
Overexpression of Neuron-Derived Orphan Receptor 1 (NOR-1) Rescues Cardiomyocytes from Cell Death and Improves Viability after Doxorubicin Induced Stress


Abstract
:1. Introduction
2. Methods and Materials
2.1. Cells and Cell Treatment
2.2. Transfection of AC16 CMs
2.3. Doxorubicin Treatment
2.4. Protein Concentration Measurements by Bradford Assay
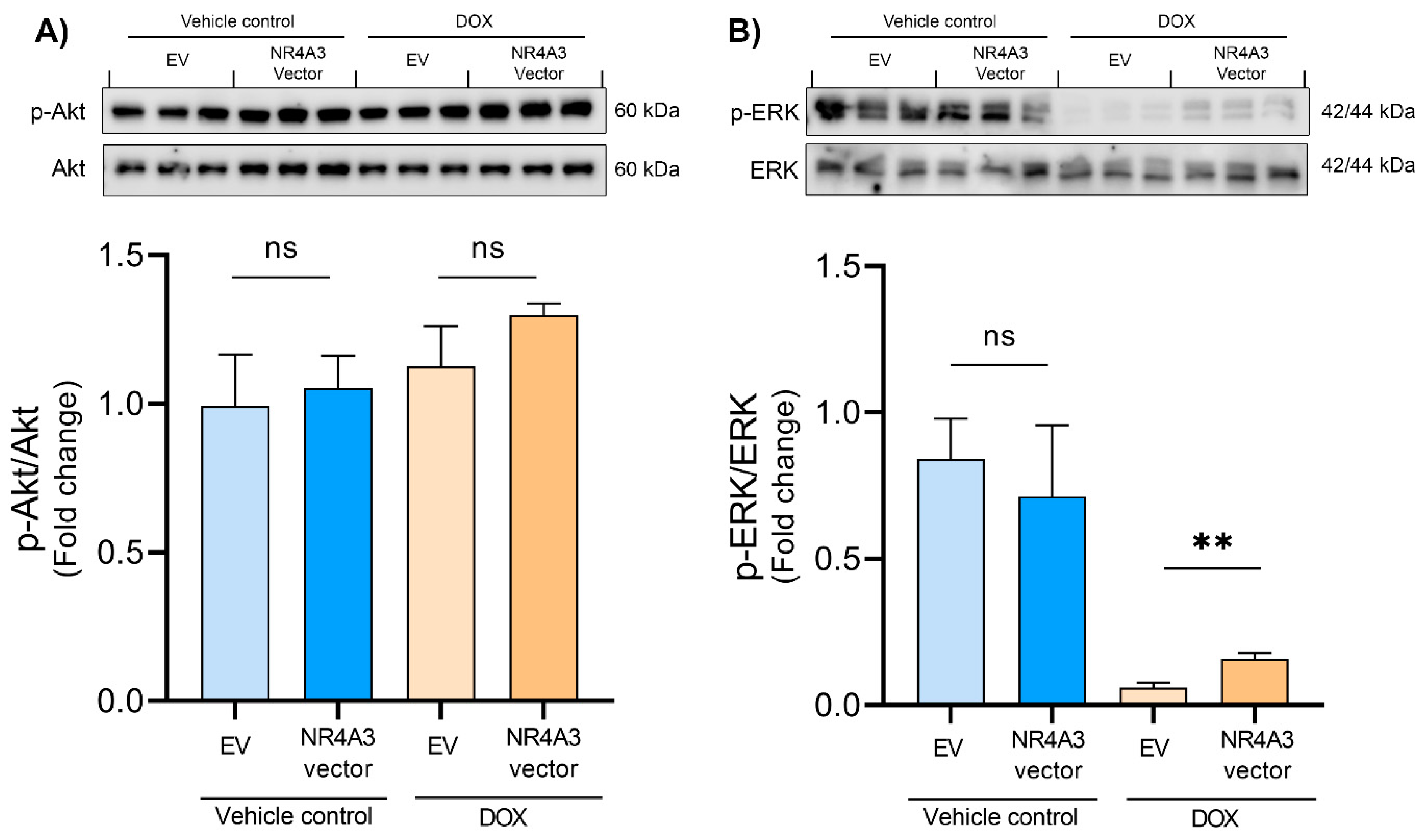
2.5. Western Blotting
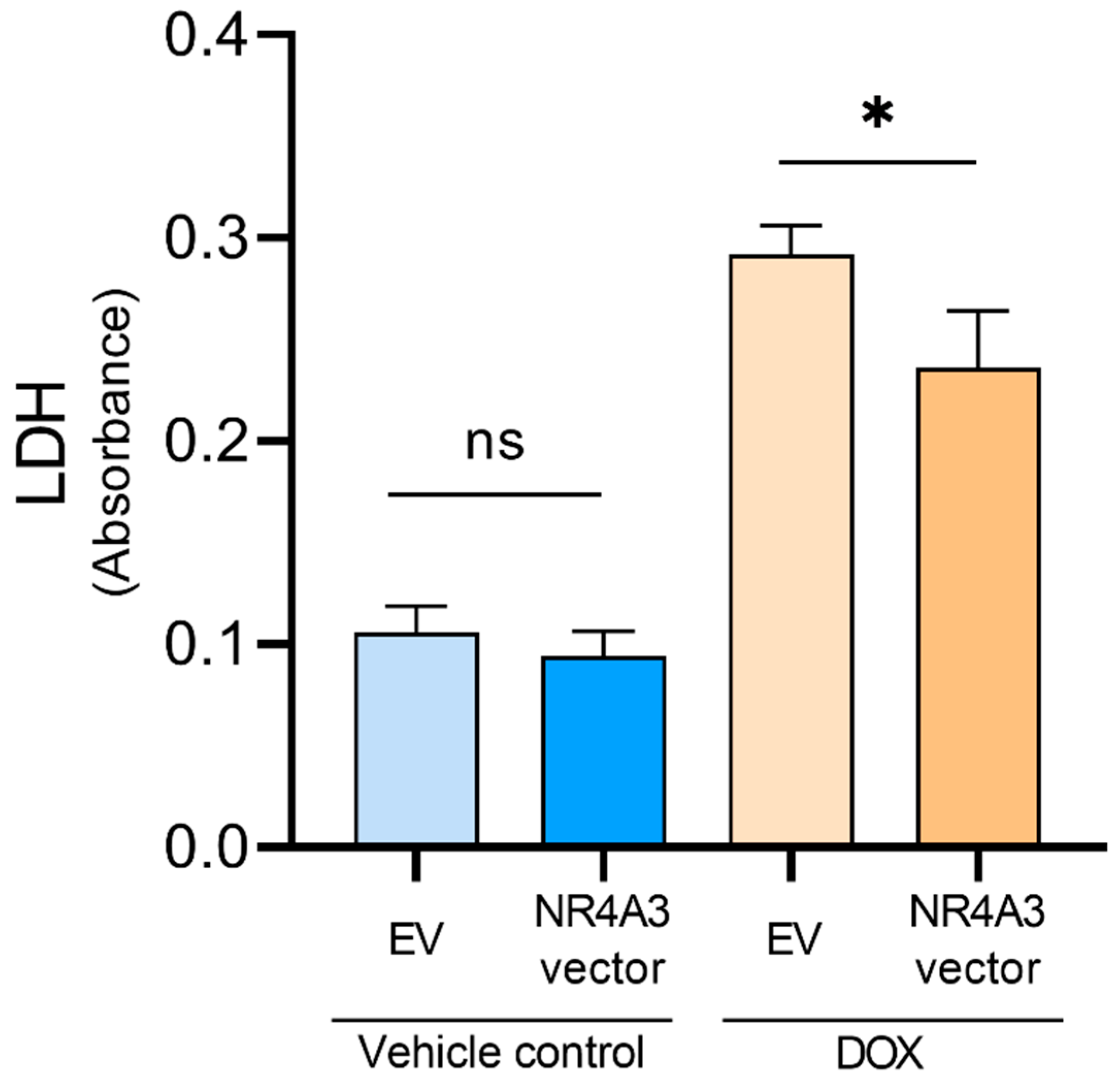
2.6. LDH Assay
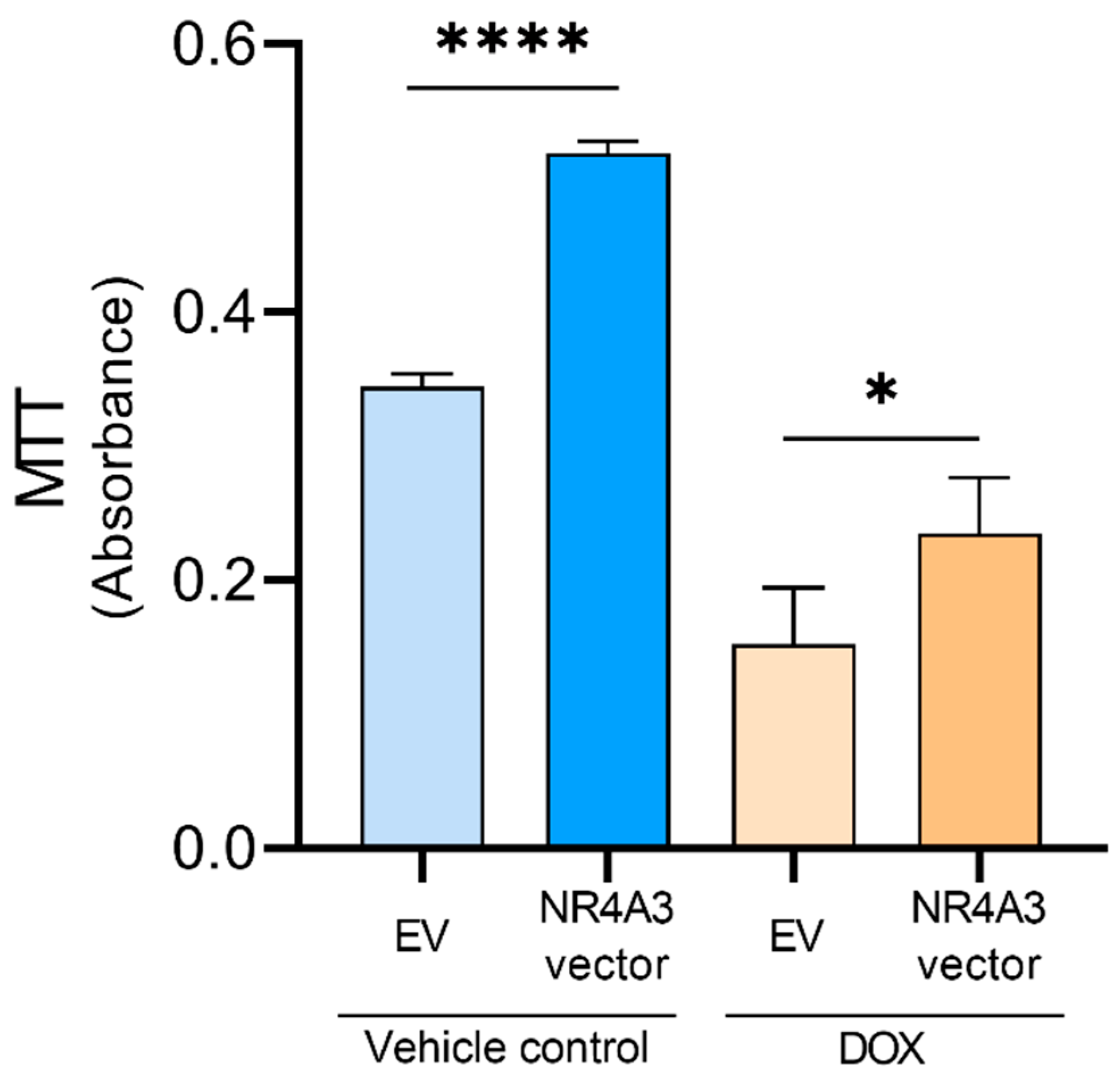
2.7. MTT Assay
2.8. Caspase-3 Assay
2.9. Statistical Analysis
3. Results
4. Discussion
5. Conclusions
6. Limitations
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Nallamothu, B.K.; Bradley, E.H.; Krumholz, H.M. Time to Treatment in Primary Percutaneous Coronary Intervention. N. Engl. J. Med. 2007, 357, 1631–1638. [Google Scholar] [CrossRef]
- Fröhlich, G.M.; Meier, P.; White, S.K.; Yellon, D.M.; Hausenloy, D.J. Myocardial reperfusion injury: Looking beyond primary PCI. Eur. Heart J. 2013, 34, 1714–1722. [Google Scholar] [CrossRef]
- Perrelli, M.-G.; Pagliaro, P.; Penna, C. Ischemia/reperfusion injury and cardioprotective mechanisms: Role of mitochondria and reactive oxygen species. World J. Cardiol. 2011, 3, 186–200. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Li, F.; Chi, Y.; Xiang, J. Potential relationship among three antioxidant enzymes in eliminating hydrogen peroxide in penaeid shrimp. Cell Stress Chaperones 2012, 17, 423–433. [Google Scholar] [CrossRef] [PubMed]
- Birben, E.; Sahiner, U.M.; Sackesen, C.; Erzurum, S.; Kalayci, O. Oxidative stress and antioxidant defense. World Allergy Organ. J. 2012, 5, 9–19. [Google Scholar] [CrossRef]
- McCully, J.D.; Wakiyama, H.; Hsieh, Y.J.; Jones, M.; Levitsky, S. Differential contribution of necrosis and apoptosis in myocardial ischemia-reperfusion injury. Am. J. Physiol. Heart Circ. Physiol. 2004, 286, H1923–H1935. [Google Scholar] [CrossRef] [PubMed]
- Carvalho, C.; Santos, R.X.; Cardoso, S.; Correia, S.; Oliveira, P.J.; Santos, M.S.; Moreira, P. Doxorubicin: The good, the bad and the ugly effect. Curr. Med. Chem. 2009, 16, 3267–3285. [Google Scholar] [CrossRef] [PubMed]
- Shan, K.; Lincoff, A.M.; Young, J.B. Anthracycline-induced cardiotoxicity. Ann. Intern. Med. 1996, 125, 47–58. [Google Scholar] [CrossRef]
- Chatterjee, K.; Zhang, J.; Honbo, N.; Karliner, J.S. Doxorubicin Cardiomyopathy. Cardiology 2010, 115, 155–162. [Google Scholar] [CrossRef]
- Singal, P.K.; Deally, C.M.; Weinberg, L.E. Subcellular effects of adriamycin in the heart: A concise review. J. Mol. Cell. Cardiol. 1987, 19, 817–828. [Google Scholar] [CrossRef]
- Kalyanaraman, B.; Perez-Reyes, E.; Mason, R.P. Spin-trapping and direct electron spin resonance investigations of the redox metabolism of quinone anticancer drugs. Biochim. Biophys. Acta 1980, 630, 119–130. [Google Scholar] [CrossRef]
- Doroshow, J.H. Effect of anthracycline antibiotics on oxygen radical formation in rat heart. Cancer Res. 1983, 43, 460–472. [Google Scholar]
- Murry, C.E.; Jennings, R.B.; Reimer, K.A. Preconditioning with ischemia: A delay of lethal cell injury in ischemic myocardium. Circulation 1986, 74, 1124–1136. [Google Scholar] [CrossRef]
- He, Q.; Wang, F.; Ryan, T.D.; Chalasani, M.; Redington, A.N. Repeated Remote Ischemic Conditioning Reduces Doxorubicin-Induced Cardiotoxicity. JACC CardioOncol. 2020, 2, 41–52. [Google Scholar] [CrossRef]
- Hausenloy, D.J.; Ng, C.T.; Chong, J.H. Repeated Remote Ischemic Conditioning Protects against Doxorubicin Cardiotoxicity: Never Too Much of a Good Thing. JACC CardioOncol. 2020, 2, 53–55. [Google Scholar] [CrossRef]
- Hausenloy, D.J.; Tsang, A.; Mocanu, M.M.; Yellon, D.M. Ischemic preconditioning protects by activating prosurvival kinases at reperfusion. Am. J. Physiol. Heart Circ. Physiol. 2005, 288, H971–H976. [Google Scholar] [CrossRef]
- Kalakech, H.; Hibert, P.; Prunier-Mirebeau, D.; Tamareille, S.; Letournel, F.; Macchi, L.; Pinet, F.; Furber, A.; Prunier, F. RISK and SAFE signaling pathway involvement in apolipoprotein A-I-induced cardioprotection. PLoS ONE 2014, 9, e107950. [Google Scholar] [CrossRef]
- Walsh, K.; Lee, D.P.; Hiatt, B.L.; Carter, A.J.; Huston, M.; Schreiber, D.; DiBattiste, P.M.; Herity, N.A.; Fearon, W.F.; Rezaee, M.; et al. Akt signaling and growth of the heart. Circulation 2006, 113, 2032–2034. [Google Scholar] [CrossRef] [PubMed]
- Gallo, S.; Vitacolonna, A.; Bonzano, A.; Comoglio, P.; Crepaldi, T. ERK: A Key Player in the Pathophysiology of Cardiac Hypertrophy. Int. J. Mol. Sci. 2019, 20, 2164. [Google Scholar] [CrossRef] [PubMed]
- Hausenloy, D.J.; Tsang, A.; Yellon, D.M. The Reperfusion Injury Salvage Kinase Pathway: A Common Target for Both Ischemic Preconditioning and Postconditioning. Trends Cardiovasc. Med. 2005, 15, 69–75. [Google Scholar] [CrossRef] [PubMed]
- Hafeez, S.; Urooj, M.; Saleem, S.; Gillani, Z.; Shaheen, S.; Qazi, M.H.; Naseer, M.I.; Iqbal, Z.; Ansari, S.A.; Haque, A.; et al. BAD, a Proapoptotic Protein, Escapes ERK/RSK Phosphorylation in Deguelin and siRNA-Treated HeLa Cells. PLoS ONE 2016, 11, e0145780. [Google Scholar] [CrossRef]
- Ding, Q.; Xia, W.; Liu, J.-C.; Yang, J.-Y.; Lee, D.-F.; Xia, J.; Bartholomeusz, G.; Li, Y.; Pan, Y.; Li, Z.; et al. Erk Associates with and Primes GSK-3β for Its Inactivation Resulting in Upregulation of β-Catenin. Mol. Cell 2005, 19, 159–170. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.-Q.; Chen, F.; Li, L. Role of glycogen synthase kinase following myocardial infarction and ischemia–reperfusion. Apoptosis 2019, 24, 539–540. [Google Scholar] [CrossRef] [PubMed]
- Yellon, D.M.; Downey, J.M. Preconditioning the Myocardium: From Cellular Physiology to Clinical Cardiology. Physiol. Rev. 2003, 83, 1113–1151. [Google Scholar] [CrossRef] [PubMed]
- Brown, D.A.; Lynch, J.M.; Armstrong, C.J.; Caruso, N.M.; Ehlers, L.B.; Johnson, M.S.; Moore, R.L. Susceptibility of the heart to ischaemia-reperfusion injury and exercise-induced cardioprotection are sex-dependent in the rat. J. Physiol. 2005, 564 Pt 2, 619–630. [Google Scholar] [CrossRef]
- Demirel, H.A.; Powers, S.K.; Zergeroglu, M.A.; Shanely, R.A.; Hamilton, K.; Coombes, J.; Naito, H. Short-term exercise improves myocardial tolerance to in vivo ischemia-reperfusion in the rat. J. Appl. Physiol. 2001, 91, 2205–2212. [Google Scholar] [CrossRef]
- Libonati, J.R.; Gaughan, J.P.; Hefner, C.A.; Gow, A.; Paolone, A.M.; Houser, S.R. Reduced ischemia and reperfusion injury following exercise training. Med. Sci. Sports Exerc. 1997, 29, 509–516. [Google Scholar] [CrossRef] [PubMed]
- Zhang, K.-R.; Liu, H.-T.; Zhang, H.-F.; Zhang, Q.-J.; Li, Q.-X.; Yu, Q.-J.; Guo, W.-Y.; Wang, H.-C.; Gao, F. Long-term aerobic exercise protects the heart against ischemia/reperfusion injury via PI3 kinase-dependent and Akt-mediated mechanism. Apoptosis 2007, 12, 1579–1588. [Google Scholar] [CrossRef]
- Sun, X.J.; Mao, J.R. Role of Janus kinase 2/signal transducer and activator of transcription 3 signaling pathway in cardioprotection of exercise preconditioning. Eur. Rev. Med. Pharm. Sci. 2018, 22, 4975–4986. [Google Scholar]
- Gomes, M.J.; Pagan, L.U.; Lima, A.R.R.; Reyes, D.R.A.; Martinez, P.F.; Damatto, F.C.; Pontes, T.H.D.; Rodrigues, E.A.; Souza, L.M.; Tosta, I.F.; et al. Effects of aerobic and resistance exercise on cardiac remodelling and skeletal muscle oxidative stress of infarcted rats. J. Cell Mol. Med. 2020, 24, 5352–5362. [Google Scholar] [CrossRef]
- Pillon, N.J.; Gabriel, B.M.; Dollet, L.; Smith, J.A.B.; Puig, L.S.; Botella, J.; Bishop, D.J.; Krook, A.; Zierath, J.R. Transcriptomic profiling of skeletal muscle adaptations to exercise and inactivity. Nat. Commun. 2020, 11, 470. [Google Scholar] [CrossRef]
- Medzikovic, L.; de Vries, C.J.M.; de Waard, V. NR4A nuclear receptors in cardiac remodeling and neurohormonal regulation. Trends Cardiovasc. Med. 2019, 29, 429–437. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Feng, Y.-P.; Tang, L.-X.; Yan, Y.-L.; Bai, J.-W. The protective role of NR4A3 in acute myocardial infarction by suppressing inflammatory responses via JAK2-STAT3/NF-κB pathway. Biochem. Biophys. Res. Commun. 2019, 517, 697–702. [Google Scholar] [CrossRef]
- Pearen, M.A.; Goode, J.M.; Fitzsimmons, R.L.; Eriksson, N.A.; Thomas, G.P.; Cowin, G.J.; Wang, S.-C.M.; Tuong, Z.K.; Muscat, G.E.O. Transgenic Muscle-Specific Nor-1 Expression Regulates Multiple Pathways That Effect Adiposity, Metabolism, and Endurance. Mol. Endocrinol. 2013, 27, 1897–1917. [Google Scholar] [CrossRef] [PubMed]
- Harris, T.E.; Finck, B.N. Dual function lipin proteins and glycerolipid metabolism. Trends Endocrinol. Metab. 2011, 22, 226–233. [Google Scholar] [CrossRef]
- Jiang, W.; Zhu, J.; Zhuang, X.; Zhang, X.; Luo, T.; Esser, K.A.; Ren, H. Lipin1 Regulates Skeletal Muscle Differentiation through Extracellular Signal-regulated Kinase (ERK) Activation and Cyclin D Complex-regulated Cell Cycle Withdrawal. J. Biol. Chem. 2015, 290, 23646–23655. [Google Scholar] [CrossRef]
- Chio, C.-C.; Wei, L.; Chen, T.G.; Lin, C.-M.; Shieh, J.-P.; Yeh, P.-S.; Chen, R.-M. Neuron-derived orphan receptor 1 transduces survival signals in neuronal cells in response to hypoxia-induced apoptotic insults. J. Neurosurg. 2016, 124, 1654–1664. [Google Scholar] [CrossRef]
- Martorell, L.; Gentile, M.; Rius, J.; Rodriguez, C.; Crespo, J.; Badimon, L.; Martínez-González, J. The hypoxia-inducible factor 1/NOR-1 axis regulates the survival response of endothelial cells to hypoxia. Mol. Cell Biol. 2009, 29, 5828–5842. [Google Scholar] [CrossRef]
- Alonso, J.; Galán, M.; Martí-Pàmies, I.; Romero, J.M.; Camacho, M.; Rodriguez, C.; Martínez-González, J. NOR-1/NR4A3 regulates the cellular inhibitor of apoptosis 2 (cIAP2) in vascular cells: Role in the survival response to hypoxic stress. Sci. Rep. 2016, 6, 34056. [Google Scholar] [CrossRef]
- Davidson, M.M.; Nesti, C.; Palenzuela, L.; Walker, W.F.; Hernandez, E.; Protas, L.; Hirano, M.; Isaac, N.D. Novel cell lines derived from adult human ventricular cardiomyocytes. J. Mol. Cell. Cardiol. 2005, 39, 133–147. [Google Scholar] [CrossRef]
- Porter, A.G.; Jänicke, R.U. Emerging roles of caspase-3 in apoptosis. Cell Death Differ. 1999, 6, 99–104. [Google Scholar] [CrossRef]
- Bertini, I.; Chevance, S.; Del Conte, R.; Lalli, D.; Turano, P. The Anti-Apoptotic Bcl-xL Protein, a New Piece in the Puzzle of Cytochrome C Interactome. PLoS ONE 2011, 6, e18329. [Google Scholar] [CrossRef] [PubMed]
- Rossello, X.; Yellon, D.M. The RISK pathway and beyond. Basic Res. Cardiol. 2017, 113, 1–5. [Google Scholar] [CrossRef]
- Rossello, X.; Riquelme, J.A.; Davidson, S.M.; Yellon, D.M. Role of PI3K in myocardial ischaemic preconditioning: Mapping pro-survival cascades at the trigger phase and at reperfusion. J. Cell Mol. Med. 2018, 22, 926–935. [Google Scholar] [CrossRef]
- Song, Y.-H.; Cai, H.; Zhao, Z.-M.; Chang, W.-J.; Gu, N.; Cao, S.-P.; Wu, M.-L. Hydrogen sulfide protects H9c2 cardiac cells against doxorubicin-induced cytotoxicity through the PI3K/Akt/FoxO3a pathway. Int. J. Mol. Med. 2016, 37, 1661–1668. [Google Scholar]
- Song, Y.-H.; Cai, H.; Zhao, Z.-M.; Chang, W.-J.; Gu, N.; Cao, S.-P.; Wu, M.-L. Icariin attenuated oxidative stress induced-cardiac apoptosis by mitochondria protection and ERK activation. Biomed. Pharmacother. 2016, 83, 1089–1094. [Google Scholar] [CrossRef]
- Chen, Y.L.; Loh, S.H.; Chen, J.J.; Tsai, C.S. Urotensin II prevents cardiomyocyte apoptosis induced by doxorubicin via Akt and ERK. Eur. J. Pharmacol. 2012, 680, 88–94. [Google Scholar] [CrossRef] [PubMed]
- Lin, L.; White, S.A.; Hu, K. Role of p90RSK in Kidney and Other Diseases. Int. J. Mol. Sci. 2019, 20, 972. [Google Scholar] [CrossRef]
- Stambolic, V.; Woodgett, J.R. Mitogen inactivation of glycogen synthase kinase-3 beta in intact cells via serine 9 phosphorylation. Biochem. J. 1994, 303 Pt 3, 701–704. [Google Scholar] [CrossRef]
- Gough, D.J.; Koetz, L.; Levy, D.E. The MEK-ERK pathway is necessary for serine phosphorylation of mitochondrial STAT3 and Ras-mediated transformation. PLoS ONE 2013, 8, e83395. [Google Scholar] [CrossRef]
- O’Sullivan, K.E.; Breen, E.P.; Gallagher, H.C.; Buggy, D.J.; Hurley, J.P. Understanding STAT3 signaling in cardiac ischemia. Basic Res. Cardiol. 2016, 111, 27. [Google Scholar] [CrossRef]
- Ludke, A.; Akolkar, G.; Ayyappan, P.; Sharma, A.K.; Singal, P.K. Time course of changes in oxidative stress and stress-induced proteins in cardiomyocytes exposed to doxorubicin and prevention by vitamin C. PLoS ONE 2017, 12, e0179452. [Google Scholar] [CrossRef]
- Wang, Y.; Branicky, R.; Noë, A.; Hekimi, S. Superoxide dismutases: Dual roles in controlling ROS damage and regulating ROS signaling. J. Cell Biol. 2018, 217, 1915–1928. [Google Scholar] [CrossRef]
- Lin, L.; Bu, G.; Mars, W.M.; Reeves, W.B.; Tanaka, S.; Hu, K. tPA Activates LDL Receptor-Related Protein 1-Mediated Mitogenic Signaling Involving the p90RSK and GSK3β Pathway. Am. J. Pathol. 2010, 177, 1687–1696. [Google Scholar] [CrossRef]
- Yang, K.; Hitomi, M.; Stacey, D.W. Variations in cyclin D1 levels through the cell cycle determine the proliferative fate of a cell. Cell Div. 2006, 1, 1–32. [Google Scholar] [CrossRef] [PubMed]
- Hayakawa, J.; Ohmichi, M.; Kurachi, H.; Kanda, Y.; Hisamoto, K.; Nishio, Y.; Adachi, K.; Tasaka, K.; Kanzaki, T.; Murata, Y. Inhibition of BAD Phosphorylation Either at Serine 112 via Extracellular Signal-regulated Protein Kinase Cascade or at Serine 136 via Akt Cascade Sensitizes Human Ovarian Cancer Cells to Cisplatin. Cancer Res. 2000, 60, 5988. [Google Scholar] [PubMed]
- Granger, D.N.; Korthuis, R.J. Physiologic Mechanisms of Postischemic Tissue Injury. Annu. Rev. Physiol. 1995, 57, 311–332. [Google Scholar] [CrossRef]
- Octavia, Y.; Tocchetti, C.G.; Gabrielson, K.L.; Janssens, S.; Crijns, H.J.; Moens, A.L. Doxorubicin-induced cardiomyopathy: From molecular mechanisms to therapeutic strategies. J. Mol. Cell. Cardiol. 2012, 52, 1213–1225. [Google Scholar] [CrossRef] [PubMed]
- Yuan, H.; Zhang, Q.; Guo, J.; Zhang, T.; Zhao, J.; Li, J.; White, A.; Carmichael, P.L.; Westmoreland, C.; Peng, S. APGC-1α-Mediated Transcriptional Network Maintains Mitochondrial Redox Bioenergetic Homeostasis against Doxorubicin-Induced Toxicity in Human Cardiomyocytes: Implementation of TT21C. Toxicol. Sci. 2016, 150, 400–417. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Wang, J.; Zhou, Y. Long non-coding RNA SNHG1 protects human AC16 cardiomyocytes from doxorubicin toxicity by regulating miR-195/Bcl-2 axis. Biosci. Rep. 2019, 39, BSR20191050. [Google Scholar] [CrossRef]
- Yoon, C.S.; Kim, H.K.; Mishchenko, N.P.; Vasileva, E.A.; Fedoreyev, S.A.; Stonik, V.A.; Han, J. Spinochrome D Attenuates Doxorubicin-Induced Cardiomyocyte Death via Improving Glutathione Metabolism and Attenuating Oxidative Stress. Mar. Drugs 2018, 17, 2. [Google Scholar] [CrossRef]
- Kumar, P.; Nagarajan, A.; Uchil, P.D. Analysis of Cell Viability by the Lactate Dehydrogenase Assay. Cold Spring Harb. Protoc. 2018, 2018, pdb.prot095497. [Google Scholar] [CrossRef] [PubMed]
- Kurakula, K.; Koenis, D.S.; van Tiel, C.M.; de Vries, C.J. NR4A nuclear receptors are orphans but not lonesome. Biochim. Biophys. Acta 2014, 1843, 2543–2555. [Google Scholar] [CrossRef]
- Nicholson, D.W.; Ali, A.; Thornberry, N.A.; Vaillancourt, J.P.; Ding, C.K.; Gallant, M.; Gareau, Y.; Griffin, P.R.; Labelle, M.; Lazebnik, Y.A.; et al. Identification and inhibition of the ICE/CED-3 protease necessary for mammalian apoptosis. Nature 1995, 376, 37–43. [Google Scholar] [CrossRef]
- Pan, J.-A.; Tang, Y.; Yu, J.-Y.; Zhang, H.; Zhang, J.-F.; Wang, C.-Q.; Gu, J. miR-146a attenuates apoptosis and modulates autophagy by targeting TAF9b/P53 pathway in doxorubicin-induced cardiotoxicity. Cell Death Dis. 2019, 10, 668. [Google Scholar] [CrossRef] [PubMed]
- Boulghobra, D.; Coste, F.; Geny, B.; Reboul, C. Exercise training protects the heart against ischemia-reperfusion injury: A central role for mitochondria? Free Radic. Biol. Med. 2020, 152, 395–410. [Google Scholar] [CrossRef] [PubMed]
- Granger, D.N.; Kvietys, P.R. Reperfusion injury and reactive oxygen species: The evolution of a concept. Redox Biol. 2015, 6, 524–551. [Google Scholar] [CrossRef]
- Mocanu, M.M.; Bell, R.M.; Yellon, D.M. PI3 kinase and not p42/p44 appears to be implicated in the protection conferred by ischemic preconditioning. J. Mol. Cell Cardiol. 2002, 34, 661–668. [Google Scholar] [CrossRef]
- Tong, H.; Chen, W.; Steenbergen, C.; Murphy, E. Ischemic Preconditioning Activates Phosphatidylinositol-3-Kinase Upstream of Protein Kinase C. Circ. Res. 2000, 87, 309–315. [Google Scholar] [CrossRef]
- Zhang, X.; Hu, C.; Kong, C.-Y.; Song, P.; Wu, H.-M.; Xu, S.-C.; Yuan, Y.-P.; Deng, W.; Ma, Z.-G.; Tang, Q.-Z. FNDC5 alleviates oxidative stress cardiomyocyte apoptosis in doxorubicin-induced cardiotoxicity via activating AKT. Cell Death Differ. 2020, 27, 540–555. [Google Scholar] [CrossRef]
- Yu, W.; Qin, X.; Zhang, Y.; Qiu, P.; Wang, L.; Zha, W.; Ren, J. Curcumin suppresses doxorubicin-induced cardiomyocyte pyroptosis via a PI3K/Akt/mTOR-dependent manner. Cardiovasc. Diagn. Ther. 2020, 10, 752–769. [Google Scholar] [CrossRef]
- Fan, G.-C.; Zhou, X.; Wang, X.; Song, G.; Qian, J.; Nicolaou, P.; Chen, G.; Ren, X.; Kranias, E.G. Heat Shock Protein 20 Interacting with Phosphorylated Akt Reduces Doxorubicin-Triggered Oxidative Stress and Cardiotoxicity. Circ. Res. 2008, 103, 1270–1279. [Google Scholar] [CrossRef] [PubMed]
- Tran, S.E.; Holmström, T.H.; Ahonen, M.; Kähäri, V.-M.; Eriksson, J.E. MAPK/ERK overrides the apoptotic signaling from Fas, TNF, and TRAIL receptors. J. Biol. Chem. 2001, 276, 16484–16490. [Google Scholar] [CrossRef] [PubMed]
- Allan, L.A.; Morrice, N.; Brady, S.; Magee, G.; Pathak, S.; Clarke, P.R. Inhibition of caspase-9 through phosphorylation at Thr 125 by ERK MAPK. Nat. Cell Biol. 2003, 5, 647–654. [Google Scholar] [CrossRef] [PubMed]
- Zhai, P.; Sciarretta, S.; Galeotti, J.; Volpe, M.; Sadoshima, J. Differential roles of GSK-3β during myocardial ischemia and ischemia/reperfusion. Circ. Res. 2011, 109, 502–511. [Google Scholar] [CrossRef] [PubMed]
- Miura, T.; Miki, T. GSK-3β, a Therapeutic Target for Cardiomyocyte Protection. Circ. J. 2009, 73, 1184–1192. [Google Scholar] [CrossRef] [PubMed]
- Hahn, J.-Y.; Cho, H.-J.; Bae, J.-W.; Yuk, H.-S.; Kim, K.-I.; Park, K.-W.; Koo, B.-K.; Chae, I.-H.; Shin, C.-S.; Oh, B.-H.; et al. Beta-catenin overexpression reduces myocardial infarct size through differential effects on cardiomyocytes and cardiac fibroblasts. J. Biol. Chem. 2006, 281, 30979–30989. [Google Scholar] [CrossRef]
- Hanf, A.; Oelze, M.; Manea, A.; Li, H.; Münzel, T.; Daiber, A. The anti-cancer drug doxorubicin induces substantial epigenetic changes in cultured cardiomyocytes. Chem.-Biol. Interact. 2019, 313, 108834. [Google Scholar] [CrossRef] [PubMed]
- Bresciani, G.; da Cruz, I.B.; González-Gallego, J. Manganese superoxide dismutase and oxidative stress modulation. Adv. Clin. Chem. 2015, 68, 87–130. [Google Scholar] [PubMed]
- Chaiswing, L.; Cole, M.P.; Ittarat, W.; Szweda, L.I.; St Clair, D.K.; Oberley, T.D. Manganese superoxide dismutase and inducible nitric oxide synthase modify early oxidative events in acute adriamycin-induced mitochondrial toxicity. Mol. Cancer 2005, 4, 1056–1064. [Google Scholar] [CrossRef]
- Cheung, K.G.; Cole, L.K.; Xiang, B.; Chen, K.; Ma, X.; Myal, Y.; Hatch, G.M.; Tong, Q.; Dolinsky, V.W. Sirtuin-3 (SIRT3) Protein Attenuates Doxorubicin-induced Oxidative Stress and Improves Mitochondrial Respiration in H9c2 Cardiomyocytes. J. Biol. Chem. 2015, 290, 10981–10993. [Google Scholar] [CrossRef] [PubMed]
- Kavazis, A.N.; Smuder, A.J.; Min, K.; Tümer, N.; Powers, S.K. Short-term exercise training protects against doxorubicin-induced cardiac mitochondrial damage independent of HSP72. Am. J. Physiol. Heart Circ. Physiol. 2010, 299, H1515–H1524. [Google Scholar] [CrossRef]
- Alonso, J.; Cañes, L.; García-Redondo, A.B.; de Frutos, P.G.; Rodríguez, C.; Martínez-González, J. The nuclear receptor NOR-1 modulates redox homeostasis in human vascular smooth muscle cells. J. Mol. Cell Cardiol. 2018, 122, 23–33. [Google Scholar] [CrossRef] [PubMed]
- Kaur, G.; Dufour, J.M. Cell lines: Valuable tools or useless artifacts. Spermatogenesis 2012, 2, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Andrysiak, K.; Stępniewski, J.; Dulak, J. Human-induced pluripotent stem cell-derived cardiomyocytes, 3D cardiac structures, and heart-on-a-chip as tools for drug research. Pflüg. Arch. Eur. J. Physiol. 2021, 473, 1061–1085. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Protein of Interest | Amount Protein Loaded (µg) | Gel Percentage (%) |
|---|---|---|
| Beta-actin | 30 | 10 |
| NOR-1 | 40 | 10 |
| Phospho-Akt (Ser473) | 40 | 10 |
| Akt | 20 | 10 |
| Phospho-ERK1/2 (Thr202/Tyr204) | 40 | 10 |
| ERK1/2 | 20 | 10 |
| Phospho-GSK-3β (Ser9) | 50 | 10 |
| GSK-3β | 20 | 10 |
| Phospho-STAT3 (Ser727) | 60 | 10 |
| STAT3 | 20 | 10 |
| Bcl-xL | 40 | 12 |
| SOD2 | 50 | 12 |
| Cyclin D1 | 30 | 10 |
| Proteins of Interest | Host | Primary Antibody Dilution in 5 mL TBS-T | Secondary Antibody Dilution in 10 mL TBS-T |
|---|---|---|---|
| Beta-Actin | Mouse | 1:1000 | 1:3000 |
| NOR-1 | Mouse | 1:500 | 1:3000 |
| Phospho-Akt (Ser473) | Rabbit | 1:1000 | 1:2000 |
| Akt | Rabbit | 1:1000 | 1:2000 |
| Phospho-ERK1/2 (Thr202/Tyr204) | Rabbit | 1:1000 | 1:2000 |
| ERK1/2 | Rabbit | 1:1000 | 1:2000 |
| Phospho-GSK-3β (Ser9) | Rabbit | 1:1000 | 1:2000 |
| GSK-3β | Rabbit | 1:1000 | 1:2000 |
| Phospho-STAT3 (Ser727) | Rabbit | 1:1000 | 1:2000 |
| STAT3 | Mouse | 1:1000 | 1:3000 |
| Bcl-xL | Rabbit | 1:2000 | 1:2000 |
| SOD2 | Rabbit | 1:1000 | 1:2000 |
| Cyclin D1 | Rabbit | 1:300 | 1:2000 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Berg, P.-C.; Hansson, Å.M.L.; Røsand, Ø.; Marwarha, G.; Høydal, M.A. Overexpression of Neuron-Derived Orphan Receptor 1 (NOR-1) Rescues Cardiomyocytes from Cell Death and Improves Viability after Doxorubicin Induced Stress. Biomedicines 2021, 9, 1233. https://doi.org/10.3390/biomedicines9091233
Berg P-C, Hansson ÅML, Røsand Ø, Marwarha G, Høydal MA. Overexpression of Neuron-Derived Orphan Receptor 1 (NOR-1) Rescues Cardiomyocytes from Cell Death and Improves Viability after Doxorubicin Induced Stress. Biomedicines. 2021; 9(9):1233. https://doi.org/10.3390/biomedicines9091233
Chicago/Turabian StyleBerg, Per-Christian, Åse Mari Larsen Hansson, Øystein Røsand, Gurdeep Marwarha, and Morten Andre Høydal. 2021. "Overexpression of Neuron-Derived Orphan Receptor 1 (NOR-1) Rescues Cardiomyocytes from Cell Death and Improves Viability after Doxorubicin Induced Stress" Biomedicines 9, no. 9: 1233. https://doi.org/10.3390/biomedicines9091233
APA StyleBerg, P.-C., Hansson, Å. M. L., Røsand, Ø., Marwarha, G., & Høydal, M. A. (2021). Overexpression of Neuron-Derived Orphan Receptor 1 (NOR-1) Rescues Cardiomyocytes from Cell Death and Improves Viability after Doxorubicin Induced Stress. Biomedicines, 9(9), 1233. https://doi.org/10.3390/biomedicines9091233