
Theoretical Insights into the Regiodivergence in Ni-Catalyzed [2+2+2] Cycloaddition of Unsymmetric Diynes and CO2


Department of Chemistry, College of Science, Shanghai Engineering Research Center of Organ Repair, Shanghai University, Shanghai 200444, China
*
Author to whom correspondence should be addressed.
†
These authors contributed equally to this work.
Inorganics 2024, 12(2), 39; https://doi.org/10.3390/inorganics12020039
Submission received: 11 December 2023
/
Revised: 15 January 2024
/
Accepted: 17 January 2024
/
Published: 25 January 2024
(This article belongs to the Special Issue Transition Metal Photocatalysts and Catalytic Reaction Mechanistic Studies)
Abstract
:To achieve the peak of carbon dioxide emission and carbon neutrality, utilizing it as a renewable carbon unit in organic synthesis presents an effective chemical solution for sustainable development. In this study, we report a theoretical investigation into the reaction mechanism and the regiodivergence of the Ni-catalyzed [2+2+2] cycloaddition of unsymmetric diynes and CO2 by using DFT calculations. The reaction mechanisms can be classified into two types: one is related to the oxidative coupling of the C≡C moiety with CO2, and the other is related to the oxidative coupling of the two C≡C moieties of diyne. In each type, two possible paths were proposed depending upon the positions of the substituents (H and silyl). Our calculation results indicate that the oxidative coupling of the C≡C moiety and CO2 favors the positions of H-substituent, while the oxidative coupling of the two C≡C moieties is beneficial for inserting CO2 at the positions of silyl-substituent. The regiodivergence is controlled by substrate chain-length and ligand in the different reaction mechanisms.

1. Introduction
Carbon dioxide (CO2) has been recognized as a greenhouse gas causing global warming. To attain the peak of carbon dioxide emissions and carbon neutrality, employing it as a renewable carbon unit in organic synthesis stands out as an effective chemical solution for sustainable development [1,2,3,4]. However, due to its chemical inertness, the widespread application of CO2 in chemical reactions is limited by thermodynamic considerations [5,6]. Transforming CO2 into other organic compounds as a feedstock remains challenging, often requiring sluggish reactivity and harsh reaction conditions such as high temperatures and pressure [7,8,9]. Various processes and technologies for activating CO2 are under development in different research fields, and recent decades have witnessed significant contributions from organic chemists in the field of CO2 chemistry [10,11,12,13,14,15,16]. Although the fixation of CO2 was initially conducted using stoichiometric organometallic reagents under harsh reaction conditions, several transition metal catalysis methods have emerged as promising approaches for the utilization of CO2 in recent years. [17,18,19,20,21,22,23,24,25,26,27] Among these, the Ni-catalyzed oxidative coupling of CO2 and unsaturated compounds has garnered attention [28,29,30,31,32,33,34,35,36,37,38]. Notably, studies by Tsuda and Saegusa et al. demonstrated intriguing regiodivergence in the Ni-catalyzed [2+2+2] cycloaddition of unsymmetric diynes and CO2 (Scheme 1) [33]. For instance, the Ni-catalyzed coupling of CO2 and diyne 1 with monodentate ligands (PR3) regiospecifically afforded the product 2, a 6-(trimethylsilyl)-substituted bicyclic α-pyrone containing a fused cyclohexane ring [39]. In contrast, the isomeric product 3 could not be obtained. When bidentate ligands were used, the isomeric product 3 became the major product, while 2 became the minor one. For diyne 4 tethered with a longer chain, the Ni-catalyzed coupling of CO2 regiospecifically generated the product 6 using both monodentate ligands and bidentate ligands. Although plausible mechanistic paths have been proposed by experimentalists [31], more detailed mechanistic information and a deeper understanding for the catalytic reaction are still valuable. Particularly, the regiodivergence involved in the Ni-catalyzed reaction remains an intriguing and unresolved issue, as raised by Shi et al. [40] and Lin et al. [41], without further mechanistic studies. Although the mechanism of the experimentally observed formation of the five-membered nickelacarboxylate complex in the Ni-assisted oxidative coupling of CO2 and C2H4 was revealed by means of density functional calculations [42], and the mechanism of the Ni-catalyzed [2+2+2] cycloaddition of unsymmetric diynes and CO2 with N,P-bidentate ligand was theoretically studied [43], the regiodivergence with different substrates and ligands remains an unsolved debated topic in recent decades.
In this paper, we present a density functional theory (DFT) study on the Ni-catalyzed [2+2+2] cycloaddition of diynes with CO2, as reported by Tsuda and Saegusa et al., aiming to give a chemical insight into the more detailed mechanistic information and the regioselectivity inherent in the reaction. These findings should prove valuable in the design of improved catalysts and ligands for carbon dioxide activation.
2. Results and Discussion
The Ni-catalyzed [2+2+2] cycloaddition reaction mechanism of diynes with CO2 catalyzed can be categorized into two types. One is the oxidative coupling of a single C≡C bond moiety in diyne with CO2, while the other entails the coordination of NiL2 with the two C≡C bonds in the diyne, followed by oxidative coupling. To delve deeper into the reaction mechanism, we conducted DFT calculations for the [2+2+2] cyclization reaction with CO2 using diynes 1 and 4 as the model substrates.
2.1. Ni-Catalyzed Coupling of CO2 and Diyne 1 with Monodentate Ligand (PR3)
2.1.1. Oxidative Coupling of the C≡C Moiety and CO2
As depicted in Figure 1, initially, Ni(COD)2 undergoes the ligand exchange with monodentate ligand (PMe3), which is exergonic to transform to Ni(PMe3)2. Then, diyne 1 coordinates with Ni(PMe3)2, and the coordinated intermediate can undergo oxidation coupling through the pathways involving the monophosphine ligand and bisphosphine ligands. The results indicate that an activation energy of 13.1 kcal/mol is required for pathway A of the terminal alkyne through transition state TS1-1a with the monophosphine ligand (black line), while an activation energy of 14.4 kcal/mol is required through TS4-1a with the bisphosphine ligands (red line). Therefore, the oxidative coupling of terminal alkynes with the monophosphine ligand is the main route of the process. Subsequently, the silyl-substituted C≡C bond is forced to coordinate with Ni through the rotation of the C-C single bond, followed by insertion into the Ni-C(sp2) bond through TS2-1a with the energy barrier of 7.1 kcal/mol, generating a seven-membered nickelacycle intermediate INT5-1a. When the bisphosphine ligands coordinate with the Ni center, the energy of TS5-1a is 19.9 kcal/mol higher than that of TS2-1a, indicating that the Ni center prefers a four-coordinate planar structure over a five-coordinate bipyramid structure. Finally, the reductive elimination reaction through TS3-1a with the monophosphine ligand took place readily to form C(sp2)-O bond with the dissociation of the Ni(PMe3)2 complex, leading to the final product 3. The energy barrier of the reductive elimination through TS6-1a with the bisphosphine ligands is 2.7 kcal/mol higher than that of TS3-1a, indicating that the reductive elimination prefers monophosphine ligand pathway.
As defined above, pathway B for the diyne 1 at the positions of TMS-substituent was related to the oxidative coupling of CO2 and INT1-1b. The subsequent oxidative coupling through TS1-1b requires an activation energy of 26.3 kcal/mol with the monophosphine ligand, while the activation energy of 24.5 kcal/mol is required through TS4-1b with the bisphosphine ligands. Both routes on pathway B have significantly higher energy barriers than that on pathway A, attributed to the steric hindrance of the TMS group. Upon reviewing all the energy profiles, it was found that the oxidative coupling of one C≡C bond moiety in the diyne with CO2 is the rate-determining step and the H-substituted C≡C bond favors oxidative coupling with CO2 over the silyl-substituted C≡C bond in the diyne.
2.1.2. Oxidative Coupling of the Two C≡C Moieties
In an alternative mechanism, the coordination of Ni(PMe3)2 occurs initially with two C≡C moieties of diyne 1, with the release of PMe3 to form INT8-1cd (Figure 2). The five-membered nickelacycle INT9-1cd was then formed through TS7-1cd with an activation energy of 10.1 kcal/mol. The subsequent step involves the insertion of CO2 into either the unsubstituted Ni-C(sp2) bond or the TMS-substituted Ni-C(sp2) bond. Surprisingly, despite the steric hindrance with the TMS group, the calculation results show that CO2 prefers to insert into TMS-substituted Ni-C(sp2) bonds. The energy barrier of TS8-1c is 5.6 kcal/mol higher than that of TS8-1d. The formed seven-membered nickelacycle intermediates are INT5-1a and INT5-1b, identical to those in pathways A and B, followed by a final reduction elimination reaction to afford the cyclized products. Upon reviewing all the energy profiles, it becomes evident that the oxidative coupling of the two C≡C moieties with Ni(PMe3) is the rate-determining step, and the insertion of CO2 serves as the regioselectivity-control step, revealing that the formation of 2 is kinetically favorable over 3.
2.2. Ni-Catalyzed Coupling of CO2 and Diyne 1 with Bisdentate Ligand (PN-Ligand)
2.2.1. Oxidative Coupling of the C≡C Moiety and CO2
Initially, Ni(COD)2 undergoes the ligand exchange with bidentate PN-ligand (L2), which is slightly exergonic to transform to Ni(L2)2 with biphosphine coordination. There are several other Ni-complexes such as NiL2 and Ni(L2)2-1-4 with bisdentate, tridentate or tetrodentate coordination, which suffer from higher energies (Figure 3). Subsequently, the replacement of P-N ligand with the unsubstituted C≡C moiety of diyne 1 to form INT1-1e is the most stable starting complex (pathway E). Then, the NiL2-catalyzed oxidative coupling of CO2 and diyne 1 starts from INT1-1e via monodentate N-ligand, P-ligand or bisdentate PN-ligand. The calculated results showed that the PN-coordinated route is the most favorable for the oxidative coupling process and the energy barrier of the bisdentate PN-coordinated TS4-1e is 4.8–15.4 kcal/mol is lower than that of the monodentate P-coordinated TS1-1e’ or N-coordinated TS1-1e. The formed five-membered nickelacycle intermediate INT6-1e with PN coordination exhibits the lowest energy and the greatest stability. Then, the TMS-substituted C≡C bond was coordinated with the Ni center, followed by insertion into the Ni-C(sp2) bond through TS2-1e with monodentate N-ligand, TS2-1e’ with monodentate P-ligand or or TS5-1e with bisdentate PN-ligand to generate the seven-membered nickelacycle intermediates INT5-1e and INT7-1e. Surprisingly, TS2-1e’ with monodentate P-ligand is favored over TS2-1e and TS5-1e by 4.6–4.9 kcal/mol, indicating that the P-coordinated route is the most favorable for the insertion process of the second C≡C bond. The formed four-coordinate nickelacycle INT7-1e is much more stable than three-coordinate nickelacycle INT5-1e. Finally, the subsequent reductive elimination via TS6-1e is facile, forming the O-C(sp2) bond to obtain the final product 3. The rate-determining step here shifts to the insertion of the TMS-substituted C≡C moiety into a C(sp2)-Ni bond, and the formation of 3 is kinetically favorable over 2.
As mentioned above, pathway F for the diyne 1 at the positions of the TMS-substituent was related to the oxidative coupling of CO2 and INT1-1f (−11.1 kcal/mol), which is higher in energy than INT1-1e (−16.7 kcal/mol). The following oxidative coupling through TS1-1f required an activation energy of 29.9 kcal/mol with monodentate N-ligand, while the activation energy of 15.0 kcal/mol was required through TS4-1f with bisdentate PN-ligand. Both routes on pathway F have significantly higher energy barriers than that on pathway E, due to the steric hindrance of the TMS group.
2.2.2. Oxidative Coupling of the Two C≡C Moieties
In an alternative mechanism, the coordination of NiL2 with the two C≡C moieties of diyne 1 occurs, resulting in the formation of INT8-1gh with a monodentate N-ligand and INT8-1gh’ with monodentate P-ligand (Figure 4). Obviously, INT8-1gh’ favors over INT8-1gh by 7.1 kcal/mol. The five-membered nickelacycle INT9-1gh’ was then formed through TS7-1gh’ with an activation energy of 8.6 kcal/mol, which is much lower than that of TS7-1gh. Although the INT9-1gh’ has a parallel energy with INT9-1gh, the following insertion of CO2 with monodentate P-ligand is favored over that with monodentate N-ligand. CO2 is favorable for insertion into TMS-substituted Ni-C(sp2) bonds despite the steric hindrance with the TMS group. The energy barrier of TS8-1h’ with monodentate P-ligand is 6.1 kcal/mol, which is lower than that of TS8-1g’ for the insertion of CO2 at the positions of H-substituent (11.8 kcal/mol). TS8-1h and TS8-1g with monodentate N-ligand require higher energies, leading to the unfavorable pathways for the insertion of CO2 process. The formed seven-membered nickelacycle intermediates INT5-1e/INT5-1f with monodentate N-ligand are less stable than INT7-1e/INT7-1f with bisdentate PN-ligand, which are followed by final reductive elimination reaction through TS6-1e/TS6-1f with the parallel low activation energies to afford the cyclized products 2/3. Upon reviewing all the energy profiles, it is evident that the oxidative coupling of the two C≡C moieties of diyne 1 with NiL2 is the rate-determining step, and the insertion of CO2 serves as the regioselectivity-control step, revealing that the formation of 2 is kinetically favorable over 3.
2.3. Ni-Catalyzed Coupling of CO2 and Diyne 4 with Monodentate Ligand (PR3)
2.3.1. Oxidative Coupling of the C≡C Moiety and CO2
The mechanism involving the oxidative coupling of the C≡C moiety and CO2 for diyne 4 exhibits potential energy profiles similar to those of diyne 1 (Figure 5). The initial step involves the coordination of diyne 4 with Ni(PMe3)2, generating intermediate INT1-4a (pathway A), which can undergo oxidation coupling through TS1-4a (energy barrier of 13.7 kcal/mol) with monophosphine ligand (black line) and TS4-4a (energy barrier of 14.9 kcal/mol) with bisphosphine ligand (red line), indicating that the oxidative coupling with the monophosphine ligand is the main route. Subsequently, the TMS-substituted C≡C bond coordinates with the Ni center, followed by insertion into the Ni-C(sp2) bond through TS2-4a (energy barrier of 8.1 kcal/mol), affording a seven-membered nickelacycle intermediate INT5-4a. Finally, the reductive elimination reaction through TS3-4a (energy barrier of 1.6 kcal/mol) takes place, forming the C(sp2)-O bond and leading to the final product 6. Alternatively, pathway B involving the oxidative coupling of TMS-unsubstituted C≡C moiety and CO2 through TS1-4b with monophosphine ligand and TS4-4b with bisphosphine ligand requires activation energies of 24.5–26.2 kcal/mol, which are significantly higher in energy than those associated with pathway A, attributed to the steric hindrance of the TMS group. Upon reviewing all the energy profiles, it is evident that the oxidative coupling is the rate-determining step and the H-substituted C≡C bond favors oxidative coupling with CO2 over the silyl-substituted C≡C bond in the diyne 4.
2.3.2. Oxidative Coupling of the Two C≡C Moieties
The alternative mechanism commences with the coordination of Ni(PMe3)2 by two C≡C moieties of diyne 4, leading to the formation of INT8-4cd with the release of PMe3 (Figure 6). Subsequently, the five-membered nickelacycle INT9-4cd is formed through TS7-4cd with an activation energy of 15.5 kcal/mol. CO2 then preferentially inserts into TMS-substituted Ni-C(sp2) bonds despite the steric hindrance posed by the TMS group. The energy barrier of TS8-4d was 7.4 kcal/mol, which is 5.9 kcal/mol lower than that of TS8-4c. The resulting seven-membered nickelacycle intermediates, INT5-4a and INT5-4b, are identical to those observed in pathways A and B in Figure 5. The subsequent final reductive elimination reaction yields the cyclized products 5/6. Upon reviewing all the energy profiles, it is evident that the oxidative coupling of the two C≡C moieties with Ni(PMe3) is the rate-determining step, and the insertion of CO2 serves as the regioselectivity-control step, revealing that the formation of 5 is kinetically favored over that of 6.
2.4. Ni-Catalyzed Coupling of CO2 and Diyne 4 with Bisdentate Ligand (PN-Ligand)
2.4.1. Oxidative Coupling of the C≡C Moiety and CO2
The first type of the mechanism for the Ni(L2)2 (L2 = PN-ligand)-catalyzed coupling of CO2 and diyne 4 originates from the coordination of NiL2 with unsubstituted C≡C moiety of diyne 4 via monodentate N-ligand or bisdentate PN-ligand on pathway E (Figure 7). The calculated results show that the PN-coordinated route was more favorable and the energy of the bisdentate PN-coordinated TS4-1e is 5.6 kcal/mol, which is 16.0 kcal/mol lower than that of monodentate N-coordinated TS1-4e. The formed five-membered nickelacycle intermediate INT6-4e with PN coordination exhibits lower energy and greater stability. Subsequently, the TMS-substituted C≡C bond is coordinated with the Ni center, followed by insertion into the Ni-C(sp2) bond through TS2-4e with monodentate N-ligand (energy barrier of 10.8 kcal/mol), TS2-4e’ with monodentate P-ligand (energy barrier of 8.8 kcal/mol) or TS5-4e with bisdentate PN-ligand (energy barrier of 15.8 kcal/mol), generating a seven-membered nickelacycle intermediate INT7-4e. It is noteworthy that the P-coordinated route is the most favorable pathway for the insertion process of the second C≡C bond and the formed four-coordinate nickelacycle INT7-1e with bisdentate PN-ligand is more stable than three-coordinate nickelacycle with monodentate P- or N-ligand. INT7-4e then undergoes facile reductive elimination reaction via TS6-4e to form O-C(sp2) bond, resulting in the final product 6. The rate-determining step in this process is the insertion of the TMS-substituted C≡C moiety into a C(sp2)-Ni bond, and the formation of 6 is kinetically favored over that of 5. Alternatively, pathway F for the diyne 4 at the positions of TMS-substituent is associated with the oxidative coupling of CO2 and INT1-4f. The subsequent oxidative coupling through TS1-4f requires an activation energy of 34.1 kcal/mol with monodentate N-ligand, while the activation energy of 18.8 kcal/mol is required through TS4-4f with bisdentate PN-ligand. Both routes on pathway F have significantly higher energy barriers than that on pathway E, attributed to the steric hindrance of the TMS group.
2.4.2. Oxidative Coupling of the Two C≡C Moieties
Alternatively, the second type of the mechanism is initiated by the coordination of NiL2 by two C≡C moieties of diyne 4, forming INT8-4gh with monodentate N-ligand and INT8-4gh’ with monodentate P-ligand (Figure 8). Similarly, INT8-4gh’ is favored over INT8-4gh by 3.9 kcal/mol. The five-membered nickelacycle INT9-4gh’ was then formed through TS7-4gh’ with an activation energy of 13.8 kcal/mol, which is 7.7 kcal/mol lower than that of TS7-4gh. The formed INT9-4gh’ is also more stable than INT9-4gh. Subsequently, the following insertion of CO2 with monodentate P-ligand is favored over that with monodentate N-ligand, and CO2 is favorable for insertion into the TMS-substituted Ni-C(sp2) bonds. The energy barrier of TS8-4h’ with monodentate P-ligand is 5.3 kcal/mol, which is lower than that of TS8-4g’ for the insertion of CO2 at the positions of H-substituent (10.5 kcal/mol). TS8-4h and TS8-4g with monodentate N-ligand require higher energies, leading to unfavorable pathways for the insertion of CO2 process. The seven-membered nickelacycle intermediates INT7-4e/INT7-4f with bisdentate PN-ligand formed are more stable than other nickelacycle intermediates with monodentate ligand. The nickelacycle intermediates then undergo final reductive elimination reaction through TS6-4e/TS6-4f with low activation energies to afford the cyclized products 5/6. Upon reviewing all the energy profiles, it is evident that the oxidative coupling of the two C≡C moieties with NiL2 is the rate-determining step, and the insertion of CO2 serves as the regioselectivity-control step, revealing that the formation of 5 is kinetically favorable over that of 6.
2.5. The Regiodivergence in Ni-Catalyzed Coupling of CO2 and Diyne 1 and 4
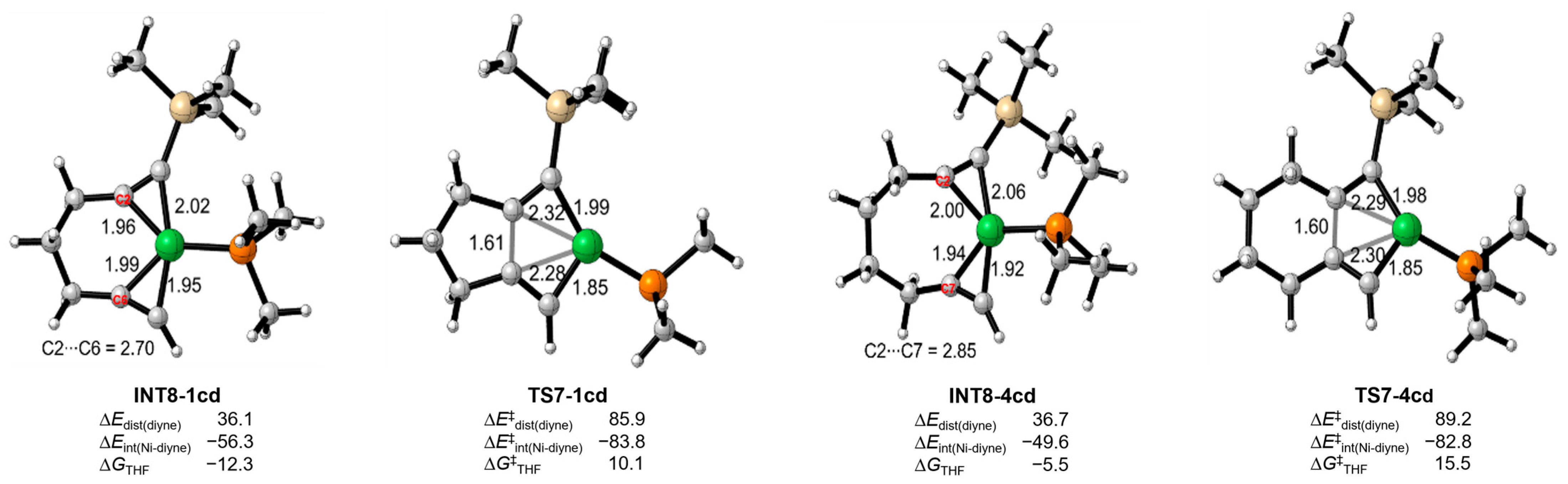
Upon reviewing all the energy profiles, the computational results demonstrate that diyne 1 preferentially undergo the oxidative coupling of the two C≡C moieties with monodentate ligand. This leads to the formation of 2, which is kinetically favored over 3 (ΔΔG‡(2–3) = −3.0 kcal/mol) (Table 1). On the other hand, the use of a bisdentate ligand (NP-ligand) is advantageous for promoting the oxidative coupling of the C≡C moiety and CO2, resulting in the formation of 3, which is both kinetically and thermodynamically favored over 2 (ΔΔG‡(2–3) = 2.0 kcal/mol), and is consistent with the experimental observation [33]. In the case of diyne 4, both monodentate and bisdentate ligands tend to facilitate the oxidative coupling of the C≡C moiety and CO2, favoring the kinetically preferred formation of 6 (ΔΔG‡(5–6) = 1.8–5.0 kcal/mol), which is once again in good agreement with the experimental results [33]. The distinct mechanisms and the regiodivergence observed for diyne 1 and 4 are actually regulated by key steps, including INT8 and TS7 (Figure 9). INT8-1cd exhibits greater stability with enhanced coordination of NiL2 by two C≡C moieties of 1 compared to that of 4. This results in a lower energy barrier through TS7-1cd, establishing the oxidative coupling of the two C≡C moieties as the dominant pathway and providing the distinct regioselectivity. Conversely, INT8-4cd, containing seven-membered ring tension, suffers from a higher energy barrier through TS7-4cd, leading to the unfavorable pathway C/D. These findings comprehensively elucidate the regiodivergence observed in the Ni-catalyzed [2+2+2] cycloaddition reaction of unsymmetric diynes and CO2.
3. Computational Methods
All of the DFT calculations were performed with the Gaussian 09 program package [44]. The geometry optimization of all the minima involved were performed at the B3LYP level of theory [45,46] with a 6-31G(d) + Lanl2DZ (for Ni) basis set (keyword 5D). The structures of the reactants, intermediates, transition states, and products were fully optimized without any restriction. The vibrational frequencies were computed at the same level to check whether each optimized structure is an energy minimum or a transition state and to evaluate its zero-point vibrational energy (ZPVE) and thermal corrections at 298 K. IRC calculations [47,48,49,50] were used to confirm that the transition states found through the optimization calculations connect the related reactants and products. Single-point solvent calculations were performed with a 6-311 + G(d,p) + SDD (for Ni) basis set at the optimized gas–phase geometries for all the intermediates and transition states, using the SMD model [51] in tetrahydrofuran (THF) as a solvent. Through the same approach, full optimization, without any restriction, was carried out for the model reactions. The reported energies are Gibbs free energies in a THF solution (ΔGTHF) (see supplementary materials). Figure 9 was prepared using CYLView, 1.0b [52]
4. Conclusions
In summary, we have carried out a theoretical study on the reaction mechanism of Ni-catalyzed [2+2+2] cycloaddition of unsymmetric diyne and CO2 by using DFT calculations. The reaction mechanisms were categorized into two types: one is related to the oxidative coupling of the C≡C moiety and CO2, and the other is related to the oxidative coupling of the two C≡C moieties of diyne. In each type, two possible paths were proposed depending upon the positions of the substituents (H and silyl). Our calculation results indicated that the oxidative coupling of the C≡C moiety and CO2 favors the positions of H-substituent, while the oxidative coupling of the two C≡C moieties is advantageous for CO2 insertion at the positions of silyl-substituent. The regioselectivity is controlled by the different reaction mechanisms. For diyne 1 containing a three-carbon linker length, it is preferable to undergo the oxidative coupling of the two C≡C moieties with monodentate ligand (PR3), resulting in the final product 2. In contrast, the bisdentate PN-ligand promotes the oxidative coupling of the C≡C moiety and CO2, leading to the formation of product 3. On the other hand, diyne 4, with a four-carbon linker length, favors the oxidative coupling of the C≡C moiety and CO2 with both monodentate ligand (PR3) and bisdentate PN-ligand, giving the final product 6. This work not only provides a chemical insight into the detailed mechanistic information and the regioselectivity involved in the reaction, but also promotes the design of better catalysts and ligands for carbon dioxide activation.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/inorganics12020039/s1, DFT-computed energies and coordinates of all stationary points.
Author Contributions
Conceptualization, X.L.; calculation investigation, K.Z., Q.H. and C.Y.; writing—original draft preparation, K.Z., Q.H. and X.L.; supervision, X.L. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the National Natural Science Foundation of China, grant number 22101168, and Shanghai Pujiang Talent Scholar, grant number 21PJ1403700.
Data Availability Statement
The data presented in this study are available on request from the corresponding author.
Acknowledgments
We appreciate the High-Performance Computing Center of Shanghai University, and Shanghai Engineering Research Center of Intelligent Computing System (No. 19DZ2252600) for providing the computing resources.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Sakakura, T.; Choi, J.; Yasuda, H. Transformation of Carbon Dioxide. Chem. Rev. 2007, 107, 2365–2387. [Google Scholar] [CrossRef] [PubMed]
- Darensbourg, D.J. Making Plastics from Carbon Dioxide: Salen Metal Complexes as Catalysts for the Production of Polycarbonates from Epoxides and CO2. Chem. Rev. 2007, 107, 2388–2410. [Google Scholar] [CrossRef] [PubMed]
- Correa, A.; Martin, R. Metal-Catalyzed Carboxylation of Organometallic Reagents with Carbon Dioxide. Angew. Chem. Int. Ed. 2009, 48, 6201–6204. [Google Scholar] [CrossRef]
- Boogaerts, I.I.F.; Nolan, S.P. Direct C–H carboxylation with complexes of the coinage metals. Chem. Commun. 2011, 47, 3021–3024. [Google Scholar] [CrossRef] [PubMed]
- Schneider, J.; Jia, H.; Muckerman, J.T.; Fujita, E. Thermodynamics and kinetics of CO2, CO, and H+ binding to the metal centre of CO2 reduction catalysts. Chem. Soc. Rev. 2012, 41, 2036–2051. [Google Scholar] [CrossRef]
- Li, Y.N.; Ma, R.; He, L.-N.; Diao, Z.-F. Homogeneous hydrogenation of carbon dioxide to methanol. Catal. Sci. Technol. 2014, 4, 1498. [Google Scholar] [CrossRef]
- Ran, C.-K.; Chen, X.-W.; Gui, Y.-Y.; Li, J.; Son, L.; Ren, K.; Yu, D.-G. Recent advances in asymmetric synthesis with CO2. Sci. China Chem. 2020, 63, 1336–1351. [Google Scholar] [CrossRef]
- Pradhan, S.; Das, S. Recent Advances on the Carboxylations of C(sp3)–H Bonds Using CO2 as the Carbon Source. Synlett 2023, 34, 1327–1342. [Google Scholar]
- Burkart, M.D.; Hazari, N.; Tway, C.L.; Zeitler, E.L. Opportunities and challenges for Catalysis in Carbon Dioxide Utilization. ACS Catal. 2019, 9, 7937–7956. [Google Scholar] [CrossRef]
- Wang, S.; Xi, C. Recent advances in nucleophile-triggered CO2-incorporated cyclization leading to heterocycles. Chem. Soc. Rev. 2019, 48, 382–404. [Google Scholar] [CrossRef]
- Zhang, Z.; Gong, L.; Zhou, X.-Y.; Yan, S.-S.; Li, J.; Yu, D.-G. Radical-Type Difunctionalization of Alkenes with CO2. Acta Chim. Sin. 2019, 77, 783–793. [Google Scholar] [CrossRef]
- Song, L.; Jiang, Y.-X.; Gui, Y.-Y.; Zhou, X.-Y.; Yu, D.-G. CO2 = CO + [O]: Recent advances in carbonylation of C–H bonds with CO2. Chem. Commun. 2020, 56, 8355–8367. [Google Scholar] [CrossRef] [PubMed]
- Cao, Y.; He, X.; Wang, N.; Li, H.-R.; He, L.-N. Photochemical and Electrochemical Carbon Dioxide Utilization with Organic Compounds. Chin. J. Chem. 2018, 36, 644–659. [Google Scholar] [CrossRef]
- Dabral, S.; Schauba, T. The Use of Carbon Dioxide (CO2) as a Building Block in Organic Synthesis from an Industrial Perspective. Adv. Synth. Catal. 2019, 361, 223–246. [Google Scholar] [CrossRef]
- Ran, C.-K.; Liao, L.-L.; Gao, T.-Y.; Gui, Y.-Y.; Yu, D.-G. Recent progress and challenges in carboxylation with CO2. Curr. Opin. Green Sust. Chem. 2021, 32, 100525. [Google Scholar] [CrossRef]
- Bertuzzi, G.; Cerveri, A.; Lombardi, L.; Bandini, M. Tandem functionalization-carboxylation reactions of πsystems with CO2. Chin. J. Chem. 2021, 39, 3116–3126. [Google Scholar] [CrossRef]
- Tortajada, A.; Juliá-Hernández, F.; Börjesson, M.; Moragas, T.; Martin, R. Transition-Metal-Catalyzed Carboxylation Reactions with Carbon Dioxide. Angew. Chem. Int. Ed. 2018, 57, 15948–15982. [Google Scholar] [CrossRef]
- Chen, Y.-G.; Xu, X.-T.; Zhang, K.; Li, Y.-Q.; Zhang, L.-P.; Fang, P.; Mei, T.-S. Transition-Metal-Catalyzed Carboxylation of Organic Halides and Their Surrogates with Carbon Dioxide. Synthesis 2018, 50, 35–48. [Google Scholar]
- Zhang, L.; Li, Z.; Takimoto, M.; Hou, Z. Carboxylation Reactions with Carbon Dioxide Using N-Heterocyclic Carbene Copper Catalysts. Chem. Rec. 2020, 20, 494–512. [Google Scholar] [CrossRef]
- Cokoja, M.; Bruckmeier, C.; Rieger, B.; Herrmann, W.A.; Kühn, F.E. Transformation of carbon dioxide with homogeneous transition-metal catalysts: A molecular solution to a global challenge? Angew. Chem. Int. Ed. 2011, 50, 8510–8557. [Google Scholar] [CrossRef]
- Tsuji, Y.; Fujihara, T. Carbon dioxide as a carbon source in organic transformation: Carbon–carbon bond forming reactions by transition-metal catalysts. Chem. Commun. 2012, 48, 9956–9964. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Hou, Z. N-Heterocyclic carbene (NHC)–copper-catalysed transformations of carbon dioxide. Chem. Sci. 2013, 4, 3395. [Google Scholar] [CrossRef]
- Song, J.; Liu, Q.; Liu, H.; Jiang, X. Recent Advances in Palladium-Catalyzed Carboxylation with CO2. Eur. J. Org. Chem. 2018, 6, 696–713. [Google Scholar] [CrossRef]
- Sang, R.; Hu, Y.; Razzaq, R.; Mollaert, G.; Atia, H.; Bentrup, U.; Sharif, M.; Neumann, H.; Junge, H.; Jackstell, R.; et al. A practical concept for catalytic carbonylations using carbon dioxide. Nat. Commun. 2022, 13, 4432. [Google Scholar] [CrossRef] [PubMed]
- Wei, D.; Sang, R.; Sponholz, P.; Junge, H.; Beller, M. Reversible hydrogenation of carbon dioxide to formic acid using a Mn-pincer complex in the presence of lysine. Nat. Energy 2022, 7, 438. [Google Scholar] [CrossRef]
- Marx, M.; Frauendorf, H.; Spannenberg, A.; Neumann, H.; Beller, M. Revisiting Reduction of CO2 to Oxalate with First-Row Transition Metals: Irreproducibility, Ambiguous Analysis, and Conflicting Reactivity. JACS Au 2022, 2, 731. [Google Scholar] [CrossRef]
- Qiao, C.; Shi, W.; Brandolese, A.; Benet-Buchholz, J.; Escudero-Adán, E.; Kleij, A.W. A Novel Catalytic Route to Polymerizable Bicyclic Cyclic Carbonate Monomers from Carbon Dioxide. Angew. Chem. Int. Ed. 2022, 61, e202205053. [Google Scholar] [CrossRef]
- Tortajada, A.; Börjesson, M.; Martin, R. Nickel-Catalyzed Reductive Carboxylation and Amidation Reactions. Acc. Chem. Res. 2021, 54, 3941–3952. [Google Scholar] [CrossRef]
- Louie, J.; Gibby, J.E.; Farnworth, M.V.; Tekavec, T.N. Efficient Nickel-Catalyzed [2+2+2] Cycloaddition of CO2 and Diynes. J. Am. Chem. Soc. 2002, 124, 15188–15189. [Google Scholar] [CrossRef]
- Tsuda, T.; Morikawa, S.; Sumiya, R.; Saegusa, T. Nickel(0)-catalyzed cycloaddition of diynes and carbon dioxide to give bicyclic α-pyrones. J. Org. Chem. 1988, 53, 3140–3145. [Google Scholar] [CrossRef]
- Tekavec, T.N.; Arif, A.; Louie, J. Regioselectivity in Nickel(0) catalyzed cycloadditions of carbon dioxide with diynes. Tetrahedron 2004, 60, 7431–7437. [Google Scholar] [CrossRef]
- Takimoto, M.; Kawamura, M.; Mori, M. Nickel(0)-Mediated Sequential Addition of Carbon Dioxide and Aryl Aldehydes into Terminal Allenes. Org. Lett. 2003, 5, 2599–2601. [Google Scholar] [CrossRef] [PubMed]
- Tsuda, T.; Morikawa, S.; Hasegawa, N.; Saegusa, T. Nickel(0)-catalyzed cycloaddition of silyl diynes with carbon dioxide to silyl bicyclic α-pyrones. J. Org. Chem. 1990, 55, 2978–2981. [Google Scholar] [CrossRef]
- Cossi, M.; Rega, N.; Scalmani, G.; Barone, V. Energies, structures, and electronic properties of molecules in solution with the C-PCM solvation model. J. Comput. Chem. 2003, 24, 669–681. [Google Scholar] [CrossRef]
- Williams, C.M.; Johnson, J.B.; Rovis, T. Nickel-Catalyzed Reductive Carboxylation of Styrenes Using CO2. J. Am. Chem. Soc. 2008, 130, 14936–14937. [Google Scholar] [CrossRef]
- Li, S.; Yuan, W.; Ma, S. Highly Regio- and Stereoselective Three-Component Nickel-Catalyzed syn-Hydrocarboxylation of Alkynes with Diethyl Zinc and Carbon Dioxide. Angew. Chem. Int. Ed. 2011, 50, 2578–2582. [Google Scholar] [CrossRef]
- Walter, D.; Braunlich, G. Aktivierung von CO2 an übergangsmetallzentren: Zum ablauf der homogen-katalytischen Bildung von 2-Pyron aus Kohlendioxid und Hex-3-in an Nickel(0)-Fragmenten. J. Org. Chem. 1992, 436, 109–119. [Google Scholar] [CrossRef]
- Diccianni, J.B.; Diao, T. Mechanisms of nickel-catalyzed cross-coupling reactions. Trends Chem. 2019, 1, 830–844. [Google Scholar] [CrossRef]
- Dieter, R.K.; Fishpaugh, J.R. A versatile synthesis of α-pyrones. J. Org. Chem. 1983, 48, 4439–4441. [Google Scholar] [CrossRef]
- Huang, K.; Sun, C.-L.; Shi, Z.-J. Transition-metal-catalyzed C–C bond formation through the fixation of carbon dioxide. Chem. Soc. Rev. 2011, 40, 2435–2452. [Google Scholar] [CrossRef]
- Fan, T.; Chen, X.; Lin, Z. Theoretical studies of reactions of carbon dioxide mediated and catalysed by transition metal complexes. Chem. Commun. 2012, 48, 10808–10828. [Google Scholar] [CrossRef] [PubMed]
- Pápai, I.; Schubert, G.; Mayer, I.; Besenyei, G.; Aresta, M. Mechanistic details of nickel(0)-assisted oxidative coupling of CO2 with C2H4. Organometallics 2004, 23, 5252–5259. [Google Scholar] [CrossRef]
- Zhao, Y.; Liu, Y.; Bi, S.; Liu, Y. Theoretical investigation on the regioselectivity of Ni(COD)2-catalyzed [2+2+2] cycloaddition of unsymmetric diynes and CO2. J. Organomet. Chem. 2014, 758, 45–54. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, Revision B.01; Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef] [PubMed]
- Fukui, K. Formulation of the reaction coordinate. J. Phys. Chem. 1970, 74, 4161–4163. [Google Scholar] [CrossRef]
- Ishida, K.; Morokuma, K.; Komornicki, A. The intrinsic reaction coordinate. An ab initio calculation for HNC → HCN and H− + CH4 → CH4 + H−. J. Chem. Phys. 1977, 66, 2153–2156. [Google Scholar] [CrossRef]
- González, C.; Schlegel, H.B. An improved algorithm for reaction path following. J. Chem. Phys. 1989, 90, 2154–2161. [Google Scholar] [CrossRef]
- González, C.; Schlegel, H.B. Reaction path following in mass-weighted internal coordinates. J. Phys. Chem. 1990, 94, 5523–5527. [Google Scholar] [CrossRef]
- Marenich, A.V.; Cramer, C.J.; Truhlar, D.G. Universal Solvation Model Based on Solute Electron Density and on a Continuum Model of the Solvent Defined by the Bulk Dielectric Constant and Atomic Surface Tensions. J. Phys. Chem. B 2009, 113, 6378–6396. [Google Scholar] [CrossRef]
- Legault, C.Y. CYLView, 1.0b; Université de Sherbrooke: Montreal, QC, Canada, 2009; Available online: http://www.cylview.org (accessed on 18 January 2024).
Scheme 1.
Reported nickel-catalyzed [2+2+2] cycloaddition of CO2 and diynes with regioselectivity.

Figure 1.
Potential energy profiles for Ni-catalyzed cycloaddition of diyne 1 and CO2 with monodentate ligand. Pathway A/B: proposed to start with the oxidative coupling of the C≡C moiety and CO2.
Figure 1.
Potential energy profiles for Ni-catalyzed cycloaddition of diyne 1 and CO2 with monodentate ligand. Pathway A/B: proposed to start with the oxidative coupling of the C≡C moiety and CO2.

Figure 2.
Potential energy profiles for Ni-catalyzed cycloaddition of diyne 1 and CO2 with monodentate ligand. Path C/D: proposed to start with the oxidative coupling between the two C≡C moieties.
Figure 2.
Potential energy profiles for Ni-catalyzed cycloaddition of diyne 1 and CO2 with monodentate ligand. Path C/D: proposed to start with the oxidative coupling between the two C≡C moieties.
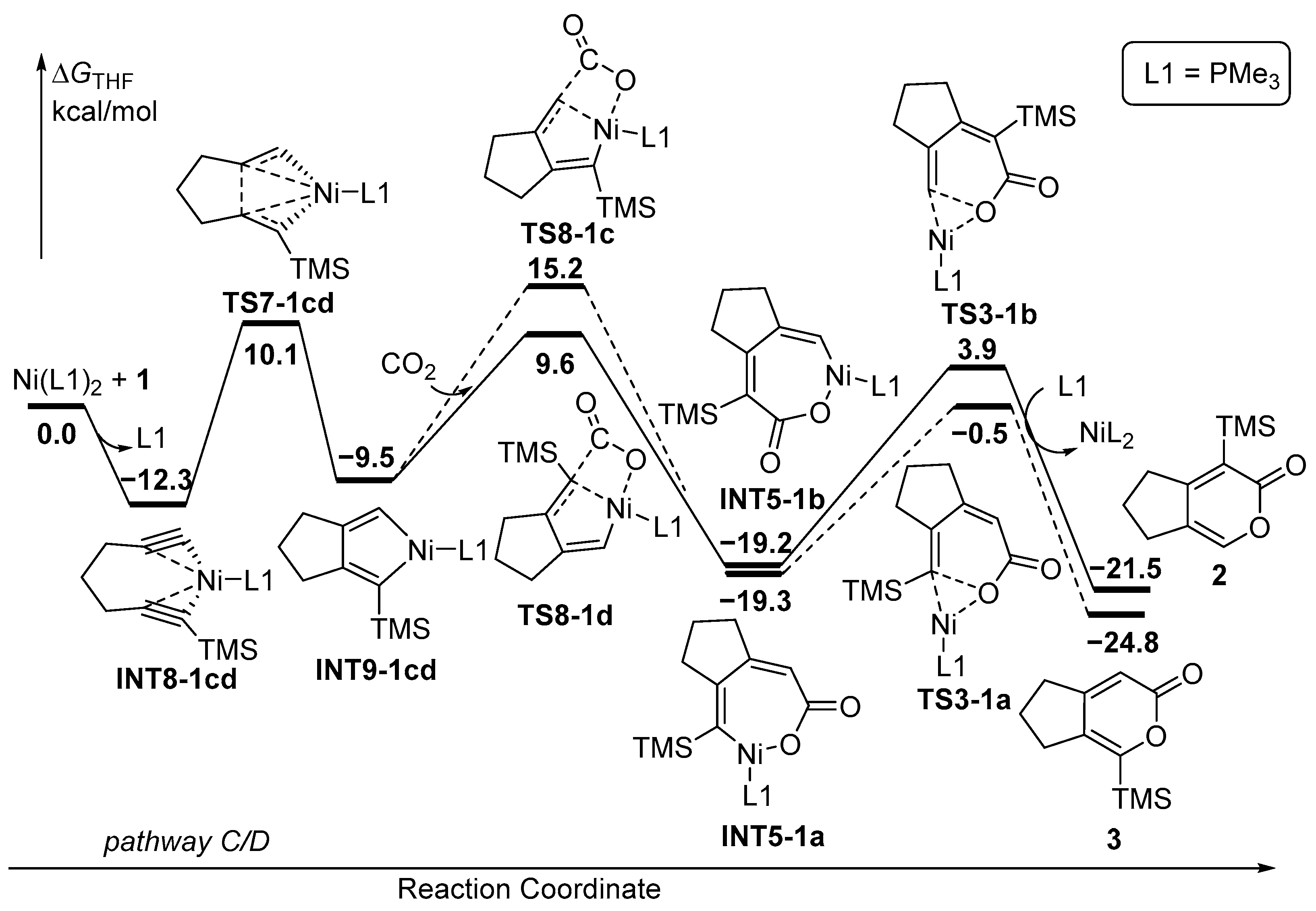
Figure 3.
Potential energy profiles for Ni-catalyzed cycloaddition of diyne 1 and CO2 with bisdentate ligand. Path E/F: proposed to start with the oxidative coupling of the C≡C moiety and CO2.
Figure 3.
Potential energy profiles for Ni-catalyzed cycloaddition of diyne 1 and CO2 with bisdentate ligand. Path E/F: proposed to start with the oxidative coupling of the C≡C moiety and CO2.
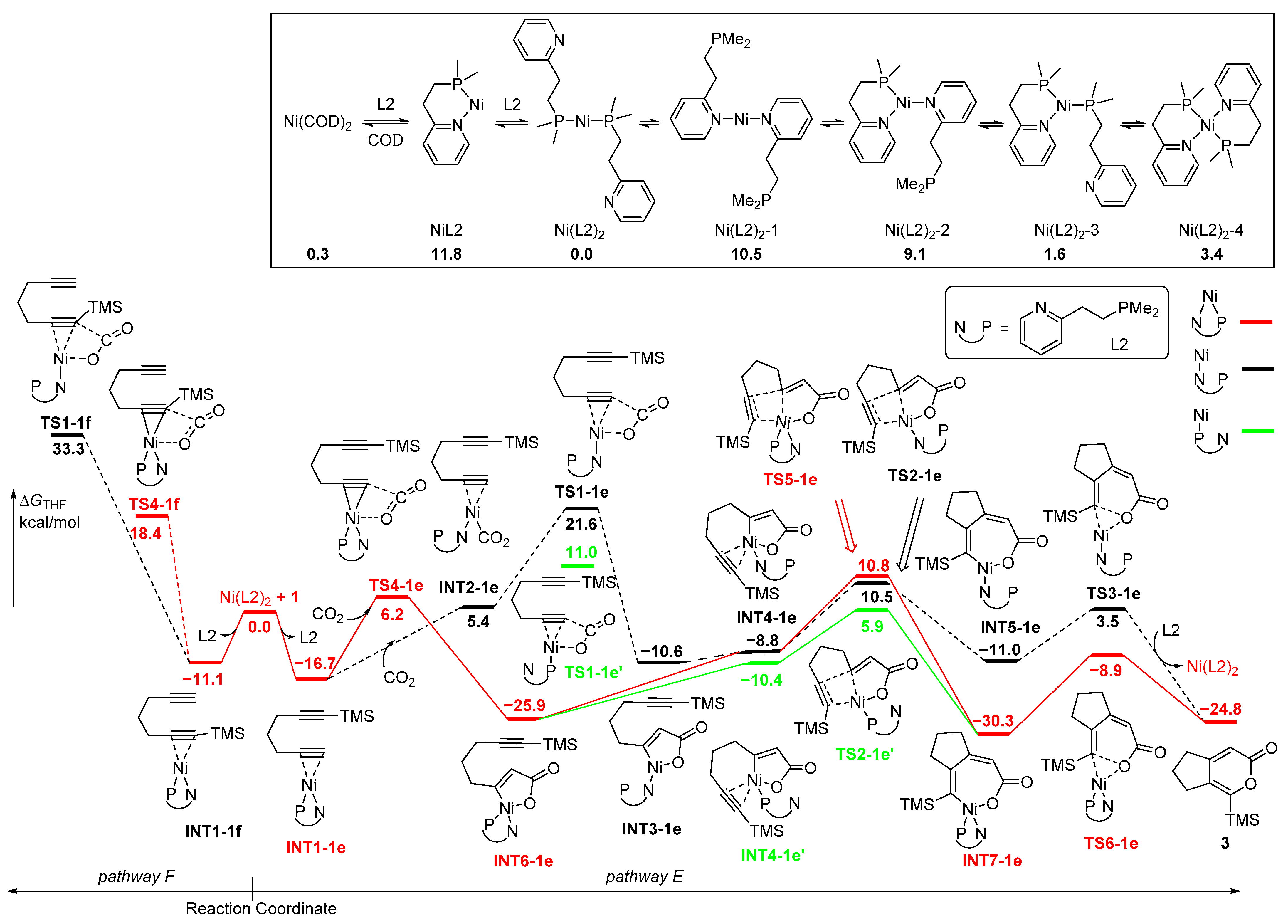
Figure 4.
Potential energy profiles for Ni-catalyzed cycloaddition of diyne 1 and CO2 with bisdentate ligand. Path G/H: proposed to start with the oxidative coupling between the two C≡C moieties.
Figure 4.
Potential energy profiles for Ni-catalyzed cycloaddition of diyne 1 and CO2 with bisdentate ligand. Path G/H: proposed to start with the oxidative coupling between the two C≡C moieties.
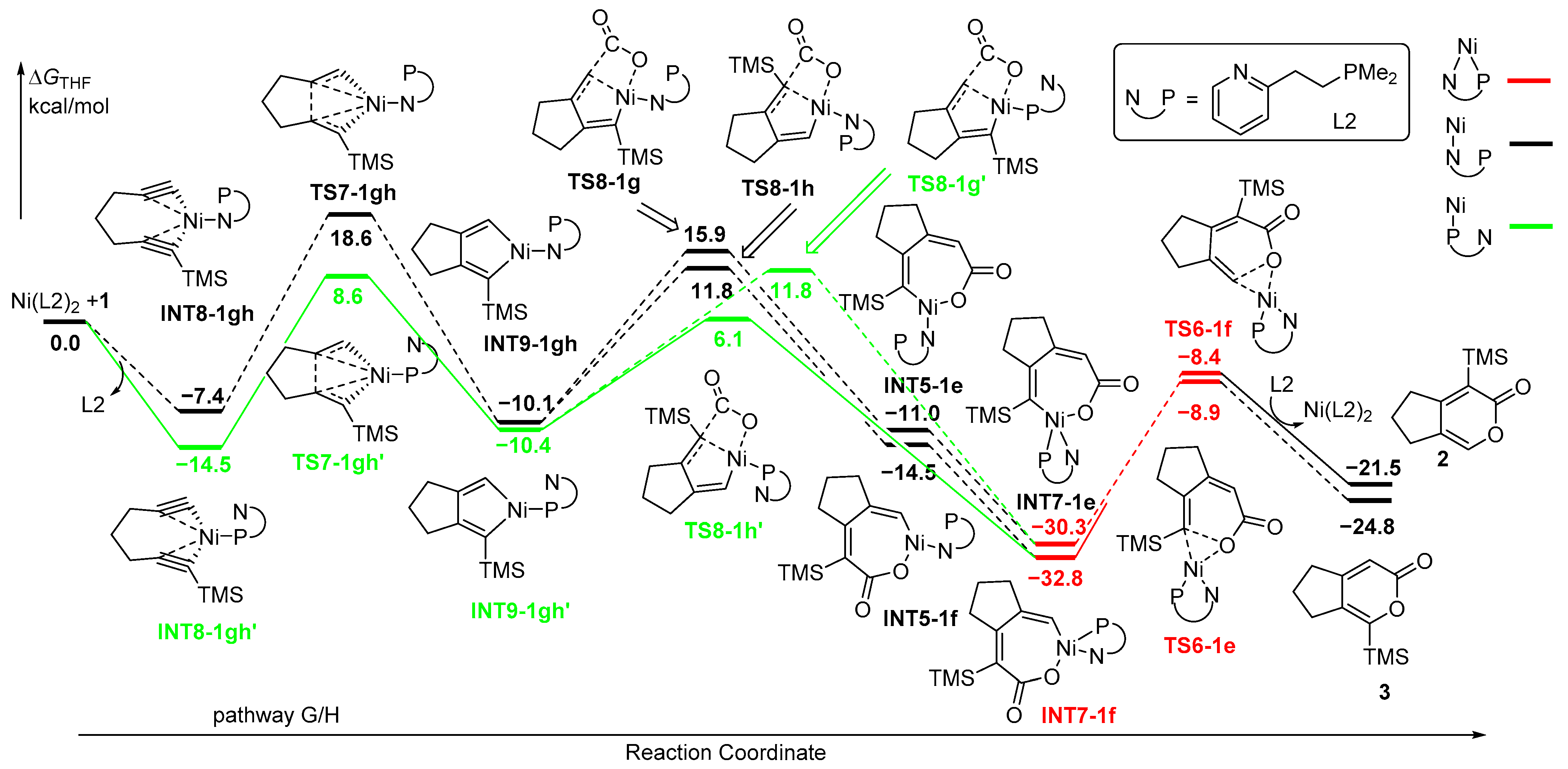
Figure 5.
Potential energy profiles for Ni-catalyzed cycloaddition of diyne 4 and CO2 with monodentate ligand. Pathway A/B: proposed to start with the oxidative coupling of the C≡C moiety and CO2.
Figure 5.
Potential energy profiles for Ni-catalyzed cycloaddition of diyne 4 and CO2 with monodentate ligand. Pathway A/B: proposed to start with the oxidative coupling of the C≡C moiety and CO2.

Figure 6.
Potential energy profiles for Ni-catalyzed cycloaddition of diyne 4 and CO2 with monodentate ligand. Path C/D: proposed to start with the oxidative coupling between the two C≡C moieties.
Figure 6.
Potential energy profiles for Ni-catalyzed cycloaddition of diyne 4 and CO2 with monodentate ligand. Path C/D: proposed to start with the oxidative coupling between the two C≡C moieties.
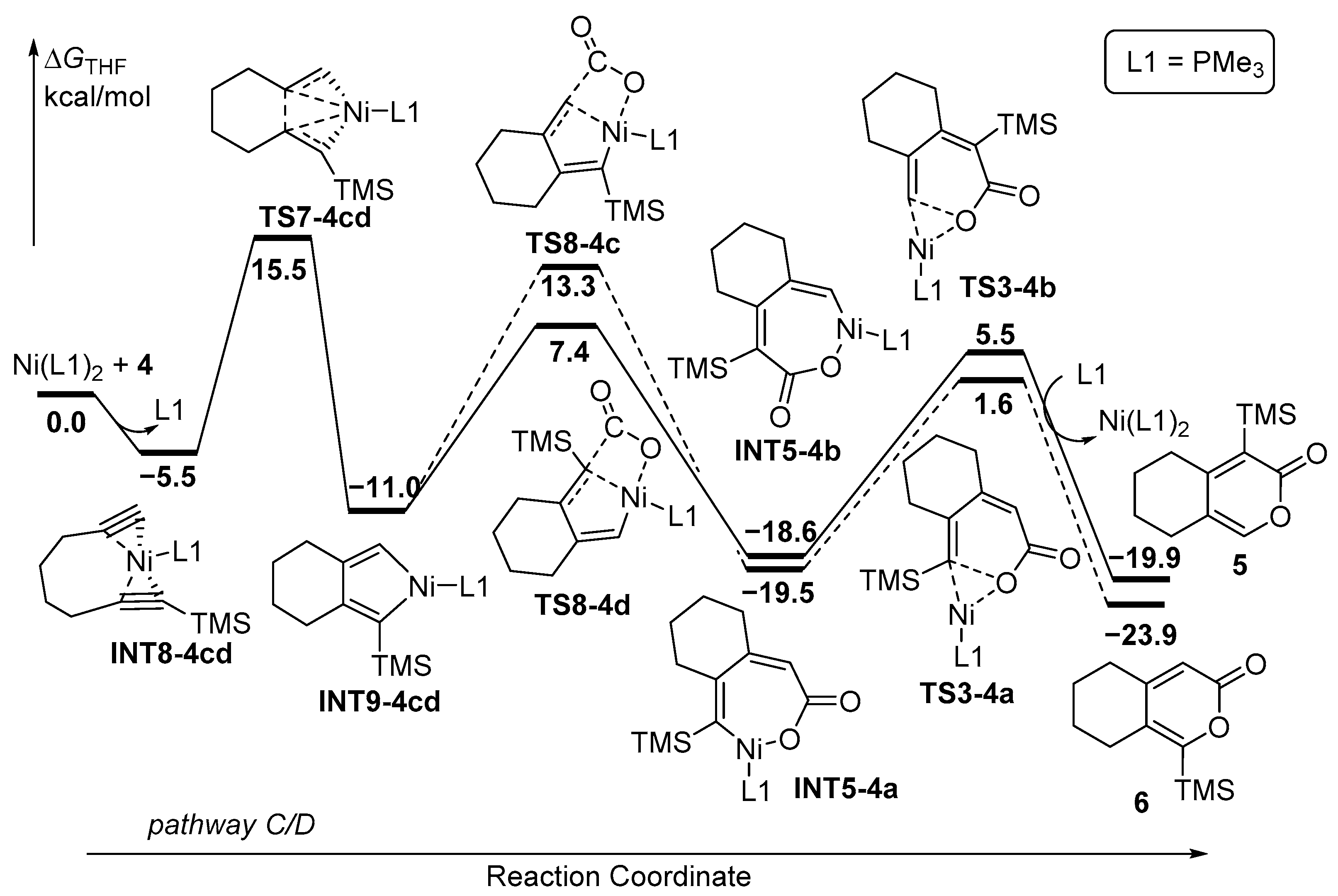
Figure 7.
Potential energy profiles for Ni-catalyzed cycloaddition of diyne 4 and CO2 with bisdentate ligand. Path E/F: proposed to start with the oxidative coupling of the C≡C moiety and CO2.
Figure 7.
Potential energy profiles for Ni-catalyzed cycloaddition of diyne 4 and CO2 with bisdentate ligand. Path E/F: proposed to start with the oxidative coupling of the C≡C moiety and CO2.
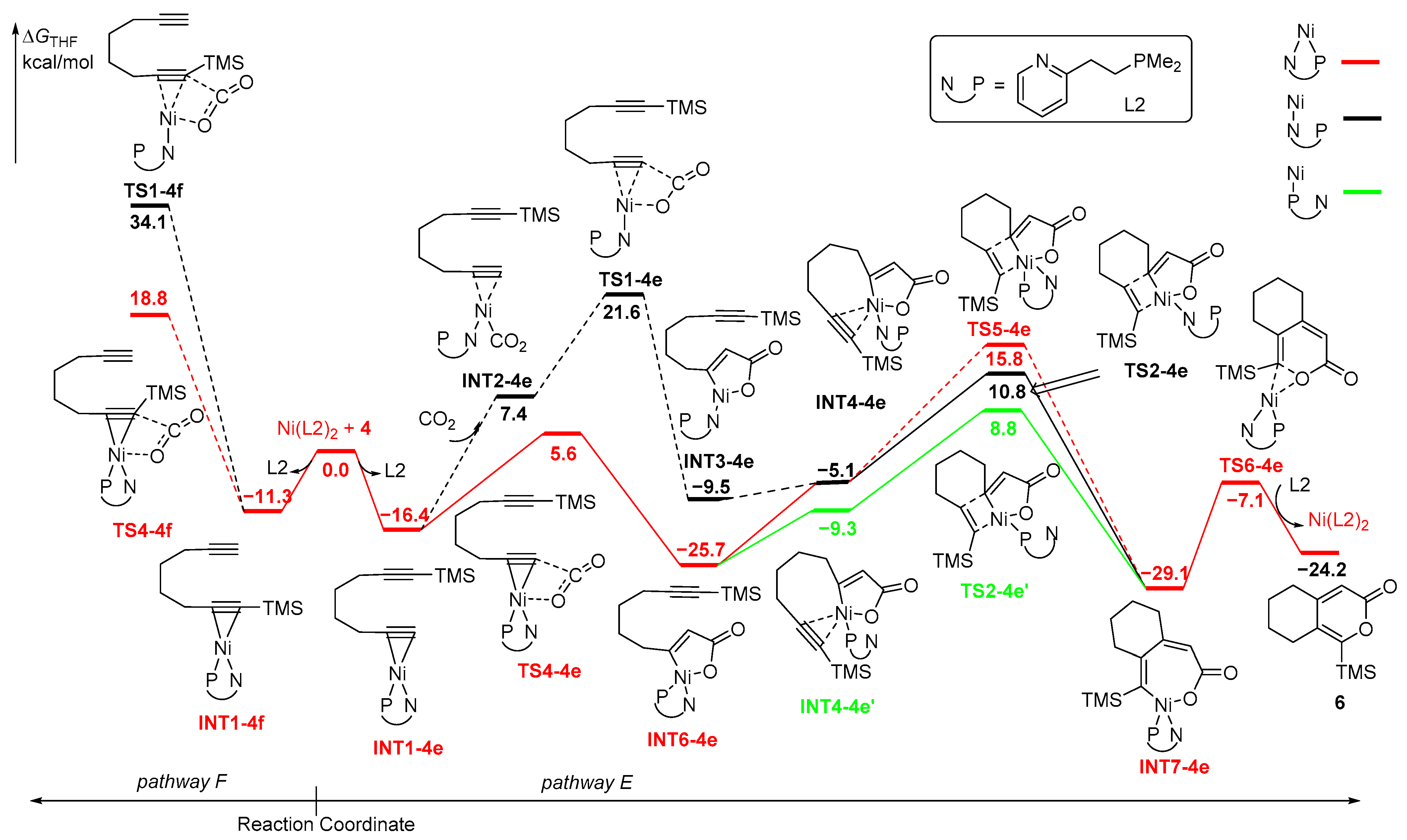
Figure 8.
Potential energy profiles for Ni-catalyzed cycloaddition of diyne 4 and CO2 with bisdentate ligand. Path G/H: proposed to start with the oxidative coupling between the two C≡C moieties.
Figure 8.
Potential energy profiles for Ni-catalyzed cycloaddition of diyne 4 and CO2 with bisdentate ligand. Path G/H: proposed to start with the oxidative coupling between the two C≡C moieties.
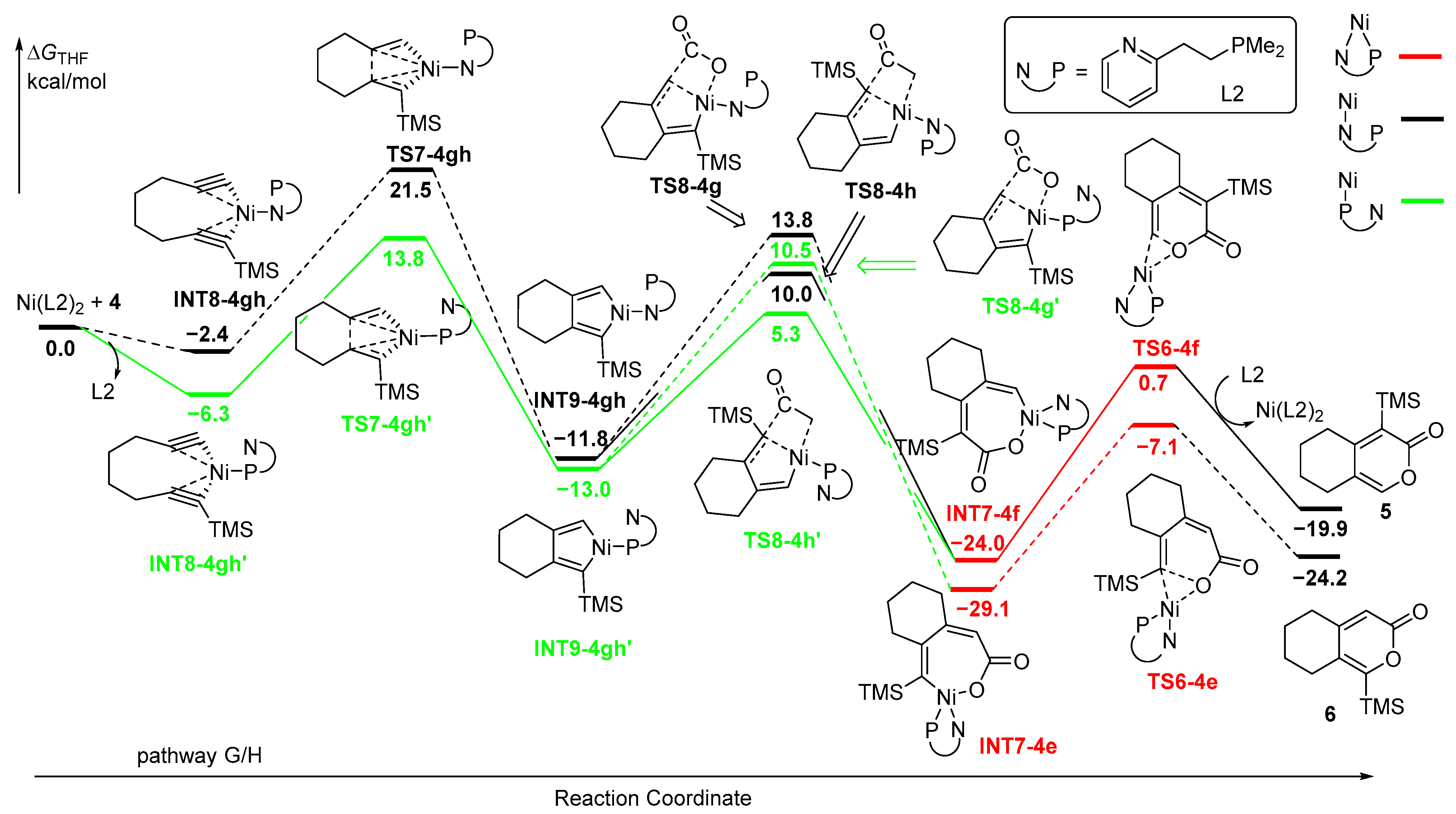
Figure 9.
Distortion-interaction analysis of the key species (energies are expressed in kcal/mol) and the bond length (Å) in the structures.
Figure 9.
Distortion-interaction analysis of the key species (energies are expressed in kcal/mol) and the bond length (Å) in the structures.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
The energy barriers and their gaps of rate-determining steps, and the ratio of products.
| Substrate | Ligand | ΔG‡ (2 or 5) (kcal/mol) | ΔG‡ (3 or 6) (kcal/mol) | ΔΔG‡ (2–3 or 5–6) (kcal/mol) | Cal. 2/3 or 5/6 | Exp. 2/3 or 5/6 | Ref. |
|---|---|---|---|---|---|---|---|
| 1 | L1 | 10.1 | 13.1 | −3.0 | >99/1 | 100/0 | [33] |
| 1 | L2 | 8.6 | 6.2 | 2.0 | 3/97 | 8/92 | [33] |
| 4 | L1 | 15.5 | 13.7 | 1.8 | 4/96 | 0/100 | [33] |
| 4 | L2 | 13.8 | 8.8 | 5.0 | <1/99 | 0/100 | [33] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Zhang, K.; Huang, Q.; Yang, C.; Li, X. Theoretical Insights into the Regiodivergence in Ni-Catalyzed [2+2+2] Cycloaddition of Unsymmetric Diynes and CO2. Inorganics 2024, 12, 39. https://doi.org/10.3390/inorganics12020039
AMA Style
Zhang K, Huang Q, Yang C, Li X. Theoretical Insights into the Regiodivergence in Ni-Catalyzed [2+2+2] Cycloaddition of Unsymmetric Diynes and CO2. Inorganics. 2024; 12(2):39. https://doi.org/10.3390/inorganics12020039
Chicago/Turabian StyleZhang, Kun, Qiwen Huang, Cun Yang, and Xinyao Li. 2024. "Theoretical Insights into the Regiodivergence in Ni-Catalyzed [2+2+2] Cycloaddition of Unsymmetric Diynes and CO2" Inorganics 12, no. 2: 39. https://doi.org/10.3390/inorganics12020039
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.