
Mimicking Hypoxic-Ischemic Encephalopathy in a Newborn with 21q Deletion Originating from Ring Chromosome 21

Abstract
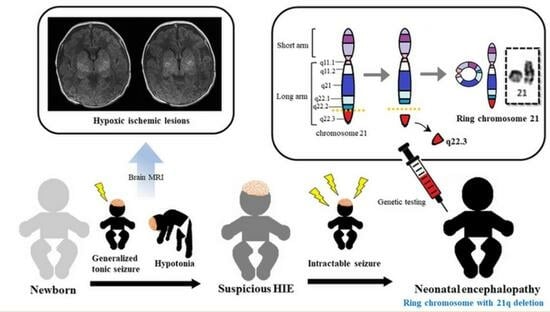
:
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Case Description
3. Discussion
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Roberson, E.D.; Wohler, E.S.; Hoover-Fong, J.E.; Lisi, E.; Stevens, E.L.; Thomas, G.H.; Leonard, J.; Hamosh, A.; Pevsner, J. Genomic analysis of partial 21q monosomies with variable phenotypes. Eur. J. Hum. Genet. 2011, 19, 235–238. [Google Scholar] [CrossRef] [PubMed]
- Burmeister, M.; Kim, S.; Price, E.R.; de Lange, T.; Tantravahi, U.; Myers, R.M.; Cox, D.R. A map of the distal region of the long arm of human chromosome 21 constructed by radiation hybrid mapping and pulsed-field gel electrophoresis. Genomics 1991, 9, 19–30. [Google Scholar] [CrossRef] [PubMed]
- Yao, G.; Chen, X.N.; Flores-Sarnat, L.; Barlow, G.M.; Palka, G.; Moeschler, J.B.; McGillivray, B.; Morse, R.P.; Korenberg, J.R. Deletion of chromosome 21 disturbs human brain morphogenesis. Genet. Med. 2006, 8, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Ronen, G.M.; Penney, S.; Andrews, W. The epidemiology of clinical neonatal seizures in Newfoundland: A population-based study. J. Pediatr. 1999, 134, 71–75. [Google Scholar] [CrossRef]
- Thayyil, S.; Montaldo, P.; Krishnan, V.; Ivain, P.; Pant, S.; Lally, P.J.; Bandiya, P.; Benkappa, N.; Kamalaratnam, C.N.; Chandramohan, R.; et al. Whole-Body Hypothermia, Cerebral Magnetic Resonance Biomarkers, and Outcomes in Neonates with Moderate or Severe Hypoxic-Ischemic Encephalopathy Born at Tertiary Care Centers vs Other Facilities: A Nested Study within a Randomized Clinical Trial. JAMA Netw. Open 2023, 6, e2312152. [Google Scholar] [CrossRef]
- Tekgul, H.; Gauvreau, K.; Soul, J.; Murphy, L.; Robertson, R.; Stewart, J.; Volpe, J.; Bourgeois, B.; du Plessis, A.J. The current etiologic profile and neurodevelopmental outcome of seizures in term newborn infants. Pediatrics 2006, 117, 1270–1280. [Google Scholar] [CrossRef]
- Douglas-Escobar, M.; Weiss, M.D. Hypoxic-Ischemic Encephalopathy: A Review for the Clinician. JAMA Pediatr. 2015, 169, 397–403. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Chen, X.; Liu, X.; Dong, X.; Ye, C.; Deng, D.; Lu, Y.; Lin, Y.; Zhou, W. Clinical features and underlying genetic causes in neonatal encephalopathy: A large cohort study. Clin. Genet. 2020, 98, 365–373. [Google Scholar] [CrossRef] [PubMed]
- Lejeune, J. The 21 Trisomy—Current Stage of Chromosomal Research. Prog. Med. Genet. 1964, 23, 144–177. [Google Scholar]
- Lindstrand, A.; Malmgren, H.; Sahlen, S.; Schoumans, J.; Nordgren, A.; Ergander, U.; Holm, E.; Anderlid, B.M.; Blennow, E. Detailed molecular and clinical characterization of three patients with 21q deletions. Clin. Genet. 2010, 77, 145–154. [Google Scholar] [CrossRef]
- Aronson, D.C.; Jansweijer, M.C.; Hoovers, J.M.; Barth, P.G. A male infant with holoprosencephaly, associated with ring chromosome 21. Clin. Genet. 1987, 31, 48–52. [Google Scholar] [CrossRef]
- Chen, C.P.; Lin, Y.H.; Chou, S.Y.; Su, Y.N.; Chern, S.R.; Chen, Y.T.; Town, D.D.; Chen, W.L.; Wang, W. Mosaic ring chromosome 21, monosomy 21, and isodicentric ring chromosome 21: Prenatal diagnosis, molecular cytogenetic characterization, and association with 2-Mb deletion of 21q21.1-q21.2 and 5-Mb deletion of 21q22.3. Taiwan J. Obstet. Gynecol. 2012, 51, 71–76. [Google Scholar] [CrossRef] [PubMed]
- Schmid, W.; Tenconi, R.; Baccichetti, C.; Caufin, D.; Schinzel, A. Ring chromosome 21 in phenotypically apparently normal persons: Report of two families from Switzerland and Italy. Am. J. Med. Genet. 1983, 16, 323–329. [Google Scholar] [CrossRef] [PubMed]
- Battaglia, A.; Guerrini, R. Chromosomal disorders associated with epilepsy. Epileptic Disord. 2005, 7, 181–192. [Google Scholar] [PubMed]
- Pardal Fernandez, J.M.; Jerez Garcia, P.; Carrascosa Romero, M.C.; Marco Giner, J. Chromosome 21 ring (r21) and epilepsy. An. Pediatr. (Barc) 2004, 60, 379–381. [Google Scholar]
- Specchio, N.; Carotenuto, A.; Trivisano, M.; Cappelletti, S.; Digilio, C.; Capolino, R.; Di Capua, M.; Fusco, L.; Vigevano, F. Ring 21 chromosome presenting with epilepsy and intellectual disability: Clinical report and review of the literature. Am. J. Med. Genet. Part A 2011, 155, 911–914. [Google Scholar] [CrossRef]
- Kunze, J.; Doose, H.; Tolksdorf, M. A dysplasia-epilepsy syndrome in a patient with ring chromosome 21. Neuropadiatrie 1975, 6, 398–402. [Google Scholar] [CrossRef]
- Palmer, C.G.; Hodes, M.E.; Reed, T.; Kojetin, J. Four new cases of ring 21 and 22 including familial transmission of ring 21. J. Med. Genet. 1977, 14, 54–60. [Google Scholar] [CrossRef]
- Serra, A.; Singh-Kahlon, D.P. 21 ring chromosome in a girl with stigmata of Down’s and G deletion I syndromes. Hum. Genet. 1976, 33, 47–53. [Google Scholar] [CrossRef]
- Pröschel, C.; Hansen, J.N.; Ali, A.; Tuttle, E.; Lacagnina, M.; Buscaglia, G.; Halterman, M.W.; Paciorkowski, A.R. Epilepsy-causing sequence variations in SIK1 disrupt synaptic activity response gene expression and affect neuronal morphology. Eur. J. Hum. Genet. 2017, 25, 216–221. [Google Scholar] [CrossRef]
- Simsek-Kiper, P.O.; Oguz, S.; Ergen, F.B.; Utine, G.E.; Alikasifoglu, M.; Haliloglu, G. A Revisited Diagnosis of Collagen VI Related Muscular Dystrophy in a Patient with a Novel COL6A2 Variant and 21q22.3 Deletion. Neuropediatrics 2020, 51, 445–449. [Google Scholar] [CrossRef] [PubMed]
- Ehling, D.; Kennerknecht, I.; Junge, A.; Prager, B.; Exeler, R.; Behre, B.; Horst, J.; Schmitt-John, T.; Bartsch, O.; Wirth, J. Mild phenotype in two unrelated patients with a partial deletion of 21q22.2-q22.3 defined by FISH and molecular studies. Am. J. Med. Genet. A 2004, 131, 265–272. [Google Scholar] [CrossRef] [PubMed]
- Chalak, L.; Ferriero, D.M.; Gressens, P.; Molloy, E.; Bearer, C. A 20 years conundrum of neonatal encephalopathy and hypoxic ischemic encephalopathy: Are we closer to a consensus guideline? Pediatr. Res. 2019, 86, 548–549. [Google Scholar] [CrossRef]
- Bruun, T.U.J.; DesRoches, C.L.; Wilson, D.; Chau, V.; Nakagawa, T.; Yamasaki, M.; Hasegawa, S.; Fukao, T.; Marshall, C.; Mercimek-Andrews, S. Prospective cohort study for identification of underlying genetic causes in neonatal encephalopathy using whole-exome sequencing. Genet. Med. 2018, 20, 486–494. [Google Scholar] [CrossRef] [PubMed]
- Sandoval Karamian, A.G.; Mercimek-Andrews, S.; Mohammad, K.; Molloy, E.J.; Chang, T.; Chau, V.; Murray, D.M.; Wusthoff, C.J.; Newborn Brain Society, G.; Publications, C. Neonatal encephalopathy: Etiologies other than hypoxic-ischemic encephalopathy. Semin. Fetal Neonatal Med. 2021, 26, 101272. [Google Scholar] [CrossRef]
- Allen, K.A.; Brandon, D.H. Hypoxic Ischemic Encephalopathy: Pathophysiology and Experimental Treatments. Newborn Infant Nurs. Rev. 2011, 11, 125–133. [Google Scholar] [CrossRef] [PubMed]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Moon, J.U.; Yum, S.K. Mimicking Hypoxic-Ischemic Encephalopathy in a Newborn with 21q Deletion Originating from Ring Chromosome 21. Children 2023, 10, 1461. https://doi.org/10.3390/children10091461
Moon JU, Yum SK. Mimicking Hypoxic-Ischemic Encephalopathy in a Newborn with 21q Deletion Originating from Ring Chromosome 21. Children. 2023; 10(9):1461. https://doi.org/10.3390/children10091461
Chicago/Turabian StyleMoon, Ja Un, and Sook Kyung Yum. 2023. "Mimicking Hypoxic-Ischemic Encephalopathy in a Newborn with 21q Deletion Originating from Ring Chromosome 21" Children 10, no. 9: 1461. https://doi.org/10.3390/children10091461