
Computing Free Energies of Hydroxylated Silica Nanoclusters: Forcefield versus Density Functional Calculations



1
Departament de Ciència de Materials i Química Física and Institut de Química Teòrica i Computacional (IQTCUB), Universitat de Barcelona, E-08028 Barcelona, Spain
2
Dipartimento di Chimica and NIS Centre, Università degli Studi di Torino, Via P. Giuria 7, I-10125 Torino, Italy
3
Institució Catalana de Recerca i Estudis Avançats (ICREA), E-08010 Barcelona, Spain
*
Author to whom correspondence should be addressed.
Inorganics 2017, 5(3), 41; https://doi.org/10.3390/inorganics5030041
Submission received: 18 May 2017
/
Revised: 19 June 2017
/
Accepted: 24 June 2017
/
Published: 29 June 2017
(This article belongs to the Special Issue Inorganic Nanoclusters: Advances in Understanding Structure and Properties)
Abstract
:We assess the feasibility of efficiently calculating accurate thermodynamic properties of (SiO2)n·(H2O)m nanoclusters, using classical interatomic forcefields (FFs). Specifically, we use a recently parameterized FF for hydroxylated bulk silica systems (FFSiOH) to calculate zero-point energies and thermal contributions to vibrational internal energy and entropy, in order to estimate the free energy correction to the internal electronic energy of these nanoclusters. The performance of FFSiOH is then benchmarked against the results of corresponding calculations using density functional theory (DFT) calculations employing the B3LYP functional. Results are reported first for a set of (SiO2)n·(H2O)m clusters with n = 4, 8 and 16, each possessing three different degrees of hydroxylation (R = m/n): 0.0, 0.25 and 0.5. Secondly, we consider five distinct hydroxylated nanocluster isomers with the same (SiO2)16·(H2O)4 composition. Finally, the free energies for the progressive hydroxylation of three nanoclusters with R = 0–0.5 are also calculated. Our results demonstrate that, in all cases, the use of FFSiOH can provide estimates of thermodynamic properties with an accuracy close to that of DFT calculations, and at a fraction of the computational cost.
1. Introduction
The energy of a chemical system standardly calculated via ab initio methods of quantum chemistry, such as density functional theory (DFT), only provides the internal energy Uelec (which includes the potential and kinetic energy contributions from electrons and their interaction with nuclei). Uelec, however, does not take into account the contribution arising from the quantum nature of the nuclear motion, i.e., the zero point energy (ZPE) for nuclei in their lowest vibrational energy level. More importantly, at finite temperatures Uelec also omits contributions from all the different microstates occupied by the system at a temperature, T, and volume, V. Using statistical mechanics, these microstates can be appropriately counted and their weighted contributions evaluated and summed to form the partition function, Q. In turn, from Q we can calculate the thermodynamic properties of the system, namely the entropy, S, and ultimately, the free energy, G. Differences in G (i.e., ∆G), often stemming from entropic factors rather than internal energy differences, determine the most likely pathways followed by (bio)chemical processes (e.g., reactions, solvation, and protein docking). Sophisticated methods, which efficiently and accurately estimate ∆G values, are thus understandably essential for the realistic computational modelling of such systems [1].
In the simplest case of a molecule in its electronic ground state residing in a local energy minimum, the estimation of free energy can be greatly simplified. Assuming that the behavior of a molecule can be represented by that of an ideal gas and that the different occupiable microstates (i.e., degrees of freedom) are independent from each other, one can obtain thermodynamic quantities by dividing Q into individual contributions from translations (qtrans), rotations (qrot), and vibrations (qvib). Computing the qtrans and qrot under these assumptions is straightforward (see below), but evaluation of qvib requires computation of the different vibrational modes. Assuming small amplitude vibrations around a minimum energy configuration, a harmonic oscillator model is a good description of the nuclear vibrations, so that one can obtain the vibrational levels provided that the Hessian matrix of the force constants (second derivative of the total energy with respect to atomic displacements) computed, either analytically or numerically, is available. In this most simple of cases, and for relatively small molecules, the estimate of free energy can be computed by efficient ab initio quantum chemical methods, such as those based on density functional theory (DFT) (thanks to efficient implementation of the analytical second derivatives of the total energy). However, the computational cost of performing these calculations quickly rises with increasing system size, becoming prohibitive for routine calculations for systems possessing more than approximately 100 atoms. This is particularly so for a system in which the Hessian matrix should be computed numerically, either by using analytical gradient or, even worse, when the gradient energy also has to be evaluated numerically. Thus, in order to be able to perform free energy calculations at a reasonable computational cost, classical interatomic forcefields (FFs) are usually employed.
Sampling the energy landscape of configurations of a (bio)molecular system using FFs is typically at least 10,000 times quicker than when done using DFT. Although allowing for faster calculations, it is essential that the FFs employed should be as accurate as possible, as not to negate the increased computational efficiency. FFs developed for modelling (bio)molecular systems have historically been parameterized using experimentally known structural properties and results from ab initio quantum chemical calculations [2]. More recently, with the increasingly widespread use of (bio)chemical free energy calculations, such FFs have used experimental free energy data in their parameterization [3,4]. In materials science, especially for inorganic materials and metals/alloys, FFs are also widely employed to study structural and mechanical properties [5]. For such materials, unlike in the biochemical world, entropic contributions to the free energy of the system are not generally thought to be very important for bulk properties. However, for small system sizes (e.g., inorganic nanoclusters) explicit consideration of all degrees of freedom can become significant for describing the structure and evolution of such species under a given set of physical conditions. Studies of inorganic nanocluster systems in a variety of contexts (e.g., nucleation [6,7,8,9], presence of a reactive atmosphere [10,11,12], solvation [13,14,15,16]) have confirmed the importance of explicitly considering the entropic contributions to the free energies. These cases have tended to look at rather small species (≤40 atoms) and, when taken into account, calculate the entropic contribution to the free energy via DFT using the harmonic approximation for vibrations. As noted above, such calculations quickly become prohibitive with increasing system size and, following the progress in the field of biochemical modelling, accurately parameterizing FFs for inorganic nanosystems would allow for huge increases in computational efficiency. FFs for inorganic materials are usually parameterized for bulk structural and mechanical properties, and thus tend not to be ideally suited for treating nanoclusters (which tend to have non-bulk-like structures and properties). Although some efforts have been made to parameterize FFs (specifically for inorganic nanoclusters) [17], or to make size-transferable FFs for inorganic materials [18], only very few FFs have explicitly included thermodynamic data in their parameterization [19,20] (in order to treat these systems more accurately) [21].
The objective of this work is to assess whether an existing FF, that was not parameterized using thermodynamic data, can be used to provide free energy estimates to a similar accuracy as that provided by DFT. Specifically, we examine the performance of FFSiOH [22], which is a polarisable ionic FF, that was parametrized with respect to the results of DFT calculations of silica bulk phases and hydroxylated silica surfaces. We have recently shown that FFSiOH can provide results in good agreement with DFT calculations (with respect to structure, relative energies, and vibrational frequencies) for a range of hydroxylated silica nanoclusters of different sizes and degrees of hydroxylation [23]. Following this work, we have selected hydroxylated silica nanoclusters as a suitable test case for the present study. Free energies in solution of these species are of particular interest for gaining insight into nucleation, growth, and dissolution of silicate materials [24,25]. The free energies of small silica nanoclusters (or oligomers) have been calculated using DFT a number of times in the literature [13,14,15,16], employing the harmonic approximation for vibrations and various solvation models. Considering that both: (i) our objective is to evaluate the performance of the FFSiOH potential (i.e., rather than different solvation models); and (ii) the most computationally expensive part of evaluating free energies in these cases is the evaluation of the vibrational entropy; we focus on the ability of FFSiOH to provide free energies in vacuum.
We calculate ZPE, vibrational entropies, and free energies (both total and of hydroxylation) of a range of hydroxylated silica clusters with a range of sizes and degrees of hydroxylation, using FFSiOH. Overall, we find good agreement with corresponding DFT calculations, indicating that FFSiOH could provide an extremely computationally efficient means through which to calculate accurate free energies of nano-silicate species.
2. Methodology
We first review how to obtain thermodynamic properties from the partition function for a silica nanocluster, residing in a local energy minimum in its electronic ground state. We will only focus on main assumptions and provide the necessary equations by which the results can be achieved (full details can be found in Reference [26]). Assuming that: (1) the nanocluster behaves as an ideal gas; and (2) that all energy levels can be grouped into either translations, rotations, vibrations, or electronic levels, and that they are uncoupled from each other, Q can be defined as:
where each q is the partition function associated with the translational, rotational, vibrational, and electronic energy, respectively. Each q can be calculated with the following expressions derived for a particle in a box of volume V, at temperature T:
where m is the mass, kB is the Boltzmann constant, h is Planck´s constant, σr is the symmetry number for rotation, and the Θ correspond to characteristic temperatures for rotations Θr and vibrations Θv. The product qvib runs over each vibrational mode K. Since the energy to reach electronically excited states is much higher than the thermal energy in our system, and we deal with closed shell molecules, the term qelec becomes 1, and does not contribute to the entropy. The rotational and vibrational characteristic temperatures are defined by:
where I is the moment of inertia and K represents each individual vibration. Assuming ideal gas behavior, we can derive the expressions of internal energy (Etrans, Erot, Evib, and Eelec), and entropy (Strans, Srot, Svib, and Selec), using the corresponding partition functions, where we assume the zero of energy to be the electronic energy:
where R is the ideal gas constant (R = N·kB). The term in Evib corresponds to the energy of vibrations at 0 K (i.e., the ZPE). In the Results section, we show the ZPE and the factor separately, with the latter defined as the thermal contribution to the total energy (Uvib). After obtaining the thermal and entropic contribution of each term, we add them to the electronic energy in the following way to obtain the zero Kelvin energy (U(T0K)), the internal energy at a given temperature (U(T)), the enthalpy (H(T)), and the free energy (G(T)):
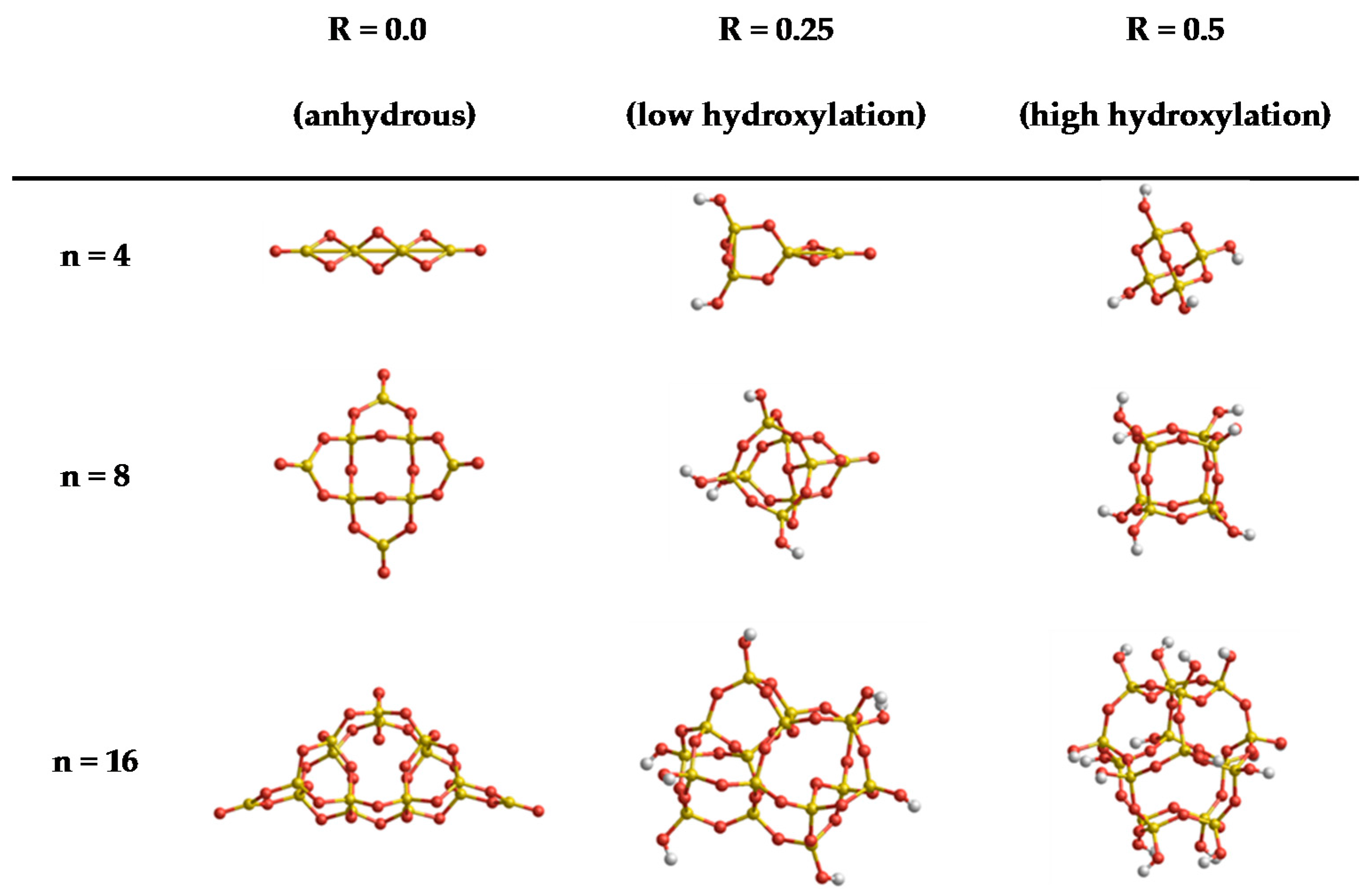
In order to give a reasonable estimate of the broad performance of FFSiOH, we perform our calculations on two sets of clusters (A and B) that provide complementary information. Set A contains nine globally minimized clusters from previous work [27,28,29,30], with stoichiometries (SiO2)n·(H2O)m with n = 4, 8, and 16 and m corresponding to different values which represent three degrees of hydroxylation (i.e., R = m/n) of: 0.0, 0.25 and 0.5. This entails that for n = 4 we have m = 0, 1, 2, for n = 8 we have m = 0, 2, 4 and for n = 16 we have m = 0, 4, 8. These ratios correspond to anhydrous, low, and high degrees of hydroxylation. The structures in set A can be seen in Figure 1. Set A allows us to assess the dependency of the FFSiOH versus DFT error in the calculated thermodynamic properties (on size and degree of hydroxylation). The contributions from Srot and Strans depend on the geometry and mass of the system, which are essentially identical for FFSiOH and DFT calculations (except for very small displacements of the atoms). The differences between the calculated values from DFT and FFSiOH should thus be very small in these cases. We will therefore focus on the global values of the thermodynamic properties and the vibrational contributions.
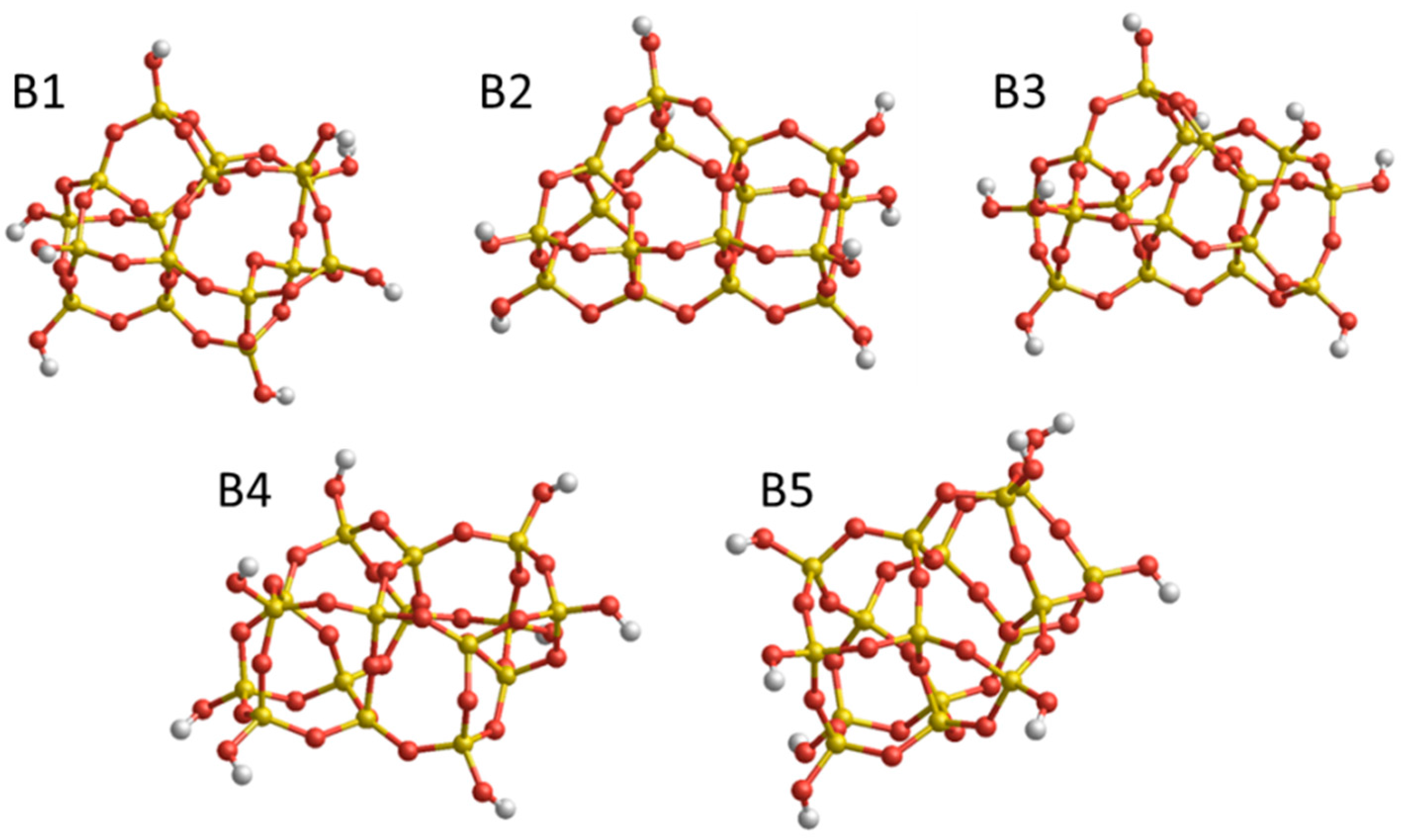
The second set (B) of structures consist of five isomers with (SiO2)16·(H2O)4 stoichiometry (see Figure 2). These structures are selected from the dataset in Reference [23] as representative low energy minima, as found by classical IP-based global optimization searches, followed by DFT refinement (see details in References [27,28,29,30]. The relative energies of the structures calculated at a DFT level using the B3LYP functional are noted in Figure 2. Moreover, this set of structures displays a wide spread of differences between the relative energies provided by FFSiOH and DFT. Set B will thus help establish whether the performance of FFSiOH with respect to reproducing DFT-calculated energy differences also propagates to the free energy corrections or not.
Finally, we compare the results for the free energy of hydroxylation (i.e., reaction with water) of clusters for each size from their anhydrous state to that of low and subsequently high hydroxylation (see Figure 1). We compare the free energy of hydroxylation using: (a) the results directly obtained from DFT calculations; and (b) the electronic energy from DFT results, and the correction to the free energy from FFSiOH. Since FFSiOH is not parameterized to describe the water molecule, we use the Gibbs free energy obtained from DFT for this quantity throughout.
All DFT calculations were performed using the Gaussian09 code [31] using the B3LYP [32] functional and 6–31g(d,p) basis set. This combination of functional and basis set has proven to provide a good compromise between computational efficiency and accuracy for hydroxylated silica systems in previous work [14,15,16,27,33]. Since our systems contain low frequency vibrational modes, we use tight cut-offs and an ultrafine integration grid for the geometry optimizations, using the Berny algorithm [34]. The FFSiOH calculations were performed using the GULP code [35], employing the rational function optimization method for geometry optimizations. All thermodynamic data were calculated at standard conditions (i.e., T = 298.15 K and 1 atm pressure). We note that all reported free energy terms are calculated using the structures optimized, using the respective method (i.e., FFSiOH-calculated free energies use FFSiOH-optimised nanocluster structures and DFT-calculated free energies use DFT-optimized nanocluster structures).
3. Results and Discussion
3.1. Thermodynamic Data with Respect to Size and Hydroxylation
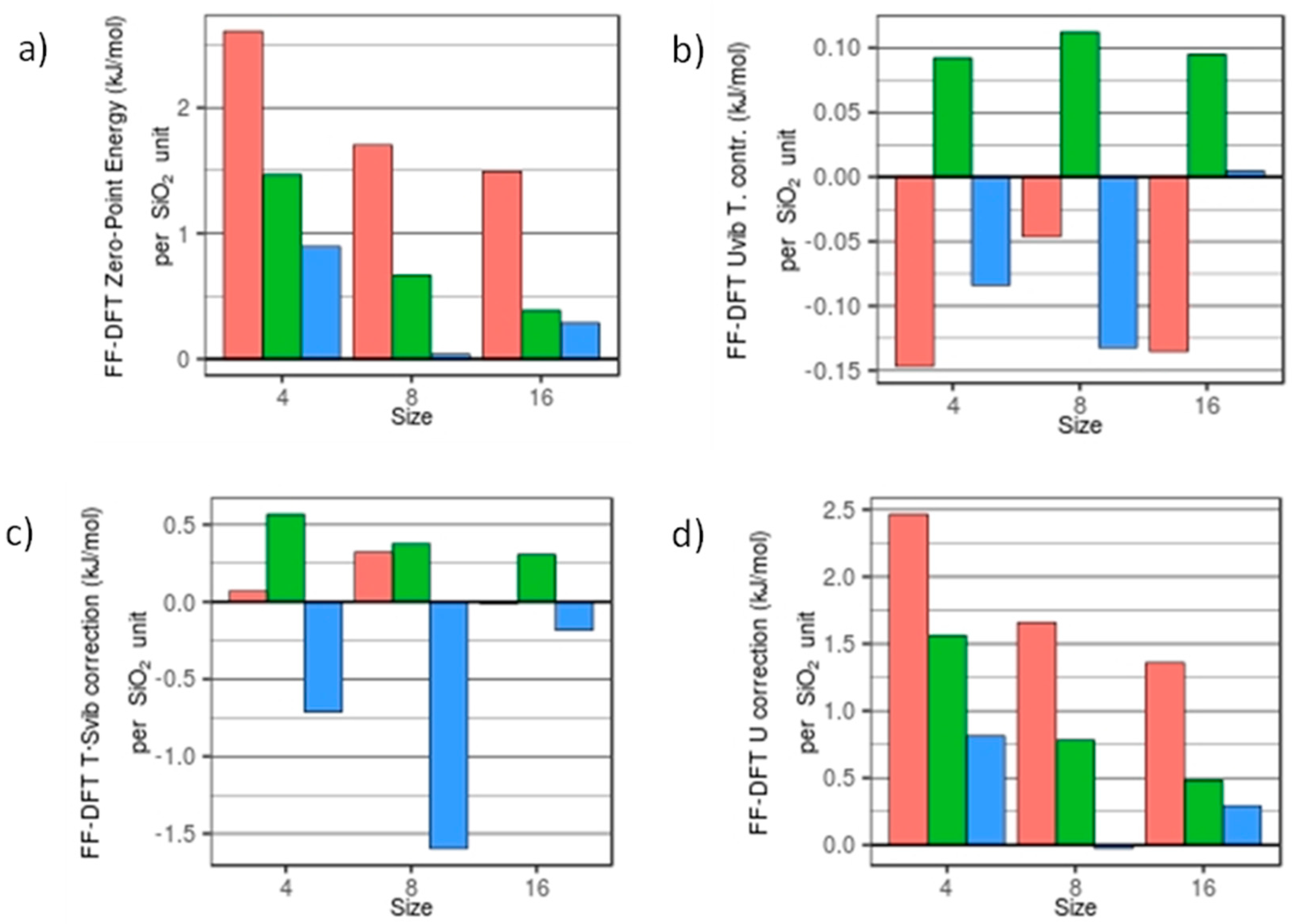
In Figure 3 we plot the FFSiOH-DFT differences in ZPE, Uvib, U correction, T·Svib, and the G correction of the nanoclusters, with respect to degree of hydroxylation and grouped by size. Energies are given reported in kJ/mol per SiO2 in order to compare quantities calculated for nanoclusters of different sizes. In the ZPE, the largest difference between the FFSiOH and DFT data is found for the anhydrous (SiO2)4 nanocluster, with a difference of 2.61 kJ/mol per SiO2 unit. The smallest difference observed is for (SiO2)8·(H2O)8, with a difference of 0.04 kJ/mol per SiO2 unit. Both increases in size and degree of hydroxylation lower the respective ZPE differences. The higher error in the anhydrous cases can be explained with the fact that at these very small sizes, the proportion of strained rings and terminal oxygens is large. These defects are not present in periodic bulk-calculations, from which the FFSiOH was parametrized, and therefore the higher errors in the anhydrous systems are not unexpected. More precisely, we identified a red-shift in the frequencies of the anhydrous systems when calculated using FFSiOH with respect to DFT (an average difference of 58.1 cm−1 for (SiO2)4, 34.5 cm−1 for (SiO2)8 and 28.9 cm−1 for (SiO2)16). When added together, these frequency differences cause an increase in the respective ZPE differences. In comparison, the nanoclusters with 25% hydroxylation have an average ZPE difference of 0.84 kJ/mol per SiO2 unit, while for 50% hydroxylation the average ZPE difference is 0.4 kJ/mol per SiO2 unit.
The thermal contribution to Uvib from the FFSiOH calculations is in very good agreement with DFT values (see Figure 3b). The FFSiOH-DFT difference averaged over all structures is 0.09 kJ/mol per SiO2 unit. The Uvib is dominated by the low frequencies, due to the negative exponential. The Uvib contribution is also smaller (from 13% to 24% the value of ZPE). The differences here do not seem to obey any relationship with either size or degree of hydroxylation. The contributions to the internal energy from translation and rotation are independent of the properties of the molecule, and therefore have the same value. Since the ZPE is five times bigger than the Uvib thermal correction, and Uvib is very close to DFT values, the total correction to U follows the same trends as the ZPE, and the differences among DFT and FFSiOH values are almost the same as for ZPE.
The T·Svib term calculated using FFSiOH, for most of the systems is, again, in very good agreement with DFT-calculated values (see Figure 3c), with an average absolute FFSiOH-DFT difference of 0.46 kJ/mol per SiO2 unit. As for the thermal contribution to Uvib, the Svib term is dominated by low frequency modes. Here, however, the largest discrepancy between DFT and FFSiOH data is for the (SiO2)8·(H2O)4 structure, where the DFT-calculated entropy is 1.60 kJ/mol SiO2 per unit higher than that calculated using FFSiOH. The average absolute FFSiOH-DFT difference of the T·Svib term (without taking into account the (SiO2)8·(H2O)4 species) is 0.32 kJ/mol per SiO2 unit.
The average absolute differences between FFSiOH results and DFT for the rotational and translational entropies are 0.05 kJ/mol per SiO2 unit and 0.03 kJ/mol prer SiO2 unit respectively. These results are not surprising, since Srot and Strans depend only on the optimized geometric structure of the respective nanocluster, which is very similar when calculated with DFT and FFSiOH (see Reference [23]).
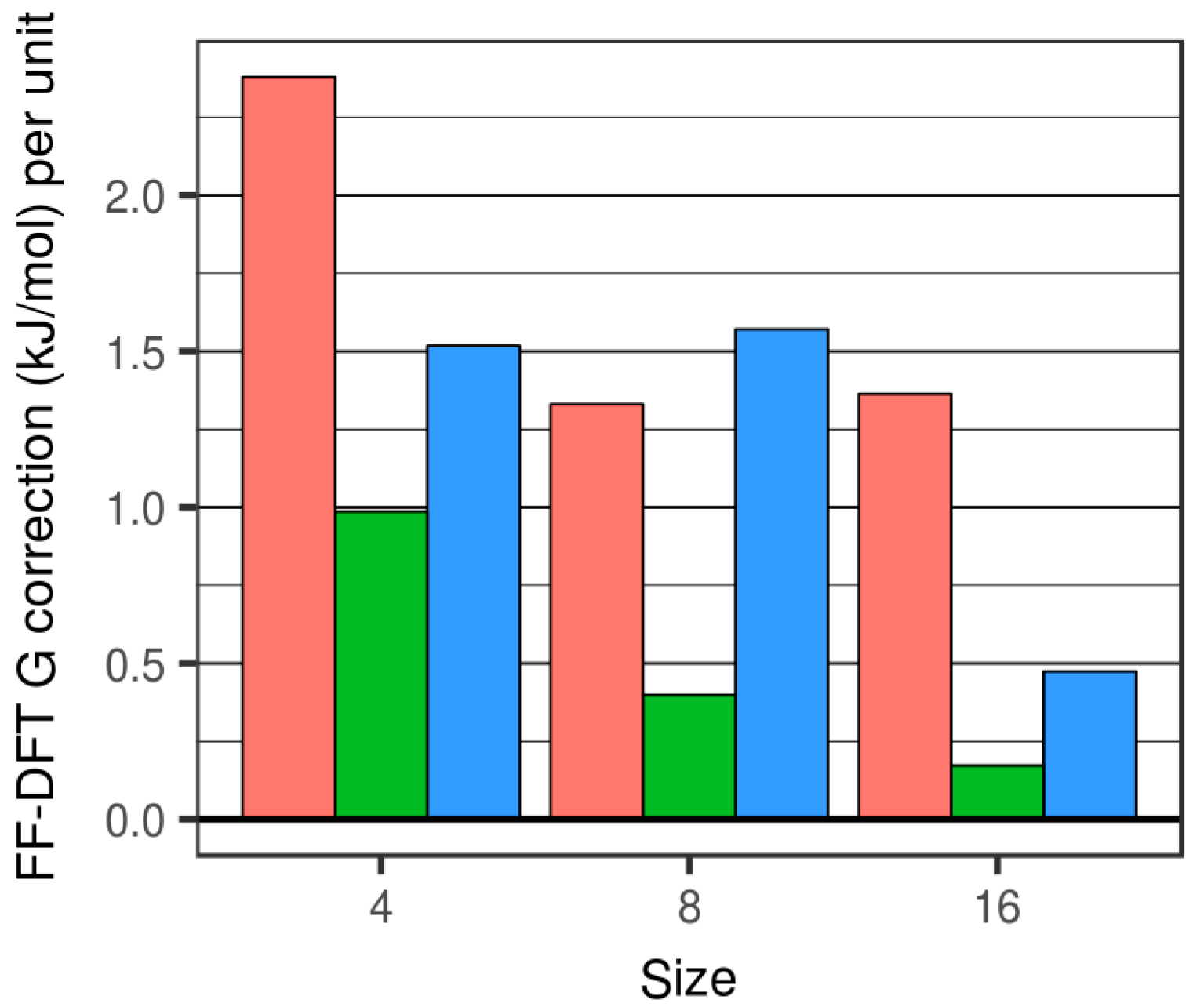
The differences between free energies calculated using DFT and FFSiOH fall in the range 0.25 kJ/mol to 2.68 kJ/mol per SiO2 unit (see Figure 4). Generally, as size increases, all the FFSiOH-DFT differences in the predicted G correction become smaller, but to a different extent with respect to the degree of hydroxylation. The percentage difference between FFSiOH and DFT results for the G correction are generally found to be <15%. We note that for the smallest cluster considered, the percentage difference is found to be relatively large due to very small DFT-calculated values of G (with respect to the fairly constant FFSiOH-DFT difference). The G correction of the highly hydroxylated (SiO2)8·(H2O)4 particle is found to be relatively large, due to the overestimation of the Svib contribution (see above). Apart from in this case, the G correction is more accurately calculated for hydroxylated systems than for anhydrous systems. However, we find that nanoclusters with lower (25%) hydroxylation are those for which FFSiOH provides the best match with the DFT-calculated G corrections. This result is in line with the capability of FFSiOH to better estimate relative Uelec values (with respect to DFT values) for this degree of hydroxylation [23].
3.2. Free Energy in a Set of Isomers
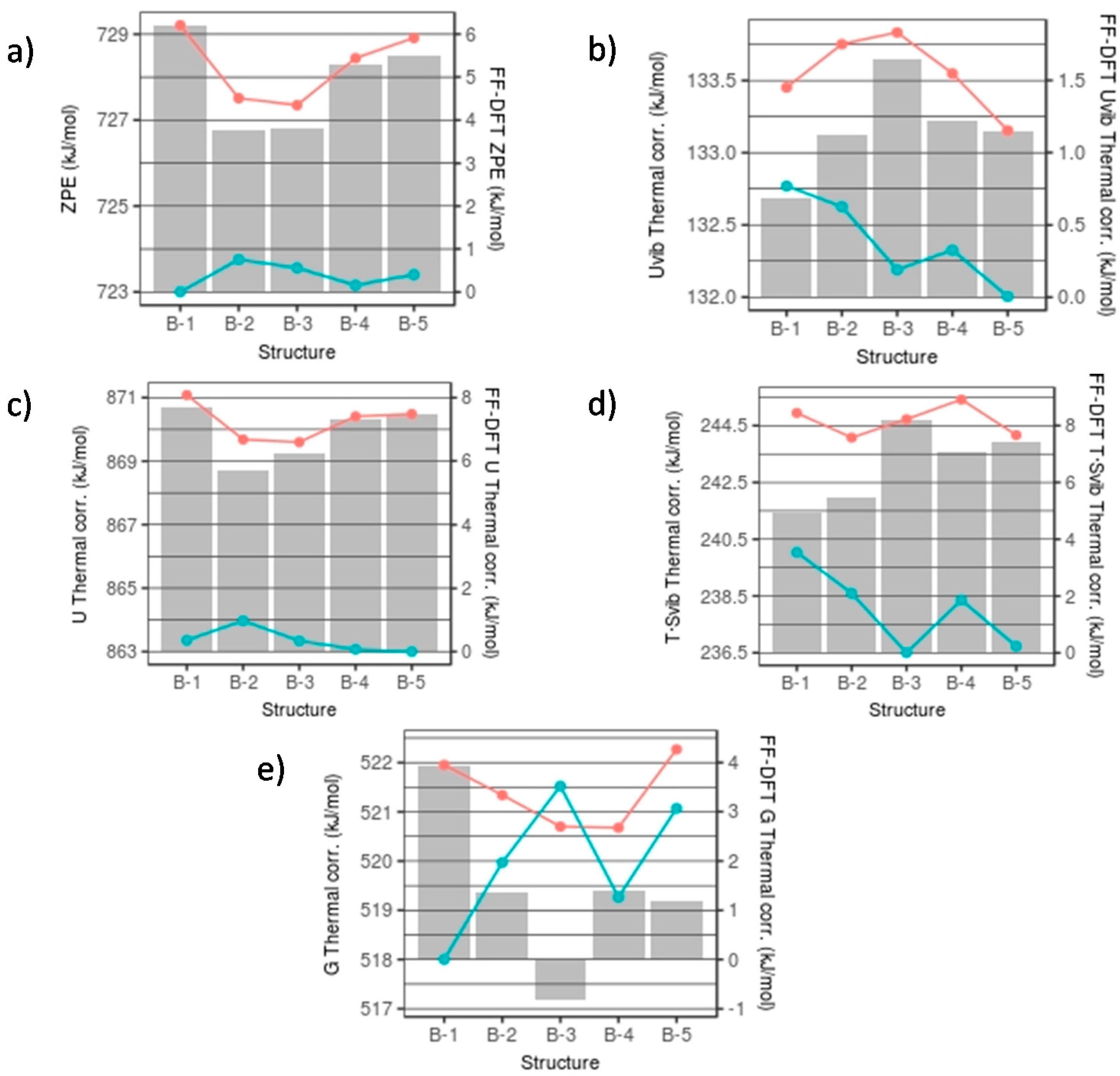
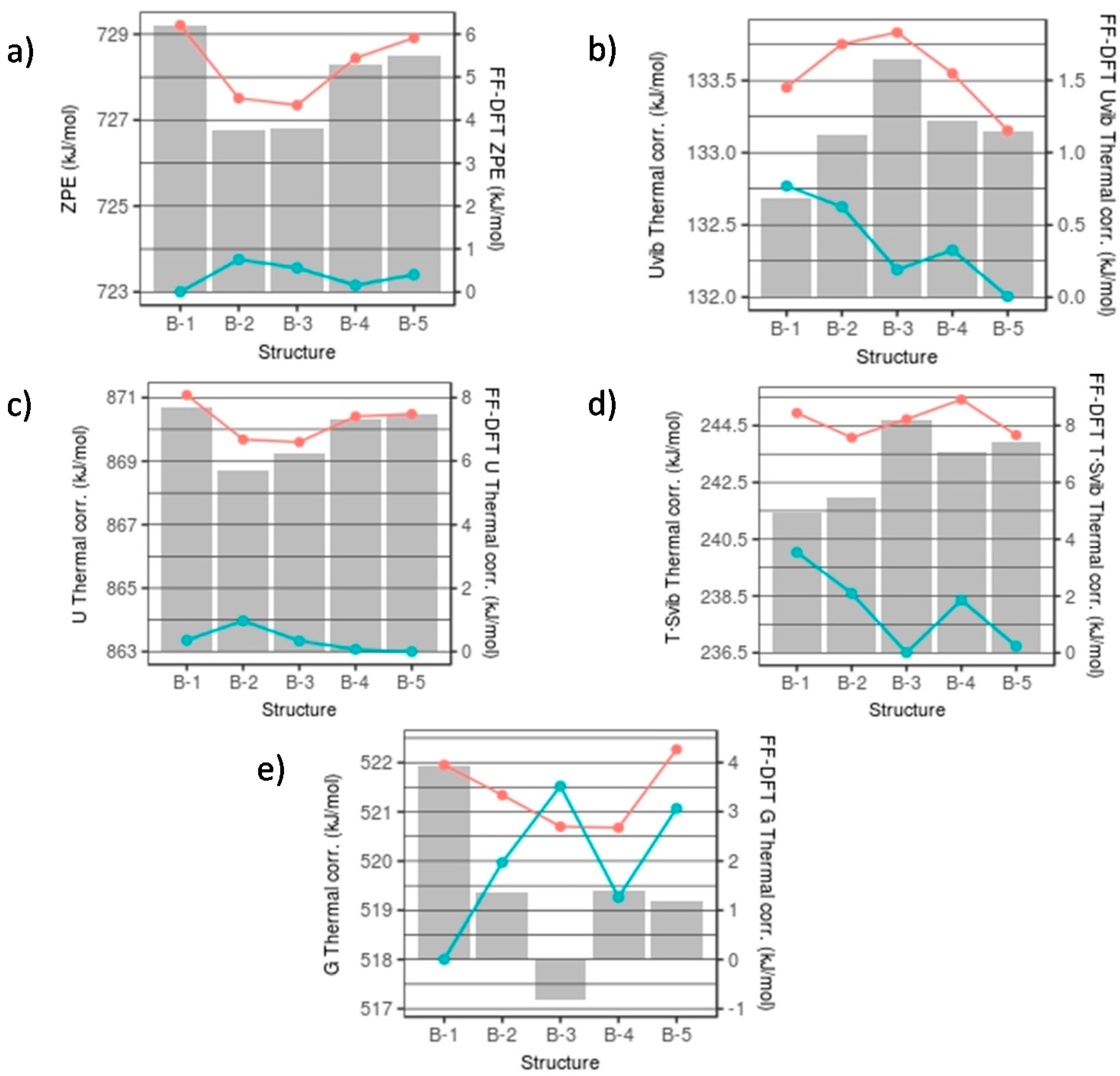
The FFSiOH-DFT differences in calculated thermodynamic properties among the (SiO2)16·(H2O)4 nanocluster isomers in set B are very low, as can be seen in Figure 5 (note that energies are given in kJ/mol). The largest difference is found for the T·Svib correction, with a value of 3.5 kJ/mol. This is due to the fact that, although different in structure, throughout this set the number of SiO4 tetrahedra is invariant and the hydroxyl groups are in very similar environments for each isomer. The vibration frequencies are therefore quite similar (standard deviation of 49 cm−1), and the vibrational partition functions are roughly the same, implying very similar thermodynamic properties. The differences between FFSiOH and DFT for the ZPE are between 3.7–6.2 kJ/mol, with an average of 4.9 kJ/mol (see Figure 5a). For the Uvib thermal contribution, the differences generally are of the order 1 kJ/mol (see Figure 5b). The total U thermal correction, as it is the sum of the ZPE and the Uvib thermal contribution, results in an increased difference between DFT and FFSiOH (see Figure 5c). The T·Svib values show the largest variation in the differences between DFT and FFSiOH over the set of isomers, which can be as high as 3.3 kJ/mol (see Figure 5d). Although all FFSiOH-DFT differences for all these individual thermodynamic components are fairly constant and positive, the size of the FFSIOH-DFT differences of the total G corrections is largely offset by the T·Svib term in the equation for G (see above and Figure 5e). Overall, the FFSiOH-DFT differences in free energies for this set are very small (<4 kJ/mol), and vary in a manner that is independent of the pattern of FFSiOH-DFT electronic energy differences (Uelec—see Figure 2). In terms of stability of nanoclusters, this means that only if the energy difference among isomers of this size would be of the order 4 kJ/mol in Uelec, the free energy ordering of structures obtained using FFSiOH could be different from the one obtained from DFT calculations. Considering that the Uelec energy differences between the low energy isomers in set B are all >13 kJ/mol, the error in the FFSiOH-predicted free energies will not alter the stability ordering.
3.3. Free Energy of Hydroxylation
The progressive free energies of hydroxylation of all nanoclusters (considered as calculated using both FFSiOH and DFT) are shown in Table 1. Specifically, for each cluster size we consider two hydroxylation processes: firstly, from the respective anhydrous (SiO2)n nanocluster to a (SiO2)n·(H2O)m nanocluster with low hydroxylation (i.e., R = 0.25); and secondly, we then consider the hydroxylation of this latter nanocluster to a (SiO2)n·(H2O)m nanocluster with a high degree of hydroxylation (i.e., R = 0.5). These processes correspond to the evolution of the clusters going from left to right in rows 2–4 in Figure 1. Note that for nanoclusters sizes n = 8, 16 these hydroxylation energies relate to dissociative chemisorption of more than one water molecule, and hydroxylation free energies are given per water molecule to allow comparison. For hydroxylation of the smallest n = 4 anhydrous nanocluster, the free energies relate to the dissociative chemisorption of a single water molecule at each step. The DFT-calculated free energy differences are −323.0 kJ/mol for the initial hydroxylation step and a further −461.8 kJ/mol for the second hydroxylation reaction. The FFSiOH-calculated values differ by 5.5 kJ/mol and −2.1 kJ/mol, correspondingly, with respect to these DFT values. For the n = 8 anhydrous cluster the initial reaction with water to a low hydroxylation state has free energy per water molecule between that of the two reactions considered for the n = 4 case (i.e., −392.7 kJ/mol from the DFT calculations). For further hydroxylation, the free energy per water molecule decreases to −244.3 kJ/mol, according to the DFT calculations. For these reactions, the FFSiOH calculations reproduce these values to within 6 kJ/mol. For the largest n = 16 cluster considered, the initial anhydrous-low hydroxylation free energy per water molecule has a magnitude well in line with those found for smaller cluster sizes (i.e., −343.2 kJ/mol from the DFT calculations). However, for the low-to-high hydroxylation process, the free energy of hydroxylation drops to only −47.9 kJ/mol per water molecule. From the study in Reference [27] this sharp reduction in hydroxylation energy per molecule is related to the reduced energetic advantage of hydroxylation of finite clusters with increasing hydroxylation above a certain threshold. The respect free energies of these processes as calculated by FFSiOH is found to be within 5 kJ/mol of the DFT-calculated free energies. In all cases the hydroxylation free energy values calculated using FFSiOH are well within the range of typical expected accuracy of DFT calculations with respect to experiment (i.e., 5–15 kJ/mol). This confirms that hydroxylation free energies can be accurately approximated using FFSiOH.
4. Conclusions
We have benchmarked the ability of FFSiOH to reproduce thermodynamic quantities of silica nanoclusters calculated using DFT, with respect to size, degree of hydroxylation, isomer structure, and hydroxylation reactions. In terms of size and degree of hydroxylation (nanocluster set A, Figure 1), the results show that the factor which most affects the thermodynamic properties is the degree of hydroxylation. Since the FFSiOH forcefield was parametrized to reproduce properties of hydroxylated silica surfaces, it is not surprising that it performs slightly worse for the anhydrous nanoclusters, where the presence of ring-strain and defects negatively affect its ability to accurately calculate vibrations. The results also show that FFSiOH performs best for nanoclusters with 25% hydroxylation, which is also the expected average degree of hydroxylation of amorphous silica materials exposed to moisture in the air. This is important, as some of these nanoclusters can be adopted to simulate the hydroxylated silica surfaces either in their crystalline or amorphous state. In terms of energy per unit, the free energy differences between DFT and FFSiOH results are found to reduce with increasing size. These results imply that FFSiOH would be well suited for evaluating free energies of relatively large silica nanoparticles with a moderate degree of hydroxylation. In the B set of low energy (SiO2)16·(H2O)4 isomers (see Figure 2), we show that the FFSiOH-DFT differences in free energies between isomers is, in all cases, lower than 4 kJ/mol. This small error (<0.25 kJ/mol per SiO2) will not thus lead to FFSiOH free energy corrections that could change in Uelec stability orderings of isomers (as calculated using DFT), where the typical Uelec difference between isomers is >0.8 kJ/mol per SiO2. For reactions, we find that the free energy of progressive hydroxylation (ΔGreact) of the considered silica nanoclusters predicted using FFSiOH is also in very good agreement with the DFT data.
In view of the obtained results, we believe that FFSiOH could be generally employed to obtain thermodynamic corrections to Uelec values of hydroxylated nanosilica systems (calculated by DFT or other means) with an accuracy similar to that obtained with DFT methods (at a fraction of the computational cost of the latter). Due to the good performance of FFSiOH for bulk and surface systems [22,23] we also expect our specific methodology to be of use for a wide range of extended hydroxylated silica systems. Overall, given a suitably accurate forcefield, the general approach outlined herein for hydroxylated nanosilica could be promising for a number of other inorganic nanosystems.
Acknowledgments
Support from Spanish MINECO/FEDER grant CTQ2015-64618-R grant and, in part, by Generalitat de Catalunya grants 2014SGR97, XRQTC is acknowledged. We also acknowledge the NOMAD Center of Excellence project (this project has received funding from the European Union’s Horizon 2020 research and innovation programme under grant agreement No. 676580). Access to supercomputer resources was provided through grants from the Red Española de Supercomputación. We also thank Alberto Figueroba for assistance in obtaining the point group symmetries of some of the nanoclusters.
Author Contributions
Antoni Macià Escatllar performed all calculations and prepared the original version of the manuscript. Piero Ugliengo discussed the topic of the work and results and helped correct the manuscript. Stefan T. Bromley supervised the work and finalized the manuscript.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Kollman, P. Free energy calculations: Applications to chemical and biochemical phenomena. Chem. Rev. 1993, 93, 2395–2417. [Google Scholar] [CrossRef]
- Van Gunsteren, W.F.; Dolenc, J. Thirty-five years of biomolecular simulation: Development of methodology, force fields and software. Mol. Simul. 2012, 38, 1271–2181. [Google Scholar] [CrossRef]
- Oostenbrink, C.; Villa, A.; Mark, A.E.; van Gunsteren, W.F. A biomolecular force field based on the free enthalpy of hydration and solvation: The GROMOS force-field parameter sets 53A5 and 53A6. J. Comput. Chem. 2004, 25, 1656–1676. [Google Scholar] [CrossRef] [PubMed]
- Shivakumar, D.; Williams, J.; Wu, Y.; Damm, W.; Shelley, J.; Sherman, W. Prediction of absolute solvation free energies using molecular dynamics free energy perturbation and the OPLS force field. J. Chem. Theory Comput. 2010, 6, 1509–1519. [Google Scholar] [CrossRef] [PubMed]
- Becker, C.; Tavazza, F.; Trautt, Z.T.; de Macedo, R.A.B. Considerations for choosing and using force fields and interatomic potentials in materials science and engineering. Curr. Opin. Solid State Mater. Sci. 2013, 17, 277–283. [Google Scholar] [CrossRef]
- Goumans, T.P.M.; Bromley, S.T. Efficient nucleation of stardust silicates via heteromolecular homogeneous condensation. Mon. Not. R. Astron. Soc. 2012, 420, 3344–3349. [Google Scholar] [CrossRef]
- Bromley, S.T.; Martin, J.C.G.; Plane, J.M.C. Under what conditions does (SiO)N nucleation occur? A bottom-up kinetic modelling evaluation. Phys. Chem. Chem. Phys. 2016, 18, 26913–26922. [Google Scholar] [CrossRef] [PubMed]
- Zachariah, M.R.; Tsang, W. Application of ab initio molecular orbital and reaction rate theories to nucleation kinetics. Aerosol Sci. Tech. 1993, 19, 499–513. [Google Scholar] [CrossRef]
- Köhler, T.M.; Gail, H.-P.; Sedlmayr, E. MgO dust nucleation in M-Stars: Calculation of cluster properties and nucleation rates. Astron. Astrophys. 1997, 320, 553–567. [Google Scholar]
- Bhattacharya, S.; Levchenko, S.V.; Ghiringhelli, L.M.; Scheffler, M. Efficient ab initio schemes for finding thermodynamically stable and metastable atomic structures: Benchmark of cascade genetic algorithms. New J. Phys. 2014, 16. [Google Scholar] [CrossRef]
- Bhattacharya, S.; Levchenko, S.V.; Ghiringhelli, L.M.; Scheffler, M. Stability and Metastability of clusters in a Reactive Atmosphere: Theoretical Evidence for Unexpected Stoichiometries of MgMOx. Phys. Rev. Lett. 2013, 111. [Google Scholar] [CrossRef] [PubMed]
- Lepeshkin, S.; Baturin, V.; Tikhonov, E.; Matsko, N.; Uspenskii, Y.; Naumova, A.; Feya, O.; Schoonene, M.A.; Oganov, A.R. Super-oxidation of silicon nanoclusters: Magnetism and reactive oxygen species at the surface. Nanoscale 2016, 8, 18616–18620. [Google Scholar] [CrossRef] [PubMed]
- Mora-Fonz, M.J.; Catlow, C.R.A.; Lewis, D.W. Oligomerization and cyclization processes in the nucleation of microporous silicas. Angew. Chem. Int. Ed. 2005, 44, 3082–3086. [Google Scholar] [CrossRef] [PubMed]
- Trinh, T.T.; Jansen, A.P.J.; van Santen, R.A. Mechanism of oligomerization reactions of silica. J. Phys. Chem. B 2006, 110, 23099–23106. [Google Scholar] [CrossRef] [PubMed]
- Schaffer, C.L.; Thomson, K.T. Density functional theory investigation into structure and reactivity of prenucleation silica species. J. Phys. Chem. C 2008, 112, 12653–12662. [Google Scholar] [CrossRef]
- Jelfs, K.; Flikkema, E.; Bromley, S.T. Evidence for atomic mixing via multiple intermediates during the dynamic interconversion of silicate oligomers in solution. Chem. Commun. 2012, 48. [Google Scholar] [CrossRef] [PubMed]
- Flikkema, E.; Bromley, S.T. A new interatomic potential for nanoscale silica. Chem. Phys. Lett. 2003, 378, 622–629. [Google Scholar] [CrossRef]
- Aguado, A.; Madden, P.A. Fully transferable interatomic potentials for large-scale computer simulations of simple metal oxides: Application to MgO. Phys. Rev. B 2004, 70. [Google Scholar] [CrossRef]
- Raiteri, P.; Demichelis, R.; Gale, J.D. Thermodynamically consistent force field for molecular dynamics simulations of alkaline-earth carbonates and their aqueous speciation. J. Phys. Chem. C 2015, 119, 24447–24458. [Google Scholar] [CrossRef]
- Raiteri, P.; Gale, J.D.; Quigley, D.; Rodger, P.M. Derivation of an accurate force-field for simulating the growth of calcium carbonate from aqueous solution: A new model for the calcite-water interface. J. Phys. Chem. C 2010, 114, 5997–6010. [Google Scholar] [CrossRef]
- Demichelis, R.; Raiteri, P.; Gale, J.D.; Quigley, D.; Gebauer, D. Stable prenucleation mineral clusters are liquid-like ionic polymers. Nat. Commun. 2011, 2, 590. [Google Scholar] [CrossRef] [PubMed]
- Pedone, A.; Malavasi, G.; Menziani, M.C.; Segre, U.; Musso, F.; Corno, M.; Civalleri, B.; Ugliengo, P. FFSiOH: A new force field for silica polymorphs and their hydroxylated surfaces based on periodic B3LYP calculations. Chem. Mater. 2008, 20, 2522–2531. [Google Scholar] [CrossRef]
- Macia, A.; Ugliengo, P.; Bromley, S.T. Modeling hydroxylated nanosilica: Testing the performance of ReaxFF and FFSiOH forcefields. J. Chem. Phys. 2017, 146. [Google Scholar] [CrossRef]
- Iler, R.K. The Chemistry of Silica: Solubility, Polymerization, Colloid and Surface Properties; Wiley: New York, NY, USA, 1979. [Google Scholar]
- Dove, P.M.; Rimstidt, J.D. Silica: Physical Behaviour, Geochemistry and Materials Applications; Heany, P.J., Pewitt, C.T., Gibbs, G.V., Eds.; Mineralogical Society of America: Washington, DC, USA, 1994; Volume 29, pp. 259–301. [Google Scholar]
- McQuarrie, D.A.; Simons, J.D. Molecular Thermodynamics; University Science Book: Sausalito, CA, USA, 1999. [Google Scholar]
- Jelfs, K.E.; Flikkema, E.; Bromley, S.T. Hydroxylation of silica nanoclusters (SiO2)M(H2O)N, M = 4, 8, 16, 24: Stability and structural trends. Phys. Chem. Chem. Phys. 2013, 15, 20438–20443. [Google Scholar] [CrossRef] [PubMed]
- Flikkema, E.; Jelfs, K.E.; Bromley, S.T. Structure and energetics of hydroxylated silica clusters (SiO2)M(H2O)N M = 8, 16 and N = 1–4: A global optimisation study. Chem. Phys. Lett. 2012, 554, 117–122. [Google Scholar] [CrossRef]
- Flikkema, E.; Bromley, S.T. Dedicated global optimization search for ground state silica nanoclusters: (SiO2)M (N = 6–12). J. Phys. Chem. B 2004, 108, 9638–9645. [Google Scholar] [CrossRef]
- Bromley, S.T.; Flikkema, E. Columnar-to-disk structural transition in nanoscale (SiO2)N clusters. Phys. Rev. Lett. 2005, 95, 185505. [Google Scholar] [CrossRef] [PubMed]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 09; Revision D.01; Gaussian Inc.: Wallingford, CT, USA, 2013. [Google Scholar]
- Stephens, P.J.; Devlin, F.J.; Chabalowski, C.F.; Frisch, M.J. Ab initio calculation of vibrational absorption and circular dichroism spectra using density functional force fields. J. Phys. Chem. 1994, 98, 11623–11627. [Google Scholar] [CrossRef]
- Ugliengo, P.; Sodupe, M.; Musso, F.; Bush, I.J.; Orlando, R.; Dovesi, R. Realistic Models of Hydroxylated Amorphous Silica Surfaces and MCM-41 Mesoporous Material Simulated by Large-scale Periodic B3LYP calculations. Adv. Mater. 2008, 20, 1–5. [Google Scholar] [CrossRef]
- Schlegel, H.B. Optimization of Equilibrium Geometries and Transition Structures. J. Comput. Chem. 1982, 3, 214–218. [Google Scholar] [CrossRef]
- Gale, J.D.; Rohl, A.L. The General Utility Lattice Program (GULP). Mol. Simul. 2003, 29, 291–341. [Google Scholar] [CrossRef]
Figure 1.
Set A: structures of global minima (SiO2)n·(H2O)m nanoclusters with sizes (n = 4, 8, and 16) and degrees of hydroxylation (R = 0.0, 0.25 and 0.5)—taken from References [27,28,29,30].
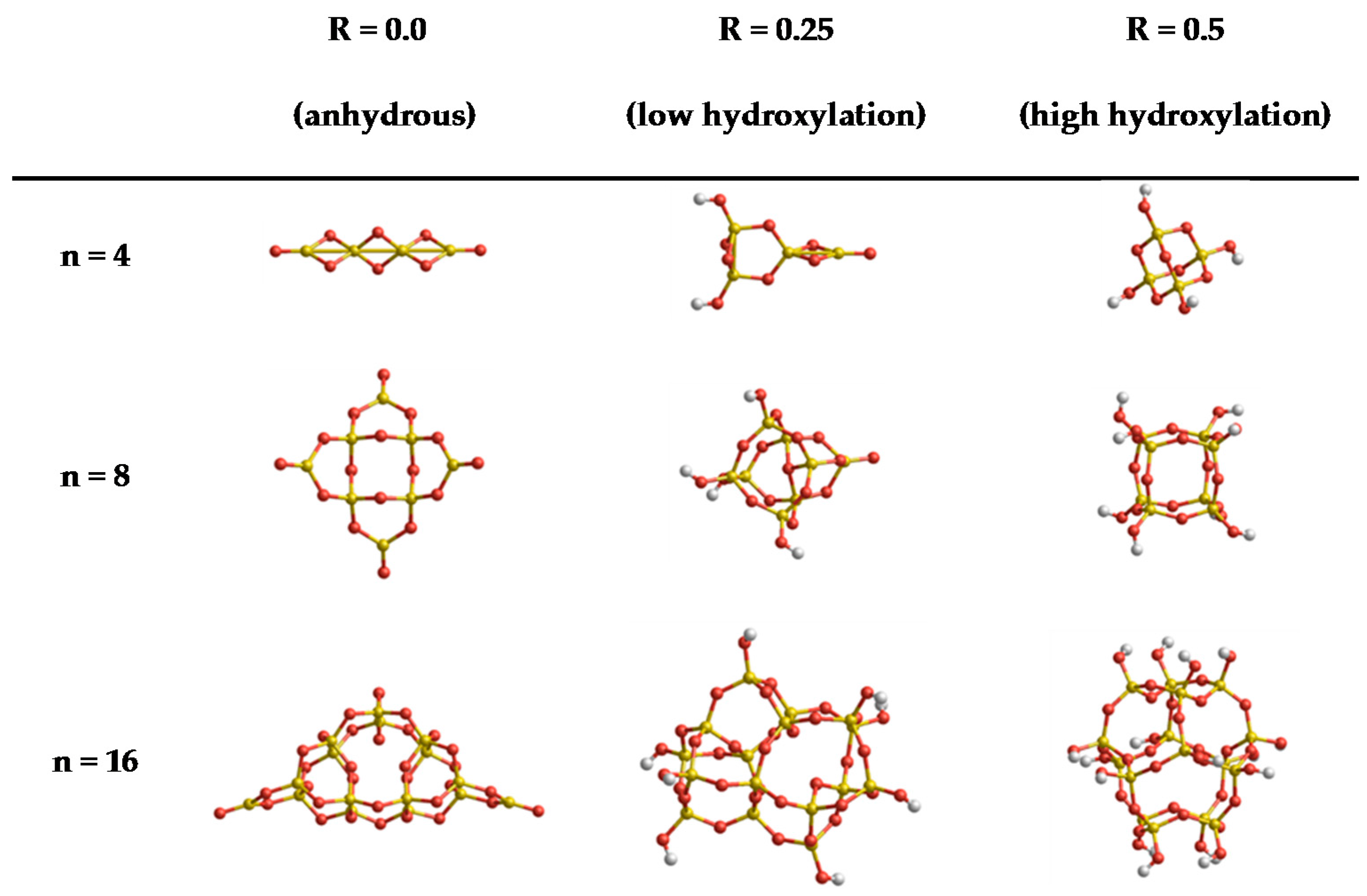
Figure 2.
Set B: low energy (SiO2)16·(H2O)4 nanoclusters. Relative Uelec values calculated using DFT in kJ/mol: B1 (global minimum)—0.0 kJ/mol, B2—13.5 kJ/mol, B3—38.6 kJ/mol, B4—48.2 kJ/mol, B5—95.5 kJ/mol. Structures were taken from the Hydroxylated Nanosilica Dataset in Ref. [23].
Figure 2.
Set B: low energy (SiO2)16·(H2O)4 nanoclusters. Relative Uelec values calculated using DFT in kJ/mol: B1 (global minimum)—0.0 kJ/mol, B2—13.5 kJ/mol, B3—38.6 kJ/mol, B4—48.2 kJ/mol, B5—95.5 kJ/mol. Structures were taken from the Hydroxylated Nanosilica Dataset in Ref. [23].
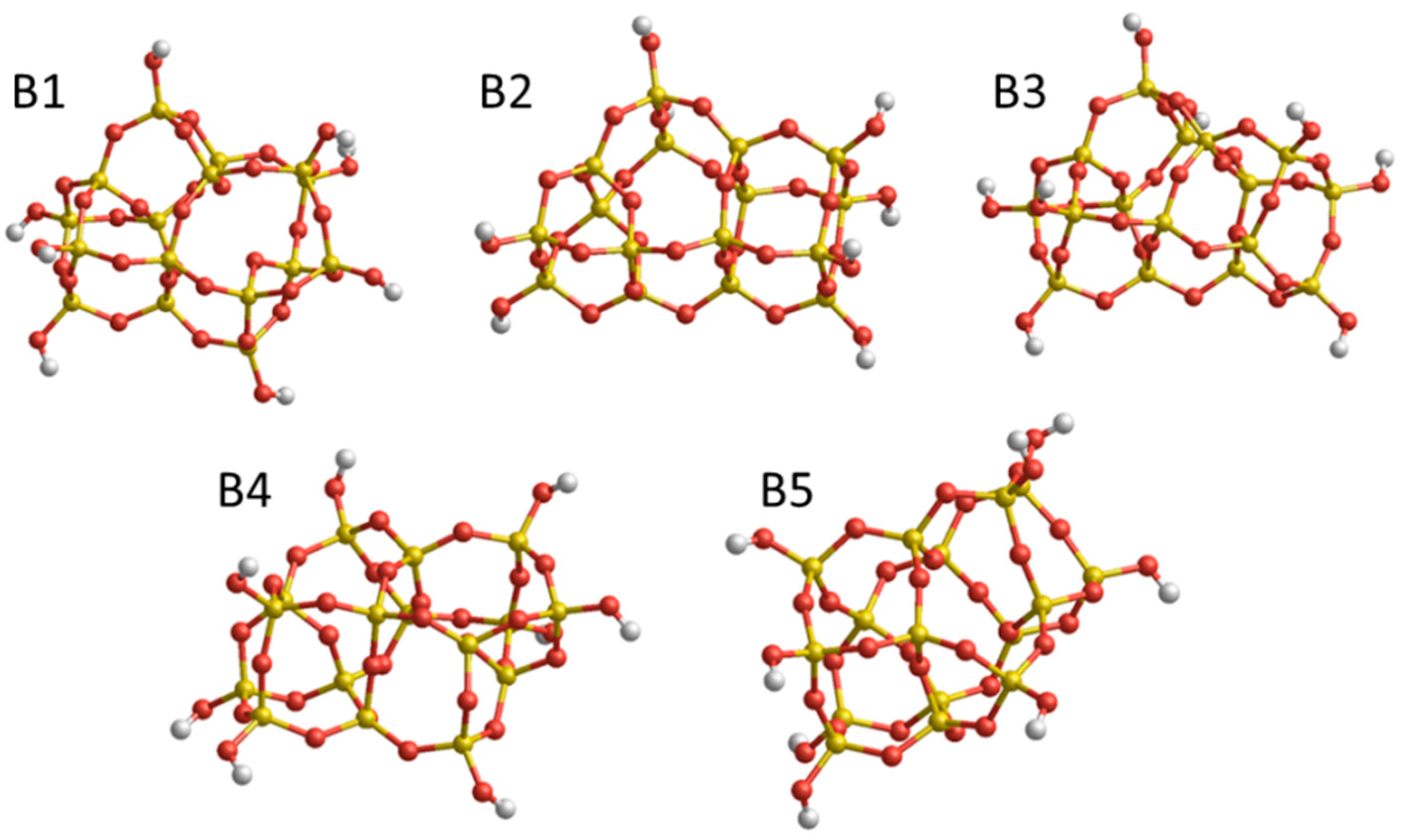
Figure 3.
FFSiOH-DFT differences in: (a) ZPE; (b) Uvib; (c) Svib; and (d) U at T = 298.15K and P = 1 atm. Red, green, and blue bars refer to the degree of hydroxylation, R = 0.0, 0.25 and 0.5, respectively.
Figure 3.
FFSiOH-DFT differences in: (a) ZPE; (b) Uvib; (c) Svib; and (d) U at T = 298.15K and P = 1 atm. Red, green, and blue bars refer to the degree of hydroxylation, R = 0.0, 0.25 and 0.5, respectively.
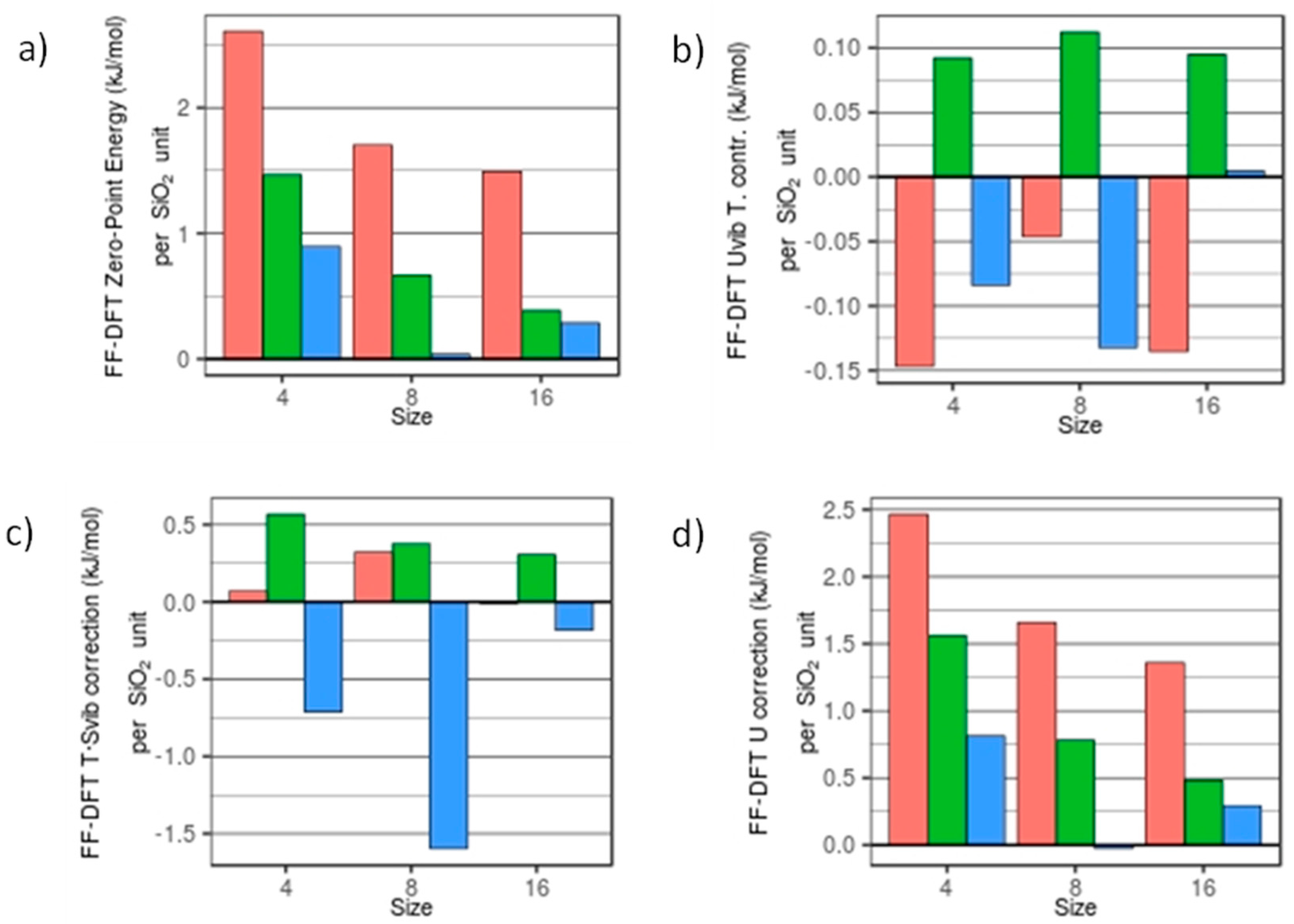
Figure 4.
FFSiOH-DFT differences for the free energy correction, G. Red, green and blue bars refers to the degree of hydroxylation R = 0.0, 0.25 and 0.50, respectively.
Figure 4.
FFSiOH-DFT differences for the free energy correction, G. Red, green and blue bars refers to the degree of hydroxylation R = 0.0, 0.25 and 0.50, respectively.
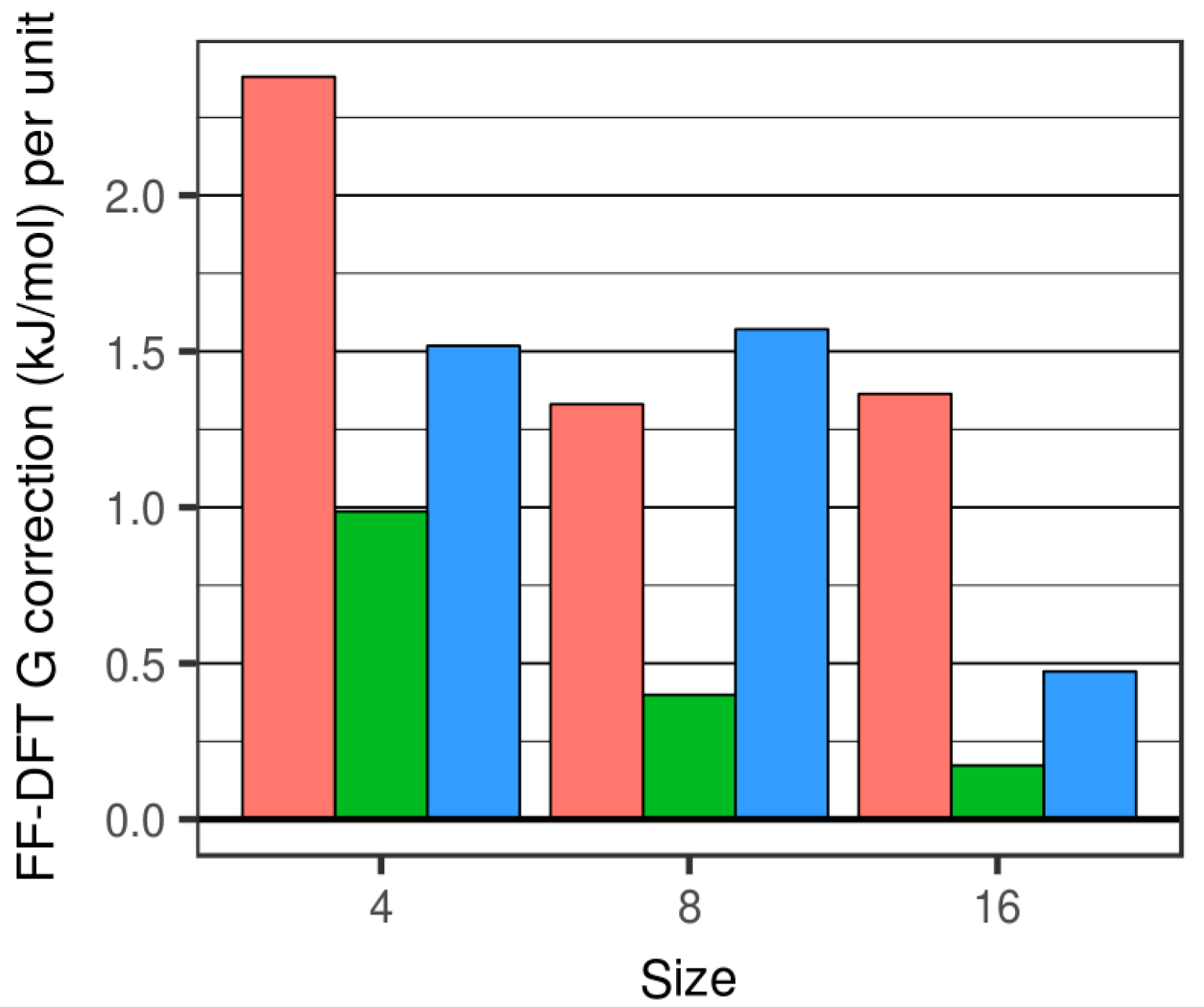
Figure 5.
FFSiOH (red line) and DFT (blue line) thermodynamic quantities and their differences (FF-DFT) for nanocluster set B for: (a) ZPE; (b) Uvib; (c) Svib; (d) U; and (e) G. Lines map the absolute values of each thermodynamic quantity. Bars refer to the FFSiOH-DFT differences.
Figure 5.
FFSiOH (red line) and DFT (blue line) thermodynamic quantities and their differences (FF-DFT) for nanocluster set B for: (a) ZPE; (b) Uvib; (c) Svib; (d) U; and (e) G. Lines map the absolute values of each thermodynamic quantity. Bars refer to the FFSiOH-DFT differences.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Progressive hydroxylation free energies (kJ/mol) of anhydrous nanoclusters calculated using DFT and FFSiOH. Note that all energies are per added water molecule.
Table 1.
Progressive hydroxylation free energies (kJ/mol) of anhydrous nanoclusters calculated using DFT and FFSiOH. Note that all energies are per added water molecule.
| Reaction | ΔGreact (DFT) | ΔGreact (FFSiOH) | ΔΔGreact (DFT-FFSiOH) |
|---|---|---|---|
| Si4O8 + H2O → H2Si4O9 | −323.0 | −328.5 | 5.5 |
| H2Si4O9 + H2O → H4Si4O10 | −461.8 | −459.7 | −2.1 |
| Si8O16 + 2H2O → H4Si8O18 | −392.7 | −396.4 | 3.7 |
| H4Si8O18 + 2H2O → H8Si8O20 | −244.3 | −239.6 | −4.7 |
| Si16O32 + 4H2O → H8Si16O36 | −343.2 | −348.0 | 4.8 |
| H8Si16O36 + 4H2O → H16Si16O40 | −47.9 | −46.6 | −1.3 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Macià Escatllar, A.; Ugliengo, P.; Bromley, S.T. Computing Free Energies of Hydroxylated Silica Nanoclusters: Forcefield versus Density Functional Calculations. Inorganics 2017, 5, 41. https://doi.org/10.3390/inorganics5030041
AMA Style
Macià Escatllar A, Ugliengo P, Bromley ST. Computing Free Energies of Hydroxylated Silica Nanoclusters: Forcefield versus Density Functional Calculations. Inorganics. 2017; 5(3):41. https://doi.org/10.3390/inorganics5030041
Chicago/Turabian StyleMacià Escatllar, Antoni, Piero Ugliengo, and Stefan T. Bromley. 2017. "Computing Free Energies of Hydroxylated Silica Nanoclusters: Forcefield versus Density Functional Calculations" Inorganics 5, no. 3: 41. https://doi.org/10.3390/inorganics5030041
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.