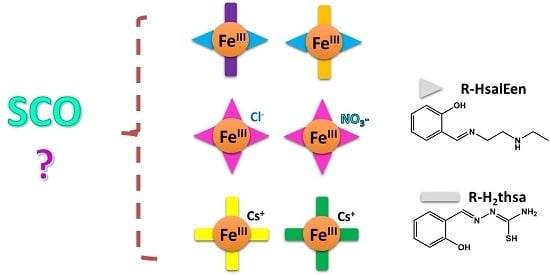
Heteroleptic and Homoleptic Iron(III) Spin-Crossover Complexes; Effects of Ligand Substituents and Intermolecular Interactions between Co-Cation/Anion and the Complex


Abstract
:
1. Introduction
2. Results and Discussion
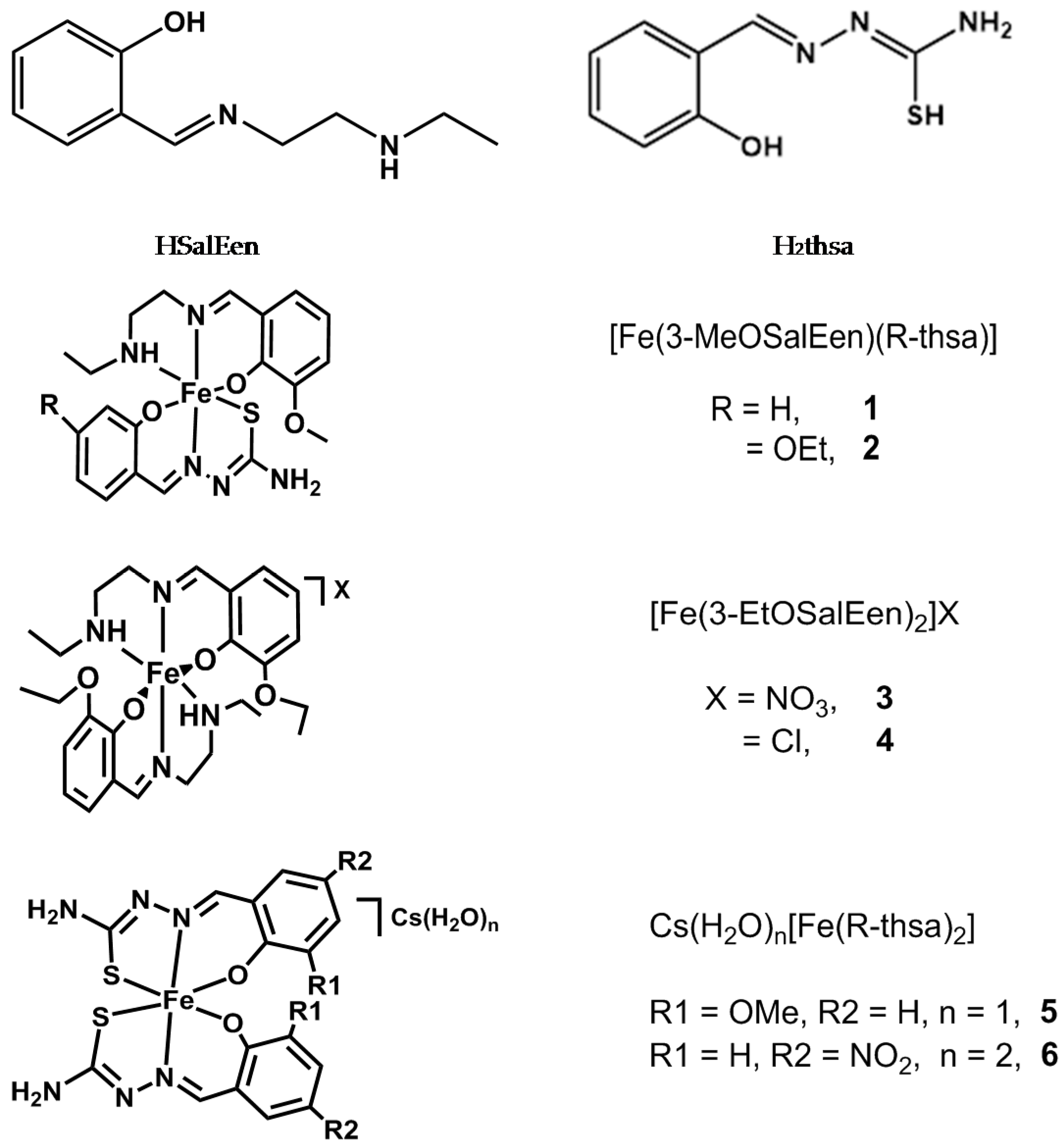
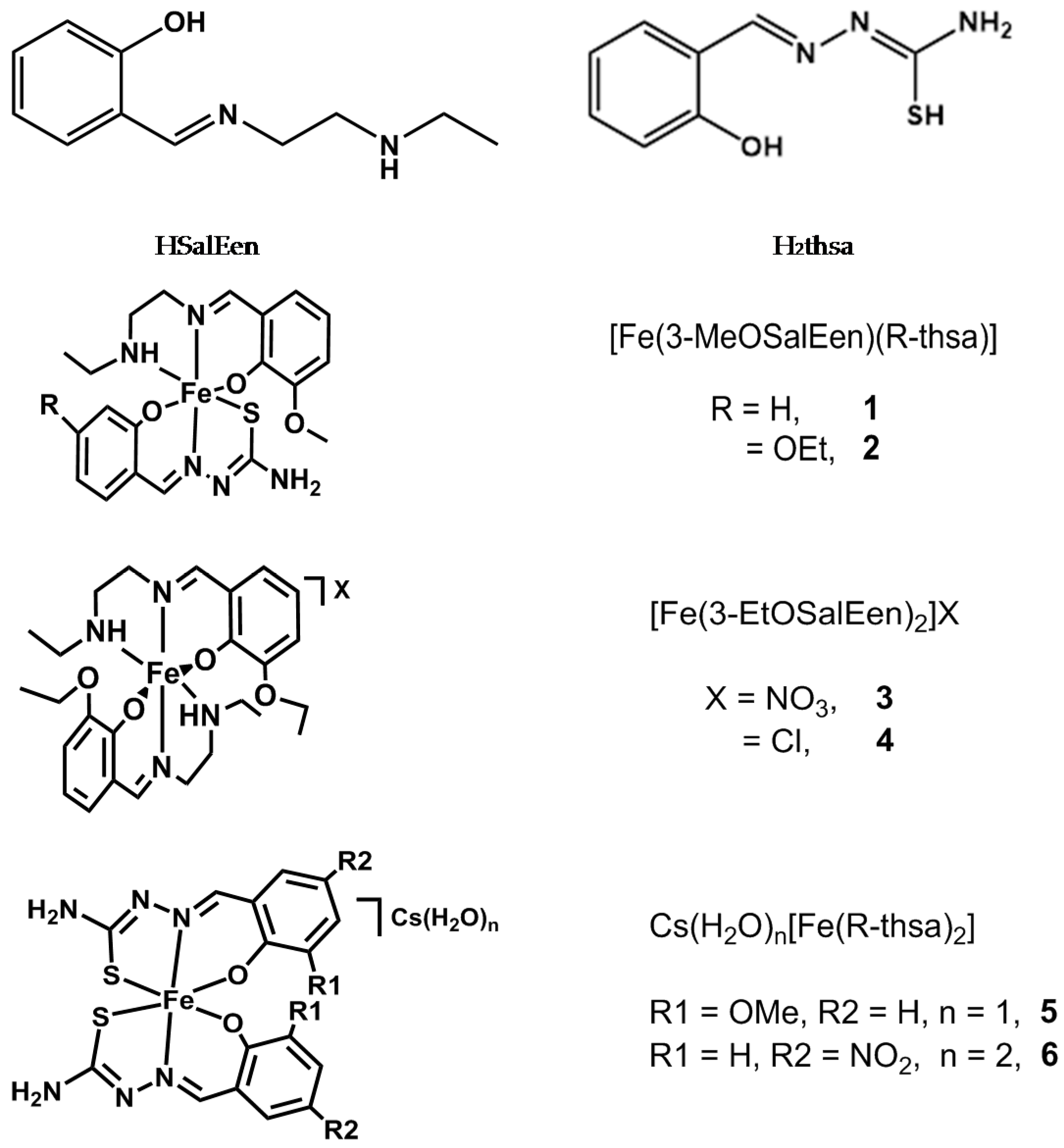
2.1. Preparation of Iron(III) Complexes
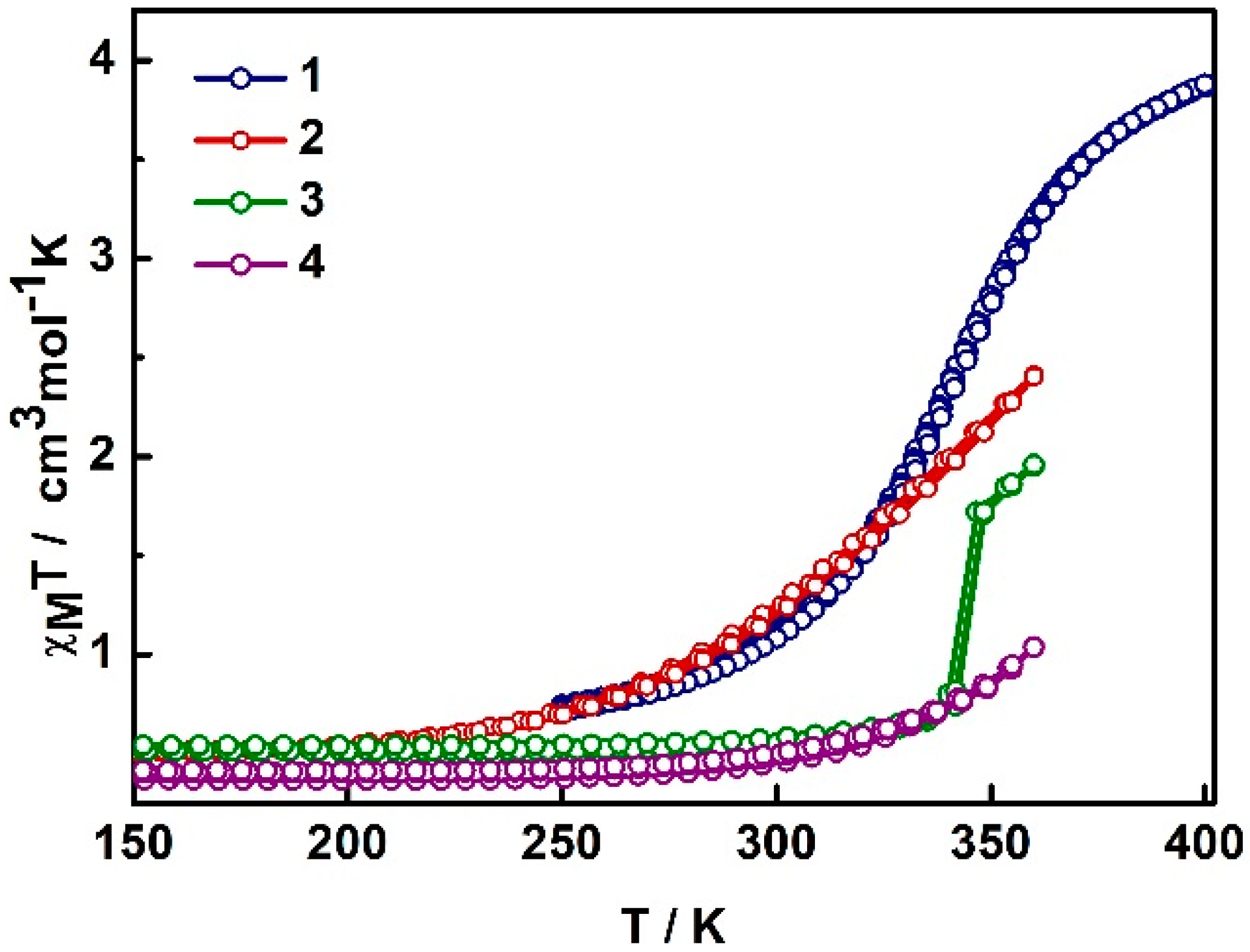
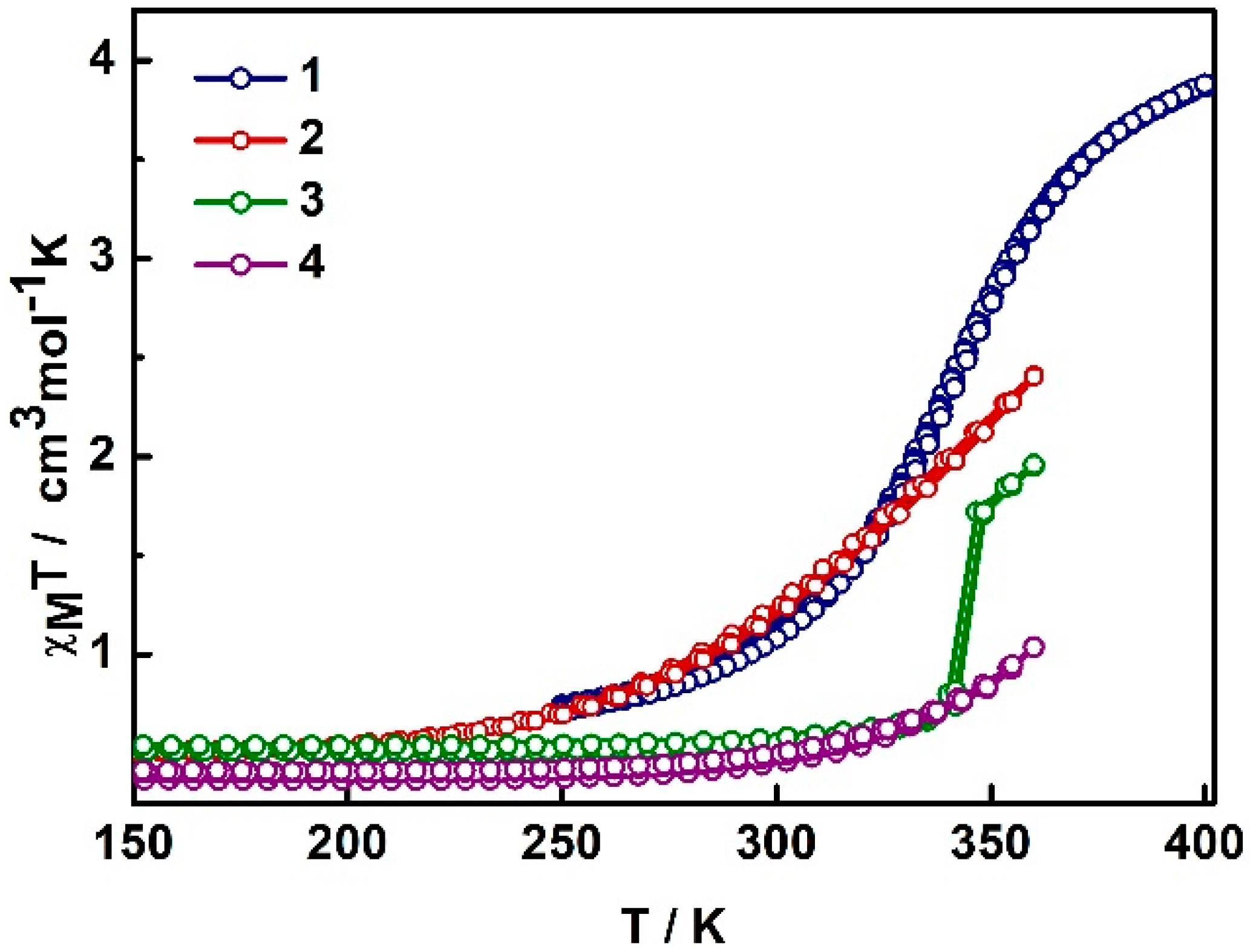
2.2. Magnetism
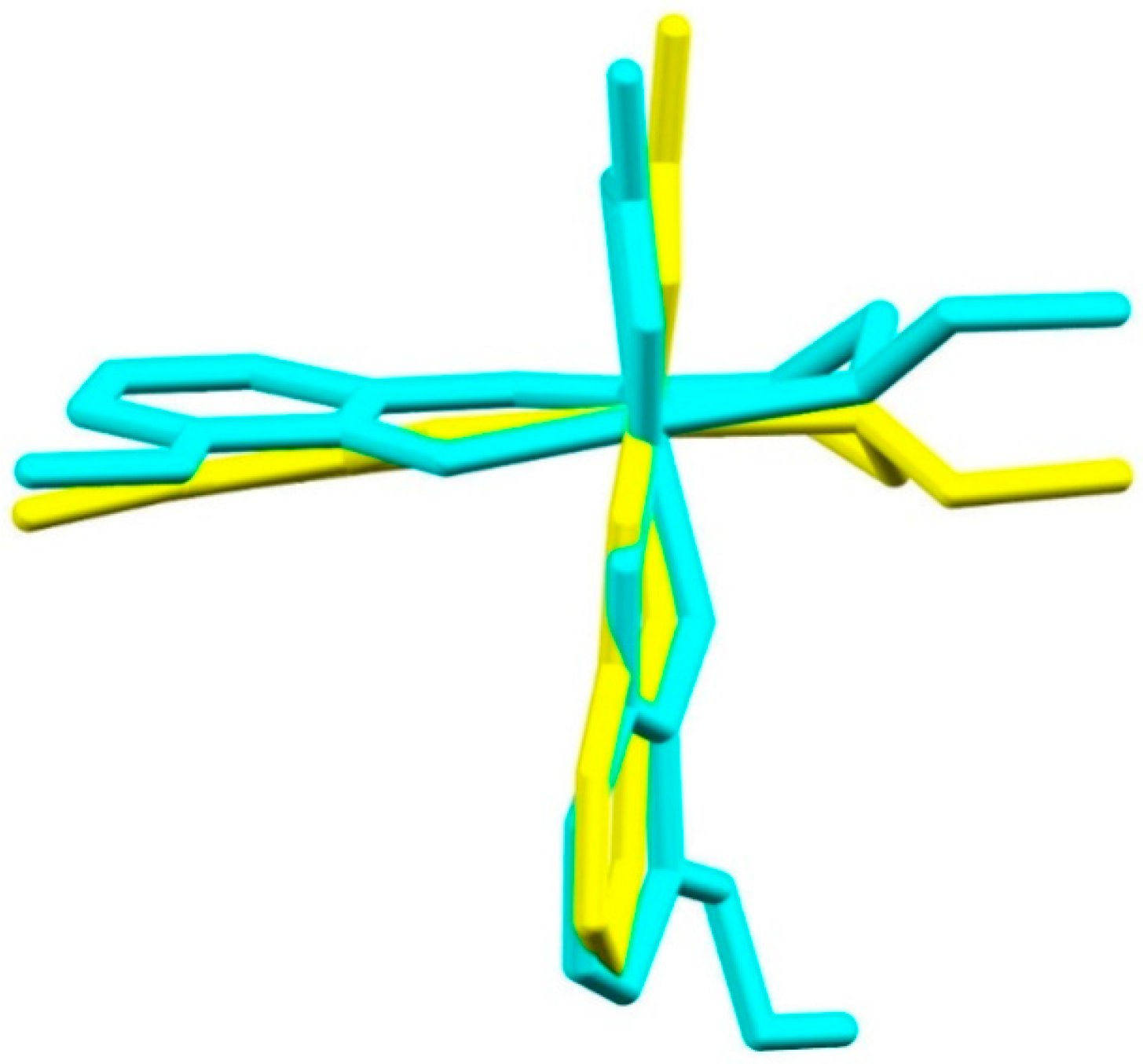
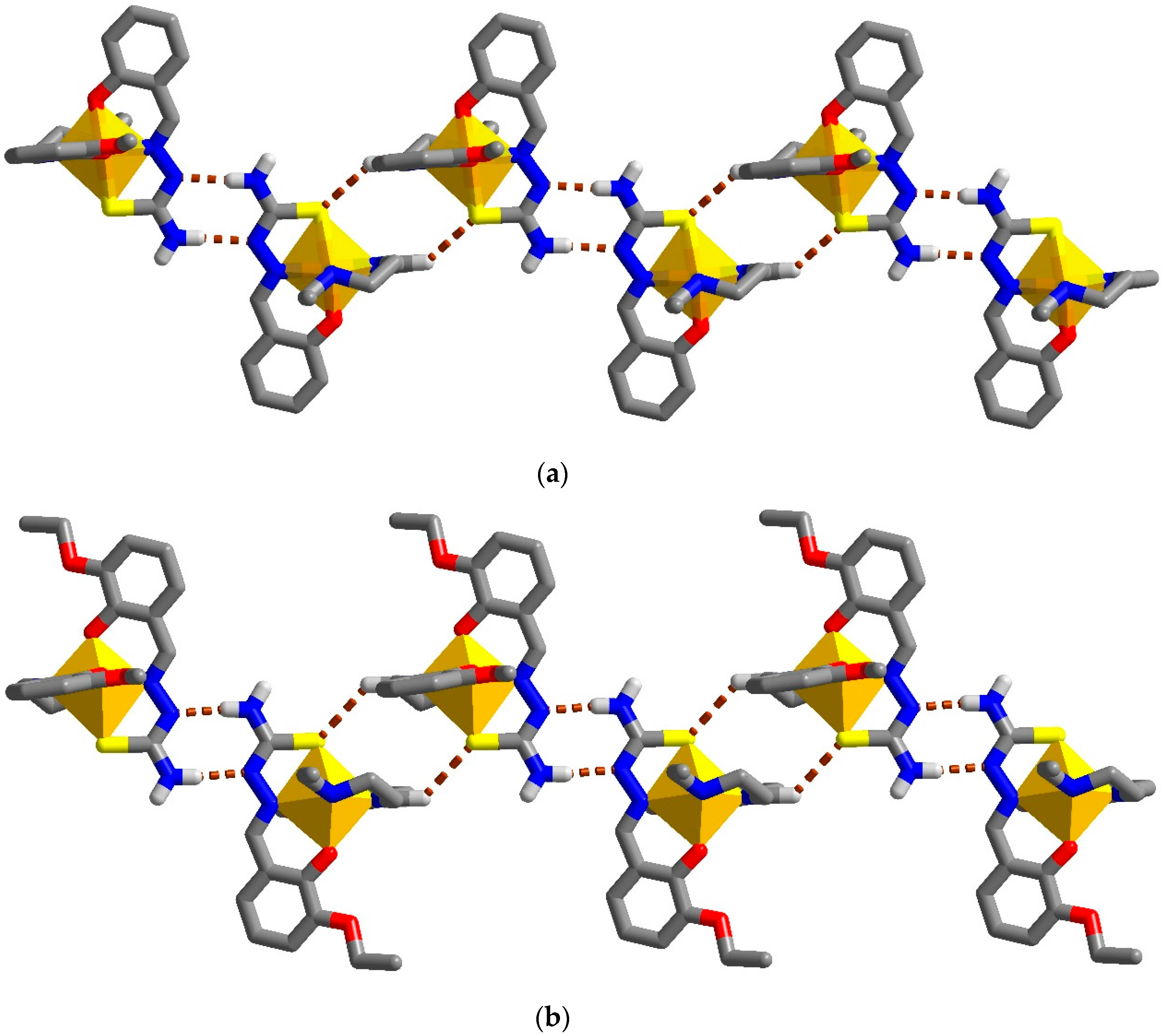
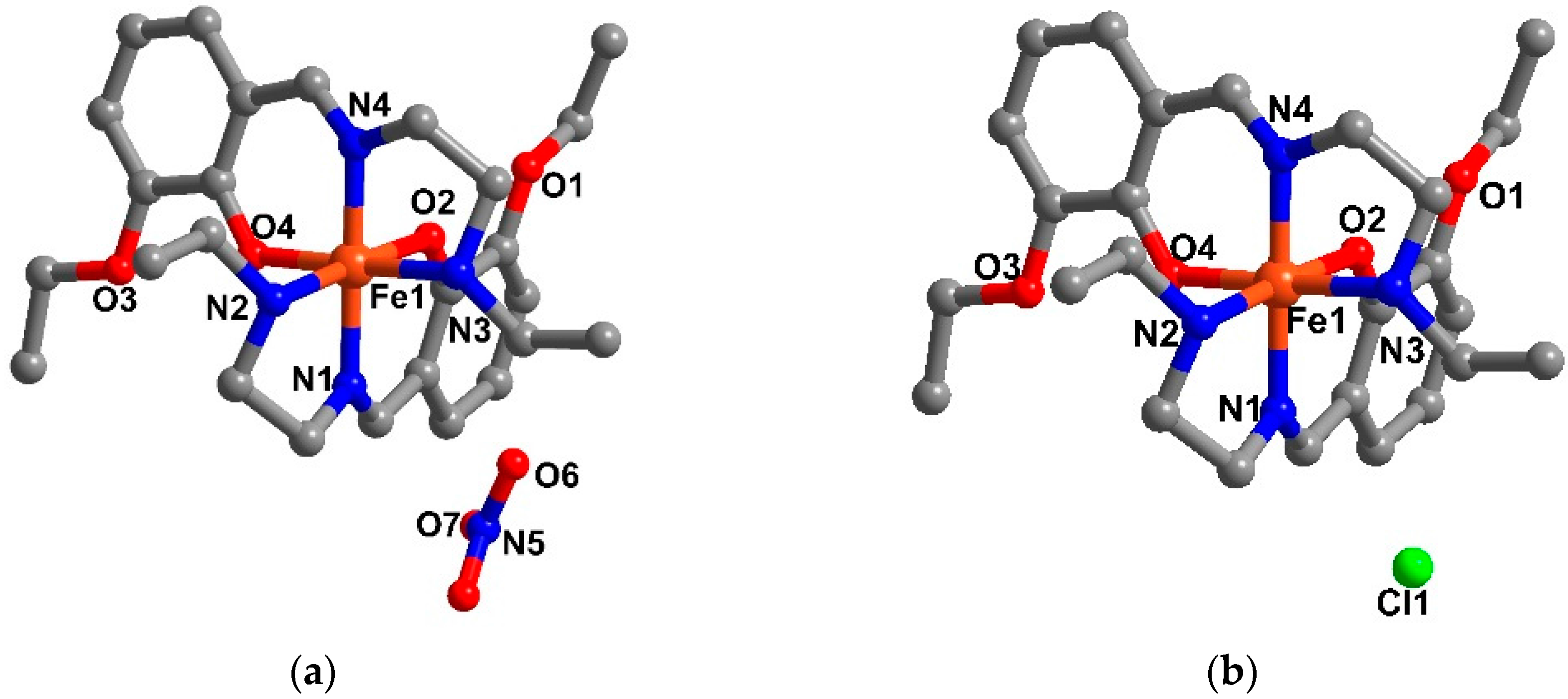
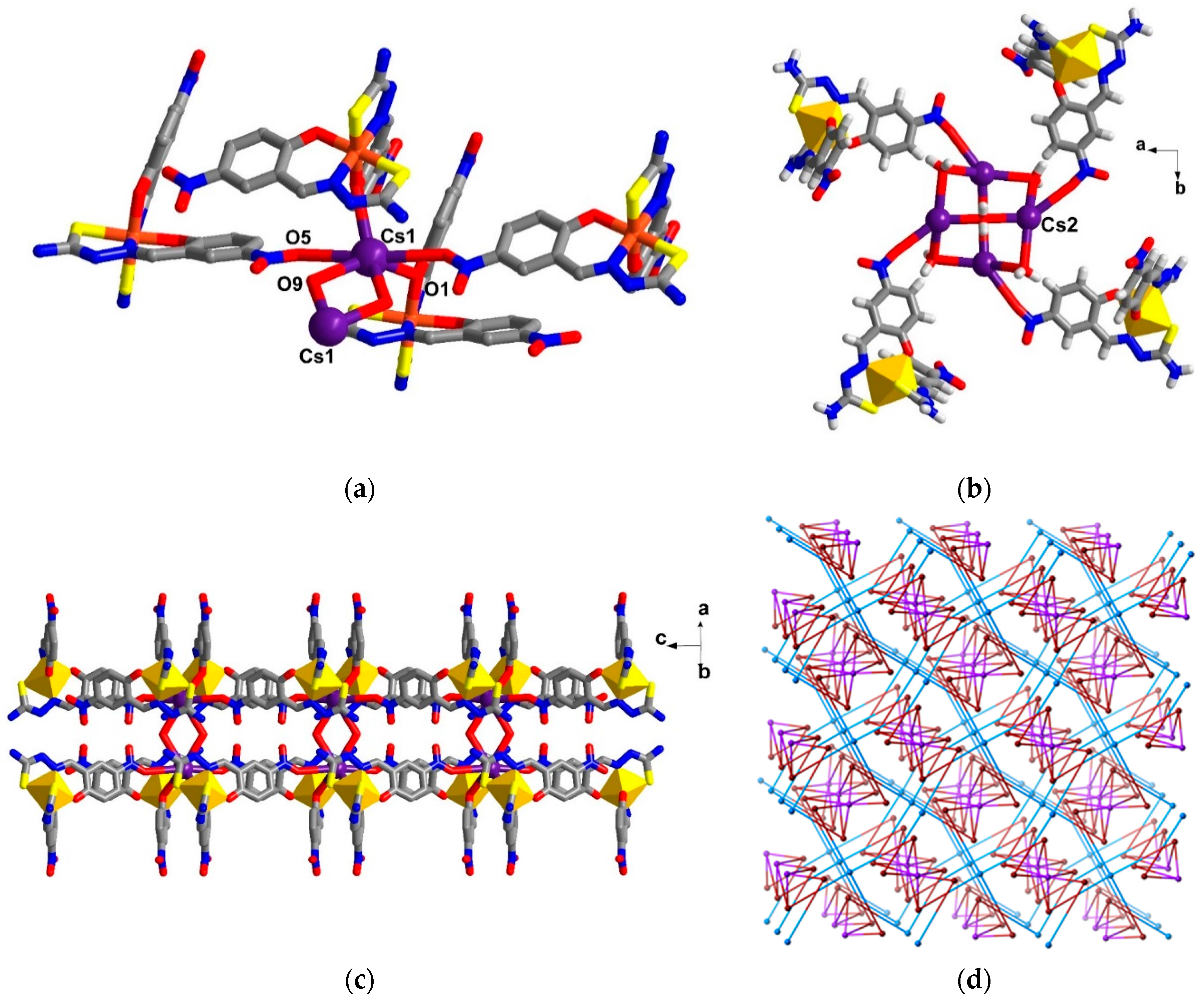
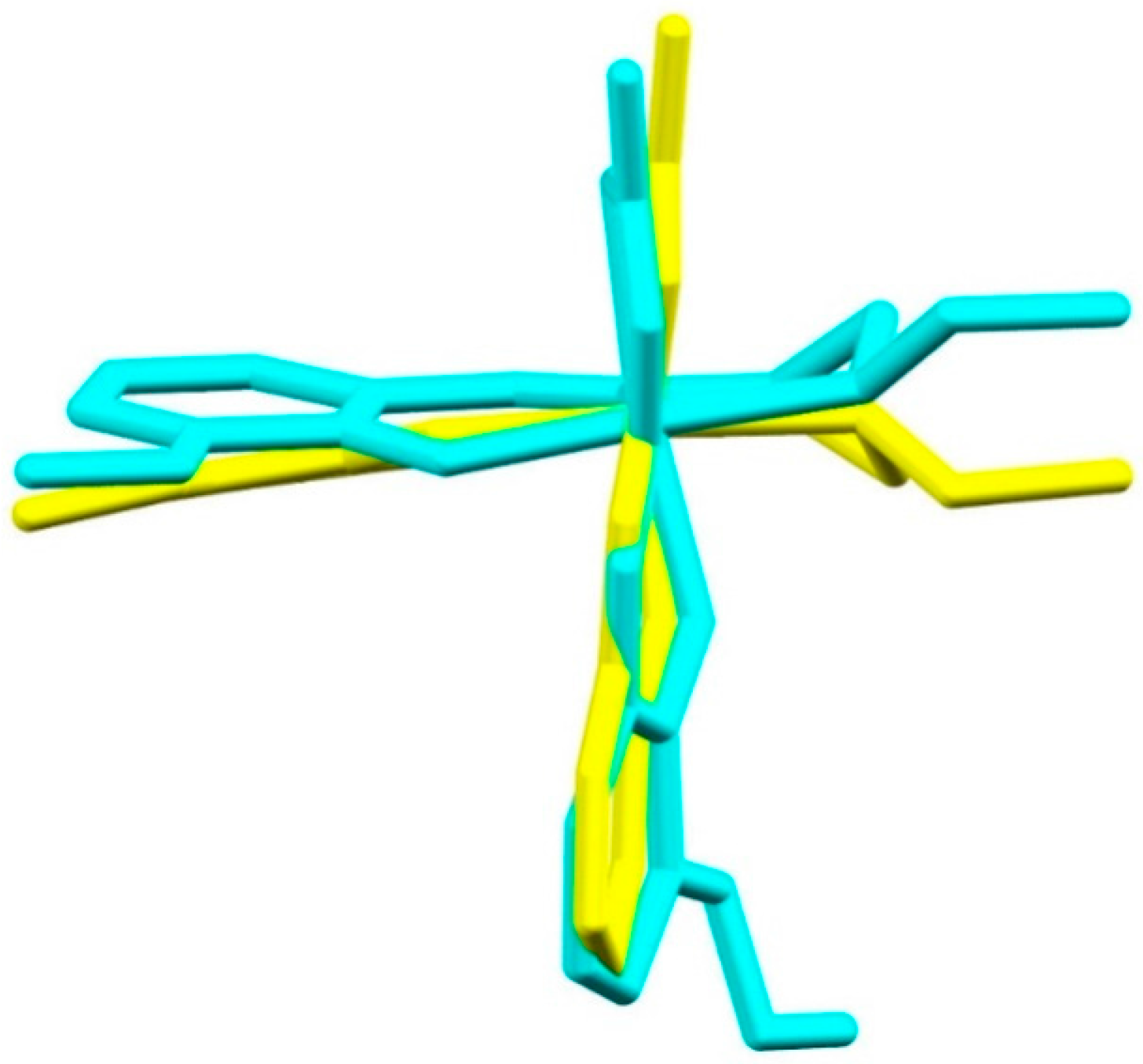
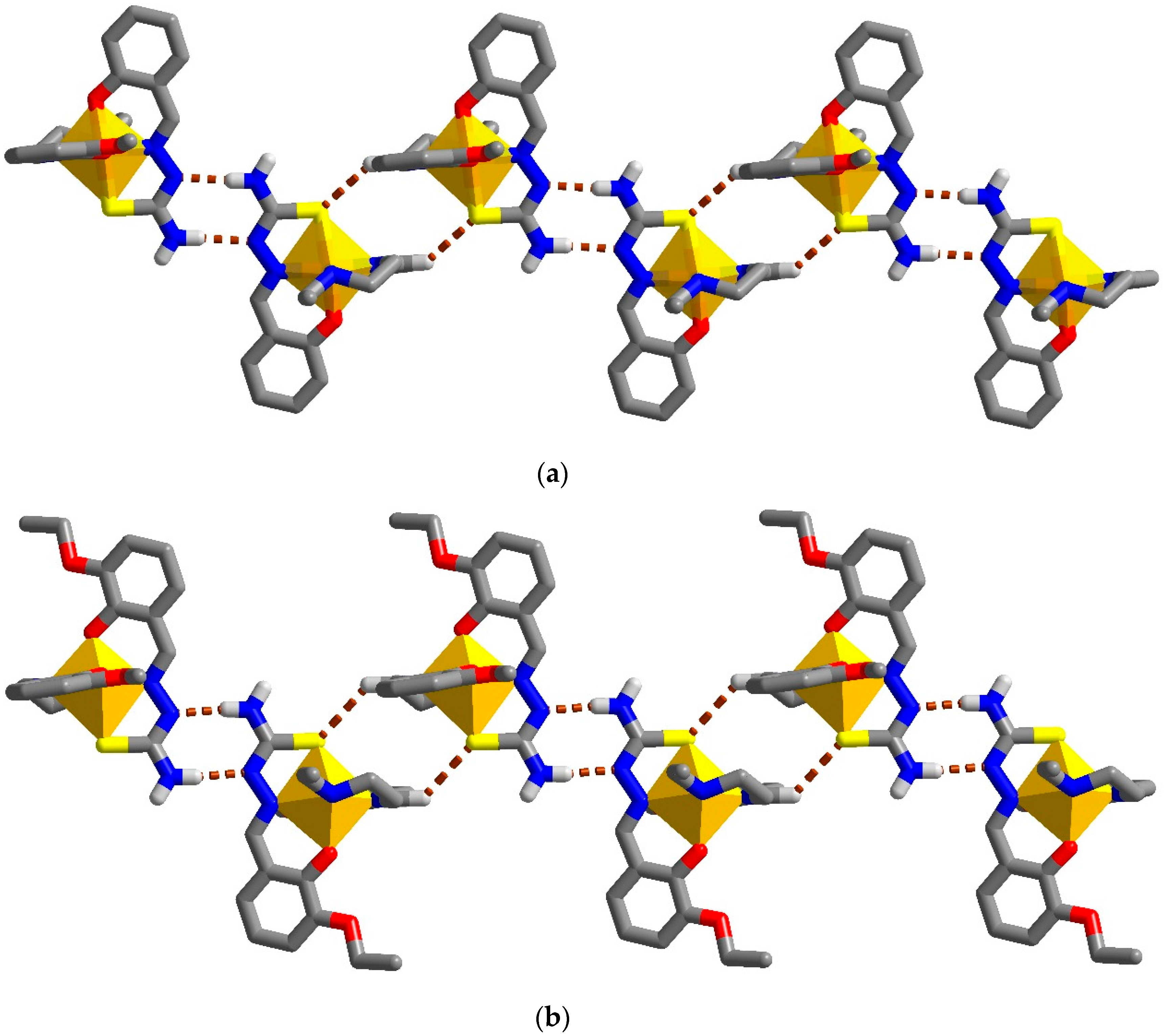
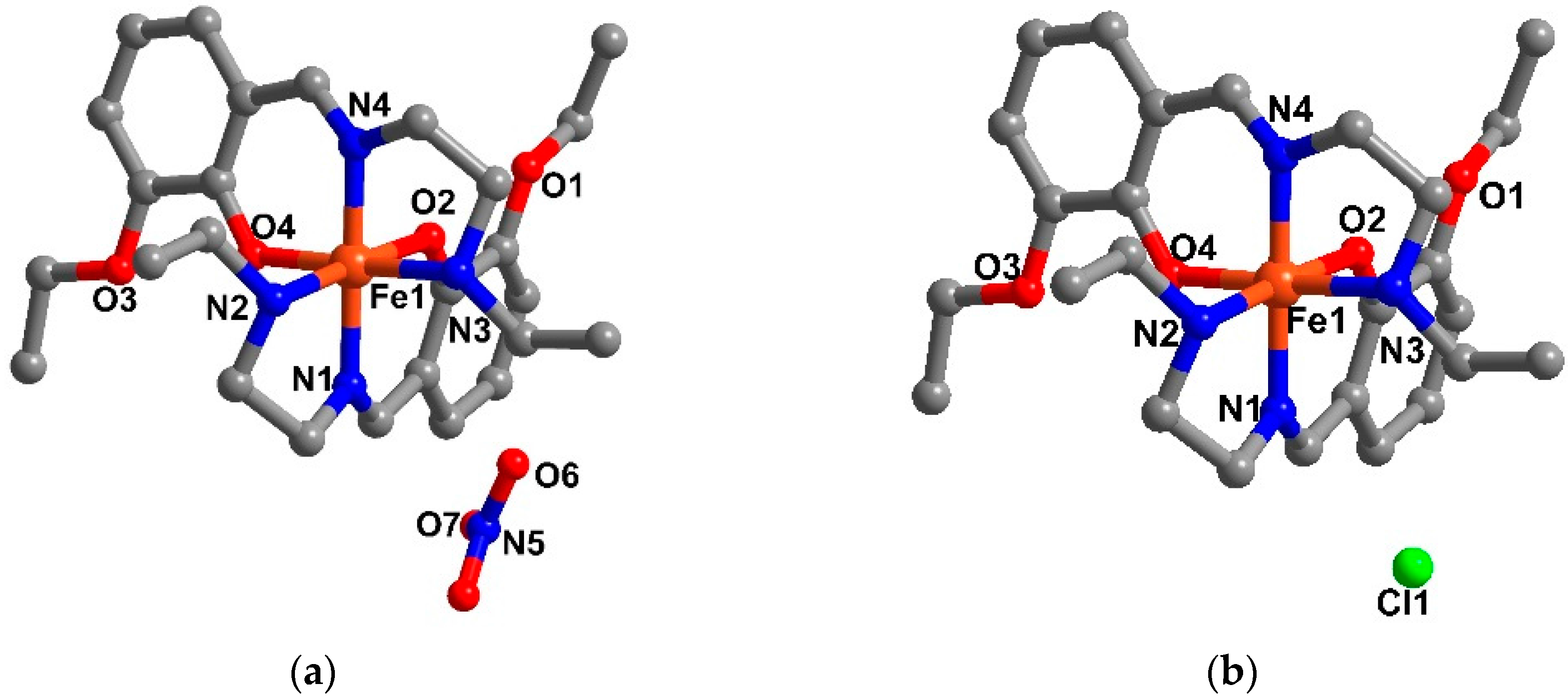
2.3. Structural Study of Heteroleptic Fe(III) Compounds 1 and 2
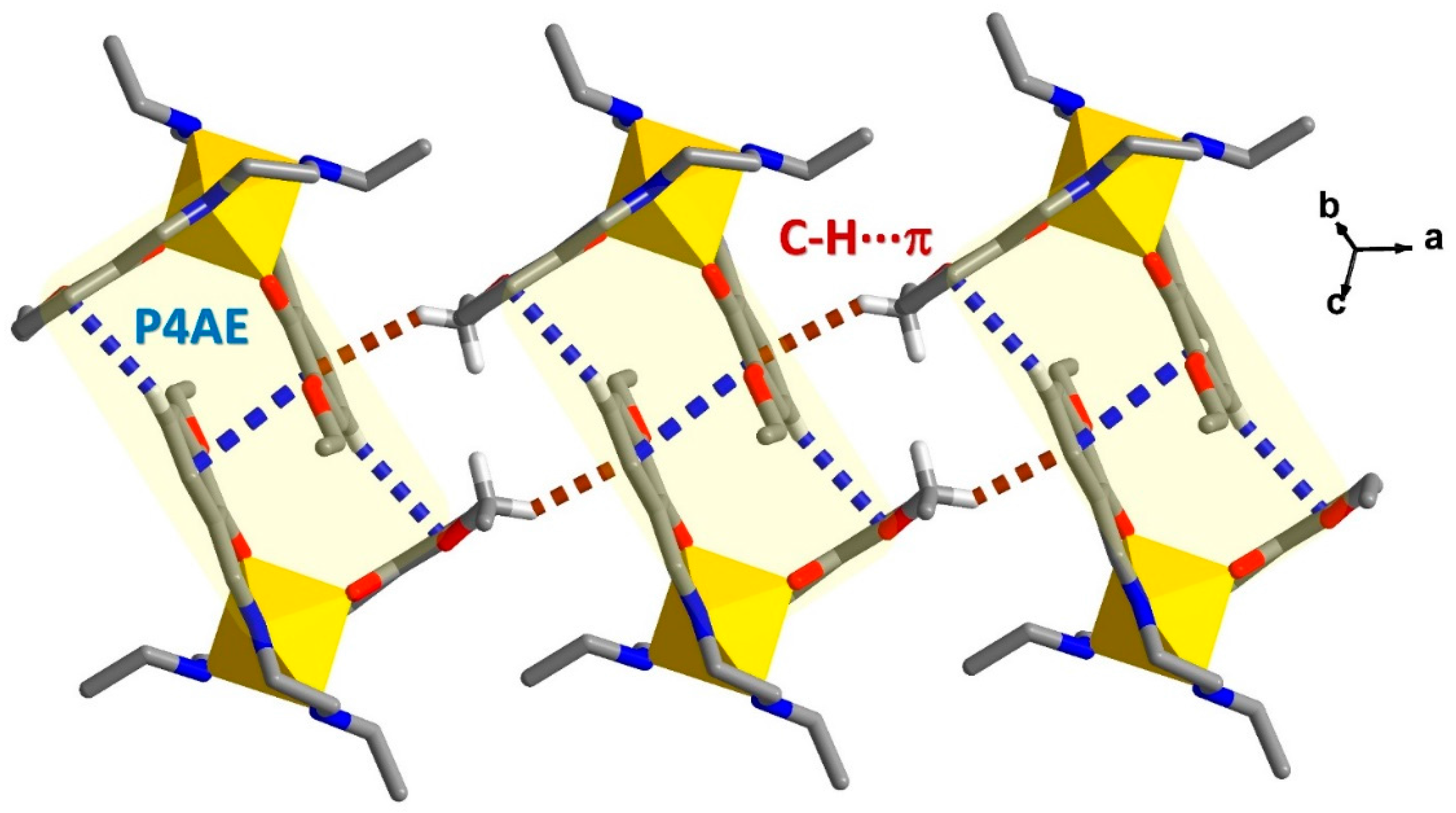
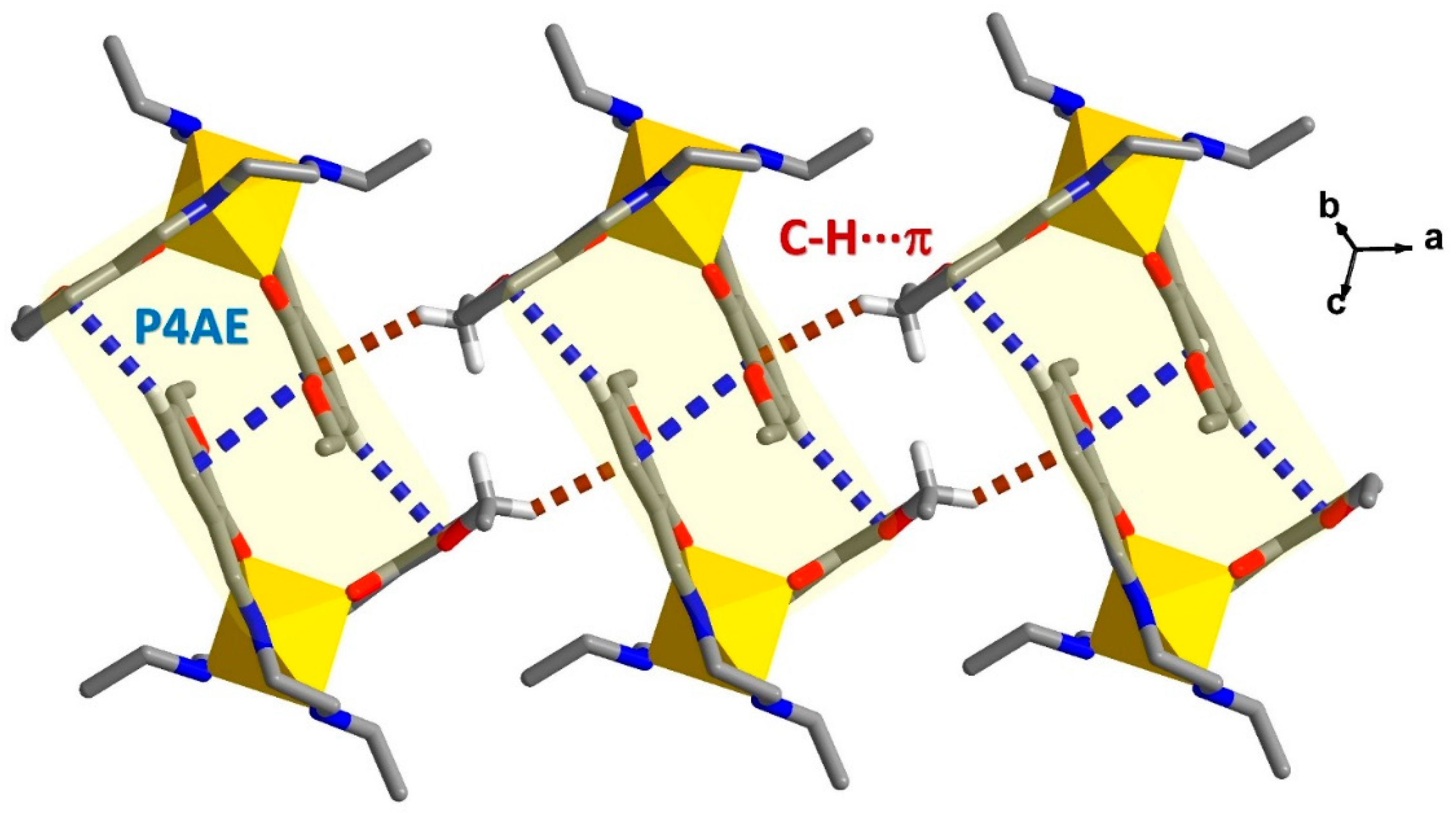
2.4. Structural Study of Homoleptic Cationic Fe(III) Compounds 3 and 4
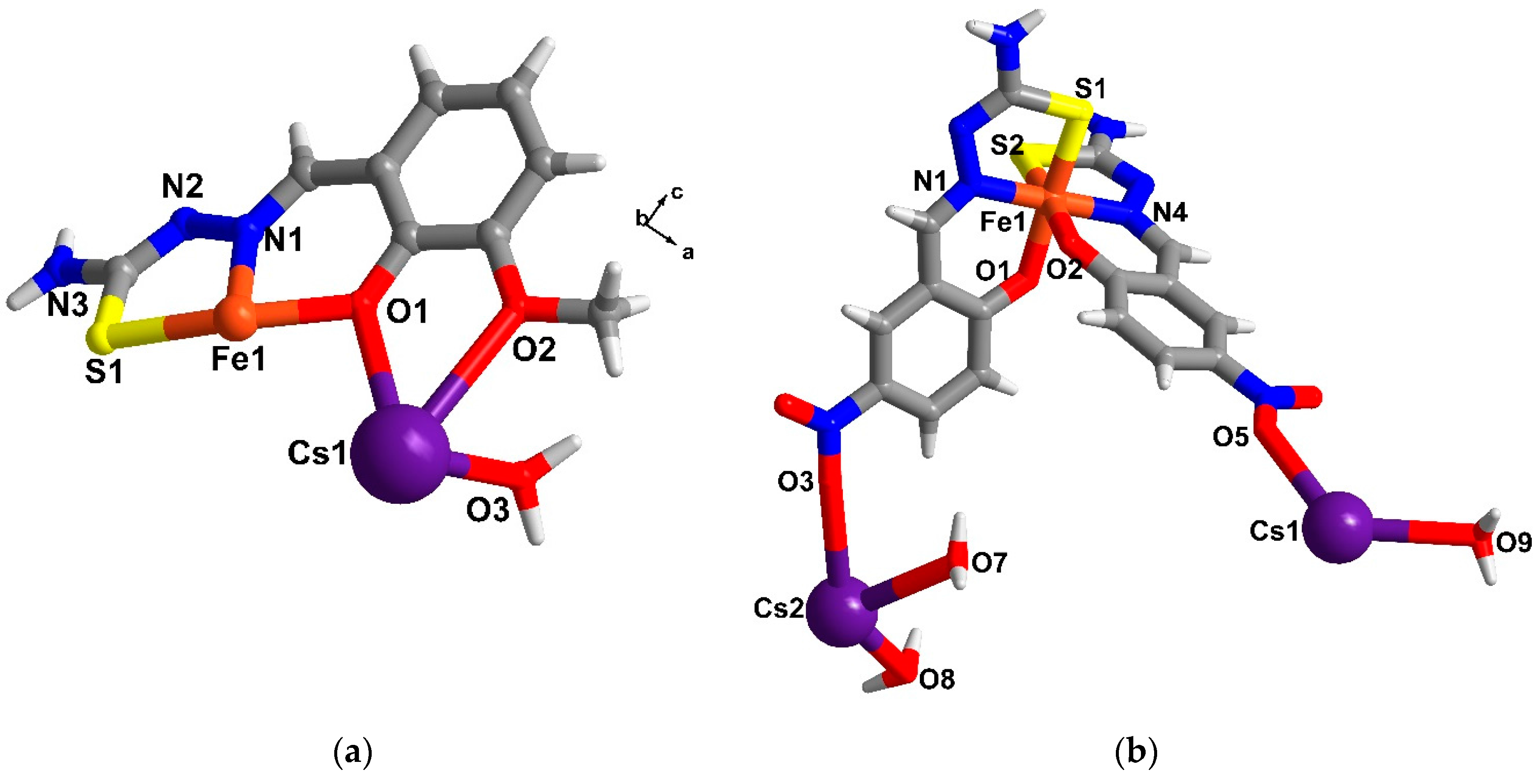
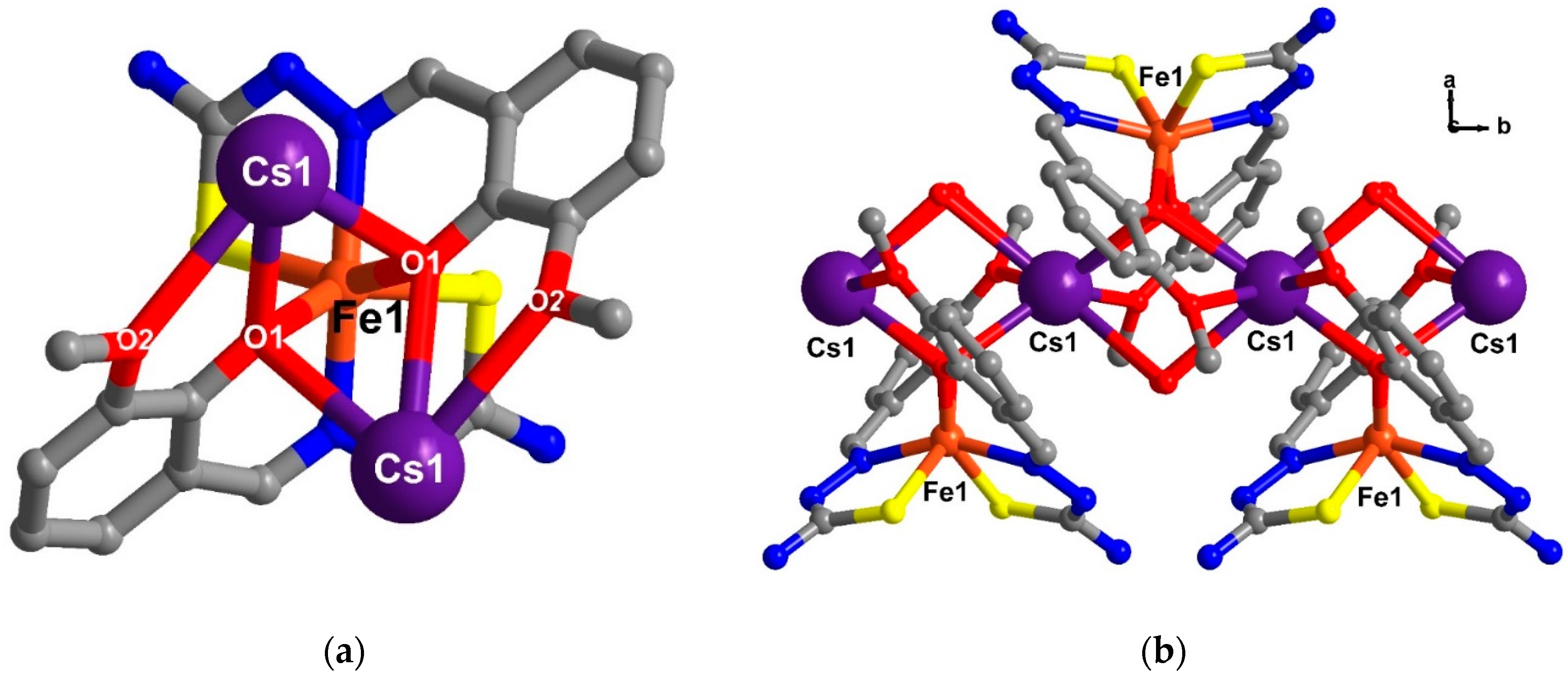
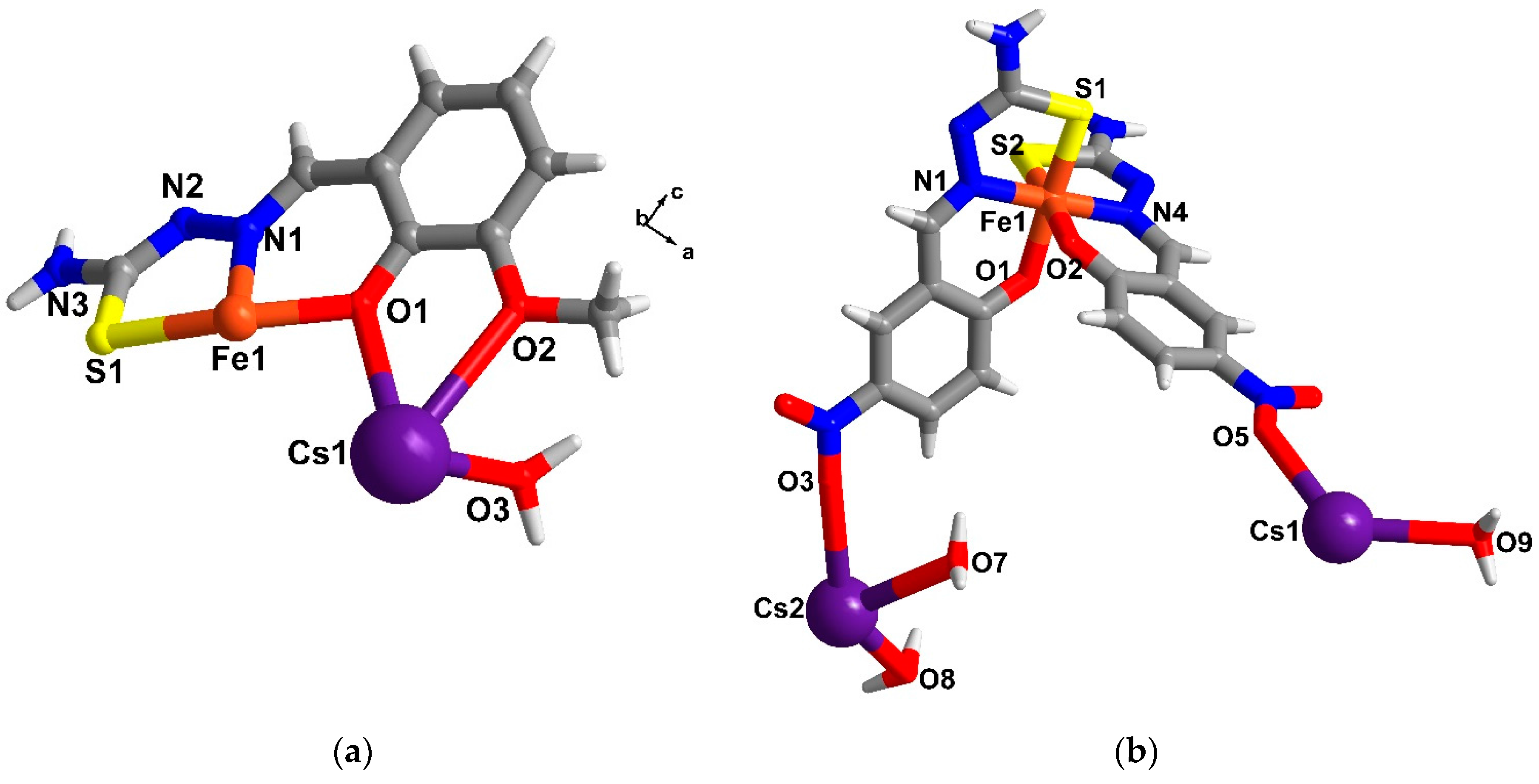
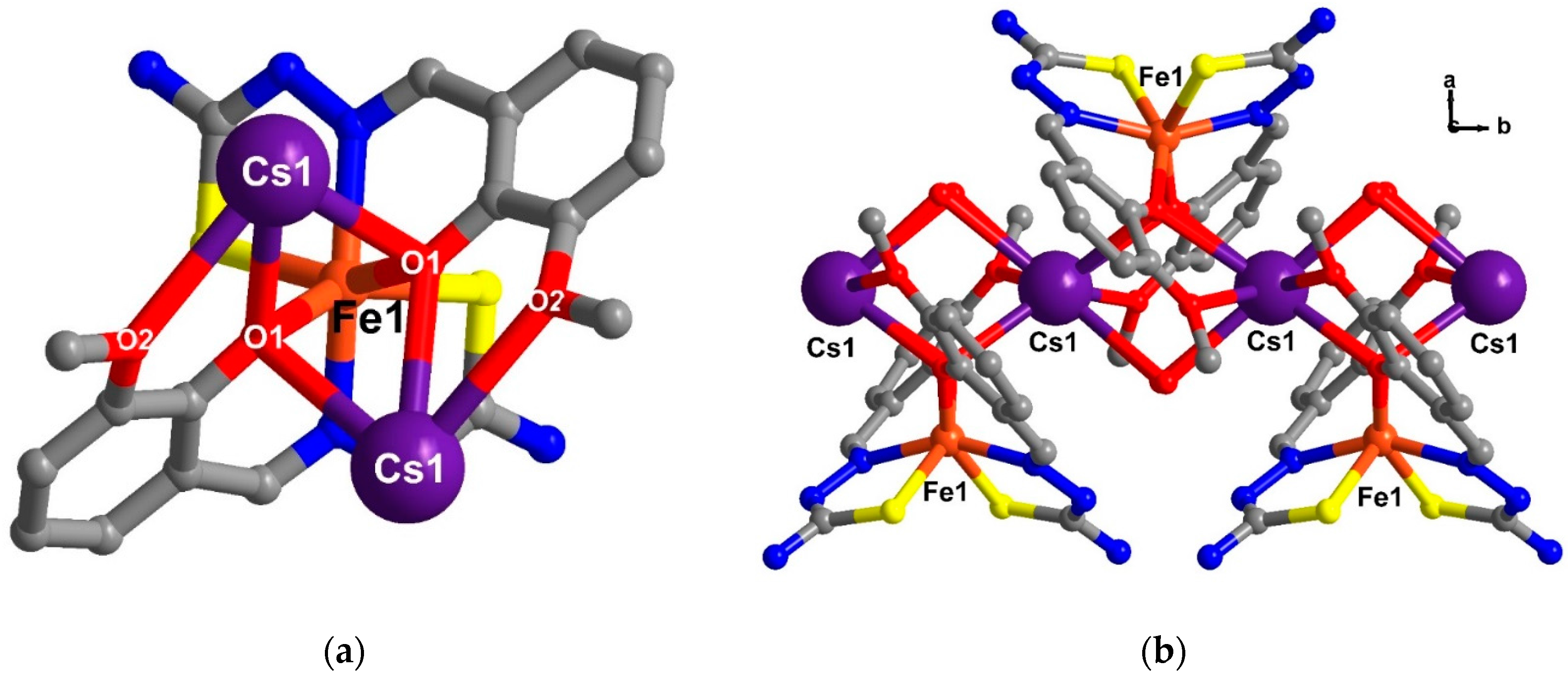
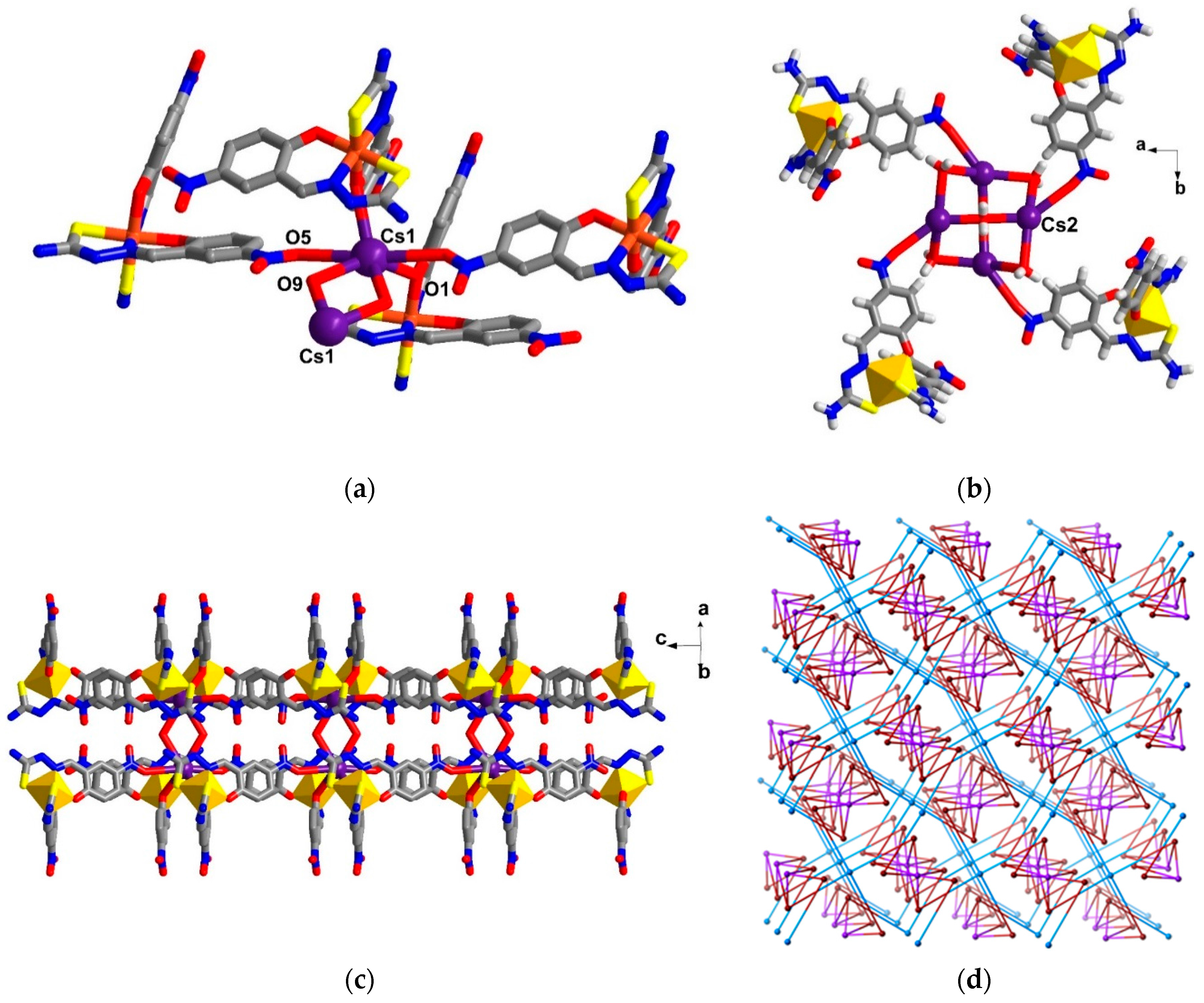
2.5. Structural Study of Homoleptic Anionic Fe(III) Compounds 5 and 6
3. Materials and Methods
3.1. General
3.2. Synthesis of Ligands
3.3. Synthesis of Iron(III) Complexes
3.3.1. Synthesis of [Fe(3-MeOSalEen)(3-EtOthsa)] 2
3.3.2. Synthesis of [Fe(3-EtOSalEen)2]NO3 3
3.3.3. Synthesis of [Fe(3-EtOSalEen)2]Cl 4
3.3.4. Synthesis of CsH2O[Fe(3-MeOthsa)2] 5
3.3.5. Synthesis of Cs(H2O)2[Fe(5-NO2-thsa)2] 6(bulk)
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Murray, K.S. The development of spin-crossover research. In Spin-Crossover Materials: Properties and Applications; Halcrow, M.A., Ed.; John Wiley & Sons Ltd.: Chichester, UK, 2013; pp. 1–54. [Google Scholar]
- Gütlich, P.; Goodwin, H.A. Spin Crossover in Transition Metal Compounds I–III; Springer: Berlin/Heidelberg, Germany, 2004; pp. 232–235. [Google Scholar]
- Harding, D.J.; Harding, P.; Phonsri, W. Spin crossover in iron(III) complexes. Coord. Chem. Rev. 2016, 313, 38–61. [Google Scholar] [CrossRef]
- Kennedy, B.J.; McGrath, A.C.; Murray, K.S.; Skelton, B.W.; White, A.H. Variable-temperature magnetic, spectral, and X-ray crystallographic studies of “spin-crossover” iron(III) schiff-base-lewis-base adducts. Influence of noncoordinated anions on spin-state interconversion dynamics in [Fe(salen)(imd)2]Y species (Y = ClO4−, BF4−, PF6−, BPh4−; imd = imidazole). Inorg. Chem. 1987, 26, 483–495. [Google Scholar]
- Phonsri, W.; Martinez, V.; Davies, C.G.; Jameson, G.N.L.; Moubaraki, B.; Murray, K.S. Ligand effects in a heteroleptic bis-tridentate iron(III) spin crossover complex showing a very high T1/2 value. Chem. Commun. 2016, 52, 1443–1446. [Google Scholar] [CrossRef] [PubMed]
- McCusker, J.K.; Rheingold, A.L.; Hendrickson, D.N. Variable-temperature studies of laser-initiated 5T2→1A1 intersystem crossing in spin-crossover complexes: Empirical correlations between activation parameters and ligand structure in a series of polypyridyl ferrous complexes. Inorg. Chem. 1996, 35, 2100–2112. [Google Scholar] [CrossRef]
- Marchivie, M.; Guionneau, P.; Letard, J.-F.; Chasseau, D. Photo-induced spin-transition: The role of the iron(II) environment distortion. Acta Crystallogr. Sect. B Struct. Sci. 2005, 61, 25–28. [Google Scholar] [CrossRef] [PubMed]
- Russell, V.; Scudder, M.; Dance, I. The crystal supramolecularity of metal phenanthroline complexes. J. Chem. Soc. Dalton Trans. 2001, 6, 789–799. [Google Scholar] [CrossRef]
- Steiner, T. The hydrogen bond in the solid state. Angew. Chem. Int. Ed. 2002, 41, 48–76. [Google Scholar] [CrossRef]
- Ryabova, N.A.; Ponomarev, V.I.; Zelentsov, V.V.; Atovmyan, L.O. Kristallografiya. Russ. Crystallogr. Rep. 1981, 26, 101. [Google Scholar]
- Powell, R.E.; Schwalbe, C.H.; Tizzard, G.J.; Koningsbruggen, P.J.V. Caesium bis(5-bromosalicylaldehyde thiosemicarbazonato-κ3 O,N,S)ferrate(III): Supramolecular arrangement of low-spin FeIII complex anions mediated by Cs+ cations. Acta Cryst. 2015, C71, 169–174. [Google Scholar]
- Kang, S.; Shiota, Y.; Kariyazaki, A.; Kanegawa, S.; Yoshizawa, K.; Sato, O. Heterometallic FeIII/K coordination polymer with a wide thermal hysteretic spin transition at room temperature. Chem. Eur. J. 2016, 22, 532–538. [Google Scholar] [CrossRef] [PubMed]
- McPhillips, T.M.; McPhillips, S.E.; Chiu, H.-J.; Cohen, A.E.; Deacon, A.M.; Ellis, P.J.; Garman, E.; Gonzalez, A.; Sauter, N.K.; Phizackerley, R.P.; et al. Blu-ice and the distributed control system: Software for data acquisition and instrument control at macromolecular crystallography beamlines. J. Synchrotron Radiat. 2002, 9, 401–406. [Google Scholar] [CrossRef] [PubMed]
- Kabsch, W. Automatic processing of rotation diffraction data from crystals of initially unknown symmetry and cell constants. J. Appl. Crystallogr. 1993, 26, 795–800. [Google Scholar] [CrossRef]
- Sheldrick, G.M. SADABS, Program for Area Detector Adsorption Correction; Institute for Inorganic Chemistry, University of Göttingen: Göttingen, Germany, 1996. [Google Scholar]
- Hui, R.-H.; Zhou, P.; You, Z.-L. Syntheses and crystal structures of two azide-bridged dinuclear zinc(II) complexes with schiff bases and halides. Synth. React. Inorg. Met. Org. Nano Met. Chem. 2012, 42, 135–139. [Google Scholar] [CrossRef]
- Mehta, B.H.; Shaikh, J.A. Synthesis, characterisation, X-ray diffraction and antimicrobial studies of Pd(II), Rh(III) and Ru(III) complexes of thiosemicarbazones. J. Ind. Counc. Chem. 2009, 26, 1–6. [Google Scholar]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Complex | 1 [5] | 2 | 3 | 4 | 5 | 6 |
|---|---|---|---|---|---|---|
| Temp. | 100 K | 100 K | 100 K | 100 K | 123 K | 123 K |
| Molecular weight/g·mol−1 | 470.35 | 514.40 | 588.45 | 561.90 | 653.28 | 701.25 |
| Crystal system | Triclinic | Monoclinic | Triclinic | Triclinic | Orthorhombic | Tetragonal |
| Space group | P | P21/c | P | P | Pnna | Pn2 |
| a/Å | 8.3940 (17) | 14.122 (3) | 9.646 (2) | 9.698 (2) | 19.0649 (7) | 20.6177 (3) |
| b/Å | 9.3500 (19) | 13.889 (3) | 10.632 (2) | 10.633 (2) | 9.1084 (4) | 20.6177 (3) |
| c/Å | 13.675 (3) | 13.470 (3) | 14.242 (3) | 13.515 (3) | 13.1902 (5) | 11.7467 (3) |
| α/o | 82.26 (3) | 90 | 100.25 (3) | 96.15 (3) | 90 | 90 |
| β/o | 73.44 (3) | 118.40 (3) | 105.21 (3) | 103.05 (3) | 90 | 90 |
| γ/o | 82.14 (3) | 90 | 102.93 (3) | 103.49 (3) | 90 | 90 |
| Cell volume/Å3 | 1013.9 (4) | 2324.1 (10) | 1329.5 (5) | 1301.3 (4) | 2290.5 (2) | 4993.4 (2) |
| Z | 2 | 4 | 2 | 2 | 4 | 8 |
| Absorption coefficient/mm−1 | 0.880 | 0.778 | 0.622 | 0.722 | 2.451 | 2.266 |
| Reflections collected | 27,497 | 23,856 | 24,637 | 21,725 | 42,177 | 188,772 |
| Independent Reflections, Rint | 5929, 0.0501 | 6297, 0.0369 | 6224, 0.0792 | 5703, 0.1664 | 3544, 0.0582 | 10084, 0.0651 |
| Max., min. transmission | 0.9913, 0.9741 | 0.9923, 0.977 | 0.9969, 0.9695 | 0.9964, 0.9857 | 0.9301, 0.7493 | 0.7727, 0.6600 |
| Restraints/parameters | 1/277 | 0/301 | 0/364 | 0/329 | 0/156 | 12/331 |
| Final R indices [I > 2σ(I)]: R1, wR2 | 0.0401, 0.1123 | 0.0408, 0.1118 | 0.0419, 0.1111 | 0.0573, 0.1518 | 0.0298, 0.0648 | 0.0747, 0.2314 |
| CCDC No. | 1420398 | 1552518 | 1552515 | 1552514 | 1552517 | 1552516 |
| Complex | 1 [5] | 2 | 3 | 4 | 5 | 6 | |||
|---|---|---|---|---|---|---|---|---|---|
| Temp. | 100 K | 100 K | 100 K | 100 K | 123 K | 123 K | |||
| Fe1–O1/Å | 1.8875 (14) | 1.882 (1) | Fe1–O2/Å | 1.877 (1) | 1.874 (2) | Fe1–O1/Å | 1.987 (2) | Fe1–O1/Å | 1.917 (6) |
| Fe1–O3/Å | 1.8937 (13) | 1.9315 (1) | Fe1–O4/Å | 1.889 (2) | 1.895 (2) | Fe1–O1 iii/Å | 1.987 (2) | Fe1–O2/Å | 1.922 (5) |
| Fe1–N1/Å | 2.2590 (8) | 2.050 (2) | Fe1–N1/Å | 1.926 (2) | 1.923 (3) | Fe1–N1/Å | 2.179 (2) | Fe1–N1/Å | 1.931 (6) |
| Fe1–N2/Å | 2.0588 (17) | 1.915 (2) | Fe1–N2/Å | 2.037 (2) | 2.041 (3) | Fe1–N1 iii/Å | 2.179 (2) | Fe1–N4/Å | 1.911 (6) |
| Fe1–N3/Å | 1.9198 (14) | 1.942 (2) | Fe1–N3/Å | 2.060 (2) | 2.046 (3) | Fe1–S1/Å | 2.416 (1) | Fe1–S1/Å | 2.214 (2) |
| Fe1–S1/Å | 1.9424 (14) | 2.248 (1) | Fe1–N4/Å | 1.923 (2) | 1.922 (3) | Fe1–S1 iii/Å | 2.416 (1) | Fe1–S2/Å | 2.237 (2) |
| Σ/° | 44 | 51 | Σ/° | 47 | 48 | Σ/° | 129 | Σ/° | 39 |
| Θ/° | 80 | 98 | Θ/° | 70 | 72 | Θ/° | 404 | Θ/° | 67 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Phonsri, W.; Darveniza, L.C.; Batten, S.R.; Murray, K.S. Heteroleptic and Homoleptic Iron(III) Spin-Crossover Complexes; Effects of Ligand Substituents and Intermolecular Interactions between Co-Cation/Anion and the Complex. Inorganics 2017, 5, 51. https://doi.org/10.3390/inorganics5030051
Phonsri W, Darveniza LC, Batten SR, Murray KS. Heteroleptic and Homoleptic Iron(III) Spin-Crossover Complexes; Effects of Ligand Substituents and Intermolecular Interactions between Co-Cation/Anion and the Complex. Inorganics. 2017; 5(3):51. https://doi.org/10.3390/inorganics5030051
Chicago/Turabian StylePhonsri, Wasinee, Luke C. Darveniza, Stuart R. Batten, and Keith S. Murray. 2017. "Heteroleptic and Homoleptic Iron(III) Spin-Crossover Complexes; Effects of Ligand Substituents and Intermolecular Interactions between Co-Cation/Anion and the Complex" Inorganics 5, no. 3: 51. https://doi.org/10.3390/inorganics5030051