
- Muggia, F.; Farrell, N. Platinum coordination compounds in cancer chemotherapy. Crit. Rev. Oncol. Hematol. 2005, 1, 1–2. [Google Scholar] [CrossRef]
- Dilruba, S.; Kalayda, G.V. Platinum-based drugs: Past, present and future. Cancer Chemother. Pharmacol. 2016, 77, 1103–1124. [Google Scholar] [CrossRef] [PubMed]
- Rabik, C.A.; Dolan, M.E. Molecular mechanisms of resistance and toxicity associated with platinating agents. Cancer Treat. Rev. 2007, 33, 9–23. [Google Scholar] [CrossRef] [Green Version]
- Siddik, Z.H. Cisplatin: Mode of cytotoxic action and molecular basis of resistance. Oncogene 2003, 22, 7265. [Google Scholar] [CrossRef] [Green Version]
- Fuertes, M.A.; Alonso, C.; Perez, J.M. Biochemical modulation of Cisplatin mechanisms of action: Enhancement of antitumor activity and circumvention of drug resistance. Chem. Rev. 2003, 103, 645–662. [Google Scholar] [CrossRef]
- Dabrowiak, J.C. Metals in Medicine, 2nd ed.; Wiley: New York, NY, USA, 2017. [Google Scholar]
- Descôteaux, C.; Provencher-Mandeville, J.; Mathieu, I.; Perron, V.; Mandal, S.K.; Asselin, E.; Bérubé, G. Synthesis of 17β-estradiol platinum(II) complexes: Biological evaluation on breast cancer cell lines. Bioorg. Med. Chem. Lett. 2003, 13, 3927–3931. [Google Scholar] [CrossRef]
- Van Themsche, C.; Parent, S.; Leblanc, V.; Descoteaux, C.; Simard, A.M.; Berube, G.; Asselin, E. VP-128, a novel oestradiol-platinum(II) hybrid with selective anti-tumour activity towards hormone-dependent breast cancer cells in vivo. Endocr. Relat. Cancer 2009, 16, 1185–1195. [Google Scholar] [CrossRef] [Green Version]
- Aris, S.M.; Farrell, N.P. Towards Antitumor Active trans-Platinum Compounds. Eur. J. Inorg. Chem. 2009, 2009, 1293. [Google Scholar] [CrossRef] [Green Version]
- Dhar, S.; Liu, Z.; Thomale, J.; Dai, H.; Lippard, S.J. Targeted single-wall carbon nanotube-mediated Pt(IV) prodrug delivery using folate as a homing device. J. Am. Chem. Soc. 2008, 130, 11467–11476. [Google Scholar] [CrossRef] [Green Version]
- Cotton, F.A.; Wilkinson, G.; Murillo, C.A.; Bochman, M. Advanced Inorganic Chemistry; John Wiley & Sons, Inc.: New York, NY, USA, 1999. [Google Scholar]
- Buettner, K.M.; Valentine, A.M. Bioinorganic chemistry of titanium. Chem. Rev. 2011, 112, 1863–1881. [Google Scholar] [CrossRef]
- Saxena, M.; Loza Rosas, S.; Gaur, K.; Sharma, S.; Perez Otero, S.C.; Tinoco, A.D. Exploring titanium(IV) chemical proximity to iron(III) to elucidate a function for Ti(IV) in the human body. Coord. Chem. Rev. 2018, 363, 109–125. [Google Scholar] [CrossRef] [PubMed]
- Valentine, A.M. Titanium: Inorganic & Coordination Chemistry. Encycl. Inorg. Chem. 2006. [Google Scholar] [CrossRef]
- Housecroft, C.E.; Sharpe, A.G. Inorganic Chemistry, 4th ed.; Pearson: San Antonio, TX, USA, 2012. [Google Scholar]
- Graetzel, M.; Rotzinger, F.P. Raman spectroscopic evidence for the existence of titanyl TiO2+ in acidic aqueous solutions. Inorg. Chem. 1985, 24, 2320–2321. [Google Scholar] [CrossRef]
- Schmidt, J.; Vogelsberger, W. Aqueous Long-Term Solubility of Titania Nanoparticles and Titanium(IV) Hydrolysis in a Sodium Chloride System Studied by Adsorptive Stripping Voltammetry. J. Solut. Chem. 2009, 38, 1267–1282. [Google Scholar] [CrossRef]
- Guo, M.; Harvey, I.; Campopiano, D.J.; Sadler, P.J. Short Oxo–Titanium(IV) Bond in Bacterial Transferrin: A Protein Target for Metalloantibiotics. Angew. Chem. 2006, 45, 2758–2761. [Google Scholar] [CrossRef] [PubMed]
- Tinoco, A.D.; Thomas, H.R.; Incarvito, C.D.; Saghatelian, A.; Valentine, A.M. Cytotoxicity of a Ti(IV) compound is independent of serum proteins. Proc. Natl. Acad. Sci. USA 2012, 109, 5016–5021. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, M.L.; Sun, H.Z.; Bihari, S.; Parkinson, J.A.; Gould, R.O.; Parsons, S.; Sadler, P.J. Stereoselective formation of seven-coordinate titanium(IV) monomer and dimer complexes of ethylenebis(o-hydroxyphenyl)glycine. Inorg. Chem. 2000, 39, 206–215. [Google Scholar] [CrossRef]
- Keppler, B.K.; Friesen, C.; Moritz, H.G.; Vongerichten, H.; Vogel, E. Tumor-inhibiting bis(β-diketonato) metal complexes. Budotitane, cis-diethoxybis(1-phenylbutane-1,3-dionato)titanium(IV)—The 1st transition-metal complex after platinum to qualfiy for clinical trials. Struct. Bond. 1991, 78, 97–127. [Google Scholar]
- Köpf, H.; Köpf-Maier, P. Titanocene dichloride—The first metallocene with cancerostatic activity. Angew. Chem. 1979, 18, 477–478. [Google Scholar] [CrossRef] [PubMed]
- Köpf-Maier, P.; Köpf, H. Transition and Main-Group Metal Cyclopentadienyl Complexes: Preclinical Studies on a Series of Antitumor Agents of Different Structural Type; Springer: Berlin, Germany, 1988; pp. 103–185. [Google Scholar]
- Harding, M.M.; Mokdsi, G. Antitumour metallocenes: Structure-activity studies and interactions with biomolecules. Curr. Med. Chem. 2000, 7, 1289–1303. [Google Scholar] [CrossRef] [PubMed]
- Olszewski, U.; Hamilton, G. Mechanisms of cytotoxicity of anticancer titanocenes. Anticancer Agents Med. Chem. 2010, 10, 302–311. [Google Scholar] [CrossRef] [PubMed]
- Toney, J.H.; Marks, T.J. Hydrolysis chemistry of the metallocene dichlorides M(η5-C5H5)2Cl2, M = titanium, vanadium, or zirconium. Aqueous kinetics, equilibria, and mechanistic implications for a new class of antitumor agents. J. Am. Chem. Soc. 1985, 107, 947–953. [Google Scholar] [CrossRef]
- Williams, J.; Moreton, K. Distribution of iron between the metal-binding sites of transferrin in human-serum. Biochem. J. 1980, 185, 483–488. [Google Scholar] [CrossRef]
- Sun, H.; Li, H.; Sadler, P.J. Transferrin as a Metal Ion Mediator. Chem. Rev. 1999, 99, 2817–2842. [Google Scholar] [CrossRef]
- Vincent, J.B.; Love, S. The binding and transport of alternative metals by transferrin. Biochim. Biophys. Acta Gen. Subj. 2012, 1820, 362–378. [Google Scholar] [CrossRef]
- Bonvin, G.; Bobst, C.E.; Kaltashov, I.A. Interaction of transferrin with non-cognate metals studied by native electrospray ionization mass spectrometry. Int. J. Mass Spectrom. 2017, 420, 74–82. [Google Scholar] [CrossRef]
- Sun, H.; Li, H.; Weir, R.A.; Sadler, P.J. The First Specific Ti(IV)-Protein Complex: Potential Relevance to Anticancer Activity of Titanocenes. Angew. Chem. 1998, 37, 1577–1579. [Google Scholar] [CrossRef]
- Messori, L.; Orioli, P.; Banholzer, V.; Pais, I.; Zatta, P. Formation of titanium(IV) transferrin by reaction of human serum apotransferrin with titanium complexes. FEBS Lett. 1999, 442, 157–161. [Google Scholar] [CrossRef]
- Guo, M.; Sun, H.; McArdle, H.J.; Gambling, L.; Sadler, P.J. Ti(IV) Uptake and Release by Human Serum Transferrin and Recognition of Ti(IV)-Transferrin by Cancer Cells: Understanding the Mechanism of Action of the Anticancer Drug Titanocene Dichloride. Biochemistry 2000, 39, 10023–10033. [Google Scholar] [CrossRef] [PubMed]
- Shen, Y.; Li, X.; Dong, D.; Zhang, B.; Xue, Y.; Shang, P. Transferrin receptor 1 in cancer: A new sight for cancer therapy. Am. J. Cancer Res. 2018, 8, 916–931. [Google Scholar] [PubMed]
- Tinoco, A.D.; Saxena, M.; Sharma, S.; Noinaj, N.; Delgado, Y.; Quiñones González, E.P.; Conklin, S.E.; Zambrana, N.; Loza-Rosas, S.A.; Parks, T.B. Unusual synergism of transferrin and citrate in the regulation of Ti(IV) speciation, transport, and toxicity. J. Am. Chem. Soc. 2016, 138, 5659–5665. [Google Scholar] [CrossRef] [PubMed]
- Köpf-Maier, P. Electron-spectroscopic imaging—A method for analysing the distribution of light elements in mammalian cells and tissues. Acta Histochem. 1991, 91, 25–37. [Google Scholar] [CrossRef]
- Guo, M.; Guo, Z.; Sadler, P.J. Titanium(IV) targets phosphoesters on nucleotides: Implications for the mechanism of action of the anticancer drug titanocene dichloride. J. Biol. Inorg. Chem. 2001, 6, 698–707. [Google Scholar] [CrossRef]
- Erxleben, A.; Claffey, J.; Tacke, M. Binding and hydrolysis studies of antitumoural titanocene dichloride and Titanocene Y with phosphate diesters. J. Inorg. Biochem. 2010, 104, 390–396. [Google Scholar] [CrossRef]
- Wang, H.; Zhong, J.; Xiao, K.; Tian, Z. Enrichment of intact phosphoproteins using immobilized titanium(IV) affinity chromatography microspheres. Sep. Sci. Plus 2018, 1, 93–99. [Google Scholar] [CrossRef]
- Christodoulou, C.V.; Eliopoulos, A.G.; Young, L.S.; Hodgkins, L.; Ferry, D.R.; Kerr, D.J. Anti-proliferative activity and mechanism of action of titanocene dichloride. Br. J. Cancer. 1998, 77, 2088–2097. [Google Scholar] [CrossRef]
- Olszewski, U.; Deally, A.; Tacke, M.; Hamilton, G. Alterations of Phosphoproteins in NCI-H526 Small Cell Lung Cancer Cells Involved in Cytotoxicity of Cisplatin and Titanocene Y. Neoplasia 2012, 14, 813–822. [Google Scholar] [CrossRef] [Green Version]
- Mokdsi, G.; Harding, M.M. Inhibition of human topoisomerase II by the antitumor metallocenes. J. Inorg. Biochem. 2001, 83, 205–209. [Google Scholar] [CrossRef]
- Fasano, M.; Curry, S.; Terreno, E.; Galliano, M.; Fanali, G.; Narciso, P.; Notari, S.; Ascenzi, P. The extraordinary ligand binding properties of human serum albumin. IUBMB Life 2005, 57, 787–796. [Google Scholar] [CrossRef] [PubMed]
- Peters Jr., T. All About Albumin: Biochemistry, Genetics, and Medical Applications; Academic Press: San Diego, CA, USA, 1996. [Google Scholar]
- Ghuman, J.; Zunszain, P.A.; Petitpas, I.; Bhattacharya, A.A.; Otagiri, M.; Curry, S. Structural basis of the drug-binding specificity of human serum albumin. J. Mol. Biol. 2005, 353, 38–52. [Google Scholar] [CrossRef]
- Merlot, A.M.; Kalinowski, D.S.; Richardson, D.R. Unraveling the mysteries of serum albumin—more than just a serum protein. Front. Physiol. 2014, 5, 299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heim, M.E.; Flechtner, H.; Keppler, B.K. Clinical Studies with Budotitane—A New Non-Platinum Metal Complex for Cancer Therapy; Ruthenium and Other Non-Platinum Metal Complexes in Cancer Chemotherapy; Baulieu, E., Forman, D.T., Ingelman-Sundberg, M., Jaenicke, L., Kellen, J.A., Nagai, Y., Springer, G.F., Träger, L., Will-Shahab, L., Wittliff, J.L., Eds.; Springer: Berlin, Germany, 1989; pp. 217–223. [Google Scholar]
- Schilling, T.; Keppler, K.B.; Heim, M.E.; Niebch, G.; Dietzfelbinger, H.; Rastetter, J.; Hanauske, A.-R. Clinical phase I and pharmacokinetic trial of the new titanium complex budotitane. Investig. New Drugs 1996, 13, 327–332. [Google Scholar] [CrossRef] [PubMed]
- Christodoulou, C.V.; Ferry, D.R.; Fyfe, D.W.; Young, A.; Doran, J.; Sheehan, T.M.; Eliopoulos, A.; Hale, K.; Baumgart, J.; Sass, G.; et al. Phase I trial of weekly scheduling and pharmacokinetics of titanocene dichloride in patients with advanced cancer. J. Clin. Oncol. 1998, 16, 2761–2769. [Google Scholar] [CrossRef] [PubMed]
- Korfel, A.; Scheulen, M.; Schmoll, H.-J.; Gründel, O.; Harstrick, A.; Knoche, M.; M Fels, L.; Skorzec, M.; Bach, F.; Baumgart, J.; et al. Phase I clinical and pharmacokinetic study of titanocene dichloride in adults with advanced solid tumors. Clin. Cancer Res. 1998, 4, 2701–2708. [Google Scholar]
- Mross, K.; Robben-Bathe, P.; Edler, L.; Baumgart, J.; Berdel, W.E.; Fiebig, H.; Unger, C. Phase I clinical trial of a day-1,-3,-5 every 3 weeks schedule with titanocene dichloride (MKT 5) in patients with advanced cancer—A study of the phase I study group of the Association for Medical Oncology (AIO) of the German Cancer Society. Onkologie 2000, 23, 576–579. [Google Scholar]
- Lümmen, G.; Sperling, H.; Luboldt, H.; Otto, T.; Rübben, H. Phase II trial of titanocene dichloride in advanced renal-cell carcinoma. Cancer Chemother. Pharmacol. 1998, 42, 415–417. [Google Scholar] [CrossRef]
- Kröger, N.; Kleeberg, U.R.; Mross, K.; Edler, L.; Hossfeld, D.K. Phase II Clinical Trial of Titanocene Dichloride in Patients with Metastatic Breast Cancer. Onkologie 2000, 23, 60–62. [Google Scholar] [CrossRef]
- Buettner, K.M.; Snoeberger, R.C.; Batista, V.S.; Valentine, A.M. Pharmaceutical formulation affects titanocene transferrin interactions. Dalton Trans. 2011, 40, 9580–9588. [Google Scholar] [CrossRef]
- Ravera, M.; Cassino, C.; Monti, E.; Gariboldi, M.; Osella, D. Enhancement of the cytotoxicity of titanocene dichloride by aging in organic co-solvent. J. Inorg. Biochem. 2005, 99, 2264–2269. [Google Scholar] [CrossRef] [PubMed]
- Pandrala, M.; Casanas-Montes, B.; Lopez Cubero, A.; Planas Fontánez, T.M.; Parham, L.R.; Vazquez, A.M.; Martinez, M.; Sharma, S.; Saxena, M.; Castro Lebron, G.; et al. Improved titanium(IV) cellular uptake alone is not sufficient to facilitate its cytotoxicity. Manuscript in preparation.
- Abeysinghe, P.M.; Harding, M.M. Antitumour bis(cyclopentadienyl) metal complexes: Titanocene and molybdocene dichloride and derivatives. Dalton Trans. 2007, 3474–3482. [Google Scholar] [CrossRef]
- Caruso, F.; Rossi, M. Antitumor Titanium Compounds. Mini Rev. Med. Chem. 2004, 4, 49–60. [Google Scholar]
- Ellahioui, Y.; Prashar, S.; Gomez-Ruiz, S. Anticancer applications and recent investigations of metallodrugs based on gallium, tin and titanium. Inorganics 2017, 5, 4. [Google Scholar] [CrossRef] [Green Version]
- de la Cueva-Alique, I.; Munoz-Moreno, L.; Benabdelouahab, Y.; Elie, B.T.; El Amrani, M.A.; Mosquera, M.E.; Contel, M.; Bajo, A.M.; Cuenca, T.; Royo, E. Novel enantiopure cyclopentadienyl Ti(IV) oximato compounds as potential anticancer agents. J. Inorg. Biochem. 2016, 156, 22–34. [Google Scholar] [CrossRef] [Green Version]
- Top, S.; Kaloun, E.B.; Vessieres, A.; Laios, I.; Leclercq, G.; Jaouen, G. The first titanocenyl dichloride moiety vectorised by a selective estrogen receptor modulator (SERM). Synthesis and preliminary biochemical behaviour. J. Organomet. Chem. 2002, 643, 350–356. [Google Scholar] [CrossRef]
- Vessieres, A.; Plamont, M.A.; Cabestaing, C.; Claffey, J.; Dieckmann, S.; Hogan, M.; Muller-Bunz, H.; Strohfeldt, K.; Tacke, M. Proliferative and anti-proliferative effects of titanium- and iron-based metallocene anti-cancer drugs. J. Organomet. Chem. 2009, 694, 874–879. [Google Scholar] [CrossRef]
- Gao, L.M.; Vera, J.L.; Matta, J.; Meléndez, E. Synthesis and cytotoxicity studies of steroid-functionalized titanocenes as potential anticancer drugs: Sex steroids as potential vectors for titanocenes. J. Biol. Inorg. Chem. 2010, 15, 851–859. [Google Scholar] [CrossRef] [Green Version]
- Gao, L.; Maldonado, W.; Narváez-Pita, X.; Carmona-Negrón, J.; Olivero-Verbel, J.; Meléndez, E. Steroid-Functionalized Titanocenes: Docking Studies with Estrogen Receptor Alpha. Inorganics 2016, 4, 38. [Google Scholar] [CrossRef] [Green Version]
- Pérez-Quintanilla, D.; Gómez-Ruiz, S.; Žižak, Ž.; Sierra, I.; Prashar, S.; del Hierro, I.; Fajardo, M.; Juranić, Z.D.; Kaluđerović, G.N. A new generation of anticancer drugs: Mesoporous materials modified with titanocene complexes. Chem. Eur. J. 2009, 15, 5588–5597. [Google Scholar] [CrossRef] [PubMed]
- Kaluđerović, G.N.; Tayurskaya, V.; Paschke, R.; Prashar, S.; Fajardo, M.; Gómez-Ruiz, S. Synthesis, characterization and biological studies of alkenyl-substituted titanocene(IV) carboxylate complexes. Appl. Organomet. Chem. 2010, 24, 656–662. [Google Scholar] [CrossRef]
- García-Peñas, A.; Gómez-Ruiz, S.; Pérez-Quintanilla, D.; Paschke, R.; Sierra, I.; Prashar, S.; del Hierro, I.; Kaluđerović, G.N. Study of the cytotoxicity and particle action in human cancer cells of titanocene-functionalized materials with potential application against tumors. J. Inorg. Biochem. 2012, 106, 100–110. [Google Scholar] [CrossRef] [PubMed]
- Ceballos-Torres, J.; Virag, P.; Cenariu, M.; Prashar, S.; Fajardo, M.; Fischer-Fodor, E.; Gómez-Ruiz, S. Anti-cancer Applications of Titanocene-Functionalised Nanostructured Systems: An Insight into Cell Death Mechanisms. Chem. Eur. J. 2014, 20, 10811–10828. [Google Scholar] [CrossRef]
- Wani, W.A.; Prashar, S.; Shreaz, S.; Gomez-Ruiz, S. Nanostructured materials functionalized with metal complexes: In search of alternatives for administering anticancer metallodrugs. Coord. Chem. Rev. 2016, 312, 67–98. [Google Scholar] [CrossRef]
- Ceballos-Torres, J.; del Hierro, I.; Prashar, S.; Fajardo, M.; Mijatović, S.; Maksimović-Ivanić, D.; Kaluđerović, G.N.; Gómez-Ruiz, S. Alkenyl-substituted titanocene dichloride complexes: Stability studies, binding and cytotoxicity. J. Organomet. Chem. 2014, 769, 46–57. [Google Scholar] [CrossRef]
- Cuffe, S.; Dowling, C.M.; Claffey, J.; Pampillón, C.; Hogan, M.; Fitzpatrick, J.M.; Carty, M.P.; Tacke, M.; Watson, R.W.G. Effects of titanocene dichloride derivatives on prostate cancer cells, specifically DNA damage-induced apoptosis. Prostate 2011, 71, 111–124. [Google Scholar] [CrossRef]
- Bannon, J.H.; Fichtner, I.; O’Neill, A.; Pampillón, C.; Sweeney, N.J.; Strohfeldt, K.; Watson, R.W.; Tacke, M.; Mc Gee, M.M. Substituted titanocenes induce caspase-dependent apoptosis in human epidermoid carcinoma cells in vitro and exhibit antitumour activity in vivo. Br. J. Cancer 2007, 97, 1234–1241. [Google Scholar] [CrossRef] [Green Version]
- Schur, J.; Manna, C.M.; Deally, A.; Köster, R.W.; Tacke, M.; Tshuva, E.Y.; Ott, I. A comparative chemical–biological evaluation of titanium(IV) complexes with a salan or cyclopentadienyl ligand. Chem. Commun. 2013, 49, 4785–4787. [Google Scholar] [CrossRef]
- Berners-Price, S.J.; Sadler, P.J. Phosphines and Metal Phosphine Complexes: Relationship of Chemistry to Anticancer and Other Biological Activity; Springer: Berlin, Germany, 1988; pp. 27–102. [Google Scholar]
- Kenny, S.L. Gold: A Cultural Encyclopedia; ABC-CLIO: Santa Barbara, CA, USA, 2011. [Google Scholar]
- Kean, W.F.; Kean, I.R. Clinical pharmacology of gold. Inflammopharmacology 2008, 16, 112–125. [Google Scholar] [CrossRef]
- Pricker, S.P. Medical uses of gold compounds: Past, present and future. Gold Bull. 1996, 29, 53–60. [Google Scholar] [CrossRef] [Green Version]
- Sutton, B.M.; McGusty, E.; Walz, D.T.; DiMartino, M.J. Oral gold. Antiarthritic properties of alkylphosphinegold coordination complexes. J. Med. Chem. 1972, 15, 1095–1098. [Google Scholar] [CrossRef] [PubMed]
- Finkelstein, A.E.; Walz, D.T.; Batista, V.; Mizraji, M.; Roisman, F.; Misher, A. Auranofin. New oral gold compound for treatment of rheumatoid arthritis. Ann. Rheum. Dis. 1976, 35, 251–257. [Google Scholar] [CrossRef] [Green Version]
- Schattenkirchner, M.; Kaik, B.; Muller-Fassbender, H.; Rau, R.; Zeidler, H. Auranofin and sodium aurothiomalate in the treatment of rheumatoid arthritis. A double-blind, comparative multicenter study. J. Rheumatol. Suppl. 1982, 8, 184–189. [Google Scholar]
- Ward, J.R.; Williams, H.J.; Boyce, E.; Egger, M.J.; Reading, J.C.; Samuelson, C.O. Comparison of auranofin, gold sodium thiomalate, and placebo in the treatment of rheumatoid arthritis: Subsets of responses. Am. J. Med. 1983, 75, 133–137. [Google Scholar] [CrossRef]
- Schattenkirchner, M.; Bröll, H.; Kaik, B.; Müller-Faßbender, H.; Rau, R.; Zeidler, H. Auranofin and gold sodium thiomalate in the treatment of rheumatoid arthritis: A one-year, double-blind, comparative multicenter study. Klin. Wochenschr. 1988, 66, 167–174. [Google Scholar] [CrossRef]
- Telleria, C.M. Drug Repurposing for Cancer Therapy. J. Cancer Sci. Ther. 2012, 4, ix–xi. [Google Scholar] [CrossRef]
- Roder, C.; Thomson, M.J. Auranofin: Repurposing an old drug for a golden new age. Drugs R&D 2015, 15, 13–20. [Google Scholar]
- Yeo, C.I.; Ooi, K.K.; Tiekink, E.R.T. Gold-Based Medicine: A Paradigm Shift in Anti-Cancer Therapy? Molecules 2018, 23, 1410. [Google Scholar] [CrossRef] [Green Version]
- Simon, T.M.; Kunishima, D.H.; Vibert, G.J.; Lorber, A. Cellular antiproliferative action exerted by auranofin. J. Rheumatol. Suppl. 1979, 5, 91–97. [Google Scholar]
- Mirabelli, C.K.; Johnson, R.K.; Sung, C.M.; Faucette, L.; Muirhead, K.; Crooke, S.T. Evaluation of the in vivo antitumor activity and in vitro cytotoxic properties of auranofin, a coordinated gold compound, in murine tumor models. Cancer Res. 1985, 45, 32–39. [Google Scholar]
- Mirabell, C.K.; Johnson, R.K.; Hill, D.T.; Faucette, L.F.; Girard, G.R.; Kuo, G.Y.; Sung, C.M.; Crooke, S.T. Correlation of the in vitro cytotoxic and in vivo antitumor activities of gold(I) coordination complexes. J. Med. Chem. 1986, 29, 218–223. [Google Scholar] [CrossRef]
- Rigobello, M.P.; Scutari, G.; Boscolo, R.; Bindoli, A. Induction of mitochondrial permeability transition by auranofin, a gold(I)-phosphine derivative. Br. J. Pharmacol. 2002, 136, 1162–1168. [Google Scholar] [CrossRef] [Green Version]
- Nobili, S.; Mini, E.; Landini, I.; Gabbiani, C.; Casini, A.; Messori, L. Gold compounds as anticancer agents: Chemistry, cellular pharmacology, and preclinical studies. Med. Res. Rev. 2010, 30, 550–580. [Google Scholar] [CrossRef]
- Rigobello, M.P.; Scutari, G.; Folda, A.; Bindoli, A. Mitochondrial thioredoxin reductase inhibition by gold(I) compounds and concurrent stimulation of permeability transition and release of cytochrome c. Biochem. Pharmacol. 2004, 67, 689–696. [Google Scholar] [CrossRef]
- Chrysouli, M.P.; Banti, C.N.; Kourkoumelis, N.; Panayiotou, N.; Markopoulos, G.S.; Tasiopoulos, A.J.; Hadjikakou, S.K. Chloro(triphenylphosphine)gold(I) a forefront reagent in gold chemistry as apoptotic agent for cancer cells. J. Inorg. Biochem. 2018, 179, 107–120. [Google Scholar] [CrossRef]
- Snyder, R.M.; Mirabelli, C.K.; Crooke, S.T. Cellular association, intracellular distribution, and efflux of auranofin via sequential ligand exchange reactions. Biochem. Pharmacol. 1986, 35, 923–932. [Google Scholar] [CrossRef]
- Snyder, R.M.; Mirabelli, C.K.; Crooke, S.T. Cellular interactions of auranofin and a related gold complex with RAW 264.7 macrophages. Biochem. Pharmacol. 1987, 36, 647–654. [Google Scholar] [CrossRef]
- Shaw, C.F. Gold-Based Therapeutic Agents. Chem. Rev. 1999, 99, 2589–2600. [Google Scholar] [CrossRef]
- Bryan, D.L.B.; Mikuriya, Y.; Hempel, J.C.; Mellinger, D.; Hashim, M.; Pasternack, R.F. Reactions of auranofin ((1-thio-.beta.-d-glucopyranose 2,3,4,6-tetraacetato-S)(triethylphosphine)gold(I)) in aqueous hydrochloric acid. Inorg. Chem. 1987, 26, 4180–4185. [Google Scholar] [CrossRef]
- Intoccia, A.P.; Flanagan, T.L.; Walz, D.T.; Gutzait, L.; Swagzdis, J.E.; Flagiello, J., Jr.; Hwang, B.Y.; Dewey, R.H.; Noguchi, H. Pharmacokinetics of auranofin in animals. J. Rheumatol. Suppl. 1982, 8, 90–98. [Google Scholar]
- Papp, K.A.; Shear, N.H. Systemic gold therapy. Clin. Dermatol. 1991, 9, 535–551. [Google Scholar] [CrossRef]
- Zou, T.; Lum, C.T.; Lok, C.-N.; Zhang, J.-J.; Che, C.-M. Chemical biology of anticancer gold(III) and gold(I) complexes. Chem. Soc. Rev. 2015, 44, 8786–8801. [Google Scholar] [CrossRef]
- Garcia, A.; Carvalhaes, R.; Grazul, R.; Lopes, M.P.; Corrêa, C.; Santos, H.; Almeida, M.; Silva, H. Novel antitumor adamantane-azole gold(I) complexes as potential inhibitors of thioredoxin reductase. J. Biol. Inorg. Chem. 2016, 21, 275–292. [Google Scholar] [CrossRef]
- Commisso, C.; Davidson, S.M.; Soydaner-Azeloglu, R.G.; Parker, S.J.; Kamphorst, J.J.; Hackett, S.; Grabocka, E.; Nofal, M.; Drebin, J.A.; Thompson, C.B.; et al. Macropinocytosis of protein is an amino acid supply route in Ras-transformed cells. Nature 2013, 497, 633–637. [Google Scholar] [CrossRef] [Green Version]
- Chaves, J.S.D.; Neumann, F.; Francisco, T.; Corrêa, C.; Lopes, M.T.P.; Silva, H.; Fontes, A.; Almeida, M. Synthesis and cytotoxic activity of gold(I) complexes containing phosphines and 3-benzyl-1,3-thiazolidine-2-thione or 5-phenyl-1,3,4-oxadiazole-2-thione as ligands. Inorg. Chim. Acta 2014, 414, 85–90. [Google Scholar] [CrossRef]
- Dean, T.C.; Yang, M.; Liu, M.; Grayson, J.M.; DeMartino, A.W.; Day, C.S.; Lee, J.; Furdui, C.M.; Bierbach, U. Human Serum Albumin-Delivered [Au(PEt(3))]+ Is a Potent Inhibitor of T Cell Proliferation. ACS Med. Chem. Lett. 2017, 8, 572–576. [Google Scholar] [CrossRef]
- Pratesi, A.; Cirri, D.; Ciofi, L.; Messori, L. Reactions of Auranofin and Its Pseudohalide Derivatives with Serum Albumin Investigated through ESI-Q-TOF MS. Inorg. Chem. 2018, 57, 10507–10510. [Google Scholar] [CrossRef]
- Hill, D.T.; Isab, A.A.; Griswold, D.E.; DiMartino, M.J.; Matz, E.D.; Figueroa, A.L.; Wawro, J.E.; DeBrosse, C.; Reiff, W.M.; Elder, R.C.; et al. Seleno-auranofin (Et3PAuSe-tagl): Synthesis, spectroscopic (EXAFS, 197Au Mossbauer, 31P, 1H, 13C, and 77Se NMR, ESI-MS) characterization, biological activity, and rapid serum albumin-induced triethylphosphine oxide generation. Inorg. Chem. 2010, 49, 7663–7675. [Google Scholar] [CrossRef]
- Dada, O.; Sánchez, G.; Tacke, M.; Zhu, X. Synthesis and anticancer activity of novel NHC-gold(I)-sugar complexes. Tetrahedron Lett. 2018, 59, 2904–2908. [Google Scholar] [CrossRef]
- Walther, W.; Dada, O.; O’Beirne, C.; Ott, I.; Sánchez, G.; Schmidt, C.; Werner, C.; Zhu, X.; Tacke, M. In Vitro and In Vivo Investigations into the Carbene Gold Chloride and Thioglucoside Anticancer Drug Candidates NHC-AuCl and NHC-AuSR. Lett. Drug Des. Discov. 2017, 14, 125–134. [Google Scholar] [CrossRef] [Green Version]
- ClinicalTrials.Gov. Available online: https://clinicaltrials.gov (accessed on 1 December 2019).
- Jatoi, A.; Radecki Breitkopf, C.; Foster, N.R.; Block, M.S.; Grudem, M.; Wahner Hendrickson, A.; Carlson, R.E.; Barrette, B.; Karlin, N.; Fields, A.P. A mixed-methods feasibility trial of protein kinase C iota inhibition with auranofin in asymptomatic ovarian cancer patients. Oncology 2015, 88, 208–213. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saba, N.S.; Ghias, M.; Manepalli, R.; Schorno, K.; Weir, S.; Austin, C.; Maddocks, K.; Byrd, J.C.; Kambhampati, S.; Bhalla, K.; et al. Auranofin Induces a Reversible In-Vivo Stress Response That Correlates With a Transient Clinical Effect In Patients With Chronic Lymphocytic Leukemia. Blood 2013, 122, 3819. [Google Scholar] [CrossRef]
- Wenzel, M.; Bertrand, B.; Eymin, M.; Comte, V.; Harvey, J.A.; Richard, P.; Groessl, M.; Zava, O.; Amrouche, H.; Harvey, P.D.; et al. Multinuclear Cytotoxic Metallodrugs: Physicochemical Characterization and Biological Properties of Novel Heteronuclear Gold–Titanium Complexes. Inorg. Chem. 2011, 50, 9472–9480. [Google Scholar] [CrossRef] [PubMed]
- Fernández-Gallardo, J.; Elie, B.T.; Sulzmaier, F.J.; Sanaú, M.; Ramos, J.W.; Contel, M. Organometallic Titanocene–Gold Compounds as Potential Chemotherapeutics in Renal Cancer. Study of their Protein Kinase Inhibitory Properties. Organometallics 2014, 33, 6669–6681. [Google Scholar] [CrossRef]
- Fernández-Gallardo, J.; Elie, B.T.; Sadhukha, T.; Prabha, S.; Sanaú, M.; Rotenberg, S.A.; Ramos, J.W.; Contel, M. Heterometallic titanium–gold complexes inhibit renal cancer cells in vitro and in vivo. Chem. Sci. 2015, 6, 5269–5283. [Google Scholar] [CrossRef] [Green Version]
- Mui, Y.F.; Fernandez-Gallardo, J.; Elie, B.T.; Gubran, A.; Maluenda, I.; Sanau, M.; Navarro, O.; Contel, M. Titanocene-Gold Complexes Containing N-Heterocyclic Carbene Ligands Inhibit Growth of Prostate, Renal, and Colon Cancers in Vitro. Organometallics 2016, 35, 1218–1227. [Google Scholar] [CrossRef]
- Curado, N.; Contel, M. Metal-based Anticancer Agents; The Royal Society of Chemistry: Cambridge, UK, 2019; pp. 143–168. [Google Scholar]
- Curado, N.; Giménez, N.; Miachin, K.; Aliaga-Lavrijsen, M.; Cornejo, M.A.; Jarzecki, A.A.; Contel, M. Preparation of Titanocene–Gold Compounds Based on Highly Active Gold(I)-N-Heterocyclic Carbene Anticancer Agents: Preliminary in vitro Studies in Renal and Prostate Cancer Cell Lines. Chem. Med. Chem. 2019, 14, 1086–1095. [Google Scholar] [CrossRef] [Green Version]
- Elie, B.T.; Fernandez-Gallardo, J.; Curado, N.; Cornejo, M.A.; Ramos, J.W.; Contel, M. Bimetallic titanocene-gold phosphane complexes inhibit invasion, metastasis, and angiogenesis-associated signaling molecules in renal cancer. Eur. J. Med. Chem. 2019, 161, 310–322. [Google Scholar] [CrossRef]
- Tabrizi, L.; Olasunkanmi, L.O.; Fadare, O.A. De novo design of thioredoxin reductase-targeted heterometallic titanocene–gold compounds of chlorambucil for mechanistic insights into renal cancer. Chem. Commun. 2020, 56, 297–300. [Google Scholar] [CrossRef]
- Meker, S.; Braitbard, O.; Hall, M.D.; Hochman, J.; Tshuva, E.Y. Specific Design of Titanium(IV) Phenolato Chelates Yields Stable and Accessible, Effective and Selective Anticancer Agents. Chem. A Eur. J. 2016, 22, 9986–9995. [Google Scholar] [CrossRef] [PubMed]
- Tshuva, E.Y.; Ashenhurst, J.A. Cytotoxic Titanium(IV) Complexes: Renaissance. Eur. J. Inorg. Chem. 2009, 2009, 2203–2218. [Google Scholar] [CrossRef]
- Cini, M.; Bradshaw, T.D.; Woodward, S. Using titanium complexes to defeat cancer: The view from the shoulders of titans. Chem. Soc. Rev. 2017, 46, 1040–1051. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parks, T.B.; Cruz, Y.M.; Tinoco, A.D. Applying the Fe(III) Binding Property of a Chemical Transferrin Mimetic to Ti(IV) Anticancer Drug Design. Inorg. Chem. 2014, 53, 1743–1749. [Google Scholar] [CrossRef]
- Loza-Rosas, S.A.; Vazquez, A.M.; Rivero, K.I.; Negrón, L.J.; Delgado, Y.; Benjamin-Rivera, J.A.; Vazquez-Maldonado, A.L.; Parks, T.B.; Munet-Colon, C.; Tinoco, A.D. Expanding the therapeutic potential of the iron chelator deferasirox in the development of aqueous stable Ti(IV) anticancer complexes. Inorg. Chem. 2017, 56, 7788–7802. [Google Scholar] [CrossRef]



















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compounds | Cancer Cells IC50 (µM ± SD) | Non-Cancer Cell BHK21 | TrxR Inhibition (% ± SD) | ||
|---|---|---|---|---|---|
| B16-F10 Melanoma | CT26-WT Colon | 4T1 Breast | |||
| AuPPh3ATT | 5.7 ± 0.5 | 5.7 ± 0.9 | 6.6 ± 0.5 | 18.5 ± 2.9 | 40.7 ± 1.5 |
| AuPEt3ATT | 1.2 ± 0.2 | 1.8 ± 0.8 | 1.6 ± 0.5 | 5.5 ± 0.1 | 51.6 ± 1.6 |
| AuPPh3MOT | 1.8 ± 0.5 | 1.8 ± 0.3 | 3.0 ± 1.8 | 6.9 ± 0.8 | 57.3 ± 0.7 |
| AuPEt3MOT | 1.0 ± 0.1 | 0.9 ± 0.1 | 1.1 ± 0.2 | 5.8 ± 0.1 | 60.2 ± 0.7 |
| AuPPh3Cl | 6.6 ± 0.1 | 12.1 ± 2.8 | 10.3 ± 2.3 | 23.0 ± 0.3 | - |
| AuPEt3Cl | 2.3 ± 0.5 | 9.0 ± 0.7 | 9.5 ± 1.1 | 22.5 ± 0.2 | - |
| AF | 0.5 ± 0.4 | 0.5 ± 0.4 | 0.6 ± 0.2 | 1.6 ± 0.5 | 59.7 ± 1.5 |
| Compounds | A498 Kidney | UO31 Kidney | Caki-1 Kidney | HEK-293T Kidney | PC3 Prostate | DU145 Prostate |
|---|---|---|---|---|---|---|
| Cp2Ti-(OC(O)-4-C6H4PPh2AuCl)2 | 6.9 ± 2.2 | 0.3 ± 0.06 | 1.0 ± 0.29 | 20.1 ± 1.6 | 37.7 ± 7.1 | 6.6 ± 1.8 |
| HOC(O)-4-C6H4PPh2AuCl | 21 ± 2.5 | 1.2 ± 0.8 | 19.2 ± 2.9 | 31 ± 0.9 | 78 ± 18.1 | 39 ± 5.7 |
| Cp2Ti-OC(O)-4-C6H4SAuPPh3 | ND | ND | 0.12 ± 0.003 | 0.49 ± 0.008 | ND | ND |
| C6H4SAuPPh3 | ND | ND | 2.76 ± 0.35 | 1.11 ± 0.65 | ND | ND |
| Cisplatin | 37.2 ± 4.6 | 8.9 ± 2.7 | 29 ± 4.1 | 3.2 ± 0.13 | 14 ± 2.3 | 12.1 ± 3.9 |
| Cp2TiCl2 | >200 | >200 | >200 | >200 | >200 | >200 |
| Titanocene Y | 29.6 ± 2.8 | >200 | 29.4 ± 4.2 | >200 | 58.1 ± 11.2 | 55.2 ± 7.9 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fernández-Vega, L.; Ruiz Silva, V.A.; Domínguez-González, T.M.; Claudio-Betancourt, S.; Toro-Maldonado, R.E.; Capre Maso, L.C.; Sanabria Ortiz, K.; Pérez-Verdejo, J.A.; Román González, J.; Rosado-Fraticelli, G.T.; et al. Evaluating Ligand Modifications of the Titanocene and Auranofin Moieties for the Development of More Potent Anticancer Drugs. Inorganics 2020, 8, 10. https://doi.org/10.3390/inorganics8020010
Fernández-Vega L, Ruiz Silva VA, Domínguez-González TM, Claudio-Betancourt S, Toro-Maldonado RE, Capre Maso LC, Sanabria Ortiz K, Pérez-Verdejo JA, Román González J, Rosado-Fraticelli GT, et al. Evaluating Ligand Modifications of the Titanocene and Auranofin Moieties for the Development of More Potent Anticancer Drugs. Inorganics. 2020; 8(2):10. https://doi.org/10.3390/inorganics8020010
Chicago/Turabian StyleFernández-Vega, Lauren, Valeria A. Ruiz Silva, Tania M. Domínguez-González, Sebastián Claudio-Betancourt, Rafael E. Toro-Maldonado, Luisa C. Capre Maso, Karina Sanabria Ortiz, Jean A. Pérez-Verdejo, Janeishly Román González, Grecia T. Rosado-Fraticelli, and et al. 2020. "Evaluating Ligand Modifications of the Titanocene and Auranofin Moieties for the Development of More Potent Anticancer Drugs" Inorganics 8, no. 2: 10. https://doi.org/10.3390/inorganics8020010