
Synthesis, Single Crystal X-ray Structure, Spectroscopy and Substitution Behavior of Niobium(V) Complexes Activated by Chloranilate as Bidentate Ligand

 ,
,
Abstract
:1. Introduction
2. Results
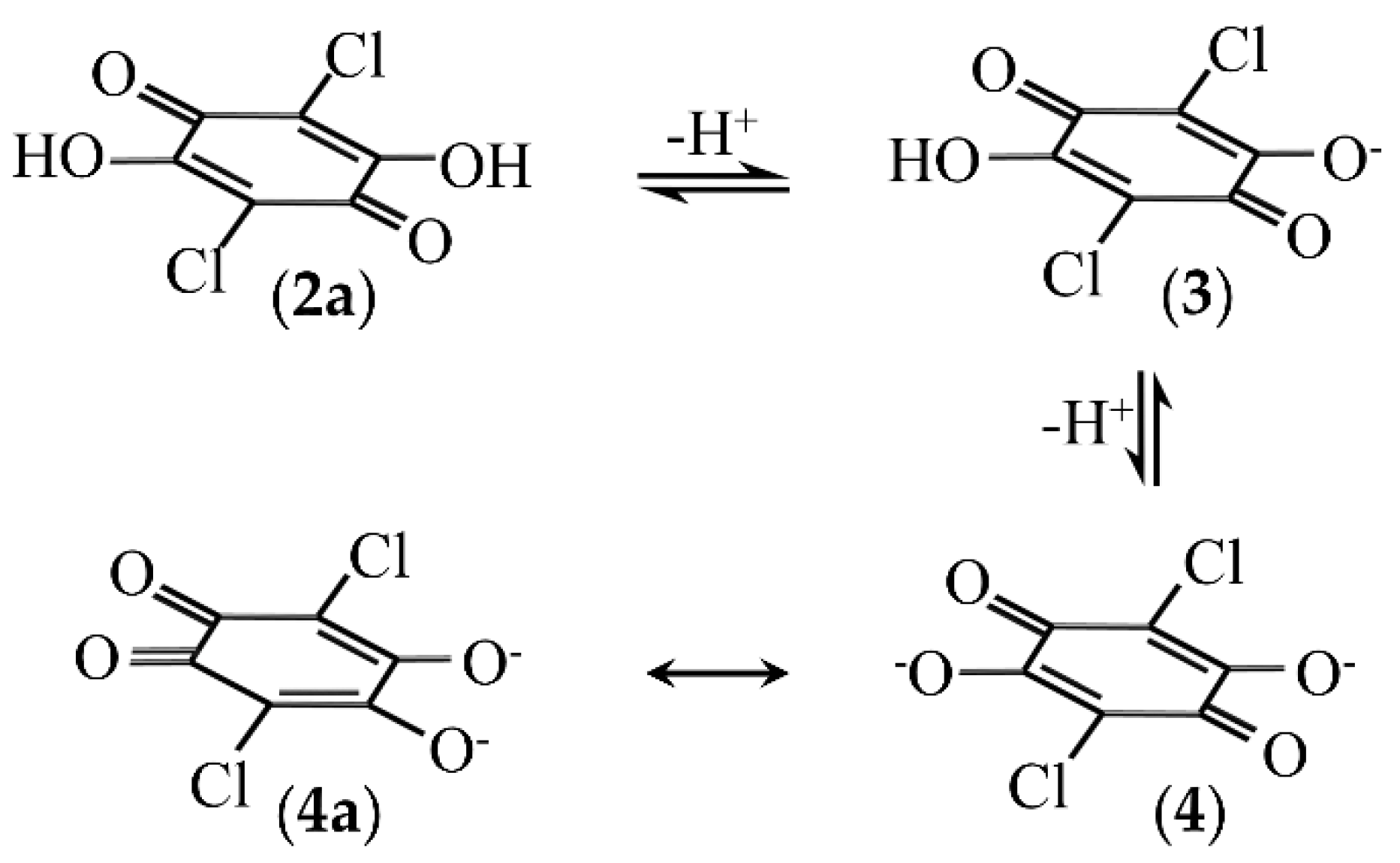
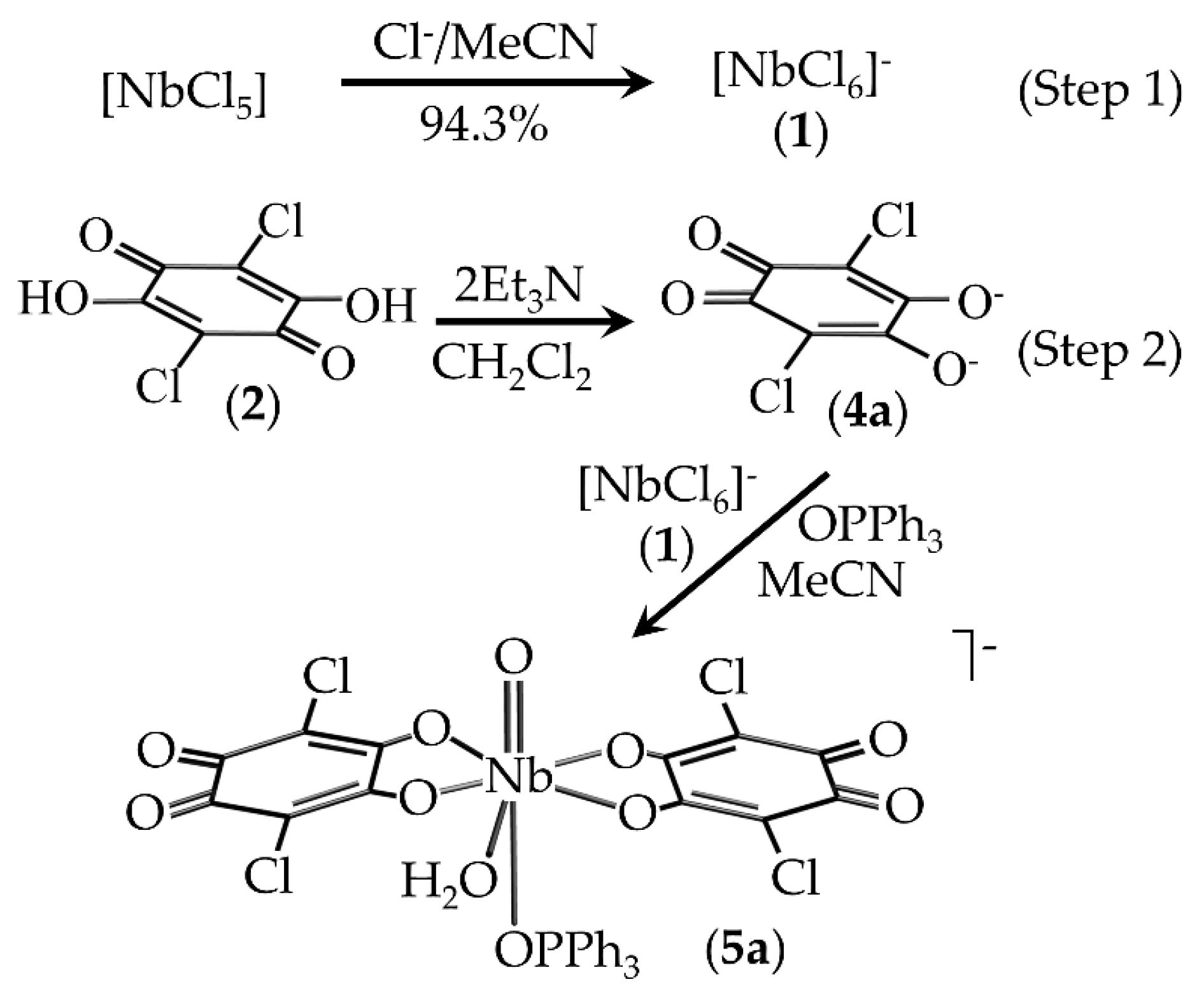
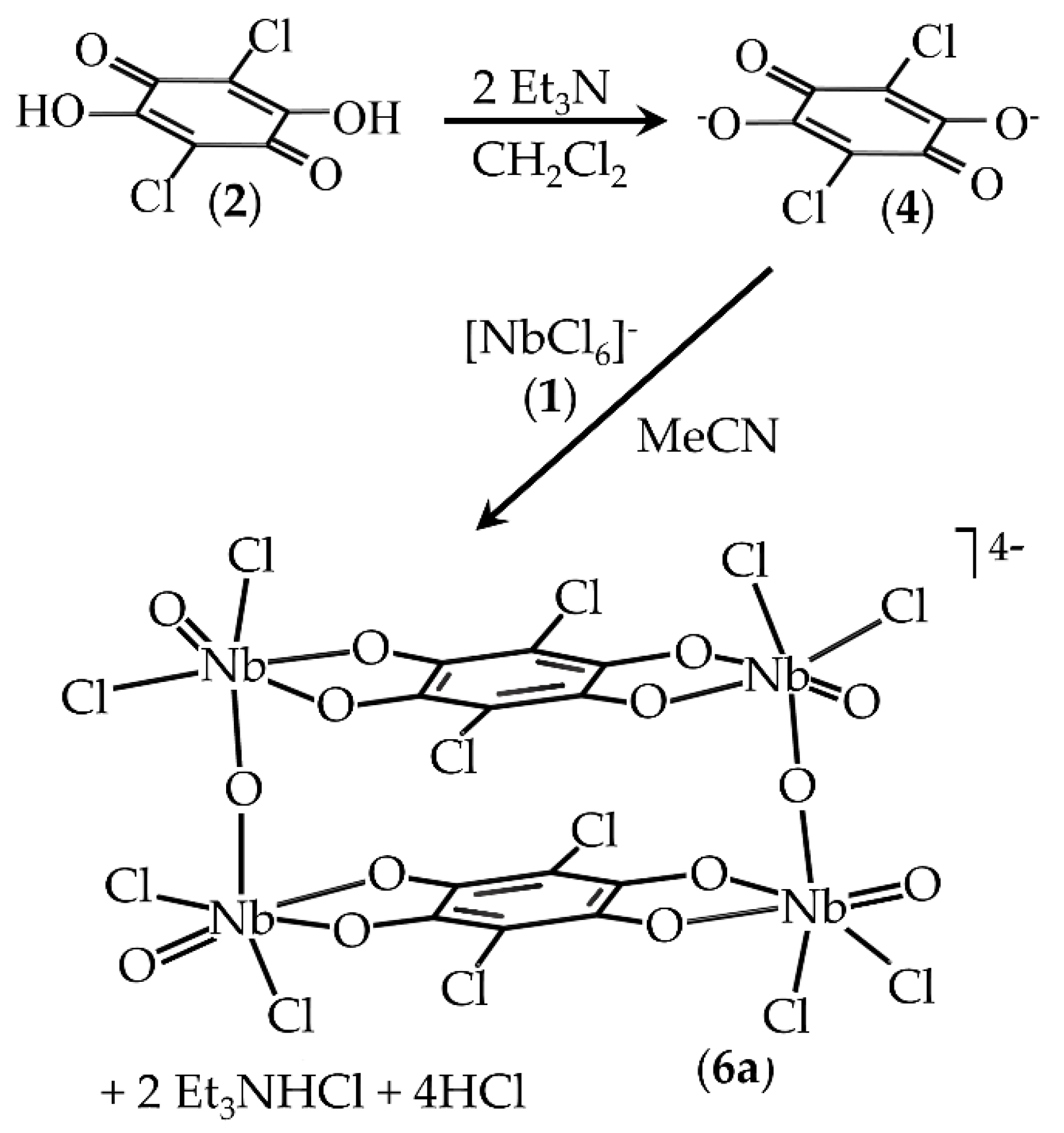
2.1. Synthesis of Compounds
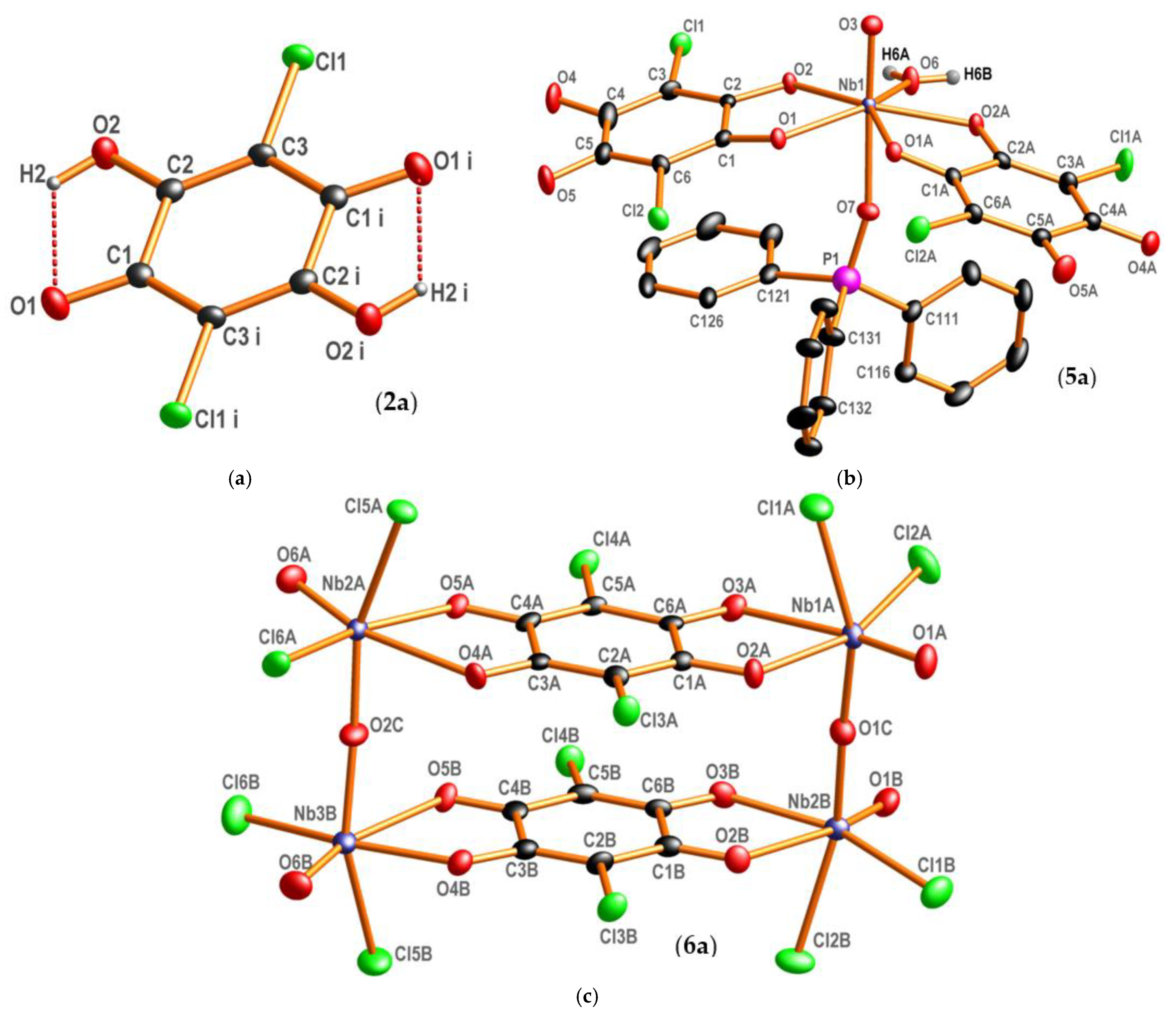
2.2. X-ray Crystallography
2.3. Kinetic Study of the OPPh3 Substitution from cis-[NbO(ca)2(H2O)OPPh3]− (5a) by Py-Type Nucleophiles
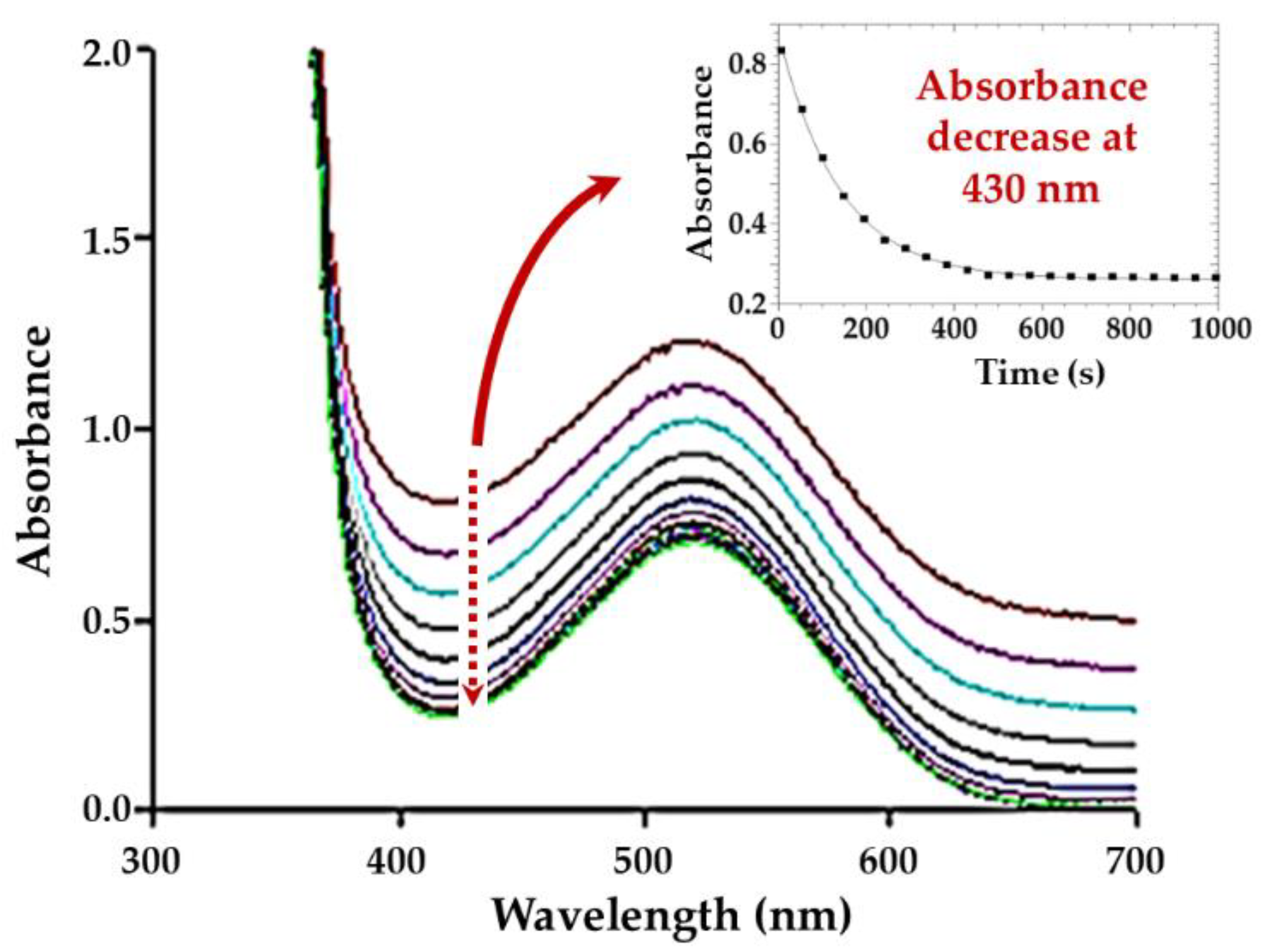
2.3.1. UV/Vis Measurements
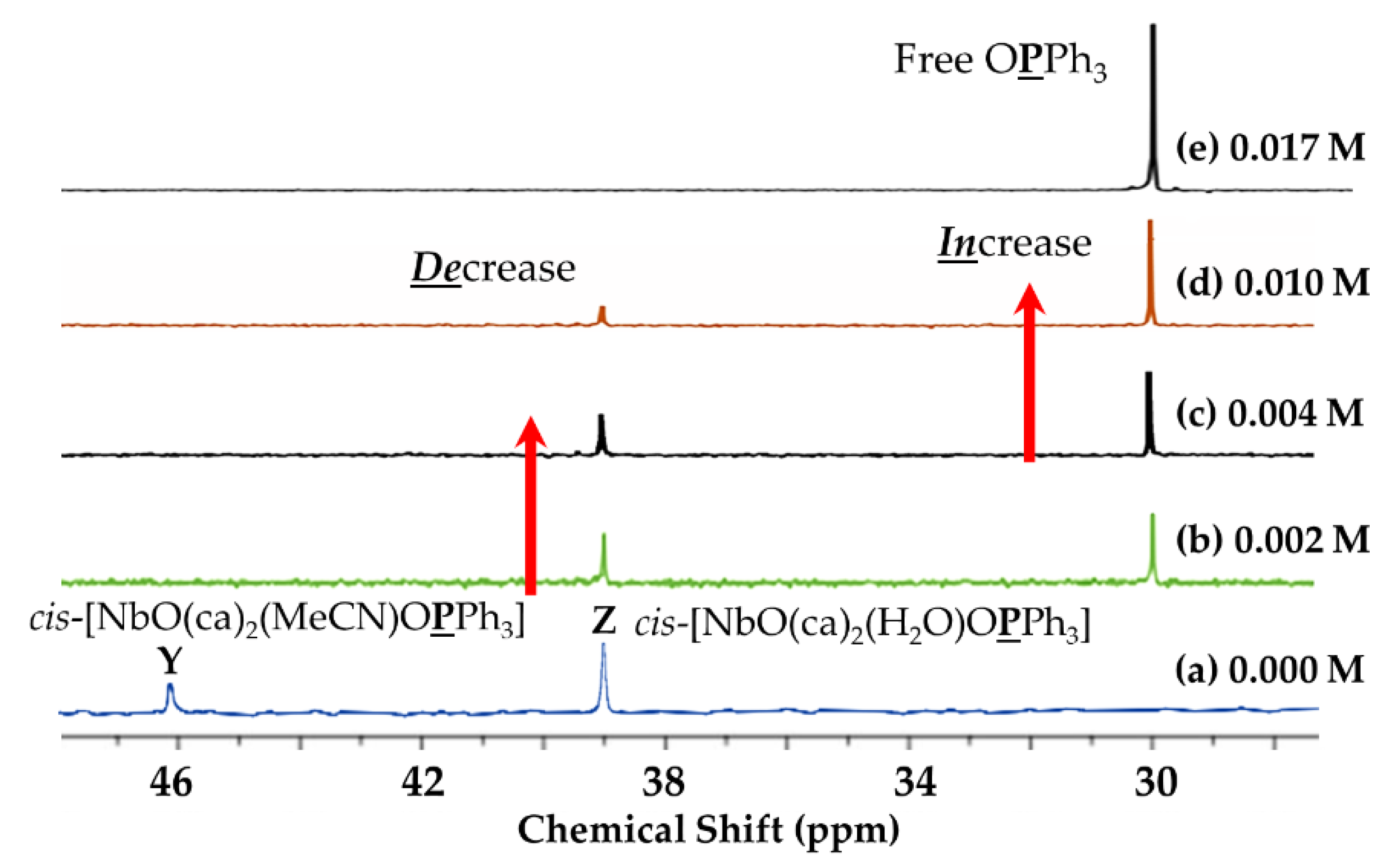
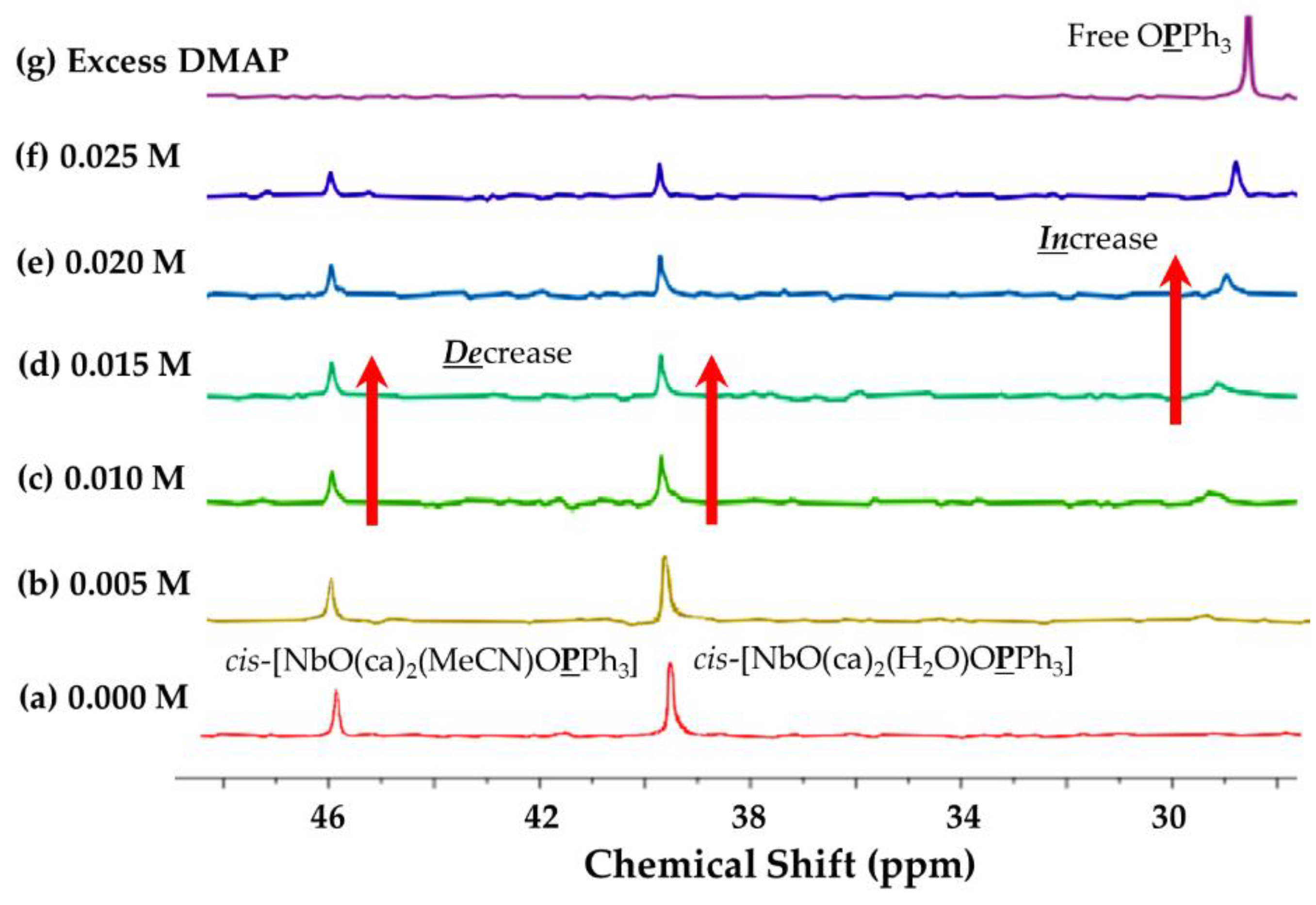
2.3.2. 31P NMR{1H} Study
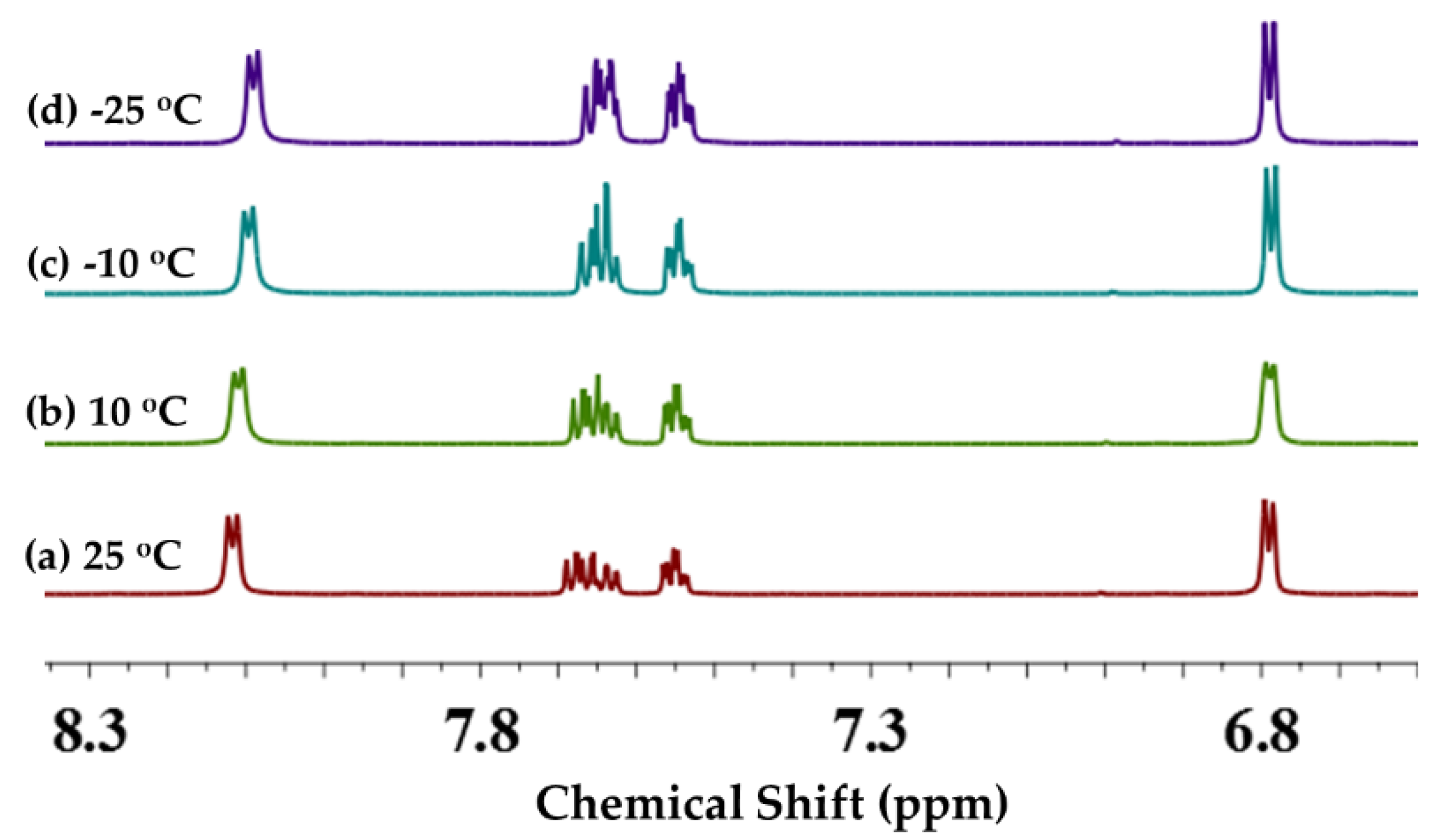
2.3.3. 1H NMR Studies
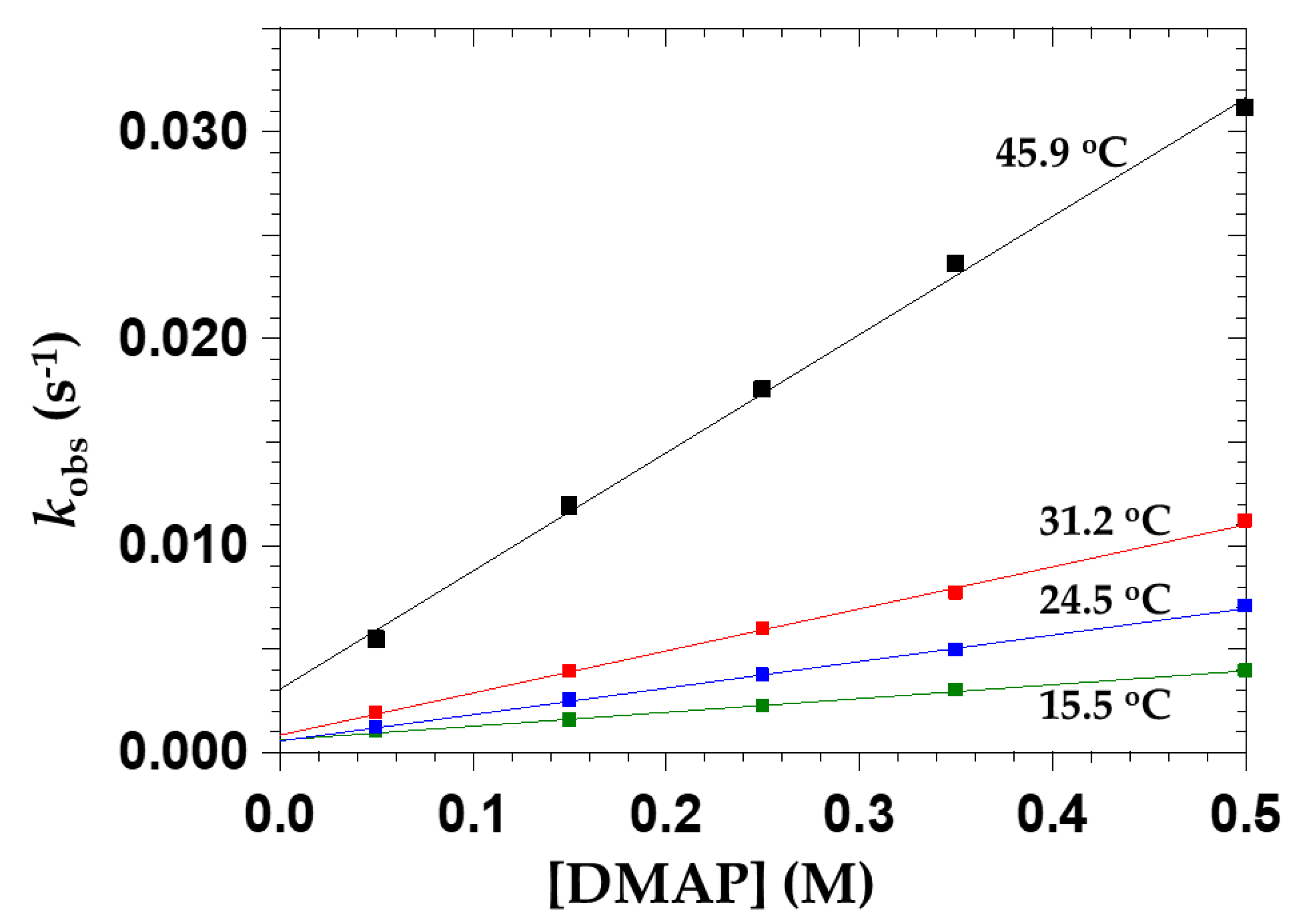
Concentration Effect of DMAP
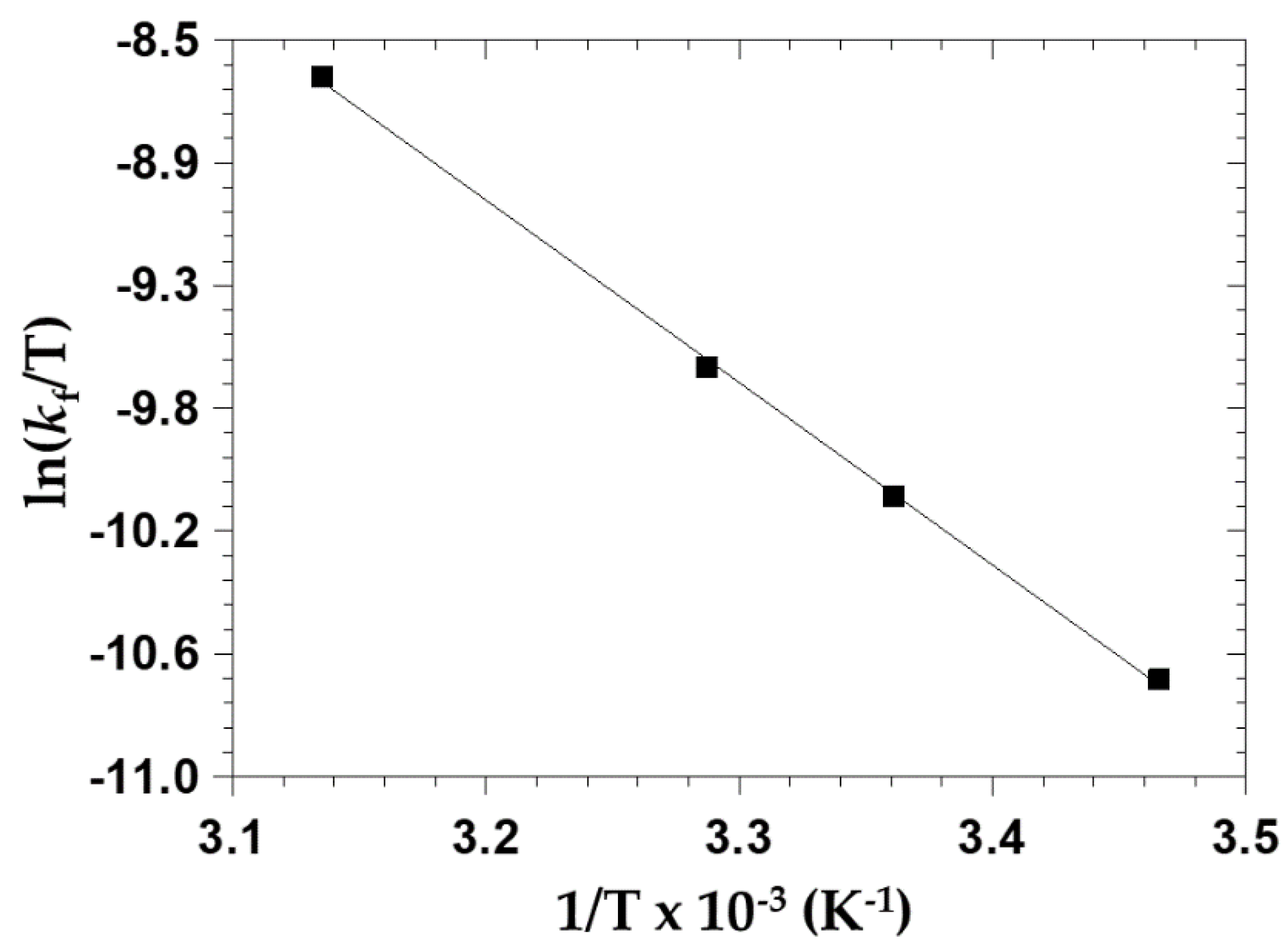
Temperature Effect
2.3.4. Mechanism of the Substitution of OPPh3 from cis-[NbO(ca)2(H2O)OPPh3]− by Pyridine-Type Ligands
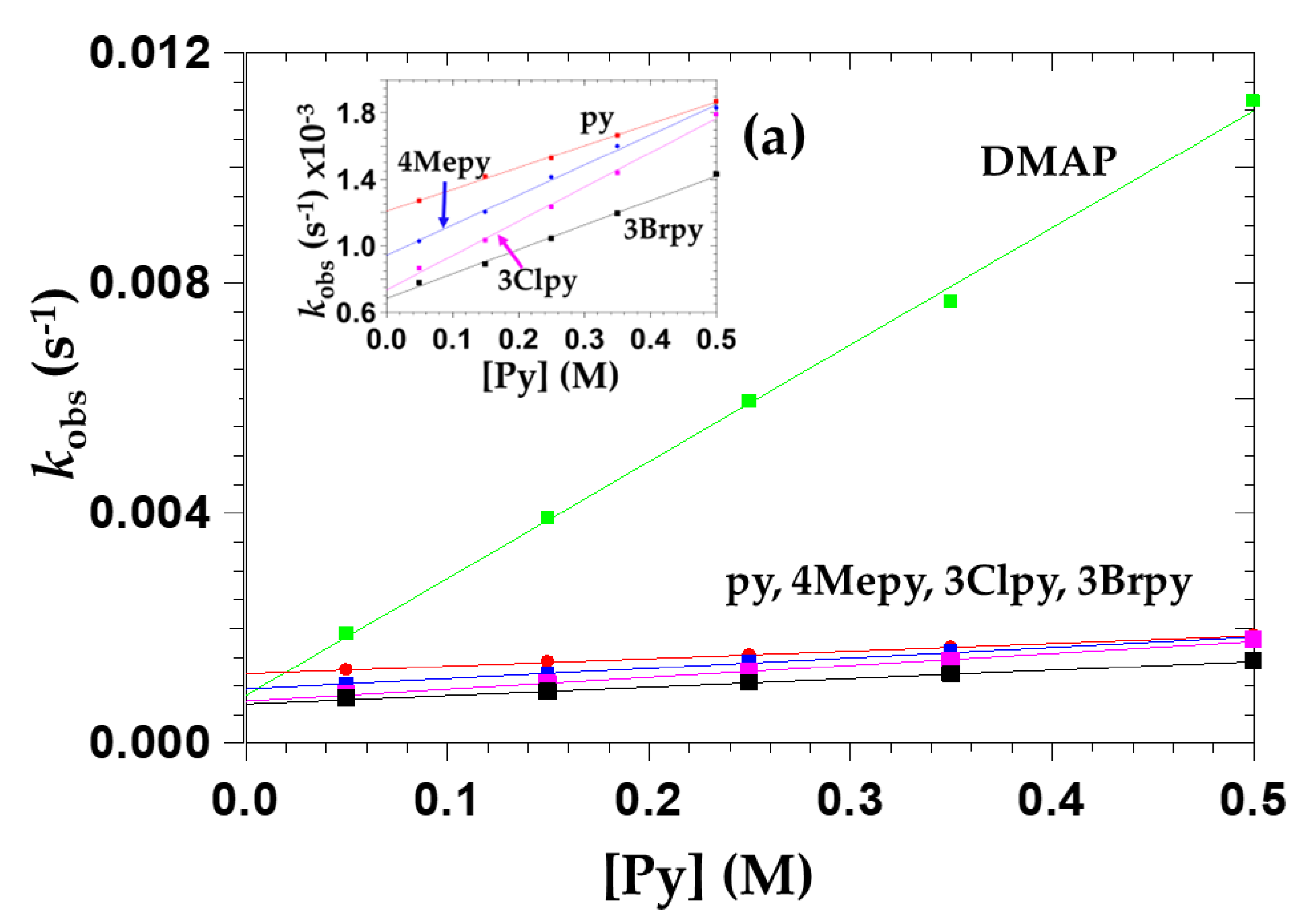
2.3.5. The Effect of Pyridine Concentration on OPPh3 Substitution from cis-[NbO(ca)2(H2O)OPPh3]− (5a)
2.3.6. The Effect of Temperature on OPPh3 Substitution from cis-[NbO(ca)2(H2O)OPPh3]−
3. Discussion
3.1. Synthesis
3.2. X-ray Crystallography
3.2.1. Correlation of Geometrical Parameters in 2, 5 and 6
3.2.2. Coordination Geometries
3.3. Solution Behavior of cis-[NbO(ca)2(H2O)OPPh3]− (5a): Kinetics and Mechanism
3.3.1. General Spectroscopic Observations
3.3.2. Mechanism of the Substitution of OPPh3 from cis-[NbO(ca)2(H2O)OPPh3]− by a Range of Py-Type Ligands
4. Materials and Methods
4.1. Materials
4.2. Physical Measurements
4.3. Synthesis
4.4. X-ray Data Collection, Reduction, and Refinement
4.5. Kinetic Experiments
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Raymond, L.P.; Michael, F. Geochemistry of Niobium and Tantalum; Government Printing Office: Washington, DC, USA, 1968; pp. 68–344.
- Belay, A.N.; Koen, R.; Venter, J.A.; Drost, R.M. Tris (N-nitroso-N-phenylhydroxylaminato-k2 O, O’) oxidoniobium(V). C18H15N6O7Nb. Z. Kristallogr. NCS 2016, 231, 513–515. [Google Scholar]
- Belay, A.N.; Venter, J.A.; Alexander, O.T. Crystal structure of 4-(dimethylamino) pyridin-1-ium-2,5-dichloro-3,6-dioxocyclohexa-1,4-diene-1,4-bis(olate) 4-dimethylaminopyridine (2:1) water undeca-solvate. Eur. J. Chem. 2020, 11, 255–260. [Google Scholar] [CrossRef]
- Herbst, L.; Visser, H.G.; Roodt, A. Redetermination of the niobium(V) precursor, [NbCl5 (NCCH3)]. Adv. Mat. Res. 2014, 1019, 412–418. [Google Scholar]
- Koen, R.; Roodt, A.; Visser, H.G. The coordination of novel, hard ligand systems to tantalum(V) and niobium(V) metal centers. Adv. Mat. Res. 2014, 1019, 426–432. [Google Scholar]
- Davies, H.O.; Leedham, T.J.; Jones Brien, P.O.; White, A.J.P.; Williams, D.J. Some tantalum(V) β-diketonate and tantalum(V) aminoalcoholate derivatives potentially important in the deposition of tantalum-containing materials. Polyhedron 1999, 18, 3165–3172. [Google Scholar] [CrossRef]
- Wendrup, P.; Kessler, V.G. Interaction of Co(acac)2 and Ta(OMe)5: Isolation and single crystal study of the products. MII2MV2(acac)2(OMe)12, MII = Co, Ni, Zn or Mg and MV = Ta or Nb: A new class of heterometallic heteroleptic alkoxide complexes. J. Chem. Soc. Dalton Trans. 2001, 574–579. [Google Scholar] [CrossRef]
- Herbst, L.; Visser, H.G.; Pretorius, C.; Roodt, A. (Acetylacetonato-κ2O, O’) dichloridobis (methanolato-κO) niobium(V). Acta Crystallogr. 2012, 68, m1392–m1394. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soria, D.B.; Grundy, J.; Coles, M.P.; Hitchcock, P.B. Stabilization of high oxidation-state niobium using ‘electron-rich’ bicyclic-guanidinates. J. Organomet. Chem. 2005, 690, 2278–2284. [Google Scholar] [CrossRef]
- Kergoat, R.; Guerchais, J.E. Contribution a l’etude stereochimique d’oxochloro-(dicetonato)niobates(V). J. Less-Common Met. 1975, 39, 91–98. [Google Scholar] [CrossRef]
- Marchetti, F.; Pampaloni, G.; Zacchini, S. Reactivity of niobium(V) and tantalum(V) halides with carbonyl compounds: Synthesis of simple coordination adducts, C-H bond activation, C=O protonation, and halide transfer. J. Chem. Soc. Dalton Trans. 2007, 4343–4351. [Google Scholar] [CrossRef] [PubMed]
- Feenan, K.; Fowles, G.W.A. Reactions of niobium (V) und tantalum (V) chlorides and bromides with some cyclic ethers and thioethers. J. Chem. Soc. 1965, 2449–2451. [Google Scholar] [CrossRef]
- Monassier, A.; D’Elia, V.; Cokoja, M.; Dong, H.; Pelletier, J.D.A.; Basset, J.-M.; Kühn, F.E. Synthesis of Cyclic Carbonates from Epoxides and CO2 under Mild Conditions Using a Simple, Highly Efficient Niobium-Based Catalyst. ChemCatChem 2013, 5, 1321–1324. [Google Scholar] [CrossRef]
- Arayachukiata, S.; Yingcharoena, P.; Vummaletib, S.V.C.; Cavallob, L.; Poaterc, A.; D’Elia, V. Cycloaddition of CO2 to challenging N-tosyl aziridines using a halogen-free niobium complex: Catalytic activity and mechanistic insights. Mol. Catal. 2017, 443, 280–285. [Google Scholar] [CrossRef]
- Belay, A.N.; Venter, J.A.; Roodt, A. The crystal structure of triphenylphosphine oxide-2, 5-dichloro-3, 6-dihydroxycyclohexa-2, 5-diene-1, 4-dione (2/1), C42H32Cl2O6P2. Z. Kristallogr. NCS 2017, 232, 163–164. [Google Scholar]
- Molcanov, K.; Kojic-Prodicand, B.; Meden, A. Unique electronic and structural properties of 1, 4-benzoquinones: Crystallochemistry of alkali chloranilate hydrates. Croat. Chem. Acta 2009, 82, 387–396. [Google Scholar]
- Nieuwenhuyzen, M.; Keyes, T.E.; Gallagher, J.F.; Vos, J.G. A 2:1 co-crystal of hydroquinone and 3, 5-bis (2-pyridyl)-1, 2, 4-triazole. Acta Crystallogr. 1997, 53, 1873–1875. [Google Scholar] [CrossRef] [Green Version]
- Fortuna, A.; Costa, P.J. Optimized Halogen Atomic Radii for PBSA Calculations Using Off-Center Point Charges. J. Chem. Inf. Modeling 2021, 61, 3361–3375. [Google Scholar] [CrossRef] [PubMed]
- Rafael Santana Nunes, R.F.; Vila-Viçosa, D.; Costa, P.J. Halogen Bonding: An Underestimated Player in Membrane–Ligand Interactions. J. Am. Chem. Soc. 2021, 143, 4253–4267. [Google Scholar] [CrossRef] [PubMed]
- Tinti, F.; Verdaguer, M.; Kahn, O.; Savariault, J.M. Interaction between copper (II) ions separated by 7.6 Å. crystal structure and magnetic properties of (μ-iodanilato)bis [(N,N,N’,N’-tetramethylethylenediamine)copper(II)] diperchlorate. Inorg. Chem. 1987, 26, 2380–2384. [Google Scholar] [CrossRef]
- Deng, C.L.; Jiang, Z.H.; Liao, D.Z.; Yan, S.P.; Wang, G.L. Synthesis and magnetism of binuclear Nd(III), Gd(III), and Dy(III) complexes using the dianions of chloranilic acid as bridging ligands. Synth. React. Inorg. Met. Org. Chem. 1993, 23, 247–256. [Google Scholar] [CrossRef]
- Kawata, S.; Kitagawa, S.; Kumagai, H.; Kudo, C.; Kamesaki, H.; Ishiyama, T.; Suzuki, R.; Konodo, M.; Katado, M. Rational design of a novel intercalation system. Layer-gap control of crystalline coordination polymers, {[Cu(CA)(H2O)m](G)}n (m = 2, G = 2, 5-dimethylpyrazine and phenazine; m = 1, G = 1, 2, 3, 4, 6, 7,8,9-octahydrophenazine). Inorg. Chem. 1996, 35, 4449–4461. [Google Scholar] [CrossRef]
- Smit, J.P.; Purcell, W.; Roodt, A.; Leipoldt, J.G. Kinetics of the substitution reaction between aquaoxotetracyanomolybdate (IV) and cyanide/hydrogen cyanide. Polyhedron 1993, 12, 2271–2277. [Google Scholar] [CrossRef]
- Muller, A.J.; Otto, S.; Roodt, A. Rapid phosphorus (III) ligand evaluation utilising potassium selenocyanate. Dalton Trans. 2008, 650–657. [Google Scholar] [CrossRef] [PubMed]
- Sahar, I.M. Complexes of 2, 5-dihydroxy-1, 4-benzoquinone and chloranilic acid with second and third-row transition elements. Transit. Met. Chem. 1999, 24, 306–310. [Google Scholar]
- Andersen, E.K. The crystal and molecular structure of hydroxyquinones and salts of hydroxyquinones. I. Chloranilic acid. Acta Crystallogr. 1967, 22, 188–191. [Google Scholar] [CrossRef]
- Kitagawa, S.; Kawata, S. Coordination compounds of 1, 4-dihydroxybenzoquinone and its homologues. Structures and properties. Coord. Chem. Rev. 2002, 224, 11–34. [Google Scholar] [CrossRef]
- Viljoen, J.A.; Visser, H.G.; Roodt, A.; Steyn, M. Tetra kis (quinolin-8-olato-κ2N, O) hafnium (IV) toluene disolvate. Acta Crystallogr. 2009, 65, m1514–m1515. [Google Scholar]
- Andersen, E.K. The crystal and molecular structure of hydroxyquinones and salts of hydroxyquinones. II. Chloranilic acid dehydrate. Acta Crystallogr. 1967, 22, 191–196. [Google Scholar] [CrossRef]
- Dutkiewicz, G.; Yathirajan, H.S.; Al-arique, Q.N.M.H.; Narayana, B.; Kubicki, M. Chloranilic acid: A redetermination at 100 K. Acta Crystallogr. 2010, 66, o497–o498. [Google Scholar] [CrossRef] [Green Version]
- Steyn, M.; Roodt, A.; Steyl, G. Tetrakis (1, 1, 1-trifluoroacetylacetonato-κ2O, O′) zirconium (IV) toluene solvate. Acta Crystallogr. 2008, 64, m827. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herbst, L.; Renier, K.; Roodt, A.; Visser, H.G. (Acetyl acetonato-κ2O, O’)chloridotrimethanolatoniobium (V). Acta Crystallogr. 2010, 66, m801–m802. [Google Scholar]
- Roodt, A.; Leipoldt, J.G.; Helm, L.; Merbach, A.E. Equilibrium behavior and proton transfer kinetics of the dioxotetracyanometalate complexes of molybdenum (IV), tungsten (IV), technetium (V), and rhenium (V): Carbon-13 and Oxygen-17 NMR Study. Inorg. Chem. 1994, 33, 140–147. [Google Scholar] [CrossRef]
- Ferreira, A.C.; Crous, R.; Bennie, L.; Meij, A.M.; Blann, K.; Bezuidenhoudt, B.C.B.; Young, D.A.; Green, M.J.; Roodt, A. Borate Esters as Alternative Acid Promoters in the Palladium-Catalyzed Methoxycarbonylation of Ethylene. Angew. Chem. Int. Ed. 2007, 119, 2323–2325. [Google Scholar] [CrossRef]
- Leipoldt, J.G.; Van Eldik, R.; Basson, S.S.; Roodt, A. Kinetics and mechanism of the reaction between trans-dioxotetracyanotungstate (IV) and azide in aqueous solution. Inorg. Chem. 1986, 25, 4639–4642. [Google Scholar] [CrossRef]
- Casanova, D.; Alemany, P.; Bofill, J.M.; Alvarez, S. Shape and Symmetry of Heptacoordinate Transition-Metal Complexes: Structural Trends. Chem. Eur. J. 2003, 9, 1281–1295. [Google Scholar] [CrossRef]
- Klingelhoever, P.; Mueller, U. Addukte des Oxo-tetrachloro-niobat(V)-Ions Bildung, Schwingungsspektren und Kristallstrukturen von PPh4[NbOCl4(OH2)] und (PPh4)2[NbOCl4(O2PCl2)]·2CH2Cl2. Z. Anorg. Allg. Chem. 1984, 516, 85–92. [Google Scholar] [CrossRef]
- Schafer, H.N.; Burzlaff, H.; Grimmeiss, A.M.H.; Weiss, R. Structure of 1, 2, 3-tris (dimethylamino) cyclopropenylium aquatetrachlorooxoniobate(V). Acta Crystallogr. 1991, 47, 1808–1811. [Google Scholar] [CrossRef] [Green Version]
- Pinnavaia, T.J.; Barnett, B.L.; Podolsky, G.; Tulinsky, A. Crystal and molecular structure of tetrakis(2,2,6,6-tetramethyl-3,5-heptanedionato)niobium(IV). Square antiprismatic M(bidentate)4 stereoisomer. J. Am. Chem. Soc. 1975, 97, 2712–2717. [Google Scholar] [CrossRef]
- Leipoldt, J.G.; Basson, S.S.; Roodt, A.; Potgieter, I.M. The crystal structure of tetraphenylarsonium tricyanooxopyridine-2-carboxylatotungstate (IV) dihydrate. Transit. Met. Chem. 1986, 11, 323–326. [Google Scholar] [CrossRef]
- Leipoldt, J.G.; Basson, S.S.; Roodt, A.; Potgieter, I.M. Kinetics and mechanism of fluoride ion substitution in trans-dioxotetracyanotungstate(IV) ions and the crystal structure of K3WOF(CN)4. S. Afr. J. Chem. 1986, 39, 179–183. [Google Scholar]
- Basson, S.S.; Leipoldt, J.G.; Potgieter, I.M.; Roodt, A. Chemical structure of dicaesium sodium azidooxotetracyanomolybdate(IV). Inorg. Chim. Acta 1985, 103, 121–125. [Google Scholar] [CrossRef]
- Perrin, D.D.; Armarego, W.L.F. Purification of Laboratory Chemicals, 4th ed.; Bath Press: Bath, UK, 1996; pp. 1–544. [Google Scholar]
- Koen, R.; Visser, H.G.; Roodt, A. Crystal structure of tetraethylammonium hexachloridotantalate(V), C8H20Cl6NTa. Z. Kristallogr. NCS. 2016, 231, 227–229. [Google Scholar]
- Bruker. Apex2, (Version 2011.4–1); Bruker AXS Inc.: Madison, WI, USA, 2011.
- Bruker. SAINT-Plus, Version 6.02 (including XPREP); Bruker AXS Inc. Area-Detector Integration Software: Madison, WI, USA, 2012.
- Bruker. SADABS, Version 2004/1; Bruker AXS Inc. Area Detector Absorption Correction Software: Madison, WI, USA, 1998.
- Altomare, A.; Burla, M.C.; Camalli, M.; Cascarano, G.L.; Giacovazzo, C.; Guagliardi, A.; Moliterni, A.G.G.; Polidori, G.; Spagna, R. SIR97: A new tool for crystal structure determination and refinement. J. Appl. Cryst. 1999, 32, 115–119. [Google Scholar] [CrossRef]
- Farrugia, L.J. WinGX and ORTEP for windows: An update. J. Appl. Cryst. 2012, 45, 849–854. [Google Scholar] [CrossRef]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Crystallogr. 2015, 71, 3–8. [Google Scholar]
- Brandenburg, K.; Putz, H. DIAMOND, Release 3.0e; Crystal Impact GbR: Bonn, Germany, 2006. [Google Scholar]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Identification Code | 2 | 5 | 6 |
|---|---|---|---|
| Empirical formula | C6H6Cl2O6 | C42H51Cl4NNbO15P | C48H86Cl12N6Nb4O14 |
| Formula weight | 122.5 | 1075.5 | 1768.3 |
| Temperature (K) | 100 (2) | 100 (2) | 100 (2) |
| Wavelength (Å) | 0.71073 | 0.71073 | 0.71073 |
| Crystal system, space group | Monoclinic, P21/c | Monoclinic, P21 | Monoclinic, P21/c |
| a (Å) | 8.5571 (15) | 10.468 (7) | 21.598 (4) |
| b (Å) | 10.3147 (16) | 18.005 (13) | 10.666 (2) |
| c (Å) | 5.135 | 12.584 (9) | 31.201 (6) |
| α (°) | 90.000 | 90.00 | 90.00 |
| β (°) | 104.420 (6) | 92.66 (3) | 92.198 (10) |
| γ (°) | 90.000 | 90.00 | 90.00 |
| Volume (Å3) | 438.97 (10) | 2369.2 (3) | 7182 (2) |
| Z | 4 | 2 | 4 |
| Densitycalc (g.cm−3) | 1.854 | 1.508 | 1.6326 |
| μ (mm−1) | 0.740 | 0.580 | 1.127 |
| Crystal size (mm) | 0.119 × 0.329 × 0.448 | 0.143 × 0.320 × 0.788 | 0.6 × 0.46 × 0.17 |
| Rint | 0.0375 | 0.0458 | 0.0710 |
| Goodness-of-fit on F2 | 1.194 | 1.026 | 1.201 |
| Final R indices (I > 2σ(I)) | R1 = 0.0205 wR2 = 0.0566 | R1 = 0.0341 wR2 = 0.0849 | R1 = 0.1117 wR2 = 0.2600 |
| ∆ρmax and ∆ρmin (e.Å−3) | 0.338, −0.278 | 1.522, −1.353 | 1.896, −3.264 |
| Bond Lengths (Å) | ||||||
| Type of Bond | 2 | (Å) | 5 | (Å) | 6 | (Å) |
| C=O | C1-O1 | 1.2256 (15) | C1-O1 | 1.316 (3) | C6b-O3b | 1.269 (12) |
| C1i-O1i | 1.2256 (15) | C2-O2 | 1.314 (3) | C1b-O2b | 1.237 (12) | |
| C1a-O1a | 1.278 (5) | C4b-O5b | 1.248 (12) | |||
| C2a-O2a | 1.288 (5) | C3b-O4b | 1.279 (12) | |||
| Average | 1.226 | 1.299 | 1.258 | |||
| C-O | C2-O2 | 1.3173 (14) | C4-O4 | 1.301 (4) | C6a-O3a | 1.241 (12) |
| C2i-O2i | 1.3173 (14) | C5-O5 | 1.285 (4) | C1a-O2a | 1.278 (11) | |
| C4a-O4a | 1.227 (5) | C4a-O5a | 1.278 (12) | |||
| C5a-O5a | 1.224 (5) | C3a-O4a | 1.253 (12) | |||
| Average | 1.317 | 1.259 | 1.263 | |||
| C-Cl | C3-Cl1 | 1.7186 (12) | C3-Cl1 | 1.799 (2) | C2b-Cl3b | 1.747 (10) |
| C3i-Cl1i | 1.7186 (12) | C3a-Cl1a | 1.725 (5) | C5b-Cl4b | 1.724 (10) | |
| C6-Cl2 | 1.782 (2) | C2a-Cl3a | 1.713 (10) | |||
| C6a-Cl2a | 1.728 (4) | C5a-Cl4a | 1.728 (9) | |||
| Average | 1.717 | 1.759 | 1.728 | |||
| Bond angles (°) | ||||||
| Type of Angle | 2 (a) | (°) (a) | 5 (b) | (°) | 6 (b) | (°) |
| O-(C-C)MC (c) | O1 C1 C2 | 118.15 (11) | Ok1-Ck2-Ck3 (d) | 115.4 | Ok1-Ck2-Ck3 (d) | 115.4 |
| O-C-COut (e) | O2 C2 C3 | 122.05 (11) | Ol1-Cl2-Cl3 (f) | 123.7 | Ol1-Cl2-Cl3 (f) | 125.3 |
| C-C-ClOut (g) | C2 C3 Cl1 | 121.12 (9) | Cm1-Cm2-Clm3 (h) | 120.0 | Cm1-Cm2-Clm3 (h) | 119.4 |
| C-C-C (i) | C3 C2 C1 | 120.18 (10) | Cn1-Cn2-Cn3 (j) | 120.0 | Cn1-Cn2-Cn3 (j) | 120.0 |
| Bond Lengths | 5 | 6 | ||||
|---|---|---|---|---|---|---|
| Type Bond | Bond | (Å) | Bond | (Å) | Bond | (Å) |
| Nb-Oca | Nb1-O1 | 2.103 (3) | Nb3b-O4b | 2.142 (7) | Nb2a-O5a | 2.129 (7) |
| Nb1-O2 | 2.116 (3) | Nb2b-O3b | 2.132 (7) | Nb1a-O2a | 2.139 (7) | |
| Nb1-O1a | 2.149 (3) | Nb3b-O5b | 2.402 (7) | Nb2a-O4a | 2.398 (7) | |
| Nb1-O2a | 2.145 (3) | Nb2b-O2b | 2.390 (7) | Nb1a-O3a | 2.381 (7) | |
| Nb-OOPPh3 | Nb1-O7 | 2.192 (3) | - | - | - | - |
| Nb-OH2 | Nb1-O6 | 2.108 (3) | - | - | - | - |
| Nb-O-Nb | - | - | Nb2b-O1c | 1.900 (8) | Nb2a-O2c | 1.896 (7) |
| - | - | Nb1a-O1c | 1.919 (8) | Nb3b-O2c | 1.914 (7) | |
| Nb=O | Nb1-O3 | 1.718 (3) | Nb3b-O6b | 1.716 (8) | Nb2a-O6a | 1.731 (8) |
| - | - | Nb2b-O1b | 1.705 (8) | Nb1a-O1a | 1.711 (8) | |
| Nb-Cl | - | - | Nb2b-Cl1b | 2.383 (3) | Nb1a-Cl1a | 2.464 (3) |
| - | - | Nb2b-Cl2b | 2.469 (3) | Nb1a-Cl2a | 2.376 (3) | |
| - | - | Nb3b-Cl6b | 2.375 (3) | Nb2a-Cl5a | 2.459 (3) | |
| - | - | Nb3b-Cl5b | 2.450 (3) | Nb2a-Cl6a | 2.386 (3) | |
| Nb-Nb | - | - | Nb2b-Nb2a | 9.131 (2) | Nb2b-Nb3b | 8.406 (2) |
| - | - | Nb3b-Nb1a | 9.265 (2) | Nb1a-Nb2a | 8.375 (2) | |
| - | - | Nb2b-Nb1a | 3.818 (6) | Nb3b-Nb2a | 3.807 (6) | |
| Bond Angles | 5 | 6 | ||||
|---|---|---|---|---|---|---|
| Type Angle | Angle | (°) | Angle | (°) | Angle | (°) |
| (O-Nb-O)bite | O1-Nb1-O2 | 73.07 (11) | O3b-Nb2b-O2b | 70.0 (3) | O3a-Nb1a-O2a | 70.9 (3) |
| O1a-Nb1-O2a | 71.93 (11) | O5b-Nb3b-O4b | 69.8 (3) | O5a-Nb2a-O4a | 70.3 (2) | |
| O1a-Nb1-O1 | 72.07 (11) | - | - | - | - | |
| O2-Nb1-O6 | 70.52 (11) | - | - | - | - | |
| O=Nb-Oca | O3-Nb1-O1 | 98.46 (13) | O1b-Nb2b-O3b | 95.0 (3) | O1a-Nb1a-O2a | 97.6 (3) |
| O1a-Nb1-O3 | 92.39 (13) | O6b-Nb3b-O4b | 94.0 (4) | O6a-Nb2a-O5a | 94.1 (3) | |
| O6-Nb1-O7 | 86.89 (12) | O1b-Nb2b-O1c | 100.3 (3) | O1a-Nb1a-O1c | 100.9 (4) | |
| O2-Nb1-O7 | 89.47 (12) | O6b-Nb3b-O2c | 102.1 (4) | O6a-Nb2a-O2c | 103.1 (4) | |
| O=Nb-Otrans | O2a-Nb1-O2 | 140.32 (12) | O1b-Nb2b-O2b | 164.8 (3) | O1a-Nb1a-O3a | 168.4 (3) |
| O3-Nb1-O7 | 174.93 (13) | O6b-Nb3b-O5b | 163.6 (3) | O6a-Nb2a-O4a | 163.4 (3) | |
| Cl-Nb-Cl | - | - | Cl1b-Nb2b-Cl2b | 88.12 (11) | Cl6b-Nb3b-Cl5b | 89.36 (12) |
| - | - | Cl2a-Nb1a-Cl1a | 88.97 (12) | Cl6a-Nb2a-Cl5a | 88.49 (10) | |
| O=Nb-Cl | - | - | O1b-Nb2b-Cl2b | 96.1 (3) | O1b-Nb2b-Cl1b | 103.0 (3) |
| - | - | O6b-Nb3b-Cl5b | 99.5 (3) | O6b-Nb3b-Cl6b | 100.5 (3) | |
| - | - | O1a-Nb1a-Cl1a | 97.5 (3) | O1a-Nb1a-Cl2a | 102.5 (3) | |
| - | - | O6a-Nb2a-Cl5a | 96.1 (3) | O6a-Nb2a-Cl6a | 102.5 (3) | |
| Entering Py Nucleophiles | pKa (a) | Keq (b) | kf | kr | |
|---|---|---|---|---|---|
| Number | Name | (M−1s−1) × 10−2 (c) | (s−1) × 10−2 (c) | ||
| 1 | DMAP | 9.60 | 24 ± 5 | 2.03 ± 0.05 | 0.08 ± 0.02 |
| 2 | 4Mepy | 6.02 | 1.91 ± 0.08 | 0.180 ± 0.006 | 0.09 ± 0.02 |
| 3 | py | 5.25 | 1.09 ± 0.02 | 0.130 ± 0.001 | 0.12 ± 0.01 |
| 4 | 3Clpy | 2.84 | 2.82 ± 0.13 | 0.210 ± 0.007 | 0.07 ± 0.02 |
| 5 | 3Brpy | 2.84 | 2.15 ± 0.09 | 0.150 ± 0.005 | 0.07 ± 0.01 |
| 15.5 °C | 24.5 °C | 31.2 °C | 45.9 °C | |
|---|---|---|---|---|
| kf (M−1s−1) × 10−3 | 6.7 ± 0.2 | 12.8 ± 0.3 | 20.3 ± 0.5 | 57.1 ± 1.5 |
| kr (s−1) × 10−3 | 0.59 ± 0.06 | 0.54 ± 0.08 | 0.83 ± 0.16 | 3.1 ± 0.5 |
| K1 (M−1) (a) | 11.3 ± 1.2 | 24 ± 3 | 24 ± 5 | 19 ± 3 |
| ΔH≠(kf) (kJ mol−1) | - | - | 51.5 ± 1.0 | - |
| ΔS≠(kf) (J K−1 mol−1) | - | - | −108 ± 3 | - |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Belay, A.N.; Venter, J.A.; Alexander, O.T.; Roodt, A. Synthesis, Single Crystal X-ray Structure, Spectroscopy and Substitution Behavior of Niobium(V) Complexes Activated by Chloranilate as Bidentate Ligand. Inorganics 2022, 10, 166. https://doi.org/10.3390/inorganics10100166
Belay AN, Venter JA, Alexander OT, Roodt A. Synthesis, Single Crystal X-ray Structure, Spectroscopy and Substitution Behavior of Niobium(V) Complexes Activated by Chloranilate as Bidentate Ligand. Inorganics. 2022; 10(10):166. https://doi.org/10.3390/inorganics10100166
Chicago/Turabian StyleBelay, Alebel Nibret, Johan Andries Venter, Orbett Teboho Alexander, and Andreas Roodt. 2022. "Synthesis, Single Crystal X-ray Structure, Spectroscopy and Substitution Behavior of Niobium(V) Complexes Activated by Chloranilate as Bidentate Ligand" Inorganics 10, no. 10: 166. https://doi.org/10.3390/inorganics10100166