
Reactivity Studies of Phosphinines: The Selenation of Diphenyl-Phosphine Substituents and Formation of a Chelating Bis(Phosphinine) Palladium(II) Complex


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
3. Conclusions
4. Materials and Methods
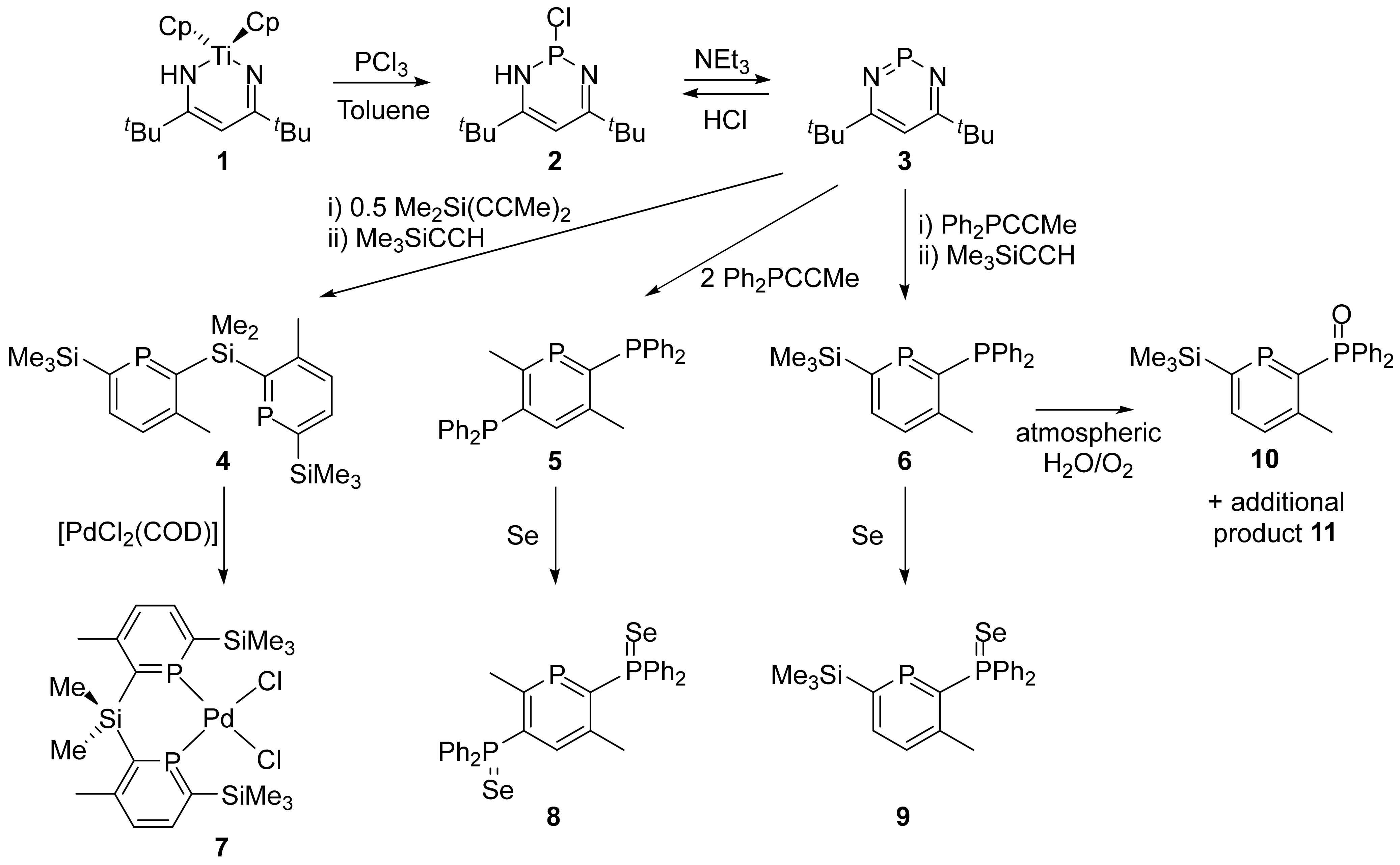
4.1. Preparation of Chlorodiazaphosphacycle (2)
4.2. Preparation of [PdCl2(κ:η1:η1-4)] (7)
4.3. Preparation of 2,5-Bis(diphenylphosphineselenide)-3,6-dimethylphosphinine (8)


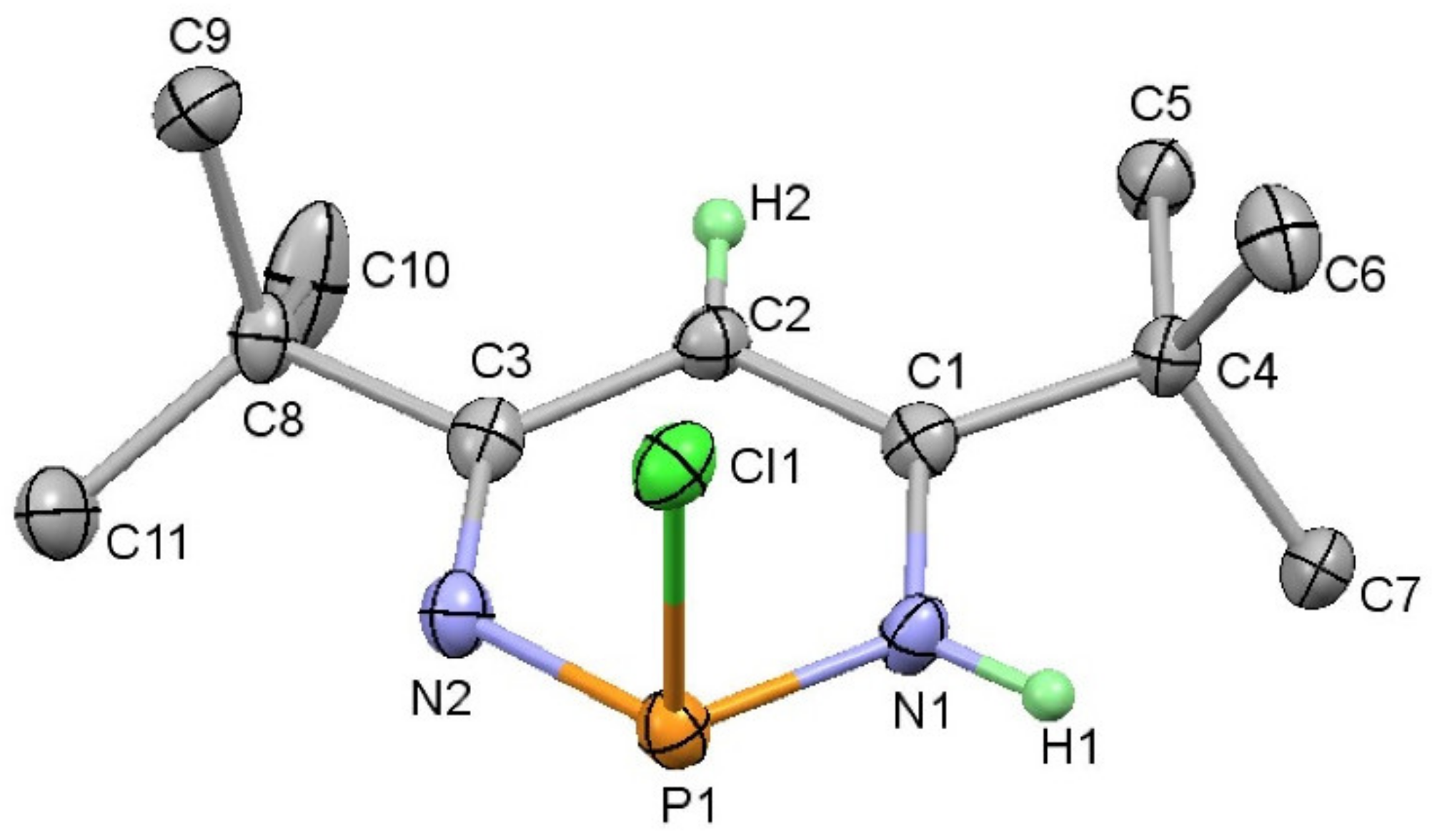
4.4. 2-Diphenylphosphineselenide-3-methyl-6-trimethylsilylphosphinine (9)

4.5. Preparation of 2-Diphenylphosphineoxide-3-methyl-6-trimethylsilylphosphinine (10)
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Märkl, G. 2,4,6-triphenylphosphabenzene. Angew. Chem. Int. Ed. 1966, 5, 846–847. [Google Scholar] [CrossRef]
- Ashe, A.J. Phosphabenzene and arsabenzene. J. Am. Chem. Soc. 1971, 93, 3293–3295. [Google Scholar] [CrossRef]
- Ashe, A.J. The route to phosphabenzene and beyond. Eur. J. Inorg. Chem. 2016, 2016, 572–574. [Google Scholar] [CrossRef] [Green Version]
- Nakajima, K.; Takata, S.; Sakata, K.; Nishibayashi, Y. Synthesis of phosphabenzenes by an iron-catalyzed [2+2+2] cycloaddition reaction of diynes with phosphaalkynes. Angew. Chem. Int. Ed. 2015, 54, 7597–7601. [Google Scholar] [CrossRef]
- Coles, N.T.; Sofie Abels, A.; Leitl, J.; Wolf, R.; Grützmacher, H.; Müller, C. Phosphinine-based ligands: Recent developments in coordination chemistry and applications. Coord. Chem. Rev. 2021, 433, 213729. [Google Scholar] [CrossRef]
- Müller, C.; Sklorz, J.A.W.; de Krom, I.; Loibl, A.; Habicht, M.; Bruce, M.; Pfeifer, G.; Wiecko, J. Recent developments in the chemistry of pyridyl-functionalized, low-coordinate phosphorus heterocycles. Chem. Lett. 2014, 43, 1390–1404. [Google Scholar] [CrossRef] [Green Version]
- Müller, C.; Broeckx, L.E.E.; de Krom, I.; Weemers, J.J.M. Developments in the coordination chemistry of phosphinines. Eur. J. Inorg. Chem. 2013, 2013, 187–202. [Google Scholar] [CrossRef]
- Müller, C. Phosphinine ligands. In Phosphorus(iii) Ligands in Homogeneous Catalysis: Design and Synthesis; John Wiley & Sons, Ltd.: Hoboken, NJ, USA, 2012; pp. 287–307. [Google Scholar]
- Müller, C.; Vogt, D. Recent developments in the chemistry of donor-functionalized phosphinines. Comptes Rendus Chim. 2010, 13, 1127–1143. [Google Scholar] [CrossRef]
- Le Floch, P. Phosphaalkene, phospholyl and phosphinine ligands: New tools in coordination chemistry and catalysis. Coord. Chem. Rev. 2006, 250, 627–681. [Google Scholar] [CrossRef]
- Müller, C.; Vogt, D. Phosphinine-based ligands in homogeneous catalysis: State of the art and future perspectives. In Phosphorus Compounds: Catalysis by Metal Complexes; Peruzzini, M., Gonsalvi, L., Eds.; Springer: Dordrecht, The Netherlands, 2011; Volume 37. [Google Scholar]
- Müller, C.; Vogt, D. Phosphinines as ligands in homogeneous catalysis: Recent developments, concepts and perspectives. Dalton Trans. 2007, 47, 5505–5523. [Google Scholar] [CrossRef]
- Kollár, L.; Keglevich, G. P-heterocycles as ligands in homogeneous catalytic reactions. Chem. Rev. 2010, 110, 4257–4302. [Google Scholar] [CrossRef]
- Weber, L. Phosphorus heterocycles: From laboratory curiosities to ligands in highly efficient catalysts. Angew. Chem. Int. Ed. 2002, 41, 563–572. [Google Scholar] [CrossRef]
- DiMauro, E.F.; Kozlowski, M.C. Phosphabenzenes as electron withdrawing phosphine ligands in catalysis. J. Chem. Soc. Perkin Trans. 1 2002, 33, 439–444. [Google Scholar] [CrossRef]
- Mansell, S.M. Catalytic applications of small bite-angle diphosphorus ligands with single-atom linkers. Dalton Trans. 2017, 46, 15157–15174. [Google Scholar] [CrossRef]
- Trodden, E.C.; Delve, M.P.; Luz, C.; Newland, R.J.; Andresen, J.M.; Mansell, S.M. A ruthenium cis-dihydride with 2-phosphinophosphinine ligands catalyses the acceptorless dehydrogenation of benzyl alcohol. Dalton Trans. 2021, 50, 13407–13411. [Google Scholar] [CrossRef]
- Newland, R.J.; Wyatt, M.F.; Wingad, R.L.; Mansell, S.M. A ruthenium(ii) bis(phosphinophosphinine) complex as a precatalyst for transfer-hydrogenation and hydrogen-borrowing reactions. Dalton Trans. 2017, 46, 6172–6176. [Google Scholar] [CrossRef] [Green Version]
- Newland, R.J.; Smith, A.; Smith, D.M.; Fey, N.; Hanton, M.J.; Mansell, S.M. Accessing alkyl- and alkenylcyclopentanes from cr-catalyzed ethylene oligomerization using 2-phosphinophosphinine ligands. Organometallics 2018, 37, 1062–1073. [Google Scholar] [CrossRef] [Green Version]
- Newland, R.J.; Lynam, J.M.; Mansell, S.M. Small bite-angle 2-phosphinophosphinine ligands enable rhodium-catalysed hydroboration of carbonyls. Chem. Commun. 2018, 54, 5482–5485. [Google Scholar] [CrossRef] [Green Version]
- Breit, B.; Winde, R.; Mackewitz, T.; Paciello, R.; Harms, K. Phosphabenzenes as monodentate pi-acceptor ligands for rhodium-catalyzed hydroformylation. Chem. Eur. J. 2001, 7, 3106–3121. [Google Scholar] [CrossRef]
- Breit, B. Highly regioselective hydroformylation under mild conditions with new classes of π-acceptor ligands. Chem. Commun. 1996, 2071–2072. [Google Scholar] [CrossRef]
- Müller, C.; López, L.G.; Kooijman, H.; Spek, A.L.; Vogt, D. Chiral bidentate phosphabenzene-based ligands: Synthesis, coordination chemistry, and application in rh-catalyzed asymmetric hydrogenations. Tetrahedron Lett. 2006, 47, 2017–2020. [Google Scholar] [CrossRef] [Green Version]
- Rigo, M.; Hettmanczyk, L.; Heutz, F.J.L.; Hohloch, S.; Lutz, M.; Sarkar, B.; Müller, C. Phosphinines versus mesoionic carbenes: A comparison of structurally related ligands in au(i)-catalysis. Dalton Trans. 2017, 46, 86–95. [Google Scholar] [CrossRef]
- Broeckx, L.E.E.; Bucci, A.; Zuccaccia, C.; Lutz, M.; Macchioni, A.; Müller, C. Cyclometalated phosphinine–iridium(iii) complexes: Synthesis, reactivity, and application as phosphorus-containing water-oxidation catalysts. Organometallics 2015, 34, 2943–2952. [Google Scholar] [CrossRef]
- Hayashi, M. Phosphine sulfides: New aspects of organophosphorus compounds. Chem. Lett. 2020, 50, 1–6. [Google Scholar] [CrossRef]
- Hua, G.; Woollins, J.D. Organic phosphorus-selenium chemistry. In Selenium and Tellurium Chemistry: From Small Molecules to Biomolecules and Materials; Woollins, J.D., Laitinen, R., Eds.; Springer: Berlin/Heidelberg, Germany, 2011; pp. 1–39. [Google Scholar]
- Benton, A.; Durand, D.J.; Copeland, Z.; Watson, J.D.; Fey, N.; Mansell, S.M.; Rosair, G.M.; Welch, A.J. On the basicity of carboranylphosphines. Inorg. Chem. 2019, 58, 14818–14829. [Google Scholar] [CrossRef]
- Allen, D.W.; Taylor, B.F. The chemistry of heteroarylphosphorus compounds. Part 15. Phosphorus-31 nuclear magnetic resonance studies of the donor properties of heteroarylphosphines towards selenium and platinum(ii). J. Chem. Soc. Dalton Trans. 1982, 13, 51–54. [Google Scholar] [CrossRef]
- Beckmann, U.; Süslüyan, D.; Kunz, P.C. Is the 1 j pse coupling constant a reliable probe for the basicity of phosphines? A 31p nmr study. Phosphorus Sulfur Silicon Relat. Elem. 2011, 186, 2061–2070. [Google Scholar] [CrossRef]
- Holah, D.G.; Hughes, A.N.; Knudsen, K.L. Formation and partial characterisation of a stable phosphinine 1-sulphide. J. Chem. Soc. Chem. Commun. 1988, 19, 493–495. [Google Scholar] [CrossRef]
- Hughes, A.N.; Knudsen, K.L. A 31p nmr study of reactions of 2,3-bis(diphenylphosphino)-6-phenyl-λ3-phosphinine with sulfur: Formation, characterization, and further reaction of a stable phosphinine 1-sulfide. Heterocycles 1990, 30, 543–550. [Google Scholar] [CrossRef]
- Moores, A.; Cantat, T.; Ricard, L.; Mézailles, N.; Le Floch, P. Experimental and theoretical study of phosphinine sulfides. New J. Chem. 2007, 31, 1493–1498. [Google Scholar] [CrossRef]
- Pfeifer, G.; Ribagnac, P.; Le Goff, X.-F.; Wiecko, J.; Mézailles, N.; Müller, C. Reactivity of aromatic phosphorus heterocycles—differences between nonfunctionalized and pyridyl-substituted 2,4,6-triarylphosphinines. Eur. J. Inorg. Chem. 2015, 2015, 240–249. [Google Scholar] [CrossRef]
- Wossidlo, F.; Frost, D.S.; Lin, J.; Coles, N.T.; Klimov, K.; Weber, M.; Böttcher, T.; Müller, C. Making aromatic phosphorus heterocycles more basic and nucleophilic: Synthesis, characterization and reactivity of the first phosphinine selenide. Chem. Eur. J. 2021, 27, 12788–12795. [Google Scholar] [CrossRef]
- Newland, R.J.; Delve, M.P.; Wingad, R.L.; Mansell, S.M. Two isomers of a bis(diphenylphosphino)-phosphinine, and the synthesis and reactivity of ru arene/cp* phosphinophosphinine complexes. New J. Chem. 2018, 42, 19625–19636. [Google Scholar] [CrossRef] [Green Version]
- Doux, M.; Bouet, C.; Mézailles, N.; Ricard, L.; Le Floch, P. Synthesis and molecular structure of a palladium complex containing a λ5-phosphinine-based sps pincer ligand. Organometallics 2002, 21, 2785–2788. [Google Scholar] [CrossRef]
- Doux, M.; Mézailles, N.; Ricard, L.; Le Floch, P. Group 10 metal complexes of sps-based pincer ligands: Syntheses, X-ray structures, and dft calculations. Eur. J. Inorg. Chem. 2003, 2003, 3878–3894. [Google Scholar] [CrossRef]
- Moores, A.; Mézailles, N.; Ricard, L.; Jean, Y.; le Floch, P. H2-palladium and platinum(ii) complexes of a λ4-phosphinine anion: Syntheses, X-ray crystal structures, and dft calculations. Organometallics 2004, 23, 2870–2875. [Google Scholar] [CrossRef]
- Blug, M.; Doux, M.; Le Goff, X.; Maître, P.; Ribot, F.; Le Floch, P.; Mézailles, N. The effect of a fourth binding site on the stabilization of cationic sps pincer palladium complexes: Experimental, dft, and mass spectrometric studies. Organometallics 2009, 28, 2020–2027. [Google Scholar] [CrossRef]
- Doux, M.; Mézailles, N.; Ricard, L.; Le Floch, P. Planar discrimination in an sps-based rhodium(i) complex. Organometallics 2003, 22, 4624–4626. [Google Scholar] [CrossRef]
- Doux, M.; Ricard, L.; Le Floch, P.; Mézailles, N. Group 11 metal complexes of sps-based pincer ligands: Syntheses, X-ray structures and reactivity. Dalton Trans. 2004, 2593–2600. [Google Scholar] [CrossRef]
- Doux, M.; Thuéry, P.; Blug, M.; Ricard, L.; Le Floch, P.; Arliguie, T.; Mézailles, N. Anions of tridentate sps ligands: Syntheses, X-ray structures and dft calculations. Organometallics 2007, 26, 5643–5653. [Google Scholar] [CrossRef]
- Arliguie, T.; Blug, M.; Le Floch, P.; Mézailles, N.; Thuéry, P.; Ephritikhine, M. Organouranium complexes with phosphinine-based sps pincer ligands. Variations with the substituent at the phosphorus atom. Organometallics 2008, 27, 4158–4165. [Google Scholar] [CrossRef]
- Dochnahl, M.; Doux, M.; Faillard, E.; Ricard, L.; Le Floch, P. A new mixed p,s-bidentate ligand featuring a λ4-phosphinine anion and a phosphanyl sulfide group—Synthesis, X-ray crystal structures and catalytic properties of its chloro(cymene)ruthenium and allylpalladium complexes. Eur. J. Inorg. Chem. 2005, 2005, 125–134. [Google Scholar] [CrossRef]
- Avarvari, N.; Le Floch, P.; Ricard, L.; Mathey, F. 1,3,2-diazaphosphinines and -diazaarsinines as precursors for polyfunctional phosphinines and arsinines. Organometallics 1997, 16, 4089–4098. [Google Scholar] [CrossRef]
- Avarvari, N.; Le Floch, P.; Mathey, F. 1,3,2-diazaphosphinines: New, versatile precursors of 1,2-azaphosphinines and polyfunctional phosphinines. J. Am. Chem. Soc. 1996, 118, 11978–11979. [Google Scholar] [CrossRef]
- Doxsee, K.M.; Farahi, J.B. Reductive coupling of nitriles via formal [2 + 2] cycloadditions to the titanium-carbon double bond. J. Am. Chem. Soc. 1988, 110, 7239–7240. [Google Scholar] [CrossRef]
- Petasis, N.A.; Fu, D.K. Reactions of dimethyltitanocene with alkynes and nitriles. Organometallics 1993, 12, 3776–3780. [Google Scholar] [CrossRef]
- Spinney, H.A.; Korobkov, I.; DiLabio, G.A.; Yap, G.P.A.; Richeson, D.S. Diamidonaphthalene-stabilized n-heterocyclic pnictogenium cations and their cation-cation solid-state interactions. Organometallics 2007, 26, 4972–4982. [Google Scholar] [CrossRef]
- Spinney, H.A.; Yap, G.P.A.; Korobkov, I.; DiLabio, G.; Richeson, D.S. Construction of a stable n-heterocyclic phosphenium cation with an electron-rich framework and its complexation to rhodium. Organometallics 2006, 25, 3541–3543. [Google Scholar] [CrossRef]
- Kozma, Á.; Rust, J.; Alcarazo, M. Bis[(dialkylamino)cyclopropenimine]-stabilized piii- and pv-centered dications. Chem.-Eur. J. 2015, 21, 10829–10834. [Google Scholar] [CrossRef] [Green Version]
- Sonnenburg, R.; Borkenhagen, F.; Neda, I.; Holger; Thönnessen; Jones, P.G.; Schmutzler, R. N-phosphorylated nitrogen mustards; preparation of 2-chloroethyl- and bis(2-chloroethyl)-amides with the benzodiazaphosphorinone ring system. Phosphorus Sulfur Silicon Relat. Elem. 1997, 126, 11–26. [Google Scholar] [CrossRef]
- Mallov, I.; Spinney, H.; Jurca, T.; Gorelsky, S.; Burchell, T.; Richeson, D. The 2,2′-diindolylmethane dianion supporting scaffold for group 15 compounds. Inorg. Chim. Acta 2012, 392, 5–9. [Google Scholar] [CrossRef]
- Mecke, E.; Frank, W. Crystal structure of 1,3-di-tert-butyl-2-chloro-1,3,2-diazaphosphorinane—A saturated six-membered phosphorus nitrogen heterocycle with a partially flattened chair conformation and a long piii-cl bond. Acta Crystallogr. Sect. E 2019, 75, 552–556. [Google Scholar] [CrossRef] [PubMed]
- Vidovic, D.; Lu, Z.; Reeske, G.; Moore, J.A.; Cowley, A.H. An n,n′-chelated phosphenium cation supported by a β-diketiminate ligand. Chem. Commun. 2006, 33, 3501–3503. [Google Scholar] [CrossRef] [PubMed]
- Lesikar, L.A.; Woodul, W.D.; Richards, A.F. The reactions of phosphorus(iii) halides with β-diketiminato ligands. Polyhedron 2007, 26, 3242–3246. [Google Scholar] [CrossRef]
- Cleaves, P.A.; Mansell, S.M. Unexpected multiple coordination modes in silyl-bridged bis(phosphinine) complexes. Organometallics 2019, 38, 1595–1605. [Google Scholar] [CrossRef]
- Clendenning, S.B.; Hitchcock, P.B.; Lawless, G.A.; Nixon, J.F.; Tate, C.W. Differences in the η1-ligating properties of 2,4,6-tritertiarybutyl-phosphabenzene, pc5h2bu3t and 2,4,6-tritertiarybutyl-1,3,5-triphosphabenzene, p3c3bu3t. J. Organomet. Chem. 2010, 695, 717–720. [Google Scholar] [CrossRef]
- Campos-Carrasco, A.; Broeckx, L.E.E.; Weemers, J.J.M.; Pidko, E.A.; Lutz, M.; Masdeu-Bultó, A.M.; Vogt, D.; Müller, C. Pdii and ptii complexes of 2-(2′-pyridyl)-4,6-diphenylphosphinine: Synthesis, structure, and reactivity. Chem.-Eur. J. 2011, 17, 2510–2517. [Google Scholar] [CrossRef]
- Chen, X.; Li, Z.; Grützmacher, H. Multidentate phosphanyl phosphinines: Synthesis and properties. Chem.-Eur. J. 2018, 24, 8432–8437. [Google Scholar] [CrossRef]
- Doux, M.; Mézailles, N.; Melaimi, M.; Ricard, L.; Le Floch, P. A σ4, λ5-phosphinine palladium complex: A new type of phosphorus ligand and catalyst. Application to the pd-catalyzed formation of arylboronic esters. Chem. Commun. 2002, 1566–1567. [Google Scholar] [CrossRef]
- Habicht, M.H.; Wossidlo, F.; Bens, T.; Pidko, E.A.; Müller, C. 2-(trimethylsilyl)-λ3-phosphinine: Synthesis, coordination chemistry, and reactivity. Chem.-Eur. J. 2018, 24, 944–952. [Google Scholar] [CrossRef]
- Ferro, V.R.; Omar, S.; González-Jonte, R.H.; García de la Vega, J.M. The σ-donating and π-accepting properties of ortho-si(ch3)3 phosphinine macrocycles. Heteroat. Chem. 2003, 14, 160–169. [Google Scholar] [CrossRef]
- Avarvari, N.; Mézailles, N.; Ricard, L.; Floch, P.L.; Mathey, F. Silacalix-[n]-phosphaarenes: Macrocyclic ligands based on dicoordinate phosphorus centers. Science 1998, 280, 1587–1589. [Google Scholar] [CrossRef] [PubMed]
- Payack, J.F.; Hughes, D.L.; Cai, D.; Cottrell, I.F.; Verhoeven, T.R. Dimethyltitanocene. Org. Synth. 2002, 79, 19. [Google Scholar]
- Sheldrick, G. Shelxt-integrated space-group and crystal-structure determination. Acta Crystallogr. A 2015, 71, 3–8. [Google Scholar] [CrossRef] [Green Version]
- Sheldrick, G. A short history of shelx. Acta Crystallogr. A 2008, 64, 112–122. [Google Scholar] [CrossRef] [Green Version]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. Olex2: A complete structure solution, refinement and analysis program. J. Appl. Crystallogr. 2009, 42, 339–341. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cleaves, P.A.; Gourlay, B.; Newland, R.J.; Westgate, R.; Mansell, S.M. Reactivity Studies of Phosphinines: The Selenation of Diphenyl-Phosphine Substituents and Formation of a Chelating Bis(Phosphinine) Palladium(II) Complex. Inorganics 2022, 10, 17. https://doi.org/10.3390/inorganics10020017
Cleaves PA, Gourlay B, Newland RJ, Westgate R, Mansell SM. Reactivity Studies of Phosphinines: The Selenation of Diphenyl-Phosphine Substituents and Formation of a Chelating Bis(Phosphinine) Palladium(II) Complex. Inorganics. 2022; 10(2):17. https://doi.org/10.3390/inorganics10020017
Chicago/Turabian StyleCleaves, Peter A., Ben Gourlay, Robert J. Newland, Robert Westgate, and Stephen M. Mansell. 2022. "Reactivity Studies of Phosphinines: The Selenation of Diphenyl-Phosphine Substituents and Formation of a Chelating Bis(Phosphinine) Palladium(II) Complex" Inorganics 10, no. 2: 17. https://doi.org/10.3390/inorganics10020017