Mono- and Hexanuclear Zinc Halide Complexes with Soft Thiopyridazine Based Scorpionate Ligands

 and
and
Abstract
:
1. Introduction
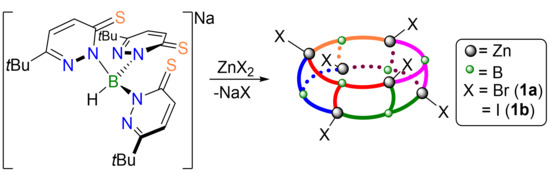
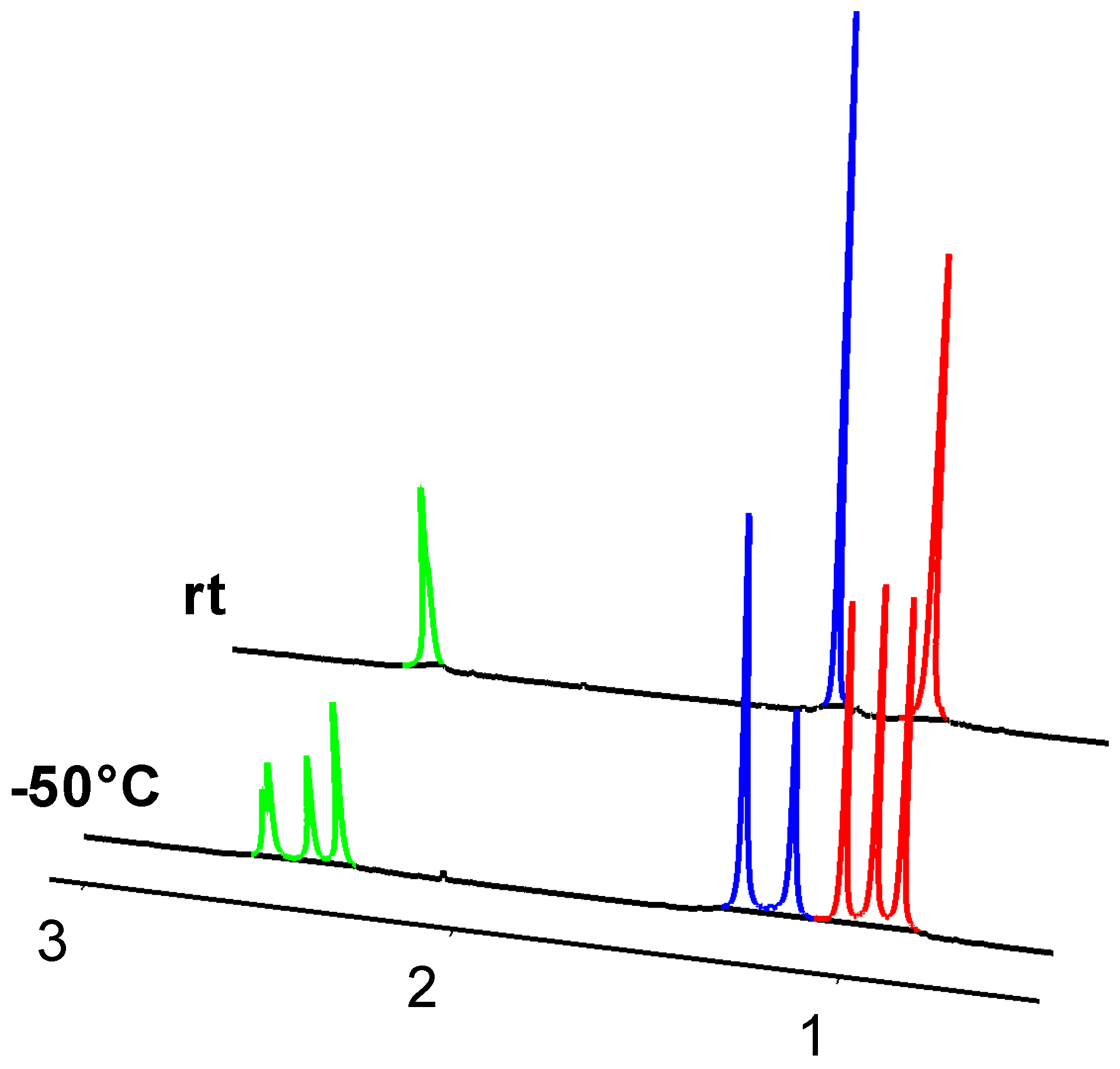
2. Results and Discussion
2.1. Complex Synthesis
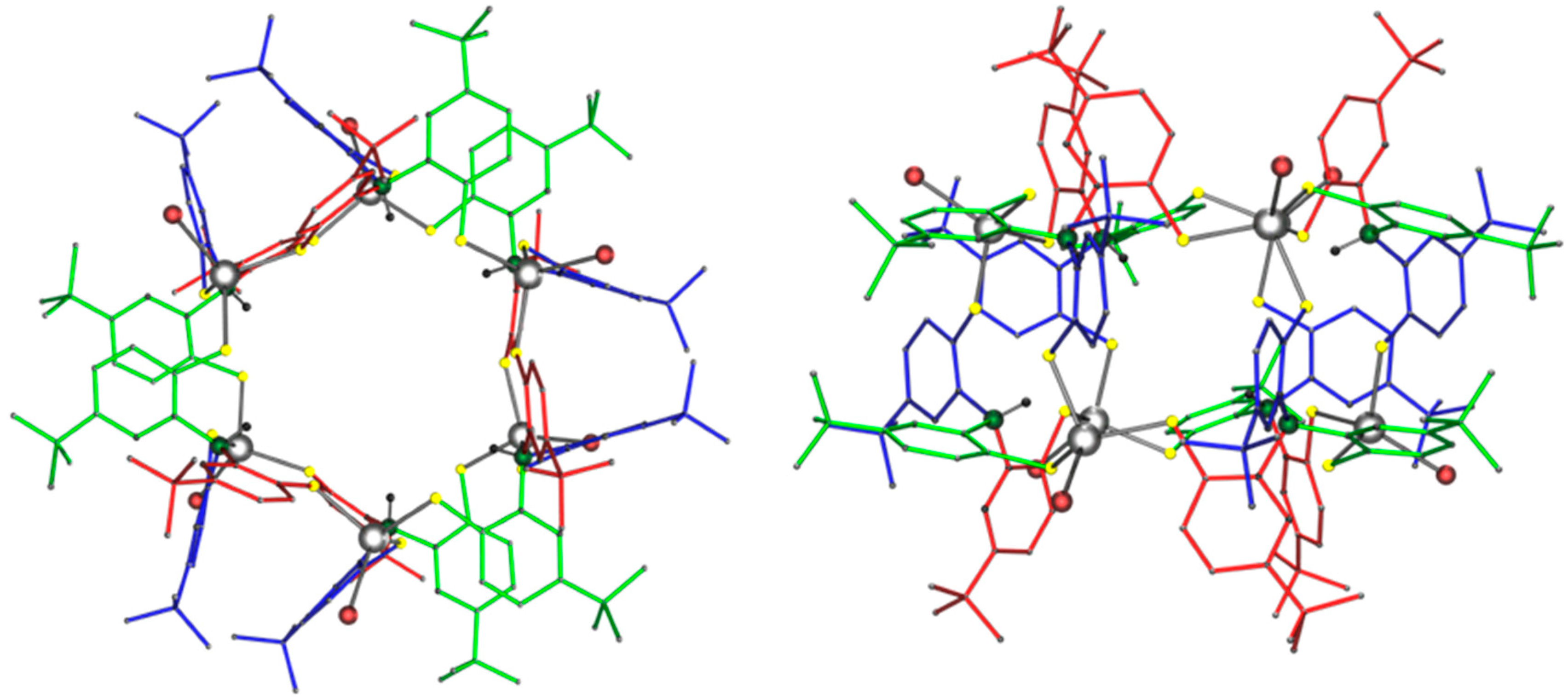
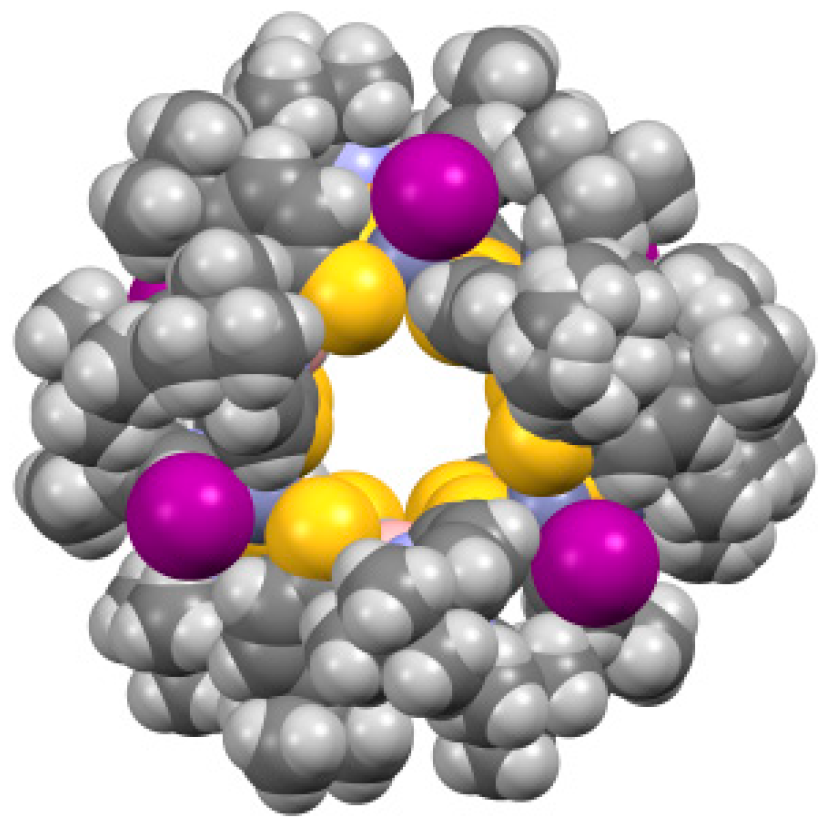
2.2. Molecular Structures
3. Experimental Section
3.1. General Information
3.2. Synthetic Procedures
4. Conclusions
Supplementary Materials
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Trofimenko, S. Scorpionates: Genesis, milestones, prognosis. Polyhedron 2004, 23, 197–203. [Google Scholar] [CrossRef]
- Trofimenko, S. Boron-Pyrazole Chemistry. J. Am. Chem. Soc. 1966, 3, 1842–1844. [Google Scholar] [CrossRef]
- Trofimenko, S. Recent Advances in Poly(pyrazoly1) borate (Scorpionate) Chemistry. Chem. Rev. 1993, 93, 943–980. [Google Scholar] [CrossRef]
- Parkin, G. Synthetic analogues relevant to the structure and function of zinc enzymes. Chem. Rev. 2004, 104, 699–767. [Google Scholar] [CrossRef]
- Costas, M.; Chen, K.; Que, L. Biomimetic nonheme iron catalysts for alkane hydroxylation. Coord. Chem. Rev. 2000, 202, 517–544. [Google Scholar]
- Spicer, M.D.; Reglinski, J. Soft Scorpionate Ligands Based on Imidazole-2-thione Donors. Eur. J. Inorg. Chem. 2009, 2009, 1553–1574. [Google Scholar] [CrossRef]
- Dyson, G.; Hamilton, A.; Mitchell, B.; Owen, G.R. A new family of flexible scorpionate ligands based on 2-mercaptopyridine. Dalton Trans. 2009, 6120. [Google Scholar] [CrossRef]
- Nuss, G.; Saischek, G.; Harum, B.N.; Volpe, M.; Gatterer, K.; Belaj, F.; Mösch-Zanetti, N.C. Novel pyridazine based scorpionate ligands in cobalt and nickel boratrane compounds. Inorg. Chem. 2011, 50, 1991–2001. [Google Scholar] [CrossRef] [PubMed]
- Garner, M.; Reglinski, J.; Cassidy, I.; Spicer, M.D.; Kennedy, A.R. Hydrotris(methimazolyl)borate, a soft analogue of hydrotris(pyrazolyl)borate. Preparation and crystal structure of a novel zinc complex. Chem. Commun. 1996, 355, 1975–1976. [Google Scholar] [CrossRef]
- Nuss, G.; Saischek, G.; Harum, B.N.; Volpe, M.; Belaj, F.; Mösch-Zanetti, N.C. Pyridazine based scorpionate ligand in a copper boratrane compound. Inorg. Chem. 2011, 50, 12632–12640. [Google Scholar] [CrossRef] [PubMed]
- Holler, S.; Tüchler, M.; Belaj, F.; Veiros, L.F.; Kirchner, K.; Mösch-Zanetti, N.C. Thiopyridazine-Based Copper Boratrane Complexes Demonstrating the Z-type Nature of the Ligand. Inorg. Chem. 2016, 55, 4980–4991. [Google Scholar] [CrossRef]
- Tüchler, M.; Belaj, F.; Raber, G.; Neshchadin, D.; Mösch-Zanetti, N.C. Photoinduced Reactivity of the Soft Hydrotris(6-tert-butyl-3-thiopyridazinyl)borate Scorpionate Ligand in Sodium, Potassium, and Thallium Salts. Inorg. Chem. 2015, 54, 8168–8170. [Google Scholar] [CrossRef] [PubMed]
- Tüchler, M.; Holler, S.; Schachner, J.A.; Belaj, F.; Mösch-Zanetti, N.C. Unusual C–N Coupling Reactivity of Thiopyridazines: Efficient Synthesis of Iron Diorganotrisulfide Complexes. Inorg. Chem. 2017, 56, 8159–8165. [Google Scholar] [CrossRef]
- Parkin, G. The bioinorganic chemistry of zinc: Synthetic analogues of zinc enzymes that feature tripodal ligands. Chem. Commun. 2000, 1971–1985. [Google Scholar] [CrossRef]
- Sattler, W.; Parkin, G. Structural characterization of zinc bicarbonate compounds relevant to the mechanism of action of carbonic anhydrase. Chem. Sci. 2012, 3, 2015–2019. [Google Scholar] [CrossRef]
- Alsfasser, R.; Trofimenko, S.; Looney, A.G.; Parkin, G.; Vahrenkamp, H. A mononuclear zinc hydroxide complex stabilized by a highly substituted tris(pyrazolyl)hydroborato ligand: Analogies with the enzyme carbonic anhydrase. Inorg. Chem. 1991, 30, 4098–4100. [Google Scholar] [CrossRef]
- Looney, A.G.; Han, R.; Mcneill, K.; Parkin, G. Tris (pyrazolyl) hydroboratozinc Hydroxide Complexes as Functional Models for Carbonic Anhydrase: On the Nature of the Bicarbonate Intermediate. J. Am. Chem. Soc. 1993, 115, 4690–4697. [Google Scholar] [CrossRef]
- Bridgewater, B.M.; Parkin, G. A zinc hydroxide complex of relevance to 5-aminolevulinate dehydratase: The synthesis, structure and reactivity of the tris(2-mercapto-1-phenylimidazolyl) hydroborato complex [TmPh]ZnOH. Inorg. Chem. Commun. 2001, 4, 126–129. [Google Scholar] [CrossRef]
- Kimblin, C.; Bridgewater, B.M.; Churchill, D.G.; Parkin, G. Mononuclear tris(2-mercapto-1-arylimidazolyl) hydroborato complexes of zinc, [TmAr]ZnX: Structural evidence that a sulfur rich coordination environment promotes the formation of a tetrahedral alcohol complex in a synthetic analogue of LADH. Chem. Commun. 1999, 993, 2301–2302. [Google Scholar] [CrossRef]
- Ibrahim, M.M.; Olmo, C.P.; Tekeste, T.; Seebacher, J.; He, G.; Maldonado Calvo, J.A.; Böhmerle, K.; Steinfeld, G.; Brombacher, H.; Vahrenkamp, H. Zn–OH2 and Zn–OH Complexes with Hydroborate-Derived Tripod Ligands: A Comprehensive Study. Inorg. Chem. 2006, 45, 7493–7502. [Google Scholar] [CrossRef]
- Tüchler, M.; Holler, S.; Huber, E.; Fischer, S.; Boese, A.D.; Belaj, F.; Mösch-Zanetti, N.C. Synthesis and Characterization of a Thiopyridazinylmethane-Based Scorpionate Ligand: Formation of Zinc Complexes and Rearrangement Reaction. Organometallics 2017, 36, 3790–3798. [Google Scholar] [CrossRef]
- Tüchler, M.; Gärtner, L.; Fischer, S.; Boese, A.D.; Belaj, F.; Mösch-Zanetti, N.C. Efficient CO2 Insertion and Reduction Catalyzed by a Terminal Zinc Hydride Complex. Angew. Chem. Int. Ed. 2018, 57, 6906–6909. [Google Scholar] [CrossRef] [PubMed]
- Tüchler, M.; Holler, S.; Rendl, S.; Stock, N.; Belaj, F.; Mösch-Zanetti, N.C. Zinc Scorpionate Complexes with a Hybrid (Thiopyridazinyl)(thiomethimidazolyl)borate Ligand. Eur. J. Inorg. Chem. 2016, 2016, 2609–2614. [Google Scholar] [CrossRef]
- Fulmer, G.R.; Miller, A.J.M.; Sherden, N.H.; Gottlieb, H.E.; Nudelman, A.; Stoltz, B.M.; Bercaw, J.E.; Goldberg, K.I. NMR Chemical Shifts of Trace Impurities: Common Laboratory Solvents, Organics, and Gases in Deuterated Solvents Relevant to the Organometallic Chemist. Organometallics 2010, 29, 2176–2179. [Google Scholar] [CrossRef] [Green Version]
- Gibbs, S.J.; Johnson, C.S. A PFG NMR experiment for accurate diffusion and flow studies in the presence of eddy currents. J. Magn. Reson. 1991, 93, 395–402. [Google Scholar] [CrossRef]
- Li, D.; Kagan, G.; Hopson, R.; Williard, P.G. Formula weight prediction by internal reference diffusion-ordered NMR spectroscopy (DOSY). J. Am. Chem. Soc. 2009, 131, 5627–5634. [Google Scholar] [CrossRef] [PubMed]
- Parkin, G. Applications of Tripodal [S3] and [Se3] L2X Donor Ligands to Zinc, Cadmium and Mercury Chemistry: Organometallic and Bioinorganic Perspectives. New J. Chem. 2007, 31, 1996–2014. [Google Scholar] [CrossRef]
- Seebacher, J.; Vahrenkamp, H. A new bonding mode of tripodal sulfur ligands: Synthesis and structure of tetranuclear [Ttt-BuZn]ClO4. J. Mol. Struct. 2003, 656, 177–181. [Google Scholar] [CrossRef]
- Schneider, A.; Vahrenkamp, H. Ein dreikerniger Zinkkomplex mit ZnS4-, ZnS3O- und ZnS2NO-Koordinationen. Z. Anorg. Allg. Chem. 2004, 630, 1059–1061. [Google Scholar] [CrossRef]
- Paul, R.L.; Amoroso, A.J.; Jones, P.L.; Couchman, S.M.; Reeves, Z.R.; Rees, L.H.; Jeffery, J.C.; McCleverty, J.A.; Ward, M.D. Effects of metal co-ordination geometry on self-assembly: A monomeric complex with trigonal prismatic metal co-ordination vs. tetrameric complexes with octahedral metal co-ordination. J. Chem. Soc. Dalton Trans. 1999, 1563–1568. [Google Scholar] [CrossRef]
- Zhang, D.-X.; Zhang, H.-X.; Wen, T.; Zhang, J. Targeted design of a cubic boron imidazolate cage with sensing and reducing functions. Dalton Trans. 2015, 44, 9367–9369. [Google Scholar] [CrossRef]
- Cassidy, I.; Garner, M.; Kennedy, A.R.; Potts, G.B.S.; Reglinski, J.; Slavin, P.A.; Spicer, M.D. The Preparation and Structures of Group 12 (Zn, Cd, Hg) Complexes of the Soft Tripodal Ligand Hydrotris(methimazolyl)borate (Tm). Eur. J. Inorg. Chem. 2002, 2002, 1235–1239. [Google Scholar] [CrossRef]
- Yurkerwich, K.; Yurkerwich, M.; Parkin, G. Synthesis and structural characterization of tris(2-mercapto-1-adamantylimidazolyl)hydroborato complexes: A sterically demanding tripodal [S3] donor ligand. Inorg. Chem. 2011, 50, 12284–12295. [Google Scholar] [CrossRef] [PubMed]
- Melnick, J.G.; Docrat, A.; Parkin, G. Methyl, hydrochalcogenido, and phenylchalcogenolate complexes of zinc in a sulfur rich coordination environment: Syntheses and structural characterization of the tris(2-mercapto-1-tert-butylimidazolyl)hydroboratozinc complexes [TmBut]ZnMe, [TmBut]ZnEH (E = S, Se) and [TmBut]ZnEPh (E = O, S, Se, Te). Chem. Commun. 2004, 2870–2871. [Google Scholar] [CrossRef]
- Barth, B.E.K.; Leusmann, E.; Harms, K.; Dehnen, S. Towards the installation of transition metal ions on donor ligand decorated tin sulfide clusters. Chem. Commun. 2013, 49, 6590–6592. [Google Scholar] [CrossRef] [PubMed]
- Márquez, C.; Hudgins, R.R.; Nau, W.M. Mechanism of host-guest complexation by cucurbituril. J. Am. Chem. Soc. 2004, 126, 5806–5816. [Google Scholar] [CrossRef] [PubMed]
- Jeon, Y.J.; Kim, S.-Y.; Ko, Y.H.; Sakamoto, S.; Yamaguchi, K.; Kim, K. Novel molecular drug carrier: Encapsulation of oxaliplatin in cucurbit[7]uril and its effects on stability and reactivity of the drug. Org. Biomol. Chem. 2005, 3, 2122–2125. [Google Scholar] [CrossRef] [PubMed]
- Lagona, J.; Mukhopadhyay, P.; Chakrabarti, S.; Isaacs, L. The cucurbitnuril family. Angew. Chem. Int. Ed. 2005, 44, 4844–4870. [Google Scholar] [CrossRef] [PubMed]
- Bakbak, S.; Bhatia, V.K.; Incarvito, C.D.; Rheingold, A.L.; Rabinovich, D. Synthesis and characterization of two new bulky tris(mercaptoimidazolyl)borate ligands and their zinc and cadmium complexes. Polyhedron 2001, 20, 3343–3348. [Google Scholar] [CrossRef]
- White, J.L.; Tanski, J.M.; Rabinovich, D. Bulky tris(mercaptoimidazolyl)borates: Synthesis and molecular structures of the Group 12 metal complexes [TmtBu]MBr (M = Zn, Cd, Hg). J. Chem. Soc. Dalton Trans. 2002, 2987–2991. [Google Scholar] [CrossRef]
- Coates, W.J.; Mckillop, A. One-Pot Preparation of 6-Substituted 3(2H)-Pyridazinones from Ketones. Synthesis 1993, 1993, 334–342. [Google Scholar] [CrossRef]
- Wu, D.H.; Chen, A.D.; Johnson, C.S. An Improved Diffusion-Ordered Spectroscopy Experiment Incorporating Bipolar-Gradient Pulses. J. Magn. Reson. 1995, 115, 260–264. [Google Scholar] [CrossRef]
- Jerschow, A.; Müller, N. 3D Diffusion-Ordered TOCSY for Slowly Diffusing Molecules. J. Magn. Reson. 1996, 123, 222–225. [Google Scholar] [CrossRef]
- Jerschow, A.; Müller, N. Suppression of Convection Artifacts in Stimulated-Echo Diffusion Experiments. Double-Stimulated-Echo Experiments. J. Magn. Reson. 1997, 125, 372–375. [Google Scholar] [CrossRef]
- Sheldrick, G.M. A short history of SHELX. Acta Crystallogr. A 2008, 64, 112–122. [Google Scholar] [CrossRef] [PubMed]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
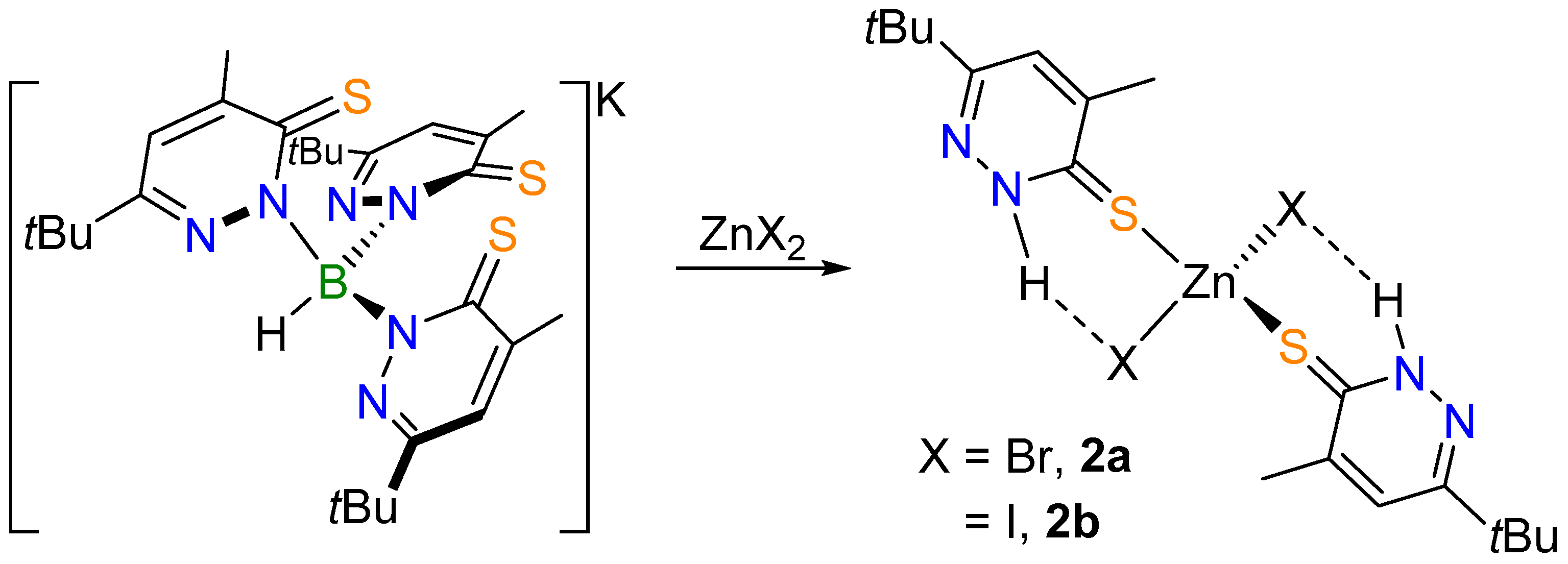{kind=link}
{kind=link}
| Compound | D (10−10 m2/s) | RH (Å) |
|---|---|---|
| 1a | 4.12 | 9.8 |
| PPh3 | 7.96 | 5.1 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tüchler, M.; Ramböck, M.; Glanzer, S.; Zangger, K.; Belaj, F.; Mösch-Zanetti, N.C. Mono- and Hexanuclear Zinc Halide Complexes with Soft Thiopyridazine Based Scorpionate Ligands. Inorganics 2019, 7, 24. https://doi.org/10.3390/inorganics7020024
Tüchler M, Ramböck M, Glanzer S, Zangger K, Belaj F, Mösch-Zanetti NC. Mono- and Hexanuclear Zinc Halide Complexes with Soft Thiopyridazine Based Scorpionate Ligands. Inorganics. 2019; 7(2):24. https://doi.org/10.3390/inorganics7020024
Chicago/Turabian StyleTüchler, Michael, Melanie Ramböck, Simon Glanzer, Klaus Zangger, Ferdinand Belaj, and Nadia C. Mösch-Zanetti. 2019. "Mono- and Hexanuclear Zinc Halide Complexes with Soft Thiopyridazine Based Scorpionate Ligands" Inorganics 7, no. 2: 24. https://doi.org/10.3390/inorganics7020024