
Hetero- and Homoleptic Magnesium Triazenides


Institut für Anorganische und Analytische Chemie, Johannes Gutenberg-Universität Mainz, Duesbergweg 10-14, 55128 Mainz, Germany
*
Author to whom correspondence should be addressed.
Inorganics 2017, 5(2), 33; https://doi.org/10.3390/inorganics5020033
Submission received: 3 April 2017
/
Revised: 23 April 2017
/
Accepted: 24 April 2017
/
Published: 1 May 2017
(This article belongs to the Special Issue s-Block Metal Complexes)
Abstract
:Using monoanionic triazenide ligands derived from biphenyl and m-terphenyl substituted triazenes Dmp(Tph)N3H (1a), (Me4Ter)2N3H (1b) or Dmp(Mph)N3H (1c) (Dmp = 2,6-Mes2C6H3 with Mes = 2,4,6-Me3C6H2; Me4Ter = 2,6-(3,5-Me2C6H3)2C6H3; Mph = 2-MesC6H4; Tph = 2-TripC6H4 with Trip = 2,4,6-i-Pr3C6H2), several magnesium triazenides were synthesized. Heteroleptic complexes [Mg(N3Ar2)I(OEt2)] (Ar2 = Dmp/Tph (2a), (Me4Ter)2 (2b) were obtained from metalation of the corresponding triazenes with di-n-butylmagnesium followed by reaction with iodine in diethyl ether as the solvent in high yields. Replacing diethyl ether by n-heptane afforded trinuclear compounds [Mg3(N3Ar2)2I4] (3a, 3b) in low yields in which a central MgI2 fragment is coordinated by two iodomagnesium triazenide moieties. Two unsolvated homoleptic magnesium compounds [Mg(N3Ar2)2] (4b, 4c) were obtained from di-n-butylmagnesium and triazenes 1b or 1c in a 1:2 ratio. Depending on the nature of the substituents, the magnesium center either shows the expected tetrahedral or a rather unusual square planar coordination.
1. Introduction
The quest for suitable ligand systems that are able to stabilize unsolvated monomeric metal complexes is one of the most intensely-studied fields of coordination and organometallic chemistry [1]. Exploration of this area is motivated by potential applications of these reactive complexes in catalysis and organic synthesis. Well-known examples of monoanionic chelating N-donor ligands that have been used extensively include the β-diketiminate [2] and amidinate [3] ligand systems. Much less attention has been given to the closely-related triazenides [4]. During the last decade, we reported the preparation of derivatives of diaryl-substituted, sterically-crowded triazenido ligands that are bulky enough to prevent undesirable ligand redistribution reactions [5,6,7,8,9,10,11,12]. These ligands allowed structurally characterizing the first examples of aryl compounds of the heavier alkaline earth metals Ca, Sr and Ba [5] and unsolvated pentafluorophenyl organyls of the divalent lanthanides Yb and Eu [6]. The different degree of metal···π-arene interactions to pending aromatic substituents accounts for the unusual “inverse” aggregation behavior of alkali metal triazenides in their solid-state structures [7]. A series of homologous potassium and thallium triazenides crystallizes in isomorphous cells and represents the first examples of isostructural molecular species reported for these elements [8]. Recently, using the same type of ligands, a spectacular series of pnicogen(I) triazenides for the elements P, As and Sb was published by Schulz et al. [13].
In this paper, we describe the synthesis and characterization of several heteroleptic and homoleptic magnesium triazenides. The latter are the first examples of unsolvated magnesium triazenides, whereas the former are potential precursors for magnesium(I) triazenides. A small number of magnesium triazenides, mainly using less bulky substituents, has been reported before [11,14,15,16]. With relatively small p-tolyl and slightly bigger mesityl substituents, two additional THF molecules are required to provide electronic and steric saturation of the Lewis acetic metal centers in the six-coordinate magnesium complexes [Mg(N3Ar2)2(thf)2] (Ar = p-Tol [14], Mes [15]) published by the groups of Walsh and Westerhausen, respectively. The use of 2,6-di-iso-propylphenyl (Dip) substituted triazenide by Gibson et al. afforded the five-coordinate magnesium etherate [Mg(N3Dip2)2(OEt2)] [16], which was prepared as the aforementioned compounds by metalation of the corresponding triazene with di-n-butylmagnesium. For the Dip derivative, attempts to synthesize a monosubstituted triazenide were not successful. Even in the presence of an excess of di-n-butylmagnesium, the bis-triazenido complex was obtained as a result of ligand redistribution reactions. However, using the dimesityl substituted triazene and the chelating donor 1,2-bis(dimethylamino)ethane (TMEDA), Westerhausen et al. succeeded at isolating a heteroleptic complex of the composition [Mes2N3MgnBu(tmeda)] [15]. Another heteroleptic magnesium triazenide [Dmp(Tph)N3MgI(thf)] was obtained in our group by an alternative synthetic approach via redox transmetallation between the iodomercury triazenide [Dmp(Tph)N3HgI] and magnesium metal [11].
2. Results and Discussion
2.1. Syntheses and Spectroscopic Characterization
The heteroleptic iodomagnesium triazenides 2a and 2b are accessible in diethyl ether as the solvent via metalation of the diaryltriazenes Dmp(Tph)N3H (1a) or (Me4Ter)2N3H (1b) (Dmp = 2,6-Mes2C6H3 with Mes = 2,4,6-Me3C6H2; Me4Ter = 2,6-(3,5-Me2C6H3)2C6H3; Tph = 2-TripC6H4 with Trip = 2,4,6-i-Pr3C6H2) with one equivalent of di-n-butylmagnesium, followed by addition of iodine (Scheme 1a). After crystallization, the complexes [Mg(N3Ar2)I(OEt2)] (Ar2 = Dmp/Tph (2a), (Me4Ter)2 (2b)) are isolated in good yields. Repeating the same reactions in the non-coordinating solvent n-heptane afforded trinuclear donor-free complexes [Mg3(N3Ar2)2I4] (Ar2 = Dmp/Tph (3a), (Me4Ter)2 (3b)) as the least soluble compounds in low isolated yields. Heteroleptic complexes [Mg(N3Ar2)I] (Ia, Ib) (Scheme 1b) are possible intermediates that might rearrange via Schlenk-type equilibria and ligand redistribution reactions to 3a and 3b. Analysis of the better soluble fractions in the mother liquor by NMR experiments showed the presence of other moieties, most probably a mixture of homo- and hetero-leptic compounds. However, it was not possible to separate these main products by crystallization. A more rational synthetic approach to homoleptic magnesium triazenides consists of the reaction of di-n-butylmagnesium with the corresponding triazene in a 1:2 ratio to give [Mg{N3(Me4Ter)2}2] (4b) or [Mg{N3(Dmp)Mph}2] (4c) in good to excellent yields (Scheme 1c). The corresponding homoleptic magnesium triazenide derived from triazene 1a could not be obtained by this route. This is in accordance with earlier observations that homoleptic alkaline earth metal triazenides with the [N3(Dmp)Tph] ligand are accessible for the heavier elements strontium and barium only, due to steric crowding [10].
The pale yellow (3a, 3b, 4b) or deep yellow (2a, 2b, 4c) complexes are moisture-sensitive and, with the exception of 3b, possess good or moderate solubility in aromatic or aliphatic hydrocarbons. They show considerable thermal stability, but decompose, presumably with N2 evolution, at higher temperature. The most thermally-stable compound is the homoleptic complex 4c, which decomposes above 300 °C. The IR spectra show strong νas N3 absorptions in the range of 1255–1282 cm−1, which is indicative of the triazenido groups acting as chelating ligands. In the 1H NMR spectra of 2b and 3a, the expected sets of signals are observed at ambient temperature. However, more complex temperature-dependent spectra are found for 2a, 4b and 4c. For heteroleptic complex 2a at 273 K, five and three well-separated resonances are observed for the methyl groups of the 2,4,6-tri-iso-propylphenyl and 2,4,6-trimethylphenyl substituents, respectively. Warming of the NMR sample results in broadening, coalescence and finally resharpening to three and two resonances at 373 K. This behavior can be explained by hindered rotation around the N–C(aryl) bonds (cf. Figures S1 and S2 in the Supplementary Materials). For the homoleptic complexes 4b and 4c, the high-temperature 1H NMR data indicate free (for 4c at 373 K) or almost free (for 4b at 338 K) rotation around the N–C(aryl) bonds since some broadening of the resonances is still observed (cf. Figures S3b and S4 in the Supplementary Materials). For 4b, an interesting feature in the 1H NMR spectrum at ambient temperature is a low-field shifted resonance at 7.64 ppm that moves to higher field at elevated temperatures. It has been noted before [7] that the presence of low-field shifted signals in biphenyl-substituted triazenes indicates short intermolecular C–H···N contacts at the NNN backbone of the ligands and therefore is a very sensitive probe for conformational preferences in solution. In the case of 4b, a C–H···N interaction of 2.48 Å between the central nitrogen atom N2 and a hydrogen atom of the ortho-C6H3Me2 ring in the solid-state structure correlates with the observed low-field resonance in solution.
2.2. Structural Studies
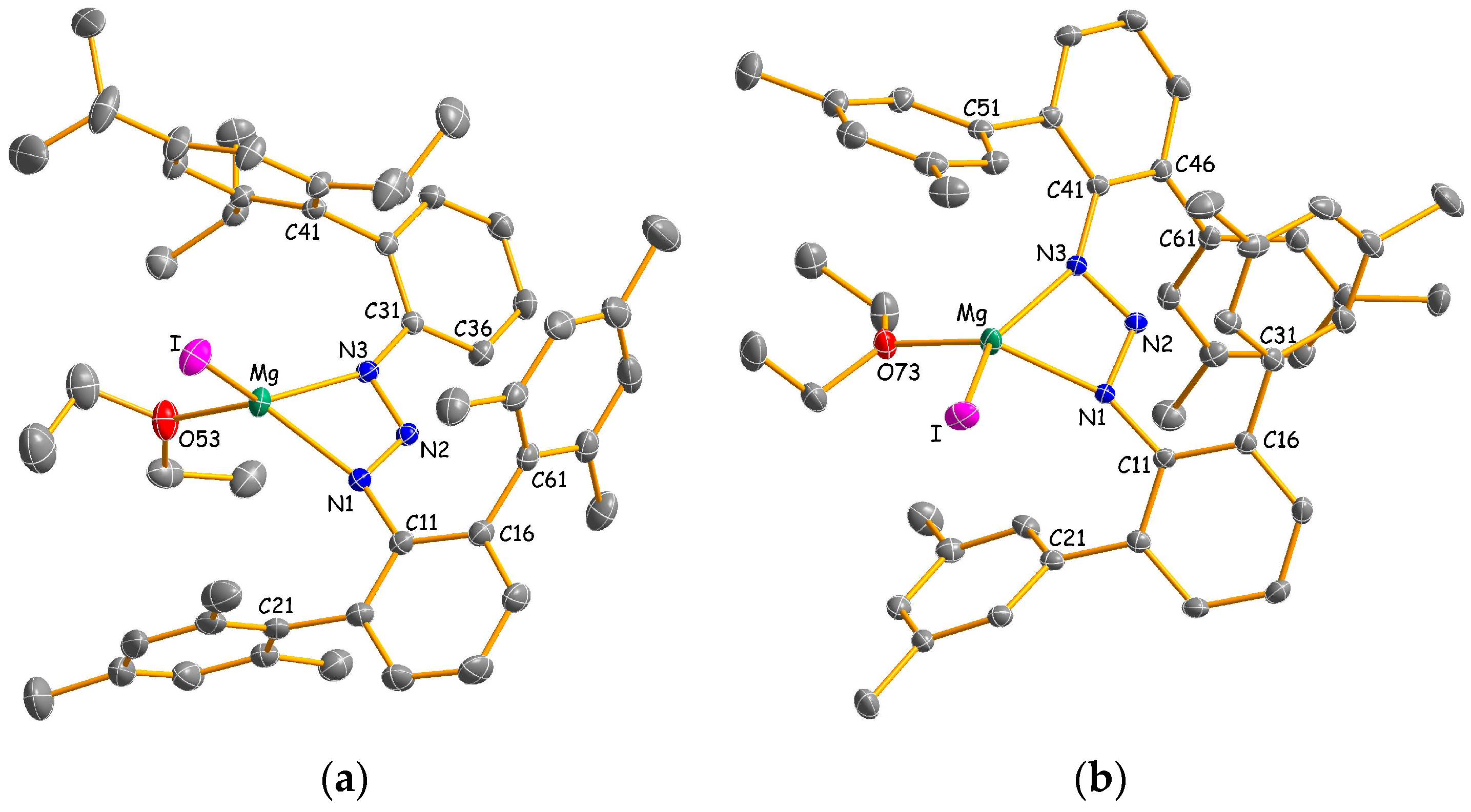
All compounds were examined by X-ray crystallography, and their molecular structures and selected bond parameters are shown in Figure 1, Figure 2 and Figure 3. In the heteroleptic iodomagnesium triazenides 2a and 2b, the magnesium atoms possess a very distorted tetrahedral coordination by two nitrogen atoms N1 and N3 of a η2-bonded triazenide ligand, an iodine atom I and the oxygen atom O53 (2a) or O73 (2b) of a diethyl ether molecule (Figure 1). The degree of distortion is reflected by interligand angles in the wide range 61.45(6)°–132.64(5)° (2a) and 61.04(7)°–143.56(7)° (2b), respectively. In an alternative and possibly more appropriate description that assigns only one coordination site, represented by the central nitrogen atom N2, to the small-bite triazenido ligand, the metal atoms show trigonal planar coordination with corresponding angles of 105.39(5)°–128.51(4)° (2a) and 108.36(6)°–125.04(5)° (2b), respectively. The relatively small variation of the N1–N2 and N2–N3 distances (2a: 1.317(2)/1.307(2) Å; 2b: 1.312(2)/1.312(3) Å) is consistent with delocalized bonding. Nonetheless, coordination of the triazenide ligand is slightly asymmetric for 2a with Mg–N bond lengths of 2.1151(16) Å and 2.0880(16) Å. A more symmetric coordination with Mg–N distances of 2.101(2) Å and 2.0958(19) Å is observed for the magnesium atom in 2b. Interestingly, the Mg–N bond length correlates with the conformation of the triazenide ligand. Thus, a coplanar arrangement of the substituted arene rings with respect to the central triazaallyl fragment as reflected by a CCNN torsion angle close to 0° increases the basicity of the bonded nitrogen atom. Therefore, for 2a, the shortest Mg–N distance to the biphenyl substituted nitrogen atom N3 of 2.0880(16) Å corresponds to the smallest torsion angle N2–N3–C31–C36 of 18.0(3)°.
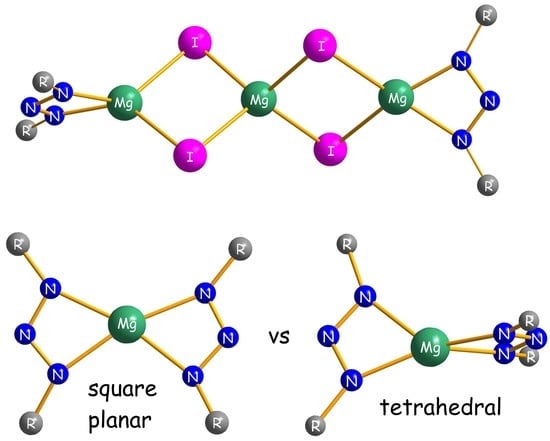
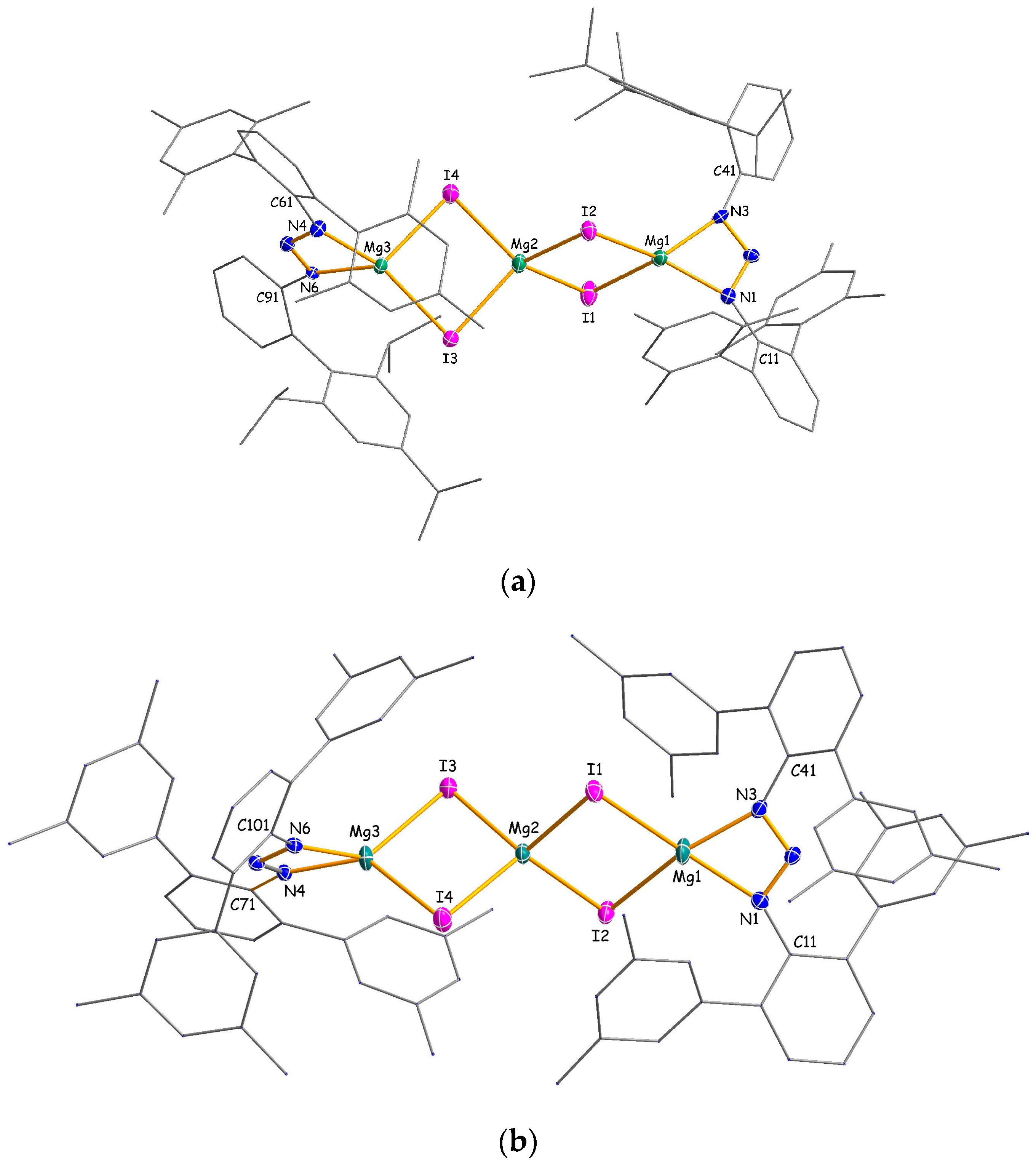
In the rather unusual trinuclear MgI2 addition compounds 3a and 3b, a central four-coordinate magnesium atom Mg2 is bridged by four iodine atoms to two terminal magnesium centers Mg1 and Mg3 (Figure 2). Each of the latter is additionally coordinated via two nitrogen atoms by a chelating triazenido ligand. The three metal atoms form a nearly perfect linear arrangement with an angle of 178.5° for 3a and 179.1° for 3b. Notably, there appear to be no previous reports on molecular compounds that contain such a trinuclear Mg3I42+ or even an MgI42− fragment [17]. However, the terminal [(Ar2N3)MgI2]2− fragments may be compared with related dimeric complexes of the general composition [(L)Mg-μ-I2Mg(L)] where L represents bulky amido, diketiminato, diiminophosphinato or guanidinato ligands [18,19,20,21,22]. In 3a and 3b, the coordination spheres of the central magnesium atoms feature distorted tetrahedral geometries with I–Mg2–I angles in the range 96.24(6)°–120.55(8)° (3a) and 98.09(4)°–116.70(5)° (3b), respectively. As expected, the average Mg2–I distance of 2.741(2) Å (3a) and 2.7209(13) Å (3b) is shorter than the corresponding value of 2.9183(5) Å in the solid state structure of MgI2 [23] that adopts the CdI2 type of structure with hexa-coordinate magnesium atoms. For the gas phase structure of molecular di-coordinate magnesium diiodide, the Mg–I distance was determined by electron diffraction to 2.52 ± 0.03 Å [24]. Moreover, if only one coordination site is assigned to the small-bite angle triazenido ligands, a distorted trigonal planar coordination results for the terminal magnesium atoms as can be judged by the sum of the angles around Mg1 and Mg3 in the range of 358.2°–360.0°. Alternatively, if the triazenido ligands are viewed as bidentate, the resulting four-coordination of Mg1 and Mg3 is somehow intermediate between tetrahedral and square planar geometry. A more precise description of these distortions uses the τ4 parameter [25]:
It is defined as the sum of angles α and β, the two largest angles in the four-coordinate species, subtracted from 360° and all divided by 141°. The values of τ4 will range from zero for a perfect square planar to 1.00 for a perfect tetrahedral geometry. Intermediate structures fall within the range of 0–1.00. By using Equation (1), τ4 parameters of 0.62/0.87/0.64 (0.40/0.91/0.62) are calculated for Mg1/Mg2/Mg3 in complex 3a (3b), respectively. Therefore, a transition from tetrahedral to square planar coordination is evident for Mg1 in 3b. This is also reflected by the interplanar angle of 38.2° between the Mg1/I1/I2 and Mg1/N1/N3 planes.
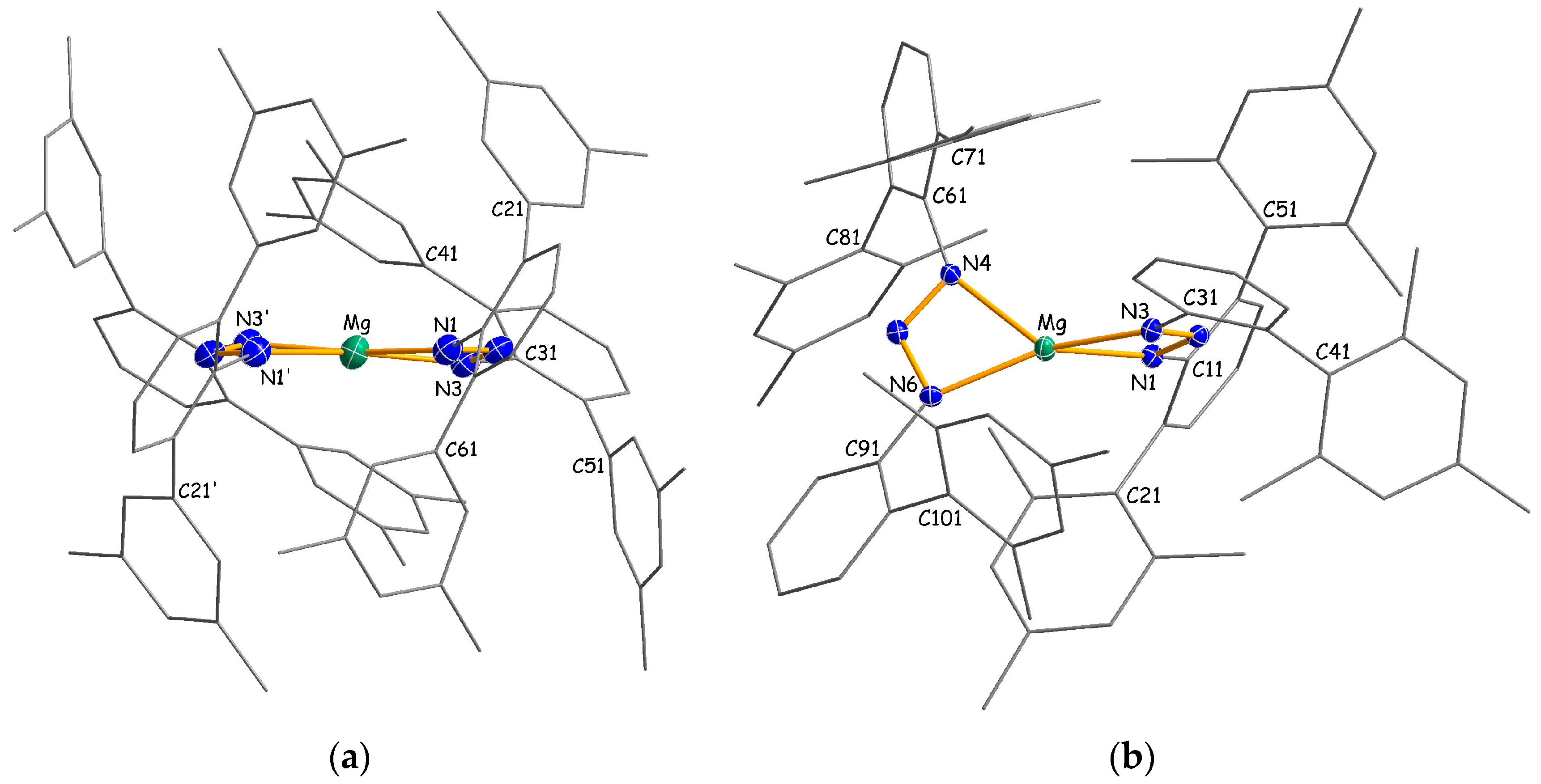
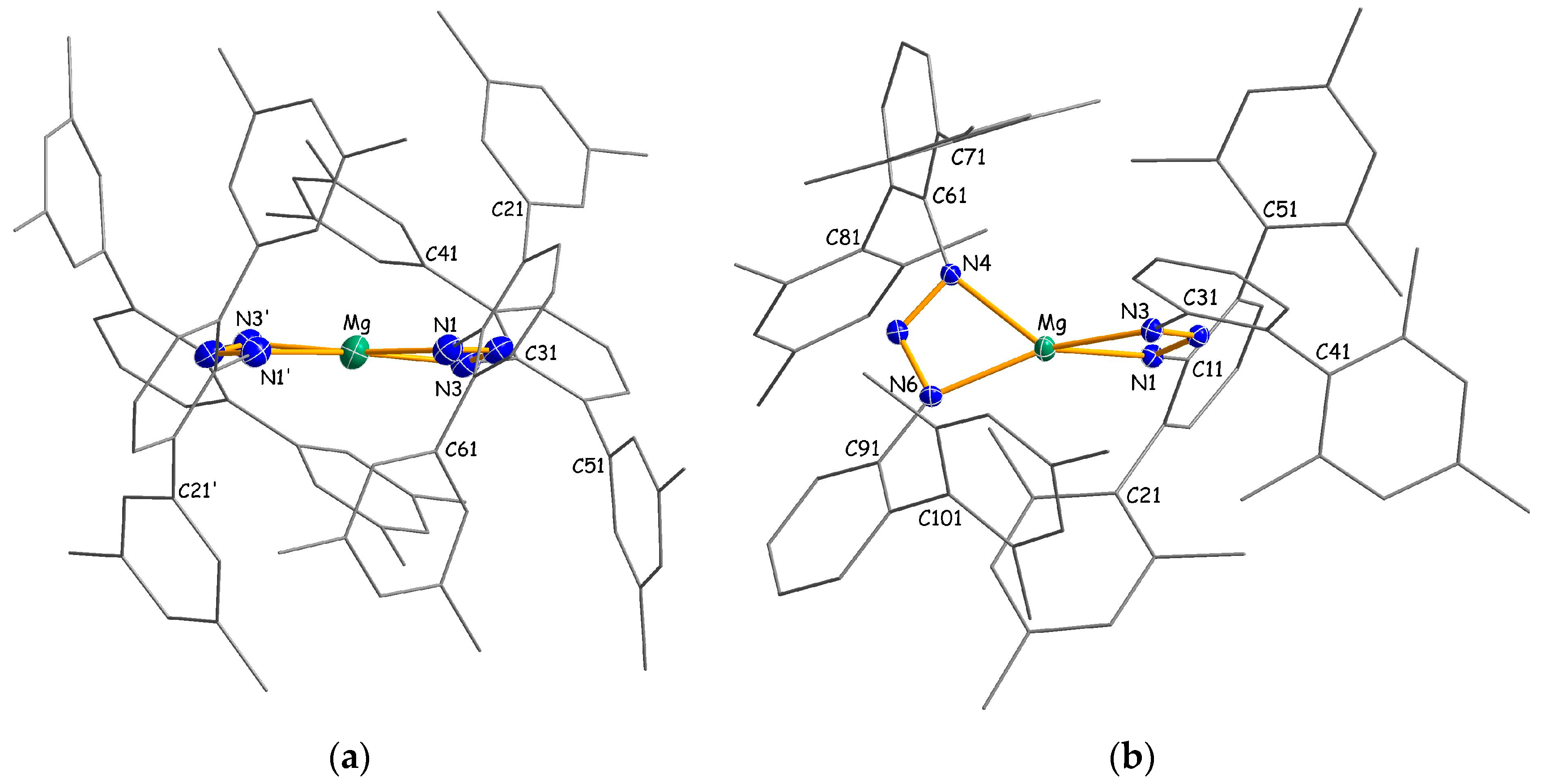
Homoleptic packing complexes 4b·(C7H16) and 4c·(C7H8)0.5 crystallize as monomers with four-coordinate metal atoms in which the triazenide ligands are coordinated in a chelating η2-fashion (Figure 3). There are no significant interactions between the complexes and the co-crystallized n-heptane or toluene solvent molecules. In C2-symmetric 4b, the two-fold axis runs almost parallel to the NNN plane through the magnesium atom, whereas C1-symmetric 4c has no additional crystallographically-imposed symmetry. Interestingly, the magnesium atom in 4c shows a distorted tetrahedral coordination with an average Mg–N distance of 2.086(2) Å, whereas a distorted square planar coordination around the magnesium center with a significant longer average Mg–N distance of 2.128(2) Å is observed for 4b. The different coordination is reflected by the interplanar angle γ, which is defined as the angle between the two MgNN planes (e.g., for 4c, angle between the plane normals through the atoms Mg/N1/N3 and Mg/N4/N6), of 83.6° (4c) and 9.6° (4b), or alternatively, by the τ4 parameter of 0.51 (4c) and 0.20 (4b). These values may be compared with the corresponding parameters in previously-published homoleptic magnesium amidinates [16,26,27,28,29,30], guanidinates [20] and β-diketiminates [31,32,33], as summarized in Table 1. For the six known magnesium amidinates, considered to possess tetrahedral metal coordination, γ angles and τ4 parameters are observed in the range of 54.1°–89.5° and 0.40°–0.60°, respectively. The relatively small values for τ4, compared to the ideal value of 1.00, can be rationalized by the small bite angles of the amidinate and triazenide ligands that enforce “flattened tetrahedral” geometries. In contrast, higher values in the range of 0.83–0.92 are found for β-diketiminates that have larger bite angles with more separated N donor atoms.
Magnesium complexes with square planar coordinated metal atoms are quite uncommon and usually restricted to ligands with rigid geometry, such as porphyrins [34,35,36,37,38]. Rare examples of planar magnesium compounds with non-rigid ligands are Lappert’s 1-azallyl complex [Mg(Me3SiNC(tBu)C(H)SiMe3)2] [33], Junk’s amidinate [Mg{DipN{C(pTol)}NDip}2] [26] and Kays’ guanidinate [Mg{MesN{C(NcHex)}NMes}2] [20]. It has been argued that interligand repulsion between peripheric substituents is responsible for the square planar coordination in these compounds. Moreover, it is known that attractive dispersion forces may contribute to unusual coordination geometries [39,40,41]. Therefore, it is reasonable to assume that a combination of repulsive and attractive interligand interactions accounts for the different metal coordination in 4b and 4c. Notably, the propensity of the [(Me4Ter)2N3]− ligand to support square planar coordination is not limited to magnesium. A similar complex with a square planar coordinated Yb(II) center was characterized in our group [42].
In order to shed some light on the relative energetic levels of tetrahedral or square planar coordinated magnesium triazenides, DFT calculations were performed for suitable model compounds. Unfortunately, we did not succeed to locate stationary points for both geometries with the same ligand systems. Therefore, simple phenyl substituted model complexes 5T and 5SP were calculated using the B3LYP functional and 6-311+G* basis sets. The experimentally-determined geometries of 4b and 4c were taken as the starting point, after replacing the bulky biphenyl and terphenyl substituents by phenyl groups. A minimum on the potential energy surface with S4 symmetry corresponds to the tetrahedral isomer 5T (γ = 90°, τ4 = 0.60). Since it was at first not possible to locate a stationary point for a square planar isomer, the conformation of the starting geometry was partly frozen by fixing NNMgN and NNCC torsion angles to the experimentally-determined values. The resulting energy-minimized C1-symmetric isomer 5SP (γ = 9.7°, τ4 = 0.07) is energetically disfavored over 5T by +60.7 KJ·mol−1.
Table 2 summarizes some pertinent bond parameters in structurally-characterized magnesium triazenides. Overall, the expected correlation between coordination number and Mg–N bond length is observed. However, two exceptions are noteworthy. Firstly, in distorted square planar coordinated 4b, the Mg–N distance of 2.128 Å is significantly longer than the corresponding values in distorted tetrahedral coordinated metal complexes that fall within the range of 2.070–2.102 Å. Secondly, in Westerhausen’s heteroleptic five-coordinate magnesium complex [Mg(nBu){N3(Mes)2}(tmeda)] [15], the Mg–N bond length is longer than the average values in Gibson’s five-coordinate magnesium compound [Mg{N3(Dip)2}2(OEt2)] [16] and in the six-coordinate metal bis(THF) adducts [Mg{N3(Ar)2}2(thf)2] (Ar = pTol [14], Mes [15]). The elongated bond may be attributed to the competition of the moderate nucleophilic triazenide ligand with the powerful carbanionic ligand. In addition, there appears to be some correlation between the N–Mg–N angle and the coordination number. Slightly more acute angles are observed for higher coordinated magnesium atoms. In contrast, there seems to be no clear correlation between steric crowding inside the complexes and the size of the average N–Mg–N or Mg–N–C angle.
3. Materials and Methods
3.1. General Procedures
All manipulations were performed by using standard Schlenk techniques under an inert atmosphere of purified argon. Solvents were dried and purified using an MBraun 800 solvent purification system. The triazenes Dmp(Tph)N3H [5], (Me4Ter)2N3H [8] or Dmp(Mph)N3H [5] were synthesized as previously described. NMR spectra were recorded on Bruker AM200, AM400 or Biospin DRX 400 instruments (Karlsruhe, Germany) and referenced to solvent resonances. IR spectra have been obtained in the range of 4000–200 cm−1 with a Varian 3100 FT-IR spectrometer (Palo Alto, CA, USA). Melting points were determined under Ar atmosphere in sealed glass tubes.
3.2. Syntheses
3.2.1. Experimental Procedure for [Mg{N3(Dmp)Tph}I(OEt2)] (2a)
To a stirred solution of triazene 1a (1.27 g, 2.0 mmol) in 60 mL of diethyl ether, a 1.0 M solution of di-n-butylmagnesium in n-heptane (2.0 mL, 2.0 mmol) was added, and stirring was continued for 30 min. To the resulting bright yellow solution, iodine (0.51 g, 2.0 mmol) was added. The solution was stirred for another 3 h until the typical iodine color disappeared. The volume of the obtained yellow solution was reduced to incipient crystallization under reduced pressure. Storage at room temperature overnight afforded 2a as yellow needles. Yield: 1.6 g (1.86 mmol, 93%); m.p.: 175 °C (dec.); 1H NMR (200.1 MHz, [D8]toluene, 373 K): δ 0.69 (t, 3JHH = 7.1 Hz, 6H, (CH3CH2)2O), 0.95 (d, 3JHH = 6.6 Hz, 6H, CH(CH3)2), 1.07 (d, 3JHH = 6.8 Hz, 6H, CH(CH3)2), 1.27 (d, 3JHH = 7,1 Hz, 6H, CH(CH3)2), 2.05 (s, 12H, o-CH3), 2.15 (s, 6H, p-CH3), 2.50 (sep, 3JHH = 6.8 Hz, 2H, o-CH(CH3)2), 2.83 (sep, 1H, p-CH(CH3)2), 3.14 (q, 4H, 3JHH = 7.1 Hz, (CH3CH2)2O), 6.3–7.0 (m, 13H, various Aryl-H). 13C NMR (62.9 MHz, [D6]benzene): δ 13.7 ((CH3CH2)2O), 21.3 (o-CH3), 21.9 (br, p-CH3), 24.3, 24.5, 25.4 (o+p-CH(CH3)2), 30.7 (br, o-CH(CH3)2), 34.9 (p-CH(CH3)2), 66.5 ((CH3CH2)2O), 120.9 (m-Mes), 123.9, 124.8, 127.6, 130.7, 132.1 (aromatic CH), 131.7, 133.4, 134.7, 136.1, 139.5, 143.7, 147.1 (aromatic C). IR (Nujol, cm−1) = 1664w, 1609m, 1595sh, 1583w, 1564m, 1509w, 1415s, 1362m, 1261vs, 1184m, 1106m, 1093m, 1080w, 1056w, 1032s, 1016m, 977w, 938m, 901m, 884w, 872m, 853s, 834m, 803m, 787s, 762s, 750s, 724m, 690m, 653s, 602w, 589m, 576w, 562w, 538m, 520m, 491m, 475m, 440m, 382s, 290m. Anal. Calcd. for C49H62N3MgIO: C, 68.41; H, 7.26; N, 4.88. Found: C, 67.73; H, 6.99; N, 4.92.
3.2.2. Experimental Procedure for [Mg{N3(Me4Ter)2}I(OEt2)] (2b)
The synthesis was accomplished in a manner similar to the preparation of 2a using triazene 1b (0.61 g, 1.0 mmol), a 1.0 M solution of di-n-butylmagnesium in n-heptane (1.0 mL, 1.0 mmol) and iodine (0.25 g, 1.0 mmol). Storage of the obtained solution at room temperature overnight afforded 2b as yellow blocks. Yield: 0.74 g (0.88 mmol, 88%); m.p.: 170 °C (dec.); 1H NMR (400.1 MHz, [D6]benzene): δ 0.51 (br s, 6H, (CH3CH2)2O), 2.27 (s, 24H, m-CH3), 2.95 (q, 3JHH = 6.7 Hz, 4H, (CH3CH2)2O), 6.70 (s, 4H, p-C6H3Me2), 6.86 (t, 3JHH = 7.6 Hz, 2H, p-C6H3), 6.98 (s, 8H, o-C6H3Me2), 7.10 (d, 3JHH = 7.6 Hz, 4H, o-C6H3). 13C NMR (100.6 MHz, [D6]benzene): δ 14.0 ((CH3CH2)2O), 21.8 (m-CH3), 66.0 ((CH3CH2)2O), 123.4 (p-C6H3), 128.0 (o-C6H3Me2), 128.5 (p-C6H3Me2), 130.5 (m-C6H3), 136.0 (o-C6H3), 137.4 (m-C6H3Me2), 142.3 (i-C6H3Me2), 143.1 (i-C6H3) ppm. IR (Nujol, cm−1) = 1684w, 1602s, 1558m, 1541m, 1490s, 1398m, 1280m, 1176m, 1036m, 849s, 795m, 761m, 704s, 681s, 668s. Anal. Calcd. for C48H52IMgN3O: C, 68.78; H, 6.25; N, 5.01. Found: C, 68.24; H, 6.02; N, 5.12.
3.2.3. Experimental Procedure for [Mg3{N3(Dmp)Tph}2I4] (3a)
To a stirred solution of triazene 1a (1.27 g, 2 mmol) in 60 mL of n-heptane, a 1.0 M solution of di-n-butylmagnesium in n-heptane (2 mL, 2 mmol) was added. After 30 min, the reaction mixture was treated with iodine (0.51 g, 2 mmol), and stirring was continued overnight. The volume of the resulting solution was reduced to incipient crystallization under reduced pressure, and the obtained precipitate was redissolved by slight warming. Storage at ambient temperature overnight afforded 3a as a pale yellow crystalline material. Yield: <10%, m.p.: 200 °C (dec.); 1H NMR (400.1 MHz, [D6]benzene): δ 1.03 (d, 3JHH = 6.7 Hz, 12H, o-CH(CH3)2), 1.11 (d, 3JHH = 6.7 Hz, 12H, o-CH(CH3)2), 1.21 (d, 3JHH = 7.0 Hz, 12H, p-CH(CH3)2), 2.17 (s, 12H, p-CH3), 2.36 (s, 24H, o-CH3), 2.65 (sept, 3JHH = 6.7 Hz, 4H, o-CH(CH3)2), 2.76 (sept, 3JHH = 7.0 Hz, 2H, p-CH(CH3)2), 6.51 (d, 3JHH = 8.2 Hz, 2H, 6-C6H4), 6.78–7.11 (m, 24H, various aryl-H). 13C NMR (100.6 MHz, [D6]benzene): δ 21.2 (p-CH3), 22.6 (o-CH3), 23.9, 24.2, 25.6 (o+p-CH(CH3)2), 30.7 (o-CH(CH3)2), 34.4 (p-CH(CH3)2), 120.8 (m-Trip), 128.5 (m-Mes), 130.5 (m-C6H3), 123.2, 123.5, 125.6, 127.6, 132.6 (aromatic CH), 121.5, 131.5, 135.7, 136.2, 136.8, 137.1, 139.5, 145.5, 147.2, 147.7, 149.3 (aromatic C). Anal. Calcd. for C90H104I4Mg3N6: C, 58.42; H, 5.67; N, 4.54. Found: C, 58.28; H, 5.69; N, 4.50.
3.2.4. Experimental Procedure for [Mg3{N3(Me4Ter)2}2I4] (3b)
The synthesis was accomplished in a manner similar to the preparation of 3a using triazene 1b (0.61 g, 1.0 mmol), 1 mmol of di-n-butylmagnesium and iodine (0.25 g, 1.0 mmol). The packing complex 3b·(C7H16)1.5 was crystallized from n-heptane at ambient temperature. Yield: <10%, m.p.: 200 °C (dec.); IR (Nujol, cm−1) = 1746w, 1601s, 1557sh, 1403m, 1284sh, 1255s, 1215m, 1200m, 1171w, 1127w, 1037m, 1008w, 893m, 851s, 795m, 763m, 757sh, 706s, 683m, 669w, 602w, 529w, 472w, 417m. No satisfactory CHN analysis could be obtained due to the co-crystallized solvent.
3.2.5. Experimental Procedure for [Mg{N3(Me4Ter)2}2] (4b)
To triazene 1b (0.61 g, 1 mmol) in 50 mL of n-heptane a 1.0 M solution of di-n-butylmagnesium in n-heptane (0.5 mL, 0.5 mmol) was added, and the mixture was stirred overnight. The obtained precipitate was dissolved by slight warming, and the resulting solution slowly cooled to ambient temperature to give pale yellow crystals of the packing complex 4b·(C7H16). The material used for characterization was dried under reduced pressure to remove co-crystallized solvent. Yield: 0.46 g (0.37 mmol, 74%); m.p.: >300 °C; 1H NMR (400, 1 MHz, [D6]benzene, 333 K): δ 1.96 (s, 48H, CH3), 6.10 (s, vbr, 16H, o-C6H3Me2), 6.68 (s, 8H, p-C6H3Me2), 6.96 (t, 3JHH = 7.6 Hz, 4H, p-C6H3N), 7.10 (d, 3JHH = 7.1 Hz, 8H, m-C6H3N). 13C NMR (100,6 MHz, [D6]benzene): δ 22.3 (vbr, CH3), 123.9 (p-C6H3N), 129.1 (p-C6H3Me2), 131.1 (m-C6H3N), 132.5 (o-C6H3Me2), 136.4 (o-C6H3N), 138.2 (m-C6H3Me2), 142.2 (i-C6H3Me2), 144.7 (i-C6H3N). IR (Nujol, cm−1) = 1748w, 1600s, 1400m, 1321s, 1282s, 1171w, 1125m, 1076w, 1038m, 905w, 850s, 816m, 797s, 764s, 705s, 672m, 652m, 605w, 520w, 507w, 483w, 444m. Anal. Calcd. for C88H84MgN6: C, 84.56; H, 6.77; N, 6.72. Found: C, 84.03; H, 6.49; N, 6.82.
3.2.6. Experimental Procedure for [Mg{N3(Dmp)Mph}2] (4c)
The synthesis was accomplished in a manner similar to the preparation of 4b using triazene 1c (1.1 g, 2.0 mmol) and 1 mmol of di-n-butylmagnesium. The yellow packing complex 4c·(C7H8)0.5 was crystallized from a mixture of n-heptane and toluene at −17 °C. Yield: 1.04 g (0.89 mmol, 89%); m.p.: 220 °C (dec.); 1H NMR (400.1 MHz, [D8]toluene, 373 K): δ 1.59 (s, 12H, p-CH3), 1.74 (s, 24H, o-CH3), 2.01 (s, 6H, p-CH3), 2.06 (s, 12 H, o-CH3), 5.58 (d, 3JHH = 7.8 Hz, 2H, 6-C6H4), 6.51 (s, 4H, m-Mes), 6.55 (s, 8H, m-Mes), 6.53–7.00 (m, 12H, var. aryl-H). 13C NMR (62.9 MHz, [D6]benzene): δ 19.9 (p-CH3, Mph), 21.0 (p-CH3, Dmp), 21.1 (o-CH3, Mph), 21.3 (CH3, toluene), 21.4 (o-CH3, Dmp), 123.9 (6-C6H4), 124.7 (4-C6H4), 126.0 (5’-C6H3), 126.0 (p-CH, toluene), 127.6 (m-Mes, Mph), 128.2 (m-Mes, Dmp), 128.3 (3-C6H4), 128.7 (m-CH, toluene), 128.8 (5-C6H4), 129.4 (4’/6’-C6H3), 129.7 (o-CH, toluene), 130.1 (br), 133.1, 134.8, 135.6, 135.9, 137.5, 138.7 (aromatic C), 151.8, 153.6 (1-C6H4, 2’-C6H3). IR (Nujol, cm−1) = 1734m, 1717m, 1700m, 1695m, 1684m, 1675w, 1670w, 1653m, 1635m, 1616m, 1609m, 1576m, 1570m, 1559m, 1539m, 1521w, 1506m, 1419sh, 1308s, 1272s, 1032m, 851s, 804m, 777m, 755s, 730s, 694w, 668m, 646m, 595m, 578m, 565m, 547w, 521m, 464m, 431m, 411m. Anal. Calcd. for C78H76N6Mg···0.5 C7H8: C, 83.53; H, 7.22; N, 7.17. Found: C, 83.14; H, 7.43; N, 7.16.
3.3. X-Ray Crystallography
X-ray-quality crystals were obtained as described in the syntheses section. Crystals were removed from Schlenk tubes and immediately covered with a layer of viscous hydrocarbon oil (Paratone N, Exxon). A suitable crystal was selected, attached to a nylon loop, and instantly placed in a low temperature N2-stream. All data were collected at 173 K with MoKα radiation using either a Siemens P4 (2b, 4c·(C7H8)0.5) or a Bruker Smart Apex II (2a, 3a, 3b·(C7H16)1.5, 4b·(C7H16)) diffractometer. Calculations were performed with the SHELXTL PC 5.03a and SHELXL-97 program system [43]. The structures were solved by direct methods and refined on Fo2 by full-matrix least-squares refinement. Crystal and refinement data are given below. For the iodo complexes, absorption corrections were applied by using semiempirical ψ-scans or the multi-scan method. For 3b·(C7H16)1.5, co-crystallized solvent molecules were located in accessible cavities of the structure. Since they were severely disordered, their contribution was eliminated from the reflection data, using the BYPASS method [44] as implemented in the SQUEEZE routine of the PLATON98 [45] package. Values in brackets refer to the refinement that includes the contributions from the solvent. Crystallographic data (excluding structure factors) for the structures reported in this paper have been deposited with the Cambridge Crystallographic Data Centre. CCDC-1541009 {2a}, -1541010 {2b}, -1541011 {3a}, -1541012 {3b·(C7H16)1.5}, -1541013 {4b·(C7H16)} and -1541014 {4c·(C7H8)0.5} contain the supplementary crystallographic data for this paper. These data can be obtained free of charge via http://www.ccdc.cam.ac.uk/conts/retrieving.html (or from the CCDC, 12 Union Road, Cambridge CB2 1EZ, UK; Fax: +44-1223-3360-33; E-mail: [email protected]).
Crystallographic data for 2a: C49H62IMgN3O, M = 860.2, yellow rod 0.65 × 0.35 × 0.35 mm3, monoclinic, space group P21/n, a = 8.9827(2), b = 23.6098(6), c = 22.1855(5) Å, β = 90.8180(10)°, V = 4704.62(19) Å3, Z = 4, Dcalc = 1.215 g cm−3, μ = 0.730 mm−1, 67161 collected (3.6° ≤ 2Θ ≤ 58.6°) and 12780 unique reflections (Rint = 0.053), 521 parameters, 1 restraint, R1 = 0.036 for 7680 reflections with I > 2σ(I), wR2 = 0.094 (all data), Goodness of fit (GOF) = 0.934. The methyl carbon atoms of one disordered i-propyl group were refined with split positions and side occupation factors of 0.67 (C443) and 0.33 (C444), respectively. The corresponding C441–C443 and C441–C444 distances were refined with restraints.
Crystallographic data for 2b: C48H52IMgN3O, M = 838.1, yellow prism 0.50 × 0.35 × 0.25 mm3, monoclinic, space group P21/c, a = 19.848(4), b = 9.373(2), c = 23.033(4) Å, β = 90.386(14)°, V = 4284.9(14) Å3, Z = 4, Dcalc = 1.299 g cm−3, μ = 0.800 mm−1, 10104 collected (4.7° ≤ 2Θ ≤ 55.0°) and 9823 unique reflections (Rint = 0.046), 501 parameters, 0 restraints, R1 = 0.036 for 6945 reflections with I > 2σ(I), wR2 = 0.090 (all data), GOF = 0.886.
Crystallographic data for 3a: C90H104I4Mg3N6, M = 1850.3, pale yellow prism 0.30 × 0.20 × 0.15 mm3, orthorhombic, space group Pbca, a = 20.4142(4), b = 22.2202(4), c = 39.3523(7) Å, V = 17,850.5(6) Å3, Z = 8, Dcalc = 1.377 g cm−3, μ = 1.462 mm−1, 188607 collected (3.4° ≤ 2Θ ≤ 54.8°) and 21302 unique reflections (Rint = 0.254), 952 parameters, 0 restraints, R1 = 0.041 for 6128 reflections with I > 2σ(I), wR2 = 0.067 (all data), GOF = 0.653.
Crystallographic data for 3b·(C7H16)1.5: C88H84I4Mg3N6 [C98.5H108I4Mg3N6], M = 1806.1 [1956.4], pale yellow prism 0.40 × 0.35 × 0.20 mm3, triclinic, space group P, a = 16.9933(5), b = 17.5771(5), c = 17.8005(5) Å, a = 93.427(2)°, β = 99.534(2)°, γ = 109.991(2)°, V = 4888.5(2) Å3, Z = 2, Dcalc = 1.227 [1.329] g cm−3, μ = 1.334 [1.339] mm−1, 206993 collected (2.4° ≤ 2Θ ≤ 59.1°) and 27255 unique reflections (Rint = 0.064), 926 parameters, 0 restraints, R1 = 0.067 for 21930 reflections with I > 2σ(I), wR2 = 0.134 (all data), GOF = 1.921. The contribution of one and a half co-crystallized n-heptane molecules was eliminated from the reflection data (see above).
Crystallographic data for 4b·(C7H16): C95H100MgN6, M = 1350.1, pale yellow prism 0.35 × 0.25 × 0.20 mm3, monoclinic, space group P2/n, a = 15.0589(12), b = 13.0937(10), c = 20.3232(16) Å, β = 99.013(3)°, V = 3957.8(5) Å3, Z = 2, Dcalc = 1.133 g cm−3, μ = 0.073 mm−1, 67630 collected (3.1° ≤ 2Θ ≤ 55.0°) and 9087 unique reflections (Rint = 0.248), 464 parameters, 8 restraints, R1 = 0.067 for 3669 reflections with I > 2σ(I), wR2 = 0.188 (all data), GOF = 0.887. The co-crystallized n-heptane molecule is disordered over a center of inversion and was refined with a side occupation factor of 0.5 and isotropic displacement parameters. The 1,2-C–C and 1,3-C–C distances were restrained.
Crystallographic data for 4c·(C7H8)0.5: C81.5H84MgN6, M = 1171.9, yellow prism 0.50 × 0.40 × 0.30 mm3, monoclinic, space group P21/n, a = 13.302(2), b = 21.531(3), c = 24.332(4) Å, β = 101.877(12)°, V = 6819.8(17) Å3, Z = 4, Dcalc = 1.141 g cm−3, μ = 0.075 mm−1, 12550 collected (4.1° ≤ 2 Θ ≤ 50.0°) and 11996 unique reflections (Rint = 0.074), 837 parameters, 3 restraints, R1 = 0.044 for 5484 reflections with I > 2σ(I), wR2 = 0.098 (all data), GOF = 0.727. The arene ring of the co-crystallized toluene molecule, which is disordered over a center of inversion, was constrained to a regular hexagon. Additional restraints were applied regarding distances and angles to the toluene methyl carbon atom.
3.4. Computational Details
The Gaussian 09 package [46] was used for all energy and frequency calculations. The energies of the model compounds 5T and 5SP were minimized using density functional theory (DFT) with the functional B3LYP [47,48], starting from the crystallographically-determined or from other derived geometries and assuming S4 symmetry for 5T. The sum of the electronic energy and the zero-point energy was used to calculate the energy difference between both model complexes.
4. Conclusions
In summary, we have used sterically crowded diaryltriazenido ligands for the stabilization of several heteroleptic and homoleptic magnesium triazenides. The obtained iodo magnesium-triazenides are kinetically stable against ligand redistribution reactions and represent potential precursors for magnesium(I) triazenides. The synthesized homoleptic compounds are the first examples of unsolvated magnesium triazenides. Remarkably, the magnesium cations in these compounds feature different coordination geometries. Depending on the nature of the substituents, either the expected tetrahedral or a rather unusual square planar coordination is observed.
Supplementary Materials
The following are available online at www.mdpi.com/2304-6740/5/2/33/s1, 1H VT NMR spectra and supporting molecular plots for Compounds 2a, 4b and 4c (Figures S1, S2, S3b and S4), molecular structure plot showing intermolecular C–H···N contacts in 4b (Figure S3a), structural plots and coordinates for the DFT calculated model complexes 5T and 5SP (Figure S5 and Tables S1 and S2), CIF files and checkcif reports.
Acknowledgments
We thank Karl Klinkhammer for generous financial support.
Author Contributions
Denis Vinduš synthesized and characterized all compounds, Mark Niemeyer planned the research, performed the DFT calculations, collected the X-ray data and refined the crystal structures. Denis Vinduš wrote the first draft, and Mark Niemeyer wrote the final version of the manuscript.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Gibson, V.C.; Spitzmesser, S.K. Advances in Non-Metallocene Olefin Polymerization Catalysis. Chem. Rev. 2003, 103, 283–315. [Google Scholar] [CrossRef] [PubMed]
- Bourget-Merle, L.; Lappert, M.F.; Severn, J.R. The Chemistry of β-Diketiminatometal Complexes. Chem. Rev. 2002, 102, 3031–3065. [Google Scholar] [CrossRef] [PubMed]
- Coles, M.P. Application of neutral amidines and guanidines in coordination chemistry. Dalton Trans. 2006, 37, 985–1001. [Google Scholar] [CrossRef] [PubMed]
- Vrieze, K.; van Koten, G. Sulfurdiimine, Triazenido, Azabutadiene and Triatomic Hetero Anion Ligands. In Comprehensive Coordination Chemistry, 1st ed.; Wilkinson, G., Gillard, R.D., McCleverty, J., Eds.; Pergamon Press: Oxford, UK, 1987; Volume 2, pp. 195–206. [Google Scholar]
- Hauber, S.-O.; Lissner, F.; Deacon, G.B.; Niemeyer, M. Stabilization of Aryl-Calcium, -Strontium, and -Barium Compounds by Designed Steric and π-Bonding Encapsulation. Angew. Chem. Int. Ed. 2005, 44, 5871–5875. [Google Scholar] [CrossRef] [PubMed]
- Hauber, S.-O.; Niemeyer, M. Stabilization of Unsolvated Europium and Ytterbium Pentafluorophenyls by π-Bonding Encapsulation through a Sterically Crowded Triazenido Ligand. Inorg. Chem. 2005, 44, 8644–8646. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.S.; Niemeyer, M. Inverse Aggregation Behavior of Alkali-Metal Triazenides. Inorg. Chem. 2006, 45, 6126–6128. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.S.; Hauber, S.-O.; Vinduš, D.; Niemeyer, M. Isostructural Potassium and Thallium Salts of Sterically Crowded Triazenes: A Structural and Computational Study. Inorg. Chem. 2008, 47, 4401–4412. [Google Scholar] [CrossRef] [PubMed]
- Balireddi, S.; Niemeyer, M. A sterically crowded triazene: 1,3-Bis(3,5,3′′,5′′-tetramethyl-1,1′:3′;1′′-ter-phenyl-2′-yl)triazene. Acta Crystallogr. 2007, E63, o3525. [Google Scholar] [CrossRef]
- Lee, H.S.; Niemeyer, M. Homoleptic Heavy Alkaline Earth and Europium Triazenides. Inorg. Chem. 2010, 49, 730–735. [Google Scholar] [CrossRef] [PubMed]
- Hauber, S.-O.; Seo, J.W.; Niemeyer, M. Halogenomercury Salts of Sterically Crowded Triazenides—Convenient Starting Materials for Redox-Transmetallation Reactions. Z. Anorg. Allg. Chem. 2010, 636, 750–757. [Google Scholar] [CrossRef]
- Lee, H.S.; Niemeyer, M. Sterically crowded triazenides as novel ancillary ligands in copper chemistry. Inorg. Chim. Acta 2011, 374, 163–170. [Google Scholar] [CrossRef]
- Hinz, A.; Schulz, A.; Villinger, A.; Wolter, J.-M. Cyclo-Pnicta-triazanes: Biradicaloids or Zwitterions? J. Am. Chem. Soc. 2015, 137, 3975–3980. [Google Scholar] [CrossRef] [PubMed]
- Westhusin, S.; Gantzel, P.; Walsh, P.J. Synthesis and Crystal Structures of Magnesium and Calcium Triazenide Complexes. Inorg. Chem. 1998, 37, 5956–5959. [Google Scholar] [CrossRef]
- Kalden, D.; Krieck, S.; Görls, H.; Westerhausen, M. 1,3-Bis(2,4,6-trimethylphenyl)triazenides of potassium, magnesium, calcium, and strontium. Dalton Trans. 2015, 44, 8089–8099. [Google Scholar] [CrossRef] [PubMed]
- Nimitsiriwat, N.; Gibson, V.C.; Marshall, E.L.; Takolpuckdee, P.; Tomov, A.K.; White, A.J.P.; Williams, D.J.; Elsegood, M.R.J.; Dale, S.H. Mono-versus Bis-chelate Formation in Triazenide and Amidinate Complexes of Magnesium and Zinc. Inorg. Chem. 2007, 46, 9988–9997. [Google Scholar] [CrossRef] [PubMed]
- Groom, C.R.; Bruno, I.J.; Lightfoot, M.P.; Ward, S.C. The Cambridge Structural Database. Acta Crystallogr. 2016, B72, 171–179. [Google Scholar] [CrossRef] [PubMed]
- Bonyhady, S.J.; Jones, C.; Nembenna, S.; Stasch, A.; Edwards, A.J.; McIntyre, G.J. β-Diketiminate-Stabilized Magnesium(I) Dimers and Magnesium(II) Hydride Complexes: Synthesis, Characterization, Adduct Formation, and Reactivity Studies. Chem. Eur. J. 2010, 16, 938–955. [Google Scholar] [CrossRef] [PubMed]
- Stasch, A. Synthesis of a Dimeric Magnesium(I) Compound by an MgI/MgII Redox Reaction. Angew. Chem. Int. Ed. 2014, 53, 10200–10203. [Google Scholar] [CrossRef] [PubMed]
- Moxey, G.J.; Blake, A.J.; Lewis, W.; Kays, D.L. Alkaline Earth Complexes of a Sterically Demanding Guanidinate Ligand. Eur. J. Inorg. Chem. 2015, 2015, 5892–5902. [Google Scholar] [CrossRef]
- MacNeil, C.S.; Johnson, K.R.D.; Hayes, P.G.; Boeré, R.T. Crystal structure of a dimeric β-diketiminate magnesium complex. Acta Crystallogr. 2016, E72, 1754–1756. [Google Scholar] [CrossRef] [PubMed]
- Boutland, A.J.; Dange, D.; Stasch, A.; Maron, L.; Jones, C. Two-Coordinate Magnesium(I) Dimers Stabilized by Super Bulky Amido Ligands. Angew. Chem. Int. Ed. 2016, 55, 9239–9243. [Google Scholar] [CrossRef] [PubMed]
- Brogan, M.A.; Blake, A.J.; Wilson, C.; Gregory, D.H. Magnesium diiodide, MgI2. Acta Crystallogr. 2003, C59, i136–i138. [Google Scholar] [CrossRef]
- Akishin, P.A.; Spiridonov, V.P. Electron-diffraction investigation of magnesium iodide molecular structure. Zhurnal Fizicheskoi Khimii 1958, 32, 1682–1683. [Google Scholar]
- Yang, L.; Powell, D.R.; Houser, R.P. Structural variation in copper(I) complexes with pyridylmethylamide ligands: Structural analysis with a new four-coordinate geometry index, τ4. Dalton Trans. 2007, 9, 955–964. [Google Scholar] [CrossRef] [PubMed]
- Boeré, R.T.; Cole, M.L.; Junk, P.C. The syntheses and structures of some main group complexes of the sterically hindered N,N′-bis(2,6-diisopropylphenyl)-4-toluamidinate ligand. New J. Chem. 2005, 29, 128–134. [Google Scholar] [CrossRef]
- Moxey, G.J.; Ortu, F.; Goldney Sidley, L.; Strandberg, H.N.; Blake, A.J.; Lewis, W.; Kays, D.L. Synthesis and characterisation of magnesium complexes containing sterically demanding N,N′-bis(aryl)amidinate ligands. Dalton Trans. 2014, 43, 4838–4846. [Google Scholar] [CrossRef] [PubMed]
- Xia, A.; El-Kaderi, H.M.; Heeg, M.J.; Winter, C.H. Synthesis, structure, and properties of magnesium complexes containing cyclopentadienyl and amidinate ligand sets. J. Organomet. Chem. 2003, 682, 224–232. [Google Scholar] [CrossRef]
- Sadique, A.R.; Heeg, M.J.; Winter, C.H. Monomeric and Dimeric Amidinate Complexes of Magnesium. Inorg. Chem. 2001, 40, 6349–6355. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, J.A.R.; Arnold, J. Synthesis and characterization of a series of sterically-hindered amidines and their lithium and magnesium complexes. J. Chem. Soc. Dalton Trans. 2002, 2890–2899. [Google Scholar] [CrossRef]
- Sedai, B.; Heeg, M.J.; Winter, C.H. Magnesium complexes containing β-ketiminate and β-diketiminate ligands with dimethylamino substituents on the ligand core nitrogen atoms. J. Organomet. Chem. 2008, 693, 3495–3503. [Google Scholar] [CrossRef]
- El-Kaderi, H.M.; Xia, A.; Heeg, M.J.; Winter, C.H. Factors that Influence π-versus η2-Coordination of β-Diketiminato Ligands in Magnesium Complexes. Organometallics 2004, 23, 3488–3495. [Google Scholar] [CrossRef]
- Caro, C.F.; Hitchcock, P.B.; Lappert, M.F. Monomeric magnesium 1-azaallyl and β-diketiminato complexes derived from the bis(trimethylsilyl)methyl ligand: The X-ray structure of the four-coordinate planar magnesium complex [Mg{N(R)C(But)C(H)R}2] and of [Mg({N(R)C(Ph)}2CH)2]. Chem. Commun. 1999, 1433–1434. [Google Scholar] [CrossRef]
- Byrn, M.P.; Curtis, C.J.; Goldberg, I.; Hsiou, Y.; Khan, S.I.; Sawin, P.A.; Tendick, S.K.; Strouse, C.E. Porphyrin sponges: Structural systematics of the host lattice. J. Am. Chem. Soc. 1991, 113, 6549–6557. [Google Scholar] [CrossRef]
- Byrn, M.P.; Curtis, C.J.; Hsiou, Y.; Khan, S.I.; Sawin, P.A.; Tendick, S.K.; Terzis, A.; Strouse, C.E. Porphyrin sponges: Conservative of host structure in over 200 porphyrin-based lattice clathrates. J. Am. Chem. Soc. 1993, 115, 9480–9497. [Google Scholar] [CrossRef]
- Mizuguchi, J. Crystal Structure of Magnesiumphthalocyanine and Its Polarized Reflection Spectra. J. Phys. Chem. A 2001, 105, 1121–1124. [Google Scholar] [CrossRef]
- Janczak, J.; Kubiak, R. X-ray single crystal investigations of magnesium phthalocyanine. The 4+1 coordination of the Mg ion and its consequence. Polyhedron 2001, 20, 2901–2909. [Google Scholar] [CrossRef]
- Chandra, T.; Kraft, B.J.; Huffman, J.C.; Zaleski, J.M. Synthesis and Structural Characterization of Porphyrinic Enediynes: Geometric and Electronic Effects on Thermal and Photochemical Reactivity. Inorg. Chem. 2003, 42, 5158–5172. [Google Scholar] [CrossRef] [PubMed]
- Grimme, S.; Djukic, J.-P. Cation-Cation “Attraction”: When London Dispersion Attraction Wins over Coulomb Repulsion. Inorg. Chem. 2011, 50, 2619–2628. [Google Scholar] [CrossRef] [PubMed]
- Grimme, S.; Schreiner, P.R. Steric Crowding Can Stabilize a Labile Molecule: Solving the Hexaphenylethane Riddle. Angew. Chem. Int. Ed. 2011, 50, 12639–12642. [Google Scholar] [CrossRef] [PubMed]
- Harder, S.; Naglav, D.; Schwerdtfeger, P.; Nowik, I.; Herber, R.H. Metal Atom Dynamics in Superbulky Metallocenes: A Comparison of (CpBIG)2Sn and (CpBIG)2Eu. Inorg. Chem. 2014, 53, 2188–2194. [Google Scholar] [CrossRef] [PubMed]
- Franta, U. Synthese, Strukturchemie und Untersuchungen von Aluminium, Ytterbium und Lithiumkomplexen mit Sterisch Anspruchsvollen Triazenido-Liganden; Staatsexamensarbeit; Johannes Gutenberg-Universität: Mainz, Germany, 2011. [Google Scholar]
- Sheldrick, G.M. A Short History of SHELX. Acta Crystallogr. 2008, A64, 112–122. [Google Scholar] [CrossRef] [PubMed]
- van der Sluis, P.; Spek, A.L. BYPASS: An effective method for the refinement of crystal structures containing disordered solvent regions. Acta Crystallogr. 1990, A46, 194–201. [Google Scholar] [CrossRef]
- Spek, A.L. PLATON-98; Utrecht University: Utrecht, The Netherlands, 1998. [Google Scholar]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 09; revision A.02; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef]
Scheme 1.
Syntheses of Compounds 2a and 2b (a), 3a and 3b (b) and 4b and 4c (c).

Figure 1.
Molecular structures of 2a (a) and 2b (b) with thermal ellipsoids set to 30% probability. Hydrogen atoms have been omitted for clarity. Selected bond lengths (Å), angles and dihedral angles (°) for 2a (2b): Mg–N1/N3 = 2.1151(16)/2.0880(16) (2.101(2)/2.0958(19)), Mg–I = 2.6438(7) (2.6596(9)), Mg–O = 2.0157(15) (1.996(2)), N1–N2 = 1.317(2) (1.312(2)), N2–N3 = 1.307(2) (1.312(3)), N1–Mg–N3 = 61.45(6) (61.04(7)), N1–Mg–I = 113.36(5) (103.83(6)), N1–Mg–O = 125.70(7) (123.70(9)), N3–Mg–I = 132.64(5) (143.56(7)), N3–Mg–O = 114.51(6) (107.24(8)), I–Mg–O = 105.39(5) (108.36(6)), N2–N1–C11–C16 = 35.7(3) (37.8(3)), N2–N3–C31–C36 = 18.0(3) (N2–N3–C41–C46 = 27.0(3)).
Figure 1.
Molecular structures of 2a (a) and 2b (b) with thermal ellipsoids set to 30% probability. Hydrogen atoms have been omitted for clarity. Selected bond lengths (Å), angles and dihedral angles (°) for 2a (2b): Mg–N1/N3 = 2.1151(16)/2.0880(16) (2.101(2)/2.0958(19)), Mg–I = 2.6438(7) (2.6596(9)), Mg–O = 2.0157(15) (1.996(2)), N1–N2 = 1.317(2) (1.312(2)), N2–N3 = 1.307(2) (1.312(3)), N1–Mg–N3 = 61.45(6) (61.04(7)), N1–Mg–I = 113.36(5) (103.83(6)), N1–Mg–O = 125.70(7) (123.70(9)), N3–Mg–I = 132.64(5) (143.56(7)), N3–Mg–O = 114.51(6) (107.24(8)), I–Mg–O = 105.39(5) (108.36(6)), N2–N1–C11–C16 = 35.7(3) (37.8(3)), N2–N3–C31–C36 = 18.0(3) (N2–N3–C41–C46 = 27.0(3)).

Figure 2.
Molecular structures of 3a (a) and 3b (b) with thermal ellipsoids set to 30% probability. Hydrogen atoms have been omitted and carbon atoms are reduced in size for clarity. Selected bond lengths (Å) and angles (°) for 3a (3b): Mg1–I1 = 2.794(2) (2.8014(13)), Mg1–I2 = 2.7446(19) (2.7865(13)), Mg2–I1 = 2.736(2) (2.7097(12)), Mg2–I2 = 2.753(2) (2.7143(12)), Mg2–I3 = 2.738(2) (2.7302(13)), Mg2–I4 = 2.735(2) (2.7294(12)), Mg3–I3 = 2.7390(18) (2.7564(12)), Mg3–I4 = 2.7811(19) (2.7767(13)), Mg1–N1/N3 = 2.075(4)/2.073(5) (2.093(3)/2.057(3)), Mg3–N4/N6 = 2.068(5)/2.074(4) (2.057(3)/2.074(3)), av. N–N = 1.314(5) (1.311(4)) N1–Mg1–N3 = 62.54(17) (61.65(11)), N1–Mg1–I1 = 110.00(14) (146.66(10)), N1–Mg1–I2 = 143.16(16) (107.59(9)), N3–Mg1–I1 = 117.06(15) (105.10(9)), N3–Mg1–I2 = 128.89(14) (156.88(10)), I1–Mg1–I2 = 95.08(5) (94.29(4)), I1–Mg2–I2 = 96.24(6) (98.09(4)), I1–Mg2–I3 = 112.88(7) (114.83(5)), I1–Mg2–I4 = 113.90(8) (113.57(4)), I2–Mg2–I3 = 120.55(8) (115.56(4)), I2–Mg2–I4 = 117.21(7) (116.70(5)), I3–Mg2–I4 = 97.16(6) (99.07(4)), N4–Mg3–N6 = 62.28(17) (61.90(10)), N4–Mg3–I3 = 140.98(16) (122.96(9)), N4–Mg3–I4 = 109.31(15) (135.71(10)), N6–Mg3–I3 = 129.39(14) (136.28(10)), N6–Mg3–I4 = 118.75(15) (102.76(9)), I3–Mg3–I4 = 96.06(5) (97.30(3)).
Figure 2.
Molecular structures of 3a (a) and 3b (b) with thermal ellipsoids set to 30% probability. Hydrogen atoms have been omitted and carbon atoms are reduced in size for clarity. Selected bond lengths (Å) and angles (°) for 3a (3b): Mg1–I1 = 2.794(2) (2.8014(13)), Mg1–I2 = 2.7446(19) (2.7865(13)), Mg2–I1 = 2.736(2) (2.7097(12)), Mg2–I2 = 2.753(2) (2.7143(12)), Mg2–I3 = 2.738(2) (2.7302(13)), Mg2–I4 = 2.735(2) (2.7294(12)), Mg3–I3 = 2.7390(18) (2.7564(12)), Mg3–I4 = 2.7811(19) (2.7767(13)), Mg1–N1/N3 = 2.075(4)/2.073(5) (2.093(3)/2.057(3)), Mg3–N4/N6 = 2.068(5)/2.074(4) (2.057(3)/2.074(3)), av. N–N = 1.314(5) (1.311(4)) N1–Mg1–N3 = 62.54(17) (61.65(11)), N1–Mg1–I1 = 110.00(14) (146.66(10)), N1–Mg1–I2 = 143.16(16) (107.59(9)), N3–Mg1–I1 = 117.06(15) (105.10(9)), N3–Mg1–I2 = 128.89(14) (156.88(10)), I1–Mg1–I2 = 95.08(5) (94.29(4)), I1–Mg2–I2 = 96.24(6) (98.09(4)), I1–Mg2–I3 = 112.88(7) (114.83(5)), I1–Mg2–I4 = 113.90(8) (113.57(4)), I2–Mg2–I3 = 120.55(8) (115.56(4)), I2–Mg2–I4 = 117.21(7) (116.70(5)), I3–Mg2–I4 = 97.16(6) (99.07(4)), N4–Mg3–N6 = 62.28(17) (61.90(10)), N4–Mg3–I3 = 140.98(16) (122.96(9)), N4–Mg3–I4 = 109.31(15) (135.71(10)), N6–Mg3–I3 = 129.39(14) (136.28(10)), N6–Mg3–I4 = 118.75(15) (102.76(9)), I3–Mg3–I4 = 96.06(5) (97.30(3)).

Figure 3.
Molecular structures of 4b (a) and 4c (b) with thermal ellipsoids set to 30% probability. Hydrogen atoms and co-crystallized solvents have been omitted and carbon atoms are reduced in size for clarity. Selected bond lengths (Å), angles and dihedral angles (°) for 4b (symmetry operation (′): −x + 1/2, y, −z + 1/2): Mg–N1 = 2.131(2), Mg–N3 = 2.125(2), Mg–N2 = 2.579(2), N1–N2 = 1.316(3), N2–N3 = 1.318(3), N1–Mg–N3 = 60.97(8), N1–Mg–N1′ = 106.11(13), N3–Mg–N3′ = 132.43(13), N1–Mg–N3′ = 165.96(10), N1–N2–N3 = 110.1(2), N2–N1–C11 = 109.6(2), N2–N3–C31 = 112.7(2), N2–N1–C11–C12 = 54.7(3), N2–N3–C31–C36 = 54.6(3). Selected bond lengths (Å), angles and dihedral angles (°) for 4c: Mg–N1 = 2.0863(19), Mg–N3 = 2.0770(19), Mg–N2 = 2.568(2), Mg–N4 = 2.1151(19), Mg–N6 = 2.0673(19), Mg–N5 = 2.555(2), N1–N2 = 1.315(2), N2–N3 = 1.323(2), N4–N5 = 1.312(2), N5–N6 = 1.324(2), N1–Mg–N3 = 61.39(7), N4–Mg–N6 = 61.81(7), N1–Mg–N4 = 120.68(8), N1–Mg–N6 = 138.30(8), N3–Mg–N4 = 135.20(8), N3–Mg–N6 = 150.35(8), N2–N1–C11–C16 = −46.7(3), N2–N3–C31–C32 = −25.8(3), N5–N4–C61–C66 = −60.7(3), N5–N6–C91–C96 = 35.5(3).
Figure 3.
Molecular structures of 4b (a) and 4c (b) with thermal ellipsoids set to 30% probability. Hydrogen atoms and co-crystallized solvents have been omitted and carbon atoms are reduced in size for clarity. Selected bond lengths (Å), angles and dihedral angles (°) for 4b (symmetry operation (′): −x + 1/2, y, −z + 1/2): Mg–N1 = 2.131(2), Mg–N3 = 2.125(2), Mg–N2 = 2.579(2), N1–N2 = 1.316(3), N2–N3 = 1.318(3), N1–Mg–N3 = 60.97(8), N1–Mg–N1′ = 106.11(13), N3–Mg–N3′ = 132.43(13), N1–Mg–N3′ = 165.96(10), N1–N2–N3 = 110.1(2), N2–N1–C11 = 109.6(2), N2–N3–C31 = 112.7(2), N2–N1–C11–C12 = 54.7(3), N2–N3–C31–C36 = 54.6(3). Selected bond lengths (Å), angles and dihedral angles (°) for 4c: Mg–N1 = 2.0863(19), Mg–N3 = 2.0770(19), Mg–N2 = 2.568(2), Mg–N4 = 2.1151(19), Mg–N6 = 2.0673(19), Mg–N5 = 2.555(2), N1–N2 = 1.315(2), N2–N3 = 1.323(2), N4–N5 = 1.312(2), N5–N6 = 1.324(2), N1–Mg–N3 = 61.39(7), N4–Mg–N6 = 61.81(7), N1–Mg–N4 = 120.68(8), N1–Mg–N6 = 138.30(8), N3–Mg–N4 = 135.20(8), N3–Mg–N6 = 150.35(8), N2–N1–C11–C16 = −46.7(3), N2–N3–C31–C32 = −25.8(3), N5–N4–C61–C66 = −60.7(3), N5–N6–C91–C96 = 35.5(3).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Interplanar angles and τ4 parameters [25] in four-coordinate magnesium triazenides, amidinates, guanidinates and β-diketiminates.
Table 1.
Interplanar angles and τ4 parameters [25] in four-coordinate magnesium triazenides, amidinates, guanidinates and β-diketiminates.
| Compound 1 | γ (°) | τ4 | Ref. |
|---|---|---|---|
| Triazenides | |||
| [Mg{N3(Me4Ter)2}2] 4b | 9.6 | 0.20 | |
| [Mg{N3((Dmp)Mph)2}2] 4c | 83.6 | 0.51 | |
| Amidinates | |||
| [Mg{DipN{C(pTol)}NDip}2] | 13.3 | 0.10 | [26] |
| [Mg{DipN{C(Me)}NDip}2] 2 | 54.1/54.9 | 0.40/0.41 | [16] |
| [Mg{DipN{C(cHex)}NDip}2] | 61.3 | 0.45 | [27] |
| [Mg{DipN{C(3,5-Me2C6H3)}NDip}2] | 76.4 | 0.56 | [27] |
| [Mg{MesN{C(tBu)}NMes}2] | 80.3 | 0.57 | [28] |
| [Mg{tBuN{C(Ph)}NtBu}2] | 89.5 | 0.58 | [29] |
| [Mg{iPrN{C(Dmp)}NiPr}2] | 88.1 | 0.60 | [30] |
| Guanidinates | |||
| [Mg{MesN{C(NcHex)}NMes}2] | 8.6 | 0.06 | [20] |
| β-Diketiminates | |||
| [Mg(HC{C(Me)N(NiPr2)}2)2] | 89.5 | 0.83 | [31] |
| [Mg(HC{C(Me)N(iPr)}2)2] | 88.9 | 0.88 | [32] |
| [Mg(HC{C(Me)N(tBu)}2)2] | 88.4 | 0.92 | [32] |
| [Mg(HC{C(Ph)N(SiMe3)}2)2] | 89.0 | 0.92 | [33] |
1 cHex = cyclohexyl; Dip = 2,6-iPr2C6H3; Dmp = 2,6-Mes2C6H3; Mes = 2,4,6-Me3C6H2; Me4Ter = 2,6-(3,5-Me2C6H3)2C6H3; Mph = 2-MesC6H4; pTol = p-tolyl. 2 Two independent molecules.
Table 2.
Important structural parameters (av. values (Å, °)) in magnesium triazenides.
| Compound 1 | Cn | Mg–N | N–Mg–N | Mg–N–C | Ref. |
|---|---|---|---|---|---|
| [Mg{N3(Dmp)Tph}I(OEt2)] 2a | 4 | 2.102 | 61.5 | 151.4 | |
| [Mg{N3(Me4Ter)2}I(OEt2)] 2b | 4 | 2.098 | 61.0 | 147.3 | |
| [Mg3I4{N3(Dmp)Tph)}2] 3a | 4 | 2.074 | 62.4 | 152.0 | |
| [Mg3I4{N3(Me4Ter)2}2] 3b | 4 | 2.070 | 61.8 | 148.9 | |
| [Mg{N3(Me4Ter)2}2] 4b | 4 | 2.128 | 61.0 | 151.3 | |
| [Mg{N3(Dmp)Mph)}2] 4c | 4 | 2.086 | 61.6 | 145.5 | |
| [Mg{N3(Dmp)Tph}I(thf)] | 4 | 2.093 | 61.9 | 147.0 | [11] |
| [Mg{N3(Dip)2}2(OEt2)] | 5 | 2.137 | 60.2 | 150.6 | [16] |
| [Mg(nBu){N3(Mes)2}(tmeda)] | 5 | 2.202 | 58.2 | 150.3 | [15] |
| [Mg{N3(pTol)2}2(thf)2] | 6 | 2.183 | 58.8 | 149.6 | [14] |
| [Mg{N3(Mes)2}2(thf)2] | 6 | 2.181 | 59.1 | 150.2 | [15] |
1 Dip = 2,6-iPr2C6H3; Dmp = 2,6-Mes2C6H3; Mes = 2,4,6-Me3C6H2; Me4Ter = 2,6-(3,5-Me2C6H3)2C6H3; Mph = 2-MesC6H4; pTol = p-tolyl; Tph = 2-TripC6H4 with Trip = 2,4,6-i-Pr3C6H2.
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Vinduš, D.; Niemeyer, M. Hetero- and Homoleptic Magnesium Triazenides. Inorganics 2017, 5, 33. https://doi.org/10.3390/inorganics5020033
AMA Style
Vinduš D, Niemeyer M. Hetero- and Homoleptic Magnesium Triazenides. Inorganics. 2017; 5(2):33. https://doi.org/10.3390/inorganics5020033
Chicago/Turabian StyleVinduš, Denis, and Mark Niemeyer. 2017. "Hetero- and Homoleptic Magnesium Triazenides" Inorganics 5, no. 2: 33. https://doi.org/10.3390/inorganics5020033
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.