
The Role of Smoothened-Dependent and -Independent Hedgehog Signaling Pathway in Tumorigenesis


 , , and
, , and
Abstract
:1. Introduction
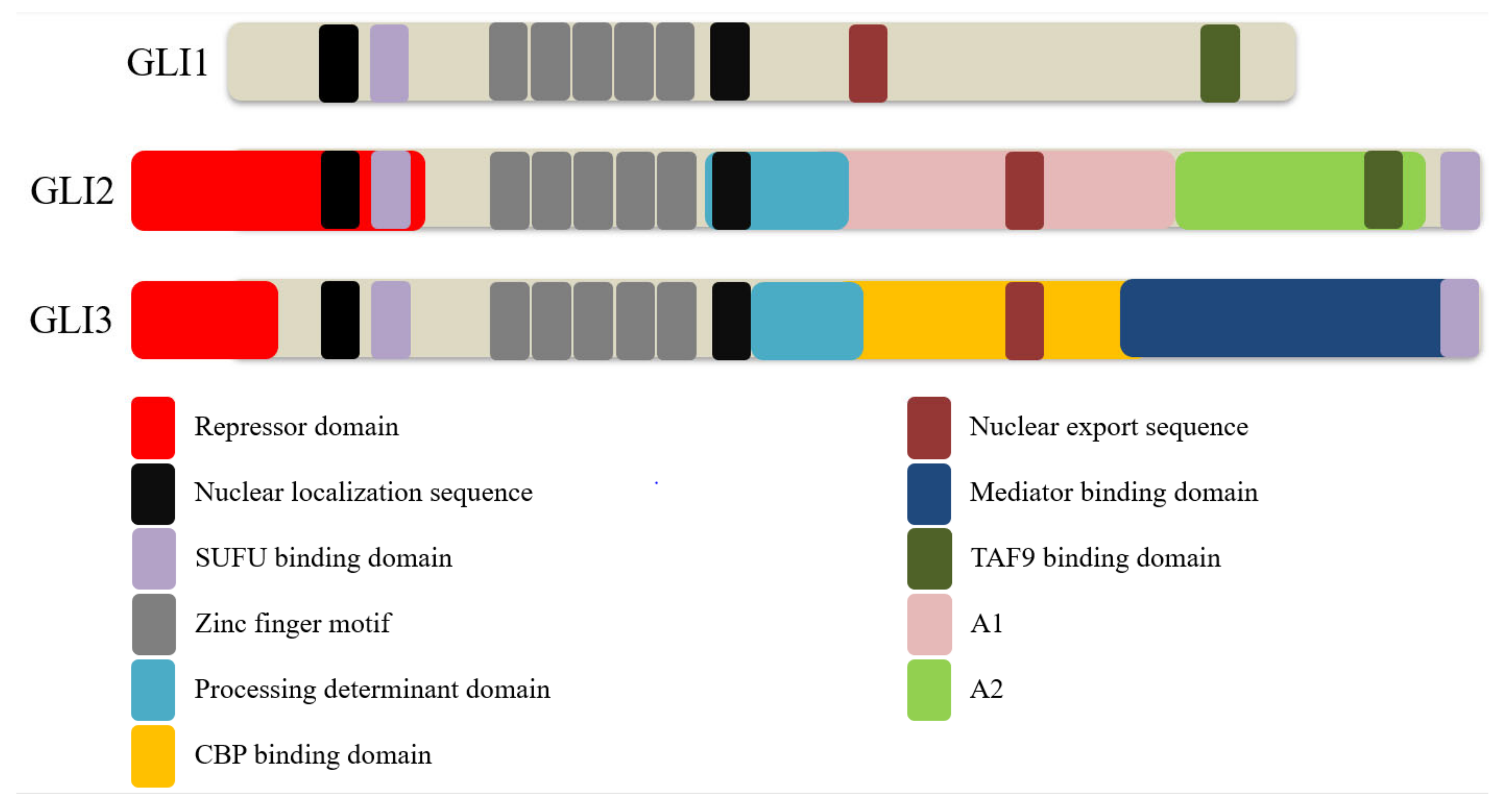
2. GLI Proteins and Their Domains
3. The Mechanism of GLI Regulation in Human Cancers
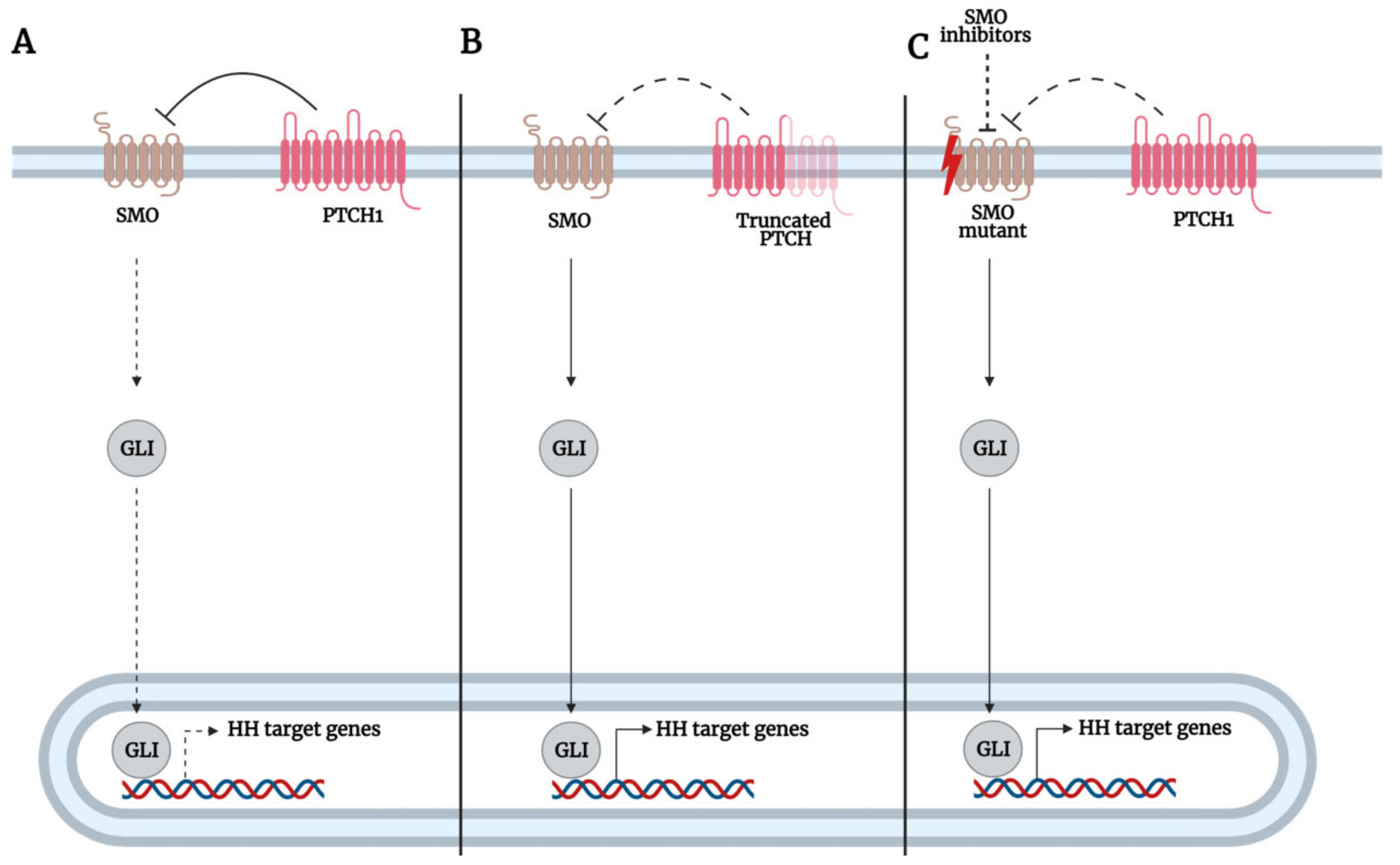
3.1. SMO-Dependent GLI Activation
3.1.1. Mutations of Hh Pathway Genes Upstream of GLI
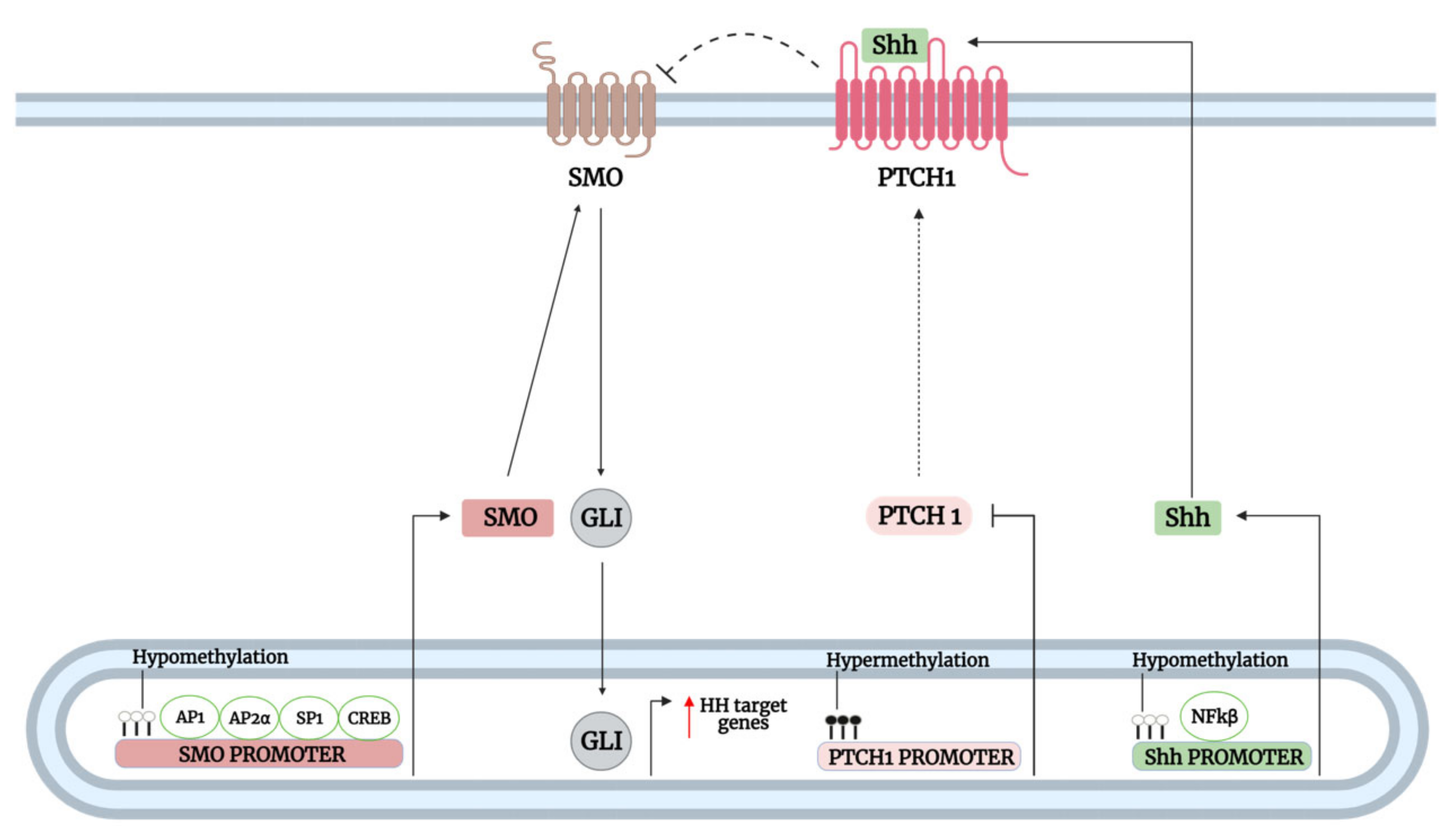
3.1.2. Transcriptional and Epigenetic Regulation of Hh Pathway Genes Upstream of GLI
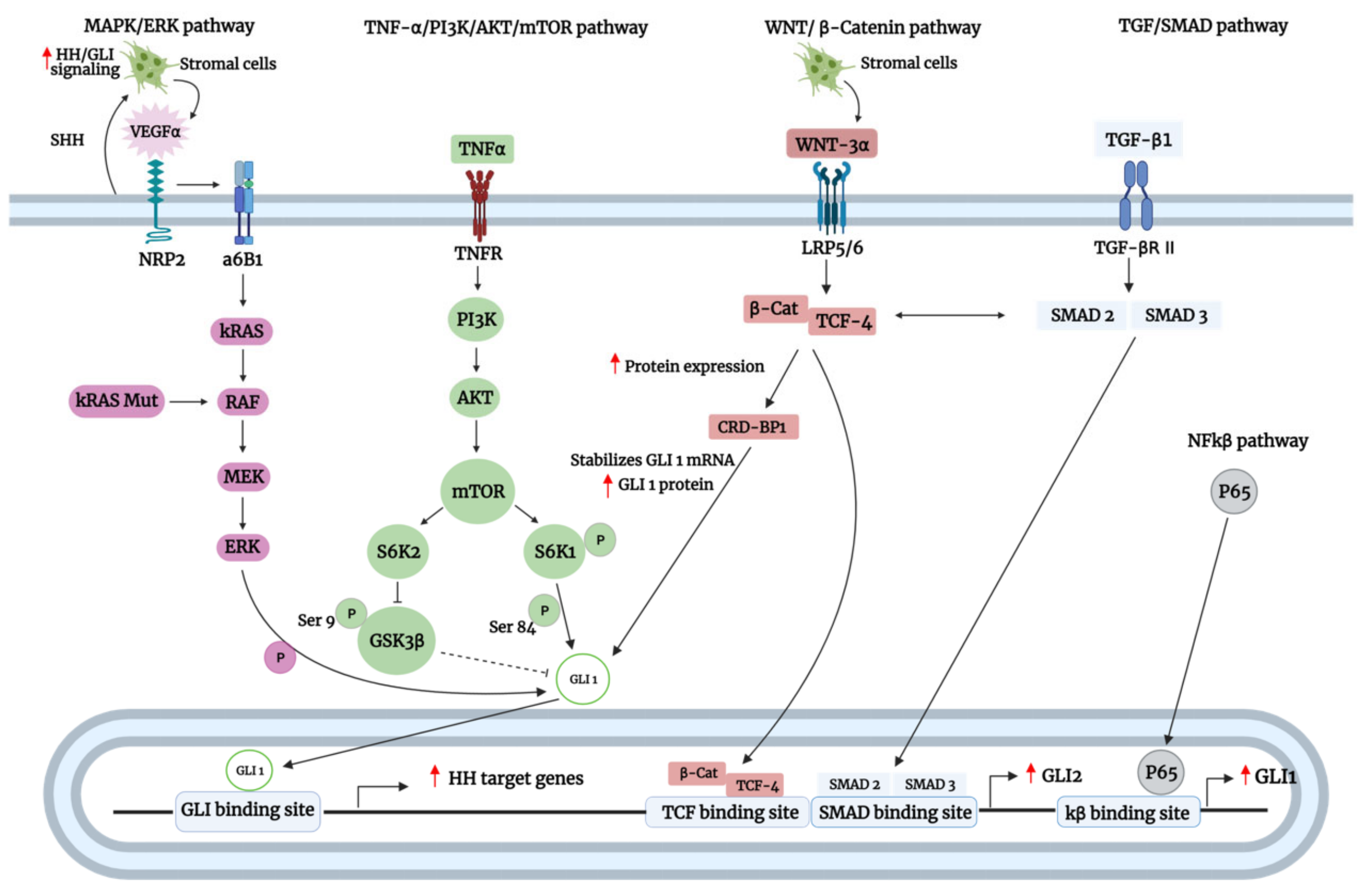
3.2. SMO-Independent GLI Activation
3.2.1. Active Crosstalk of GLI with Oncogenic Pathways
3.2.2. Active Crosstalk of GLI with Oncogenic and Tumor Suppressor Proteins
4. Hh Pathway as Therapeutic Targets in Cancer Clinical Studies
5. Current Challenges and Future Perspective for Using SMO/GLI Inhibitors in Clinical Settings
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Nüsslein-volhard, C.; Wieschaus, E. Mutations Affecting Segment Number and Polarity in Drosophila. Nature 1980, 287, 795–801. [Google Scholar] [CrossRef] [PubMed]
- Carballo, G.B.; Honorato, J.R.; De Lopes, G.P.F.; de Sampaio, E.; Spohr, T.C.L. A Highlight on Sonic Hedgehog Pathway. Cell Commun. Signal. 2018, 16, 11. [Google Scholar] [CrossRef]
- Mastronardi, F.G.; Dimitroulakos, J.; Kamel-Reid, S.; Manoukian, A.S. Co-Localization of Patched and Activated Sonic Hedgehog to Lysosomes in Neurons. Neuroreport 2000, 11, 581–585. [Google Scholar] [CrossRef] [PubMed]
- Rohatgi, R.; Milenkovic, L.; Corcoran, R.B.; Scott, M.P. Hedgehog Signal Transduction by Smoothened: Pharmacologic Evidence for a 2-Step Activation Process. Proc. Natl. Acad. Sci. USA 2009, 106, 3196–3201. [Google Scholar] [CrossRef] [Green Version]
- Hsu, S.H.C.; Zhang, X.; Yu, C.; Li, Z.J.; Wunder, J.S.; Hui, C.C.; Alman, B.A. Kif7 Promotes Hedgehog Signaling in Growth Plate Chondrocytes by Restricting the Inhibitory Function of Sufu. Development 2011, 138, 3791–3801. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Skoda, A.M.; Simovic, D.; Karin, V.; Kardum, V.; Vranic, S.; Serman, L. The Role of the Hedgehog Signaling Pathway in Cancer: A Comprehensive Review. Bosn. J. Basic Med. Sci. 2018, 18, 8–20. [Google Scholar] [CrossRef]
- Gonnissen, A.; Isebaert, S.; Haustermans, K. Targeting the Hedgehog Signaling Pathway in Cancer: Beyond Smoothened. Oncotarget 2015, 6, 13899–13913. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jia, J.; Zhang, L.; Zhang, Q.; Tong, C.; Wang, B.; Hou, F.; Amanai, K.; Jiang, J. Phosphorylation by Double-Time/CKIε and CKIα Targets Cubitus Interruptus for Slimb/β-TRCP-Mediated Proteolytic Processing. Dev. Cell 2005, 9, 819–830. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tschaikner, P.; Enzler, F.; Torres-Quesada, O.; Aanstad, P.; Stefan, E. Hedgehog and Gpr161: Regulating CAMP Signaling in the Primary Cilium. Cells 2020, 9, 118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Niewiadomski, P.; Kong, J.H.; Ahrends, R.; Ma, Y.; Humke, E.W.; Khan, S.; Teruel, M.N.; Novitch, B.G.; Rohatgi, R. Gli Protein Activity Is Controlled by Multisite Phosphorylation in Vertebrate Hedgehog Signaling. Cell Rep. 2014, 6, 168–181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Price, M.A.; Kalderon, D. Proteolysis of the Hedgehog Signaling Effector Cubitus Interruptus Requires Phosphorylation by Glycogen Synthase Kinase 3 and Casein Kinase 1. Cell 2002, 108, 823–835. [Google Scholar] [CrossRef] [Green Version]
- Shafique, S.; Rashid, S. Structural Basis of ΒTrCP1-Associated GLI3 Processing. Sci. Rep. 2019, 9, 6865. [Google Scholar] [CrossRef]
- Pan, Y.; Wang, C.; Wang, B. Phosphorylation of Gli2 by Protein Kinase A Is Required for Gli2 Processing and Degradation and the Sonic Hedgehog-Regulated Mouse Development. Dev. Biol. 2009, 326, 177–189. [Google Scholar] [CrossRef] [Green Version]
- Sabol, M.; Trnski, D.; Musani, V.; Ozretić, P.; Levanat, S. Role of GLI Transcription Factors in Pathogenesis and Their Potential as New Therapeutic Targets. Int. J. Mol. Sci. 2018, 19, 2562. [Google Scholar] [CrossRef] [Green Version]
- Zubčić, V.; Rinčić, N.; Kurtović, M.; Trnski, D.; Musani, V.; Ozretić, P.; Levanat, S.; Leović, D.; Sabol, M. GANT61 and Lithium Chloride Inhibit the Growth of Head and Neck Cancer Cell Lines Through the Regulation of GLI3 Processing by GSK3β. Int. J. Mol. Sci. 2020, 21, 6410. [Google Scholar] [CrossRef] [PubMed]
- Miyazaki, Y.; Matsubara, S.; Ding, Q.; Tsukasa, K.; Yoshimitsu, M.; Kosai, K.i.; Takao, S. Efficient Elimination of Pancreatic Cancer Stem Cells by Hedgehog/GLI Inhibitor GANT61 in Combination with MTOR Inhibition. Mol. Cancer 2016, 15, 49. [Google Scholar] [CrossRef] [Green Version]
- Pietrobono, S.; Gagliardi, S.; Stecca, B. Non-Canonical Hedgehog Signaling Pathway in Cancer: Activation of GLI Transcription Factors beyond Smoothened. Front. Genet. 2019, 10, 556. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, H.; Lo, H.-W. The Human Glioma-Associated Oncogene Homolog 1 (GLI1) Family of Transcription Factors in Gene Regulation and Diseases. Curr. Genomics 2010, 11, 238–245. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Niewiadomski, P.; Niedziółka, S.M.; Markiewicz, Ł.; Uśpieński, T.; Baran, B.; Chojnowska, K. Gli Proteins: Regulation in Development and Cancer. Cells 2019, 8, 147. [Google Scholar] [CrossRef] [Green Version]
- Pan, Y.; Wang, B. A Novel Protein-Processing Domain in Gli2 and Gli3 Differentially Blocks Complete Protein Degradation by the Proteasome. J. Biol. Chem. 2007, 282, 10846–10852. [Google Scholar] [CrossRef] [Green Version]
- Kinzler, K.W.; Vogelstein, B. The GLI Gene Encodes a Nuclear Protein Which Binds Specific Sequences in the Human Genome. Mol. Cell. Biol. 1990, 10, 634. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pavletich, N.; Pabo, C. Crystal Structure of a Five-Finger GLI-DNA Complex: New Perspectives on Zinc Fingers. Science 1993, 261, 1701–1707. [Google Scholar] [CrossRef] [PubMed]
- Hatayama, M.; Aruga, J. Gli Protein Nuclear Localization Signal. Vitam. Horm. 2012, 88, 73–89. [Google Scholar]
- Szczepny, A.; Wagstaff, K.M.; Dias, M.; Gajewska, K.; Wang, C.; Davies, R.G.; Kaur, G.; Ly-Huynh, J.; Loveland, K.L.; Jans, D.A. Overlapping Binding Sites for Importin Β1 and Suppressor of Fused (SuFu) on Glioma-Associated Oncogene Homologue 1 (Gli1) Regulate Its Nuclear Localization. Biochem. J. 2014, 461, 469–476. [Google Scholar] [CrossRef]
- Torrado, B.; Graña, M.; Badano, J.L.; Irigoín, F. Ciliary Entry of the Hedgehog Transcriptional Activator Gli2 Is Mediated by the Nuclear Import Machinery but Differs from Nuclear Transport in Being Imp-α/Β1-Independent. PLoS ONE 2016, 11, e0162033. [Google Scholar]
- Barnfield, P.C.; Zhang, X.; Thanabalasingham, V.; Yoshida, M.; Hui, C. Negative Regulation of Gli1 and Gli2 Activator Function by Suppressor of Fused through Multiple Mechanisms. Differentiation 2005, 73, 397–405. [Google Scholar] [CrossRef]
- Sheng, T.; Chi, S.; Zhang, X.; Xie, J. Regulation of Gli1 Localization by the CAMP/Protein Kinase A Signaling Axis through a Site Near the Nuclear Localization Signal. J. Biol. Chem. 2006, 281, 9–12. [Google Scholar] [CrossRef] [Green Version]
- Shi, Q.; Han, Y.; Jiang, J. Suppressor of Fused Impedes Ci/Gli Nuclear Import by Opposing Trn/Kapb2 in Hedgehog Signaling. J. Cell Sci. 2014, 127, 1092–1103. [Google Scholar] [PubMed] [Green Version]
- Han, Y.; Xiong, Y.; Shi, X.; Wu, J.; Zhao, Y.; Jiang, J. Regulation of Gli Ciliary Localization and Hedgehog Signaling by the PY-NLS/Karyopherin-Β2 Nuclear Import System. PLOS Biol. 2017, 15, e2002063. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kogerman, P.; Grimm, T.; Kogerman, L.; Krause, D.; Undén, A.B.; Sandstedt, B.; Toftgård, R.; Zaphiropoulos, P.G. Mammalian Suppressor-of-Fused Modulates Nuclear-Cytoplasmic Shuttling of GLI-1. Nat. Cell Biol. 1999, 1, 312–319. [Google Scholar] [CrossRef] [PubMed]
- Han, Y.; Shi, Q.; Jiang, J. Multisite Interaction with Sufu Regulates Ci/Gli Activity through Distinct Mechanisms in Hh Signal Transduction. Proc. Natl. Acad. Sci. USA 2015, 112, 6383–6388. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.; Shen, L.; Law, K.; Zhang, Z.; Liu, X.; Hua, H.; Li, S.; Huang, H.; Yue, S.; Hui, C.; et al. Suppressor of Fused Chaperones Gli Proteins To Generate Transcriptional Responses to Sonic Hedgehog Signaling. Mol. Cell. Biol. 2017, 37, e00421-16. [Google Scholar] [CrossRef] [Green Version]
- Akimaru, H.; Chen, Y.; Dai, P.; Hou, D.-X.; Nonaka, M.; Smolik, S.M.; Armstrong, S.; Goodman, R.H.; Ishii, S. Drosophila CBP Is a Co-Activator of Cubitus Interruptus in Hedgehog Signalling. Nature 1997, 386, 735–738. [Google Scholar] [CrossRef]
- Hughes, D.C.; Allen, J.; Morley, G.; Sutherland, K.; Ahmed, W.; Prosser, J.; Lettice, L.; Allan, G.; Mattei, M.G.; Farrall, M.; et al. Cloning and Sequencing of the Mouse Gli2 Gene: Localization to the Dominant Hemimelia Critical Region. Genomics 1997, 39, 205–215. [Google Scholar] [CrossRef] [PubMed]
- Dai, P.; Akimaru, H.; Tanaka, Y.; Maekawa, T.; Nakafuku, M.; Ishii, S. Sonic Hedgehog-Induced Activation of the Gli1Promoter Is Mediated by GLI3. J. Biol. Chem. 1999, 274, 8143–8152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, H.; Kim, S.; Ishii, S.; Boyer, T.G. Mediator Modulates Gli3-Dependent Sonic Hedgehog Signaling. Mol. Cell. Biol. 2006, 26, 8667. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoon, J.W.; Liu, C.Z.; Yang, J.T.; Swart, R.; Iannaccone, P.; Walterhouse, D. GLI Activates Transcription through a Herpes Simplex Viral Protein 16-Like Activation Domain. J. Biol. Chem. 1998, 273, 3496–3501. [Google Scholar] [CrossRef] [Green Version]
- Yoon, J.W.; Lamm, M.; Iannaccone, S.; Higashiyama, N.; Leong, K.F.; Iannaccone, P.; Walterhouse, D. P53 Modulates The Activity Of The GLI1 Oncogene Through Interactions With The Shared Coactivator TAF9. DNA Repair 2015, 34, 9. [Google Scholar] [CrossRef] [Green Version]
- Bosco-Clément, G.; Zhang, F.; Chen, Z.; Zhou, H.M.; Li, H.; Mikami, I.; Hirata, T.; Yagui-Beltran, A.; Lui, N.; Do, H.T.; et al. Targeting Gli Transcription Activation by Small Molecule Suppresses Tumor Growth. Oncogene 2014, 33, 2087–2097. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dai, P.; Shinagawa, T.; Nomura, T.; Harada, J.; Kaul, S.C.; Wadhwa, R.; Khan, M.M.; Akimaru, H.; Sasaki, H.; Colmenares, C.; et al. Ski Is Involved in Transcriptional Regulation by the Repressor and Full-Length Forms of Gli3. Genes Dev. 2002, 16, 2843. [Google Scholar] [CrossRef] [Green Version]
- Cheng, S.Y.; Bishop, J.M. Suppressor of Fused Represses Gli-Mediated Transcription by Recruiting the SAP18-MSin3 Corepressor Complex. Proc. Natl. Acad. Sci. USA 2002, 99, 5442–5447. [Google Scholar] [CrossRef] [Green Version]
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The Next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [Green Version]
- Kenney, A.M.; Rowitch, D.H. Sonic Hedgehog Promotes G1 Cyclin Expression and Sustained Cell Cycle Progression in Mammalian Neuronal Precursors. Mol. Cell. Biol. 2000, 20, 9055. [Google Scholar] [CrossRef] [Green Version]
- Kump, E.; Ji, J.; Wernli, M.; Häusermann, P.; Erb, P. Gli2 Upregulates CFlip and Renders Basal Cell Carcinoma Cells Resistant to Death Ligand-Mediated Apoptosis. Oncogene 2008, 27, 3856–3864. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Z.J.; Mack, S.C.; Mak, T.H.; Angers, S.; Taylor, M.D.; Hui, C.-C. Evasion of P53 and G 2 /M Checkpoints Are Characteristic of Hh-Driven Basal Cell Carcinoma. Oncogene 2014, 33, 2674–2680. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katoh, Y.; Katoh, M. Hedgehog Signaling, Epithelial-to-Mesenchymal Transition and MiRNA (Review). Int. J. Mol. Med. 2008, 22, 271–275. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Senga, S.S.; Grose, R.P. Hallmarks of Cancer—the New Testament. Open Biol. 2021, 11, 200358. [Google Scholar] [CrossRef] [PubMed]
- Cochrane, C.R.; Szczepny, A.; Watkins, D.N.; Cain, J.E. Hedgehog Signaling in the Maintenance of Cancer Stem Cells. Cancers 2015, 7, 1554. [Google Scholar] [CrossRef]
- Nilsson, M. Induction of Basal Cell Carcinomas and Trichoepitheliomas in Mice Overexpressing GLI-1. Proc. Natl. Acad. Sci. USA 2000, 97, 3438–3443. [Google Scholar] [CrossRef]
- Ji, J.; Kump, E.; Wernli, M.; Erb, P. Gene Silencing of Transcription Factor Gli2 Inhibits Basal Cell Carcinomalike Tumor Growthin Vivo. Int. J. Cancer 2008, 122, 50–56. [Google Scholar] [CrossRef]
- Hutchin, M.E.; Kariapper, M.S.T.; Grachtchouk, M.; Wang, A.; Wei, L.; Cummings, D.; Liu, J.; Evan Michael, L.; Glick, A.; Dlugosz, A.A. Sustained Hedgehog Signaling Is Required for Basal Cell Carcinoma Proliferation and Survival: Conditional Skin Tumorigenesis Recapitulates the Hair Growth Cycle. Genes Dev. 2005, 19, 214–223. [Google Scholar] [CrossRef] [Green Version]
- Cui, C.; Elsam, T.; Tian, Q.; Seykora, J.T.; Grachtchouk, M.; Dlugosz, A.; Tseng, H. Gli Proteins Up-Regulate the Expression of Basonuclin in Basal Cell Carcinoma. Cancer Res. 2004, 64, 5651–5658. [Google Scholar] [CrossRef] [Green Version]
- Kimura, H.; Stephen, D.; Joyner, A.; Curran, T. Gli1 Is Important for Medulloblastoma Formation in Ptc1+/- Mice. Oncogene 2005, 24, 4026–4036. [Google Scholar] [CrossRef] [Green Version]
- Romer, J.T.; Kimura, H.; Magdaleno, S.; Sasai, K.; Fuller, C.; Baines, H.; Connelly, M.; Stewart, C.F.; Gould, S.; Rubin, L.L.; et al. Suppression of the Shh Pathway Using a Small Molecule Inhibitor Eliminates Medulloblastoma in Ptc1+/-P53-/- Mice. Cancer Cell 2004, 6, 229–240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kadlub, N.; Coudert, A.; Gatibelza, M.E.; El Houmami, N.; Soufir, N.; Ruhin-Poncet, B.; L’Hermine, A.C.; Berdal, A.; Vazquez, M.P.; Descroix, V.; et al. PTCH1 Mutation and Local Aggressiveness of Odontogenic Keratocystic Tumors in Children: Is There a Relationship? Hum. Pathol. 2013, 44, 1071–1078. [Google Scholar] [CrossRef] [PubMed]
- Burns, M.A.; Liao, Z.W.; Yamagata, N.; Pouliot, G.P.; Stevenson, K.E.; Neuberg, D.S.; Thorner, A.R.; Ducar, M.; Silverman, E.A.; Hunger, S.P.; et al. Hedgehog Pathway Mutations Drive Oncogenic Transformation in High-Risk T-Cell Acute Lymphoblastic Leukemia. Blood 2017, 130, 367. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.-Y.; Chang, Y.-C.; Kuo, Y.-L.; Lee, K.-T.; Chen, P.-S.; Cheung, C.H.A.; Chang, C.-P.; Phan, N.N.; Shen, M.-R.; Hsu, H.-P. Mutation of the PTCH1 Gene Predicts Recurrence of Breast Cancer. Sci. Rep. 2019, 9, 16359. [Google Scholar] [CrossRef] [PubMed]
- Cazet, A.S.; Hui, M.N.; Elsworth, B.L.; Wu, S.Z.; Roden, D.; Chan, C.L.; Skhinas, J.N.; Collot, R.; Yang, J.; Harvey, K.; et al. Targeting Stromal Remodeling and Cancer Stem Cell Plasticity Overcomes Chemoresistance in Triple Negative Breast Cancer. Nat. Commun. 2018, 9, 2897. [Google Scholar] [CrossRef] [Green Version]
- Chakraborty, C.; Dutta, S.; Mukherjee, N.; Samadder, S.; Roychowdhury, A.; Roy, A.; Mondal, R.K.; Basu, P.; Roychoudhury, S.; Panda, C.K. Inactivation of PTCH1 Is Associated with the Development of Cervical Carcinoma: Clinical and Prognostic Implication. Tumour Biol. 2014, 36, 1143–1154. [Google Scholar] [CrossRef] [PubMed]
- Chaudary, N.; Pintilie, M.; Hedley, D.; Fyles, A.W.; Milosevic, M.; Clarke, B.; Hill, R.P.; Mackay, H. Hedgehog Pathway Signaling in Cervical Carcinoma and Outcome after Chemoradiation. Cancer 2012, 118, 3105–3115. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Wang, J.; Yang, H.; Chen, D.; Li, P. Association between FOXM1 and Hedgehog Signaling Pathway in Human Cervical Carcinoma by Tissue Microarray Analysis. Oncol. Lett. 2016, 12, 2664–2673. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grachtchouk, V.; Grachtchouk, M.; Lowe, L.; Johnson, T.; Wei, L.; Wang, A.; De Sauvage, F.; Dlugosz, A.A. The Magnitude of Hedgehog Signaling Activity Defines Skin Tumor Phenotype. EMBO J. 2003, 22, 2741–2751. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pricl, S.; Cortelazzi, B.; Dal Col, V.; Marson, D.; Laurini, E.; Fermeglia, M.; Licitra, L.; Pilotti, S.; Bossi, P.; Perrone, F. Smoothened (SMO) Receptor Mutations Dictate Resistance Tovismodegib in Basal Cell Carcinoma. Mol. Oncol. 2015, 9, 389–397. [Google Scholar] [CrossRef] [PubMed]
- Atwood, S.X.; Sarin, K.Y.; Whitson, R.J.; Li, J.R.; Kim, G.; Rezaee, M.; Ally, M.S.; Kim, J.; Yao, C.; Chang, A.L.S.; et al. Smoothened Variants Explain the Majority of Drug Resistance in Basal Cell Carcinoma. Cancer Cell 2015, 27, 342–353. [Google Scholar] [CrossRef] [Green Version]
- Xie, J.; Murone, M.; Luoh, S.M.; Ryan, A.; Gu, Q.; Zhang, C.; Bonifas, J.M.; Lam, C.W.; Hynes, M.; Goddard, A.; et al. Activating Smoothened Mutations in Sporadic Basal-Cell Carcinoma. Nature 1998, 391, 90–92. [Google Scholar] [CrossRef]
- Youssef, K.K.; Lapouge, G.; Bouvrée, K.; Rorive, S.; Brohée, S.; Appelstein, O.; Larsimont, J.C.; Sukumaran, V.; Van De Sande, B.; Pucci, D.; et al. Adult Interfollicular Tumour-Initiating Cells Are Reprogrammed into an Embryonic Hair Follicle Progenitor-like Fate during Basal Cell Carcinoma Initiation. Nat. Cell Biol. 2012, 14, 1282–1294. [Google Scholar] [CrossRef]
- Yauch, R.L.; Dijkgraaf, G.J.P.; Alicke, B.; Januario, T.; Ahn, C.P.; Holcomb, T.; Pujara, K.; Stinson, J.; Callahan, C.A.; Tang, T.; et al. Smoothened Mutation Confers Resistance to a Hedgehog Pathway Inhibitor in Medulloblastoma. Science 2009, 326, 572. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sicklick, J.K.; Li, Y.X.; Jayaraman, A.; Kannangai, R.; Qi, Y.; Vivekanandan, P.; Ludlow, J.W.; Owzar, K.; Chen, W.; Torbenson, M.S.; et al. Dysregulation of the Hedgehog Pathway in Human Hepatocarcinogenesis. Carcinogenesis 2006, 27, 748–757. [Google Scholar] [CrossRef] [Green Version]
- Kasperczyk, H.; Baumann, B.; Debatin, K.M.; Fulda, S. Characterization of Sonic Hedgehog as a Novel NF-ΚB Target Gene That Promotes NF-ΚB-Mediated Apoptosis Resistance and Tumor Growth in Vivo. FASEB J. 2009, 23, 21–33. [Google Scholar] [CrossRef]
- Nakashima, H.; Nakamura, M.; Yamaguchi, H.; Yamanaka, N.; Akiyoshi, T.; Koga, K.; Yamaguchi, K.; Tsuneyoshi, M.; Tanaka, M.; Katano, M. Nuclear Factor-ΚB Contributes to Hedgehog Signaling Pathway Activation through Sonic Hedgehog Induction in Pancreatic Cancer. Cancer Res. 2006, 66, 7041–7049. [Google Scholar] [CrossRef] [Green Version]
- Tian, H.; Callahan, C.A.; Dupree, K.J.; Darbonne, W.C.; Ahn, C.P.; Scales, S.J.; De Sauvage, F.J. Hedgehog Signaling Is Restricted to the Stromal Compartment during Pancreatic Carcinogenesis. Proc. Natl. Acad. Sci. USA 2009, 106, 4254–4259. [Google Scholar] [CrossRef] [Green Version]
- Gu, D.; Schlotman, K.E.; Xie, J. Deciphering the Role of Hedgehog Signaling in Pancreatic Cancer. J. Biomed. Res. 2016, 30, 353–360. [Google Scholar]
- Olive, K.P.; Jacobetz, M.A.; Davidson, C.J.; Gopinathan, A.; McIntyre, D.; Honess, D.; Madhu, B.; Goldgraben, M.A.; Caldwell, M.E.; Allard, D.; et al. Inhibition of Hedgehog Signaling Enhances Delivery of Chemotherapy in a Mouse Model of Pancreatic Cancer. Science 2009, 324, 1457–1461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duan, Z.H.; Wang, H.C.; Zhao, D.M.; Ji, X.X.; Song, M.; Yang, X.J.; Cui, W. Cooperatively Transcriptional and Epigenetic Regulation of Sonic Hedgehog Overexpression Drives Malignant Potential of Breast Cancer. Cancer Sci. 2015, 106, 1084–1091. [Google Scholar] [CrossRef] [PubMed]
- Cui, W.; Wang, L.H.; Wen, Y.Y.; Song, M.; Li, B.L.; Chen, X.L.; Xu, M.; An, S.X.; Zhao, J.; Lu, Y.Y.; et al. Expression and Regulation Mechanisms of Sonic Hedgehog in Breast Cancer. Cancer Sci. 2010, 101, 927–933. [Google Scholar] [CrossRef] [PubMed]
- Lou, H.; Li, H.; Huehn, A.R.; Tarasova, N.I.; Saleh, B.; Anderson, S.K.; Dean, M. Genetic and Epigenetic Regulation of the Smoothened Gene (SMO) in Cancer Cells. Cancers 2020, 12, 2219. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Lin, P.; Wang, Q.; Zheng, M.; Pang, L. Wnt3a-Regulated TCF4/β-Catenin Complex Directly Activates the Key Hedgehog Signalling Genes Smo and Gli1. Exp. Ther. Med. 2018, 16, 2101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, Y.; James, R.M.; Peter, A.; Lomas, C.; Cheung, F.; Harrison, D.J.; Bader, S.A. Functional Smoothened Is Required for Expression of GLI3 in Colorectal Carcinoma Cells. Cancer Lett. 2004, 207, 205–214. [Google Scholar] [CrossRef]
- Iwasaki, H.; Nakano, K.; Shinkai, K.; Kunisawa, Y.; Hirahashi, M.; Oda, Y.; Onishi, H.; Katano, M. Hedgehog Gli3 Activator Signal Augments Tumorigenicity of Colorectal Cancer via Upregulation of Adherence-Related Genes. Cancer Sci. 2013, 104, 328–336. [Google Scholar] [CrossRef] [PubMed]
- Kang, H.N.; Oh, S.C.; Kim, J.S.; Yoo, Y.A. Abrogation of Gli3 Expression Suppresses the Growth of Colon Cancer Cells via Activation of P53. Exp. Cell Res. 2012, 318, 539–549. [Google Scholar] [CrossRef]
- Shen, M.; Zhang, Z.; Wang, P. GLI3 Promotes Invasion and Predicts Poor Prognosis in Colorectal Cancer. Biomed Res. Int. 2021, 2021, 8889986. [Google Scholar] [CrossRef] [PubMed]
- Magistri, P.; Battistelli, C.; Strippoli, R.; Petrucciani, N.; Pellinen, T.; Rossi, L.; Mangogna, L.; Aurello, P.; D’Angelo, F.; Tripodi, M.; et al. SMO Inhibition Modulates Cellular Plasticity and Invasiveness in Colorectal Cancer. Front. Pharmacol. 2018, 8, 956. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garcia, N.; Al-Hendy, A.; Baracat, E.C.; Carvalho, K.C.; Yang, Q. Targeting Hedgehog Pathway and DNA Methyltransferases in Uterine Leiomyosarcoma Cells. Cells 2020, 10, 53. [Google Scholar] [CrossRef]
- Song, Y.; Tian, Y.; Zuo, Y.; Tu, J.C.; Feng, Y.F.; Qu, C.J. Altered Expression of PTCH and HHIP in Gastric Cancer through Their Gene Promoter Methylation: Novel Targets for Gastric Cancer. Mol. Med. Rep. 2013, 7, 1159–1168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Du, P.; Ye, H.-R.; Gao, J.; Chen, W.; Wang, Z.-C.; Jiang, H.-H.; Xu, J.; Zhang, J.-W.; Zhang, J.-C.; Cui, L. Methylation of PTCH1a Gene in a Subset of Gastric Cancers. World J. Gastroenterol. 2009, 15, 3799. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.J.; Do, I.G.; Lee, J.; Kim, K.M.; Jang, J.; Sohn, I.; Kang, W.K. Gastric Cancer (GC) Patients with Hedgehog Pathway Activation: PTCH1 and GLI2 as Independent Prognostic Factors. Target. Oncol. 2013, 8, 271–280. [Google Scholar] [CrossRef] [PubMed]
- Zuo, Y.; Song, Y.; Zhang, M.; Xu, Z.; Qian, X. Role of PTCH1 Gene Methylation in Gastric Carcinogenesis. Oncol. Lett. 2014, 8, 679. [Google Scholar] [CrossRef]
- Song, Y.; Tu, J.; Cheng, Y.; Zhou, F.; Liu, P.; Zhou, S.; Gu, Y.; Sun, Y. HHIP Overexpression Suppresses Human Gastric Cancer Progression and Metastasis by Reducing Its CpG Island Methylation. Front. Oncol. 2020, 10, 1667. [Google Scholar] [CrossRef]
- Yang, Z.; Lv, Y.; Wang, L.; Chen, Y.; Han, J.; Zhao, S.; Liu, W. Inhibition of Hedgehog Pathway Reveals the Regulatory Role of SMO in Gastric Cancer Cells. Tumour Biol. 2017, 39, 1–6. [Google Scholar] [CrossRef] [Green Version]
- Fukaya, M.; Isohata, N.; Ohta, H.; Aoyagi, K.; Ochiya, T.; Saeki, N.; Yanagihara, K.; Nakanishi, Y.; Taniguchi, H.; Sakamoto, H.; et al. Hedgehog Signal Activation in Gastric Pit Cell and in Diffuse-Type Gastric Cancer. Gastroenterology 2006, 131, 14–29. [Google Scholar] [CrossRef]
- Po, A.; Silvano, M.; Miele, E.; Capalbo, C.; Eramo, A.; Salvati, V.; Todaro, M.; Besharat, Z.M.; Catanzaro, G.; Cucchi, D.; et al. Noncanonical GLI1 Signaling Promotes Stemness Features and in Vivo Growth in Lung Adenocarcinoma. Oncogene 2017, 36, 4641–4652. [Google Scholar] [CrossRef] [Green Version]
- Kawakami, T.; Tokunaga, T.; Hatanaka, H.; Kijima, H.; Yamazaki, H.; Abe, Y.; Osamura, Y.; Inoue, H.; Ueyama, Y.; Nakamura, M. Neuropilin 1 and Neuropilin 2 Co-Expression Is Significantly Correlated with Increased Vascularity and Poor Prognosis in Nonsmall Cell Lung Carcinoma. Cancer 2002, 95, 2196–2201. [Google Scholar] [CrossRef] [PubMed]
- Goel, H.L.; Pursell, B.; Chang, C.; Shaw, L.M.; Mao, J.; Simin, K.; Kumar, P.; Vander Kooi, C.W.; Shultz, L.D.; Greiner, D.L.; et al. GLI1 Regulates a Novel Neuropilin-2/A6β1 Integrin Based Autocrine Pathway That Contributes to Breast Cancer Initiation. EMBO Mol. Med. 2013, 5, 488–508. [Google Scholar] [CrossRef] [PubMed]
- Nolan-Stevaux, O.; Lau, J.; Truitt, M.L.; Chu, G.C.; Hebrok, M.; Fernández-Zapico, M.E.; Hanahan, D. GLI1 Is Regulated through Smoothened-Independent Mechanisms in Neoplastic Pancreatic Ducts and Mediates PDAC Cell Survival and Transformation. Genes Dev. 2009, 23, 24–36. [Google Scholar] [CrossRef] [Green Version]
- Rajurkar, M.; De Jesus-Monge, W.E.; Driscoll, D.R.; Appleman, V.A.; Huang, H.; Cotton, J.L.; Klimstra, D.S.; Zhu, L.J.; Simin, K.; Xu, L.; et al. The Activity of Gli Transcription Factors Is Essential for Kras-Induced Pancreatic Tumorigenesis. Proc. Natl. Acad. Sci. USA 2012, 109, E1038–E1047. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, X.; Rumie Vittar, N.B.; Gai, X.; Fernandez-Barrena, M.G.; Moser, C.D.; Hu, C.; Almada, L.L.; McCleary-Wheeler, A.L.; Elsawa, S.F.; Vrabel, A.M.; et al. The Transcription Factor GLI1 Mediates TGFβ1 Driven EMT in Hepatocellular Carcinoma via a SNAI1-Dependent Mechanism. PLoS ONE 2012, 7, e49581. [Google Scholar] [CrossRef] [Green Version]
- Faião-Flores, F.; Alves-Fernandes, D.K.; Pennacchi, P.C.; Sandri, S.; Vicente, A.L.S.A.; Scapulatempo-Neto, C.; Vazquez, V.L.; Reis, R.M.; Chauhan, J.; Goding, C.R.; et al. Targeting the Hedgehog Transcription Factors GLI1 and GLI2 Restores Sensitivity to Vemurafenib-Resistant Human Melanoma Cells. Oncogene 2017, 36, 1849–1861. [Google Scholar] [CrossRef] [Green Version]
- Johnson, R.W.; Merkel, A.R.; Page, J.M.; Ruppender, N.S.; Guelcher, S.A.; Sterling, J.A. Wnt Signaling Induces Gene Expression of Factors Associated with Bone Destruction in Lung and Breast Cancer. Clin. Exp. Metastasis 2014, 31, 945–959. [Google Scholar] [CrossRef]
- Cannonier, S.A.; Gonzales, C.B.; Ely, K.; Guelcher, S.A.; Sterling, J.A. Hedgehog and TGFβ Signaling Converge on Gli2 to Control Bony Invasion and Bone Destruction in Oral Squamous Cell Carcinoma. Oncotarget 2016, 7, 76062–76075. [Google Scholar] [CrossRef] [Green Version]
- Noubissi, F.K.; Goswami, S.; Sanek, N.A.; Kawakami, K.; Minamoto, T.; Moser, A.; Grinblat, Y.; Spiegelman, V.S. Wnt Signaling Stimulates Transcriptional Outcome of the Hedgehog Pathway by Stabilizing GLI1 MRNA. Cancer Res. 2009, 69, 8572–8578. [Google Scholar] [CrossRef] [Green Version]
- Varnat, F.; Siegl-Cachedenier, I.; Malerba, M.; Gervaz, P.; Ruiz, I.; Altaba, A. Loss of WNT-TCF Addiction and Enhancement of HH-GLI1 Signalling Define the Metastatic Transition of Human Colon Carcinomas. EMBO Mol. Med. 2010, 2, 440–457. [Google Scholar] [CrossRef] [PubMed]
- Yao, Y.; Zhou, D.; Shi, D.; Zhang, H.; Zhan, S.; Shao, X.; Sun, K.; Sun, L.; Wu, G.; Tian, K.; et al. GLI1 Overexpression Promotes Gastric Cancer Cell Proliferation and Migration and Induces Drug Resistance by Combining with the AKT-MTOR Pathway. Biomed. Pharmacother. 2019, 111, 993–1004. [Google Scholar] [CrossRef] [PubMed]
- Chakrabarti, J.; Holokai, L.; Syu, L.; Steele, N.G.; Chang, J.; Wang, J.; Ahmed, S.; Dlugosz, A.; Zavros, Y. Hedgehog Signaling Induces PD-L1 Expression and Tumor Cell Proliferation in Gastric Cancer. Oncotarget 2018, 9, 37439. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koh, V.; Chakrabarti, J.; Torvund, M.; Steele, N.; Hawkins, J.A.; Ito, Y.; Wang, J.; Helmrath, M.A.; Merchant, J.L.; Ahmed, S.A.; et al. Hedgehog Transcriptional Effector GLI Mediates MTOR-Induced PD-L1 Expression in Gastric Cancer Organoids. Cancer Lett. 2021, 518, 59–71. [Google Scholar] [CrossRef] [PubMed]
- Kasiri, S.; Shao, C.; Chen, B.; Wilson, A.N.; Yenerall, P.; Timmons, B.C.; Girard, L.; Tian, H.; Behrens, C.; Wistuba, I.I.; et al. GLI1 Blockade Potentiates the Antitumor Activity of PI3K Antagonists in Lung Squamous Cell Carcinoma. Cancer Res. 2017, 77, 4448–4459. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kebenko, M.; Drenckhan, A.; Gros, S.J.; Jücker, M.; Grabinski, N.; Ewald, F.; Grottke, A.; Schultze, A.; Izbicki, J.R.; Bokemeyer, C.; et al. ErbB2 Signaling Activates the Hedgehog Pathway via PI3K-Akt in Human Esophageal Adenocarcinoma: Identification of Novel Targets for Concerted Therapy Concepts. Cell. Signal. 2015, 27, 373–381. [Google Scholar] [CrossRef]
- Zhu, J.; Sun, Y.; Lu, Y.; Jiang, X.; Ma, B.; Yu, L.; Zhang, J.; Dong, X.; Zhang, Q. Glaucocalyxin A Exerts Anticancer Effect on Osteosarcoma by Inhibiting GLI1 Nuclear Translocation via Regulating PI3K/Akt Pathway. Cell Death Dis. 2018, 9, 708. [Google Scholar] [CrossRef] [Green Version]
- Singh, R.; Dhanyamraju, P.K.; Lauth, M. DYRK1B Blocks Canonical and Promotes Non-Canonical Hedgehog Signaling through Activation of the MTOR/AKT Pathway. Oncotarget 2017, 8, 833–845. [Google Scholar] [CrossRef] [Green Version]
- Zhou, J.; Zhu, G.; Huang, J.; Li, L.; Du, Y.; Gao, Y.; Wu, D.; Wang, X.; Hsieh, J.T.; He, D.; et al. Non-Canonical GLI1/2 Activation by PI3K/AKT Signaling in Renal Cell Carcinoma: A Novel Potential Therapeutic Target. Cancer Lett. 2016, 370, 313–323. [Google Scholar] [CrossRef]
- Yang, H.; Hu, L.; Liu, Z.; Qin, Y.; Li, R.; Zhang, G.; Zhao, B.; Bi, C.; Lei, Y.; Bai, Y. Inhibition of Gli1-Mediated Prostate Cancer Cell Proliferation by Inhibiting the MTOR/S6K1 Signaling Pathway. Oncol. Lett. 2017, 14, 7970–7976. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Ding, Q.; Yen, C.J.; Xia, W.; Izzo, J.G.; Lang, J.Y.; Li, C.W.; Hsu, J.L.; Miller, S.A.; Wang, X.; et al. The Crosstalk of MTOR/S6K1 and Hedgehog Pathways. Cancer Cell 2012, 21, 374–387. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mizuarai, S.; Kawagishi, A.; Kotani, H. Inhibition of P70S6K2 Down-Regulates Hedgehog/GLI Pathway in Non-Small Cell Lung Cancer Cell Lines. Mol. Cancer 2009, 8, 44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stecca, B.; Mas, C.; Clement, V.; Zbinden, M.; Correa, R.; Piguet, V.; Beermann, F.; Ruiz I Altaba, A. Melanomas Require HEDGEHOG-GLI Signaling Regulated by Interactions between GLI1 and the RAS-MEK/AKT Pathways. Proc. Natl. Acad. Sci. USA 2007, 104, 5895–5900. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Colavito, S.A.; Zou, M.R.; Yan, Q.; Nguyen, D.X.; Stern, D.F. Significance of Glioma-Associated Oncogene Homolog 1 (GLI1) Expression in Claudin-Low Breast Cancer and Crosstalk with the Nuclear Factor Kappa-Light-Chain-Enhancer of Activated B Cells (NFκB) Pathway. Breast Cancer Res. 2014, 16, 444. [Google Scholar] [CrossRef] [Green Version]
- Deng, W.; Vanderbilt, D.B.; Lin, C.C.; Martin, K.H.; Brundage, K.M.; Ruppert, J.M. SOX9 Inhibits β-TrCP-Mediated Protein Degradation to Promote Nuclear GLI1 Expression and Cancer Stem Cell Properties. J. Cell Sci. 2015, 128, 1123–1138. [Google Scholar] [CrossRef] [Green Version]
- Han, B.; Qu, Y.; Jin, Y.; Yu, Y.; Deng, N.; Wawrowsky, K.; Zhang, X.; Li, N.; Bose, S.; Wang, Q.; et al. FOXC1 Activates Smoothened-Independent Hedgehog Signaling in Basal-like Breast Cancer. Cell Rep. 2015, 13, 1046–1058. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, P.; Lee, E.H.; Du, F.; Gordon, R.E.; Yuelling, L.W.; Liu, Y.; Ng, J.M.Y.; Zhang, H.; Wu, J.; Korshunov, A.; et al. Nestin Mediates Hedgehog Pathway Tumorigenesis. Cancer Res. 2016, 76, 5573–5583. [Google Scholar] [CrossRef] [Green Version]
- Chong, Y.; Tang, D.; Gao, J.; Jiang, X.; Xu, C.; Xiong, Q.; Huang, Y.; Wang, J.; Zhou, H.; Shi, Y.; et al. Galectin-1 Induces Invasion and the Epithelial-Mesenchymal Transition in Human Gastric Cancer Cells via Non-Canonical Activation of the Hedgehog Signaling Pathway. Oncotarget 2016, 7, 83611–83626. [Google Scholar] [CrossRef] [Green Version]
- Chong, Y.; Tang, D.; Xiong, Q.; Jiang, X.; Xu, C.; Huang, Y.; Wang, J.; Zhou, H.; Shi, Y.; Wu, X.; et al. Galectin-1 from Cancer-Associated Fibroblasts Induces Epithelial–Mesenchymal Transition through Β1 Integrin-Mediated Upregulation of Gli1 in Gastric Cancer. J. Exp. Clin. Cancer Res. 2016, 35, 175. [Google Scholar] [CrossRef] [Green Version]
- You, X.; Wu, J.; Wang, Y.; Liu, Q.; Cheng, Z.; Zhao, X.; Liu, G.; Huang, C.; Dai, J.; Zhou, Y.; et al. Galectin-1 Promotes Vasculogenic Mimicry in Gastric Adenocarcinoma via the Hedgehog/GLI Signaling Pathway. Aging 2020, 12, 21837–21853. [Google Scholar] [CrossRef]
- You, X.; Liu, Q.; Wu, J.; Wang, Y.; Dai, J.; Chen, D.; Zhou, Y.; Lian, Y. Galectin-1 Promotes Vasculogenic Mimicry in Gastric Cancer by Upregulating EMT Signaling. J. Cancer 2019, 10, 6286–6297. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Bosch, N.; Fernández-Barrena, M.G.; Moreno, M.; Ortiz-Zapater, E.; Munné-Collado, J.; Iglesias, M.; André, S.; Gabius, H.J.; Hwang, R.F.; Coise Poirier, F.; et al. Galectin-1 Drives Pancreatic Carcinogenesis through Stroma Remodeling and Hedgehog Signaling Activation. Cancer Res. 2014, 74, 3512–3524. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Yu, Q.; Li, R.; Luo, J.; Yuan, D.; Song, J.; Sun, Y.; Long, T.; Yang, Z. SPOP Regulates the Biological Mechanism of Ovarian Cancer Cells through the Hh Signaling Pathway. Onco Targets Ther. 2019, 12, 9239–9248. [Google Scholar] [CrossRef] [Green Version]
- Zeng, C.; Wang, Y.; Lu, Q.; Chen, J.; Zhang, J.; Liu, T.; Lv, N.; Luo, S. SPOP Suppresses Tumorigenesis by Regulating Hedgehog/Gli2 Signaling Pathway in Gastric Cancer. J. Exp. Clin. Cancer Res. 2014, 33, 75. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Pan, Y.; Wang, B. Suppressor of Fused and Spop Regulate the Stability, Processing and Function of Gli2 and Gli3 Full-Length Activators but Not Their Repressors. Development 2010, 137, 2001–2009. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tian, L.; Deng, Z.; Xu, L.; Yang, T.; Yao, W.; Ji, L.; Lu, Y.; Zhang, J.; Liu, Y.; Wang, J. Downregulation of ASPP2 Promotes Gallbladder Cancer Metastasis and Macrophage Recruitment via APKC-ι/GLI1 Pathway. Cell Death Dis. 2018, 9, 1115. [Google Scholar] [CrossRef]
- Trnski, D.; Sabol, M.; Gojević, A.; Martinić, M.; Ozretić, P.; Musani, V.; Ramić, S.; Levanat, S. GSK3β and Gli3 Play a Role in Activation of Hedgehog-Gli Pathway in Human Colon Cancer - Targeting GSK3β Downregulates the Signaling Pathway and Reduces Cell Proliferation. Biochim. Biophys. 2015, 1852, 2574–2584. [Google Scholar] [CrossRef] [Green Version]
- Muthutkumar, S.; Boyer, T.; Burleson, M.O. Med12 Mutations Promote Castration Resistant Prostate Cancer through Hyperactivated GLI3/SHH Signaling. FASEB J. 2019, 33, 647.26. [Google Scholar] [CrossRef]
- Pellegrini, C.; Maturo, M.G.; Di Nardo, L.; Ciciarelli, V.; Gutiérrez García-Rodrigo, C.; Fargnoli, M.C. Understanding the Molecular Genetics of Basal Cell Carcinoma. Int. J. Mol. Sci. 2017, 18, 2485. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Zwaan, S.E.; Haass, N.K. Genetics of Basal Cell Carcinoma. Australas. J. Dermatol. 2010, 51, 81–92. [Google Scholar] [CrossRef] [PubMed]
- Horlock, N.; Wilson, G.D.; Daley, E.M.; Richman, R.I.; Grobbelaar, A.O.; Sanders, R.; Foy, C. Cellular Proliferation Characteristics Do Not Account for the Behaviour of Horrifying Basal Cell Carcinoma. A Comparison of the Growth Fraction of Horrifying and Non Horrifying Tumours. Br. J. Plast. Surg. 1998, 51, 59–66. [Google Scholar] [CrossRef] [PubMed]
- Marcelina, P.; Mappiasse, A.; Anwar, A.I.; Ganda, I.J.; Hatta, M.; Masadah, R. Expression of Patched-1 Protein in Aggressive and Nonaggressive Basal Cell Carcinoma. Am. J. Clin. Exp. Immunol. 2016, 4, 122–128. [Google Scholar] [CrossRef] [Green Version]
- Reifenberger, J.; Wolter, M.; Knobbe, C.B.; Köhler, B.; Schönicke, A.; Scharwächter, C.; Kumar, K.; Blaschke, B.; Ruzicka, T.; Reifenberger, G. Somatic Mutations in the PTCH, SMOH, SUFUH and TP53 Genes in Sporadic Basal Cell Carcinomas. Br. J. Dermatol. 2005, 152, 43–51. [Google Scholar] [CrossRef]
- Niyaz, M.; Khan, M.S.; Mudassar, S. Hedgehog Signaling: An Achilles’ Heel in Cancer. Transl. Oncol. 2019, 12, 1334–1344. [Google Scholar] [CrossRef]
- Zheng, X.; Zeng, W.; Gai, X.; Xu, Q.; Li, C.; Liang, Z.; Tuo, H.; Liu, Q. Role of the Hedgehog Pathway in Hepatocellular Carcinoma (Review). Oncol. Rep. 2013, 30, 2020–2026. [Google Scholar] [CrossRef] [Green Version]
- Shi, Y.; Moura, U.; Opitz, I.; Soltermann, A.; Rehrauer, H.; Thies, S.; Weder, W.; Stahel, R.A.; Felley-Bosco, E. Role of Hedgehog Signaling in Malignant Pleural Mesothelioma. Clin. Cancer Res. 2012, 18, 4646–4656. [Google Scholar] [CrossRef] [Green Version]
- De la Rosa, J.; Sánchez, M.; Enguita-Germán, M.; García-López, R.; Schiapparelli, P.; Shahi, M.H.; Meléndez, B.; Rey, J.A.; Idoate, M.A.; Castresana, J.S. Inhibition of the Sonic Hedgehog Pathway by Cyclopamine or GLI1 SiRNA Reduces In Vivo Tumorigenesis of Human Medulloblastoma Cells Xenotransplanted to Immunodeficient Nude Mice. Adv. Transl. Med. Res. 2019, 1, 1–5. [Google Scholar]
- Benvenuto, M.; Masuelli, L.; De Smaele, E.; Fantini, M.; Mattera, R.; Cucchi, D.; Bonanno, E.; Di Stefano, E.; Frajese, G.V.; Orlandi, A.; et al. In Vitro and in Vivo Inhibition of Breast Cancer Cell Growth by Targeting the Hedgehog/GLI Pathway with SMO (GDC-0449) or GLI (GANT-61) Inhibitors. Oncotarget 2016, 7, 9250. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Waters, A.M.; Der, C.J. KRAS: The Critical Driver and Therapeutic Target for Pancreatic Cancer. Cold Spring Harb. Perspect. Med. 2018, 8, a031435. [Google Scholar] [CrossRef]
- Han, J.J.; Fernandez-Zapico, M.E. Crosstalk between kras and hedgehog pathways in the regulation of gli function in pancreatic cancer cells. Pancreas 2006, 33, 466. [Google Scholar] [CrossRef]
- Mills, L.D.; Zhang, Y.; Marler, R.J.; Herreros-Villanueva, M.; Zhang, L.; Almada, L.L.; Couch, F.; Wetmore, C.; di Magliano, M.P.; Fernandez-Zapico, M.E. Loss of the Transcription Factor GLI1 Identifies a Signaling Network in the Tumor Microenvironment Mediating KRAS Oncogene-Induced Transformation. J. Biol. Chem. 2013, 288, 11786. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Reyniès, A.; Javelaud, D.; Elarouci, N.; Marsaud, V.; Gilbert, C.; Mauviel, A. Large-Scale Pan-Cancer Analysis Reveals Broad Prognostic Association between TGF-β Ligands, Not Hedgehog, and GLI1/2 Expression in Tumors. Sci. Rep. 2020, 10, 14491. [Google Scholar] [CrossRef]
- Abe, Y.; Tanaka, N. The Hedgehog Signaling Networks in Lung Cancer: The Mechanisms and Roles in Tumor Progression and Implications for Cancer Therapy. Biomed Res. Int. 2016, 2016, 7969286. [Google Scholar] [CrossRef] [PubMed]
- Dennler, S.; André, J.; Verrechia, F.; Mauviel, A. Cloning of the Human GLI2 Promoter: Transcriptional Activation by Transforming Growth Factor-β via SMAD3/β-Catenin Cooperation. J. Biol. Chem. 2009, 284, 31523–31531. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.-H.; Shin, H.S.; Lee, S.H.; Lee, I.; Lee, Y.S.; Park, J.C.; Kim, Y.J.; Chung, J.B.; Lee, Y.C. Contrasting Activity of Hedgehog and Wnt Pathways According to Gastric Cancer Cell Differentiation: Relevance of Crosstalk Mechanisms. Cancer Sci. 2010, 101, 328–335. [Google Scholar] [CrossRef]
- Das, S.; Samant, R.S.; Shevde, L.A. Nonclassical Activation of Hedgehog Signaling Enhances Multidrug Resistance and Makes Cancer Cells Refractory to Smoothened-Targeting Hedgehog Inhibition. J. Biol. Chem. 2013, 288, 11824–11833. [Google Scholar] [CrossRef] [Green Version]
- Atwood, S.X.; Li, M.; Lee, A.; Tang, J.Y.; Oro, A.E. GLI Activation by Atypical Protein Kinase C ι/λ Regulates the Growth of Basal Cell Carcinomas. Nature 2013, 494, 484–488. [Google Scholar] [CrossRef] [Green Version]
- Goel, H.L.; Underwood, J.M.; Nickerson, J.A.; Hsieh, C.-C.; Languino, L.R. Β1 Integrins Mediate Cell Proliferation in Three-Dimensional Cultures by Regulating Expression of the Sonic Hedgehog Effector Protein, GLI1. J. Cell. Physiol. 2010, 224, 210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Axelson, M.; Liu, K.; Jiang, X.; He, K.; Wang, J.; Zhao, H.; Kufrin, D.; Palmby, T.; Dong, Z.; Russell, A.M.; et al. U.S. Food and Drug Administration Approval: Vismodegib for Recurrent, Locally Advanced, or Metastatic Basal Cell Carcinoma. Clin. Cancer Res. 2013, 19, 2289–2293. [Google Scholar] [CrossRef] [Green Version]
- Casey, D.; Demko, S.; Shord, S.; Zhao, H.; Chen, H.; He, K.; Putman, A.; Helms, W.; Keegan, P.; Pazdur, R. FDA Approval Summary: Sonidegib for Locally Advanced Basal Cell Carcinoma. Clin. Cancer Res. 2017, 23, 2377–2381. [Google Scholar] [CrossRef] [Green Version]
- Jamieson, C.; Martinelli, G.; Papayannidis, C.; Cortes, J.E. Hedgehog Pathway Inhibitors: A New Therapeutic Class for the Treatment of Acute Myeloid Leukemia. Blood Cancer Discov. 2020, 1, 134–145. [Google Scholar] [CrossRef]
- Cortes, J.E.; Dombret, H.; Merchant, A.; Tauchi, T.; Dirienzo, C.G.; Sleight, B.; Zhang, X.; Leip, E.P.; Shaik, N.; Bell, T.; et al. Glasdegib plus Intensive/Nonintensive Chemotherapy in Untreated Acute Myeloid Leukemia: BRIGHT AML 1019 Phase III Trials. Futur. Oncol. 2019, 15, 3531–3545. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ally, M.S.; Ransohoff, K.; Sarin, K.; Atwood, S.X.; Rezaee, M.; Bailey-Healy, I.; Kim, J.; Beachy, P.A.; Chang, A.L.S.; Oro, A.; et al. Effects of Combined Treatment With Arsenic Trioxide and Itraconazole in Patients With Refractory Metastatic Basal Cell Carcinoma. JAMA Dermatol. 2016, 152, 452. [Google Scholar] [CrossRef] [PubMed]
- Xie, H.; Paradise, B.D.; Ma, W.W.; Fernandez-Zapico, M.E. Recent Advances in the Clinical Targeting of Hedgehog/GLI Signaling in Cancer. Cells 2019, 8, 394. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Couban, S.; Benevolo, G.; Donnellan, W.; Cultrera, J.; Koschmieder, S.; Verstovsek, S.; Hooper, G.; Hertig, C.; Tandon, M.; Dimier, N.; et al. A Phase Ib Study to Assess the Efficacy and Safety of Vismodegib in Combination with Ruxolitinib in Patients with Intermediate- or High-Risk Myelofibrosis 11 Medical and Health Sciences 1103 Clinical Sciences 11 Medical and Health Sciences 1102 Cardiorespiratory Medicine and Haematology. J. Hematol. Oncol. 2018, 11, 122. [Google Scholar]
- Bixby, D.; Noppeney, R.; Lin, T.L.; Cortes, J.; Krauter, J.; Yee, K.; Medeiros, B.C.; Krämer, A.; Assouline, S.; Fiedler, W.; et al. Safety and Efficacy of Vismodegib in Relapsed/Refractory Acute Myeloid Leukaemia: Results of a Phase Ib Trial. Br. J. Haematol. 2019, 185, 595–598. [Google Scholar] [CrossRef]
- McCleary-Wheeler, A.L.; Carr, R.M.; Palmer, S.R.; Smyrk, T.C.; Allred, J.B.; Almada, L.L.; Tolosa, E.J.; Lamberti, M.J.; Marks, D.L.; Borad, M.J.; et al. Phase 1 Trial of Vismodegib and Erlotinib Combination in Metastatic Pancreatic Cancer. Pancreatology 2020, 20, 101–109. [Google Scholar] [CrossRef]
- Carr, R.M.; Duma, N.; McCleary-Wheeler, A.L.; Almada, L.L.; Marks, D.L.; Graham, R.P.; Smyrk, T.C.; Lowe, V.; Borad, M.J.; Kim, G.; et al. Targeting of the Hedgehog/GLI and MTOR Pathways in Advanced Pancreatic Cancer, a Phase 1 Trial of Vismodegib and Sirolimus Combination. Pancreatology 2020, 20, 1115–1122. [Google Scholar] [CrossRef]
- De Jesus-Acosta, A.; Sugar, E.A.; O’Dwyer, P.J.; Ramanathan, R.K.; Von Hoff, D.D.; Rasheed, Z.; Zheng, L.; Begum, A.; Anders, R.; Maitra, A.; et al. Phase 2 Study of Vismodegib, a Hedgehog Inhibitor, Combined with Gemcitabine and Nab-Paclitaxel in Patients with Untreated Metastatic Pancreatic Adenocarcinoma. Br. J. Cancer 2020, 122, 498–505. [Google Scholar] [CrossRef]
- Dréno, B.; Kunstfeld, R.; Hauschild, A.; Fosko, S.; Zloty, D.; Labeille, B.; Grob, J.J.; Puig, S.; Gilberg, F.; Bergström, D.; et al. Two Intermittent Vismodegib Dosing Regimens in Patients with Multiple Basal-Cell Carcinomas (MIKIE): A Randomised, Regimen-Controlled, Double-Blind, Phase 2 Trial. Lancet Oncol. 2017, 18, 404–412. [Google Scholar] [CrossRef]
- Mortier, L.; Bertrand, N.; Basset-Seguin, N.; Saiag, P.; Dupuy, A.; Dalac-Rat, S.; Guillot, B.; Templier, C.; Desmedt, E.; Duhamel, A.; et al. Vismodegib in Neoadjuvant Treatment of Locally Advanced Basal Cell Carcinoma: First Results of a Multicenter, Open-Label, Phase 2 Trial (VISMONEO Study). J. Clin. Oncol. 2018, 36, 9509. [Google Scholar] [CrossRef]
- Sekulic, A.; Migden, M.R.; Basset-Seguin, N.; Garbe, C.; Gesierich, A.; Lao, C.D.; Miller, C.; Mortier, L.; Murrell, D.F.; Hamid, O.; et al. Long-Term Safety and Efficacy of Vismodegib in Patients with Advanced Basal Cell Carcinoma: Final Update of the Pivotal ERIVANCE BCC Study. BMC Cancer 2017, 17, 332. [Google Scholar] [CrossRef] [PubMed]
- Fosko, S.W.; Chu, M.B.; Armbrecht, E.; Galperin, T.; Potts, G.A.; Mattox, A.; Kurta, A.; Polito, K.; Slutsky, J.B.; Burkemper, N.M.; et al. Efficacy, Rate of Tumor Response, and Safety of a Short Course (12-24 Weeks) of Oral Vismodegib in Various Histologic Subtypes (Infiltrative, Nodular, and Superficial) of High-Risk or Locally Advanced Basal Cell Carcinoma, in an Open-Label, Prospective Case Series Clinical Trial. J. Am. Acad. Dermatol. 2020, 82, 946–954. [Google Scholar]
- Basset-Seguin, N.; Hauschild, A.; Grob, J.-J.; Kunstfeld, R.; Dréno, B.; Mortier, L.; Ascierto, P.A.; Licitra, L.; Dutriaux, C.; Thomas, L.; et al. Vismodegib in Patients with Advanced Basal Cell Carcinoma (STEVIE): A Pre-Planned Interim Analysis of an International, Open-Label Trial. Lancet Oncol. 2015, 16, 729–736. [Google Scholar] [CrossRef]
- Bossi, P.; Peris, K.; Calzavara-Pinton, P.; Queirolo, P.; Alfieri, S.; Palla, M.; Rossi, M.T.; Spagnolo, F.; Tambone, S.; Astolfi, C.; et al. Cohort Analysis of Safety and Efficacy of Vismodegib in Italian Patients from the Phase II, Multicenter STEVIE Study. Futur. Oncol. 2020, 16, 1091–1100. [Google Scholar] [CrossRef] [PubMed]
- Basset-Séguin, N.; Hauschild, A.; Kunstfeld, R.; Grob, J.; Dréno, B.; Mortier, L.; Ascierto, P.A.; Licitra, L.; Dutriaux, C.; Thomas, L.; et al. Vismodegib in Patients with Advanced Basal Cell Carcinoma: Primary Analysis of STEVIE, an International, Open-Label Trial. Eur. J. Cancer 2017, 86, 334–348. [Google Scholar] [CrossRef] [Green Version]
- Ben Ishai, M.; Tiosano, A.; Fenig, E.; Ben Simon, G.; Yassur, I. Outcomes of Vismodegib for Periocular Locally Advanced Basal Cell Carcinoma from an Open-Label Trial. JAMA Ophthalmol. 2020, 138, 749–755. [Google Scholar] [CrossRef]
- Tibes, R.; Kosiorek, H.E.; Dueck, A.; Palmer, J.; Slack, J.L.; Knight, E.A.; Hashmi, S.K.; Bogenberger, J.M.; Zblewski, D.; Hogan, W.J.; et al. Phase I/IB Study of Azacitidine and Hedgehog Pathway Inhibition with Sonidegib (LDE225) in Myeloid Malignancies. Blood 2017, 130, 2629. [Google Scholar]
- Buadi, F.K.; Lacy, M.Q.; Dispenzieri, A.; Perez, G.; Gertz, M.A.; Kapoor, P.; Hayman, S.R.; Dingli, D.; Go, R.S.; Fonder, A.; et al. Phase 2 Trial of LDE225 and Lenalidomide Maintenance Post Autologous Stem Cell Transplant for Multiple Myeloma. Blood 2019, 134, 1905. [Google Scholar] [CrossRef]
- Gupta, V.; Wolleschak, D.; Hasselbalch, H.; Vannucchi, A.M.; Koschmieder, S.; Cervantes, F.; Li, Y.; Dong, T.; Wroclawska, M.; Bharathy, S.; et al. Safety and Efficacy of the Combination of Sonidegib and Ruxolitinib in Myelofibrosis: A Phase 1b/2 Dose-Finding Study. Blood Adv. 2020, 4, 3063–3071. [Google Scholar] [CrossRef]
- Sallman, D.A.; Komrokji, R.S.; Sweet, K.L.; Mo, Q.; McGraw, K.L.; Duong, V.H.; Zhang, L.; Nardelli, L.A.; Padron, E.; List, A.F.; et al. A Phase 2 Trial of the Oral Smoothened Inhibitor Glasdegib in Refractory Myelodysplastic Syndromes (MDS). Leuk. Res. 2019, 81, 56–61. [Google Scholar] [CrossRef]
- Minami, Y.; Minami, H.; Miyamoto, T.; Yoshimoto, G.; Kobayashi, Y.; Munakata, W.; Onishi, Y.; Kobayashi, M.; Ikuta, M.; Chan, G.; et al. Phase I Study of Glasdegib (PF-04449913), an Oral Smoothened Inhibitor, in Japanese Patients with Select Hematologic Malignancies. Cancer Sci. 2017, 108, 1628–1633. [Google Scholar] [CrossRef] [PubMed]
- Ottmann, O.G.; Stegelmann, F.; Breccia, M.; Steegmann, J.L.; Olavarria, E.; Aimone, P.; Lipton, J.H. Smoothened Inhibitor Erismodegib Combined with Nilotinib in Patients with Chronic Myeloid Leukemia Resistant/Intolerant to at Least One Prior Tyrosine Kinase Inhibitor: A Phase 1b Study. Leuk. Lymphoma 2021, 62, 739–742. [Google Scholar] [CrossRef] [PubMed]
- Kieran, M.W.; Chisholm, J.; Casanova, M.; Brandes, A.A.; Aerts, I.; Bouffet, E.; Bailey, S.; Leary, S.; Macdonald, T.J.; Mechinaud, F.; et al. Phase i Study of Oral Sonidegib (LDE225) in Pediatric Brain and Solid Tumors and a Phase II Study in Children and Adults with Relapsed Medulloblastoma. Neuro-Oncology 2017, 19, 1542–1552. [Google Scholar] [CrossRef] [PubMed]
- Dummer, R.; Lear, J.T.; Guminski, A.; Leow, L.J.; Squittieri, N.; Migden, M. Efficacy of Sonidegib in Histologic Subtypes of Advanced Basal Cell Carcinoma: Results from the Final Analysis of the Randomized Phase 2 Basal Cell Carcinoma Outcomes With LDE225 Treatment (BOLT) Trial at 42 Months. J. Am. Acad. Dermatol. 2020, 84, 1162–1164. [Google Scholar] [CrossRef] [PubMed]
- Lear, J.T.; Hauschild, A.; Stockfleth, E.; Squittieri, N.; Basset-Seguin, N.; Dummer, R. Efficacy and Safety of Sonidegib in Adult Patients with Nevoid Basal Cell Carcinoma Syndrome (Gorlin Syndrome): Results from a Phase 2, Double-Blind, Randomized Trial. Clin. Cosmet. Investig. Dermatol. 2020, 13, 117–121. [Google Scholar] [CrossRef] [Green Version]
- Lee, K.; Molenaar, R.J.; Klaassen, R.; Bijlsma, M.F.; Weterman, M.J.; Richel, D.J.; Wymenga, M.; van Laarhoven, H.W.M.; Wilmink, J.W. A Phase I Study of LDE225 in Combination with Gemcitabine and Nab-Paclitaxel in Patients with Metastasized Pancreatic Cancer. Ann. Oncol. 2017, 28, v260. [Google Scholar] [CrossRef]
- Pijnappel, E.N.; Klaassen, R.; van der Lee, K.S.; Pleunis - van Empel, M.; Richel, D.; Legdeur, M.; Nederveen, A.; van Laarhoven, H.W.M.; Wilmink, H.W. Phase I/II Study of LDE225 in Combination with Gemcitabine and Nab-Paclitaxel in Patients with Metastatic Pancreatic Cancer. Ann. Oncol. 2019, 30, v265. [Google Scholar] [CrossRef]
- Ruiz-Borrego, M.; Jimenez, B.; Antolín, S.; García-Saenz, J.A.; Corral, J.; Jerez, Y.; Trigo, J.; Urruticoechea, A.; Colom, H.; Gonzalo, N.; et al. A Phase Ib Study of Sonidegib (LDE225), an Oral Small Molecule Inhibitor of Smoothened or Hedgehog Pathway, in Combination with Docetaxel in Triple Negative Advanced Breast Cancer Patients: GEICAM/2012–12 (EDALINE) Study. Invest. New Drugs 2019, 37, 98–108. [Google Scholar] [CrossRef]
- Ross, A.E.; Hughes, R.M.; Glavaris, S.; Ghabili, K.; He, P.; Anders, N.M.; Harb, R.; Tosoian, J.J.; Marchionni, L.; Schaeffer, E.M.; et al. Pharmacodynamic and Pharmacokinetic Neoadjuvant Study of Hedgehog Pathway Inhibitor Sonidegib (LDE-225) in Men with High-Risk Localized Prostate Cancer Undergoing Prostatectomy. Oncotarget 2017, 8, 104182–104192. [Google Scholar] [CrossRef]
- Gerds, A.T.; Tauchi, T.; Ritchie, E.; Deininger, M.; Jamieson, C.; Mesa, R.; Heaney, M.; Komatsu, N.; Minami, H.; Su, Y.; et al. Phase 1/2 Trial of Glasdegib in Patients with Primary or Secondary Myelofibrosis Previously Treated with Ruxolitinib. Leuk. Res. 2019, 79, 38–44. [Google Scholar] [CrossRef] [PubMed]
- Savona, M.R.; Pollyea, D.A.; Stock, W.; Oehler, V.G.; Schroeder, M.A.; Lancet, J.; McCloskey, J.; Kantarjian, H.M.; Ma, W.W.; Naveed Shaik, M.; et al. Phase Ib Study of Glasdegib, a Hedgehog Pathway Inhibitor, in Combination with Standard Chemotherapy in Patients with AML or High-Risk MDS. Clin. Cancer Res. 2018, 24, 2294–2303. [Google Scholar] [CrossRef] [Green Version]
- Heuser, M.; Smith, B.D.; Fiedler, W.; Sekeres, M.A.; Montesinos, P.; Leber, B.; Merchant, A.; Papayannidis, C.; Pérez-Simón, J.A.; Hoang, C.J.; et al. Clinical Benefit of Glasdegib plus Low-Dose Cytarabine in Patients with de Novo and Secondary Acute Myeloid Leukemia: Long-Term Analysis of a Phase II Randomized Trial. Ann. Hematol. 2021, 100, 1181–1194. [Google Scholar] [CrossRef]
- Smith, B.D.; Papayannidis, C.; Heuser, M.; Montesinos, P.; Sekeres, M.A.; Oriol, A.; Schiller, G.J.; Candoni, A.; Jamieson, C.H.; Hoang, C.J.; et al. Low-Dose Cytarabine with or without Glasdegib in Newly Diagnosed Patients with Acute Myeloid Leukemia: Long-Term Analysis of a Phase 2 Randomized Trial. J. Clin. Oncol. 2019, 37, 7010. [Google Scholar] [CrossRef]
- Cortes, J.E.; Douglas Smith, B.; Wang, E.S.; Merchant, A.; Oehler, V.G.; Arellano, M.; DeAngelo, D.J.; Pollyea, D.A.; Sekeres, M.A.; Robak, T.; et al. Glasdegib in Combination with Cytarabine and Daunorubicin in Patients with AML or High-Risk MDS: Phase 2 Study Results. Am. J. Hematol. 2018, 93, 1301–1310. [Google Scholar] [CrossRef] [Green Version]
- Richards, D.A.; Stephenson, J.; Wolpin, B.M.; Becerra, C.; Hamm, J.T.; Messersmith, W.A.; Devens, S.; Cushing, J.; Schmalbach, T.; Fuchs, C.S. A Phase Ib Trial of IPI-926, a Hedgehog Pathway Inhibitor, plus Gemcitabine in Patients with Metastatic Pancreatic Cancer. J. Clin. Oncol. 2012, 30, 213. [Google Scholar] [CrossRef]
- Epstein, E.H.; Lear, J.; Saldanha, G.; Tang, J.Y.; Harwood, C. Hedgehog Pathway Inhibition by Topical Patidegib to Reduce BCC Burden in Patients with Basal Cell Nevus (Gorlin) Syndrome. J. Clin. Oncol. 2018, 36, e21626. [Google Scholar] [CrossRef]
- Ueno, H.; Kondo, S.; Yoshikawa, S.; Inoue, K.; Andre, V.; Tajimi, M.; Murakami, H. A Phase I and Pharmacokinetic Study of Taladegib, a Smoothened Inhibitor, in Japanese Patients with Advanced Solid Tumors. Invest. New Drugs 2018, 36, 647–656. [Google Scholar] [CrossRef]
- Glasspool, R.M.; Blagden, S.P.; Lockley, M.; Paul, J.; Hopkins, C.; Thomson, F.; Brown, J.; Fernandes, R.; Douglas, N.; Pou, C.; et al. A Phase I Trial of the Oral Hedgehog Inhibitor Taladegib (LY2940680) in Combination with Weekly Paclitaxel in Patients with Advanced, Solid Tumours. J. Clin. Oncol. 2017, 35, 2594. [Google Scholar] [CrossRef]
- Bendell, J.; Andre, V.; Ho, A.; Kudchadkar, R.; Migden, M.; Infante, J.; Tiu, R.V.; Pitou, C.; Tucker, T.; Brail, L.; et al. Phase i Study of Ly2940680, a Smo Antagonist, in Patients with Advanced Cancer Including Treatment-Naïve and Previously Treated Basal Cell Carcinoma. Clin. Cancer Res. 2018, 24, 2082–2091. [Google Scholar] [CrossRef] [Green Version]
- Nakamoto, Y.; Tsubamoto, H.; Sawazaki, M.; Kakuno, A.; Sonoda, T. A Phase II Study of S-1, Oxaliplatin, and Nab-Paclitaxel, and Itraconazole Aimed at Conversion Surgery for Advanced and Recurrent Gastric Cancer. J. Clin. Oncol. 2019, 37, 4026. [Google Scholar] [CrossRef]
- Lee, M.; Hong, H.; Kim, W.; Zhang, L.; Friedlander, T.W.; Fong, L.; Lin, A.M.; Small, E.J.; Wei, X.X.; Rodvelt, T.J.; et al. Itraconazole as a Noncastrating Treatment for Biochemically Recurrent Prostate Cancer: A Phase 2 Study. Clin. Genitourin. Cancer 2019, 17, e92–e96. [Google Scholar] [CrossRef] [PubMed]
- Madariaga, A.; Marastoni, S.; Colombo, I.; Mandilaras, V.; Cabanero, M.; Bruce, J.; Garg, S.; Wang, L.; Gill, S.; Dhani, N.C.; et al. Phase I/II Trial Assessing Hydroxychloroquine and Itraconazole in Women with Advanced Platinum-Resistant Epithelial Ovarian Cancer (EOC) (HYDRA-01). J. Clin. Oncol. 2020, 38, 6049. [Google Scholar] [CrossRef]
- Sekulic, A.; Migden, M.R.; Oro, A.E.; Dirix, L.; Lewis, K.D.; Hainsworth, J.D.; Solomon, J.A.; Yoo, S.; Arron, S.T.; Friedlander, P.A.; et al. Efficacy and Safety of Vismodegib in Advanced Basal-Cell Carcinoma. N. Engl. J. Med. 2012, 366, 2171–2179. [Google Scholar] [CrossRef] [Green Version]
- Migden, M.R.; Guminski, A.; Gutzmer, R.; Dirix, L.; Lewis, K.D.; Combemale, P.; Herd, R.M.; Kudchadkar, R.; Trefzer, U.; Gogov, S.; et al. Treatment with Two Different Doses of Sonidegib in Patients with Locally Advanced or Metastatic Basal Cell Carcinoma (BOLT): A Multicentre, Randomised, Double-Blind Phase 2 Trial. Lancet Oncol. 2015, 16, 716–728. [Google Scholar] [CrossRef]
- Menyhárt, O.; Győrffy, B. Principles of Tumorigenesis and Emerging Molecular Drivers of SHH-Activated Medulloblastomas. Ann. Clin. Transl. Neurol. 2019, 6, 990–1005. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Song, Q.; Day, B.W. Phase I and Phase II Sonidegib and Vismodegib Clinical Trials for the Treatment of Paediatric and Adult MB Patients: A Systemic Review and Meta-Analysis. Acta Neuropathol. Commun. 2019, 7, 123. [Google Scholar] [CrossRef] [PubMed]
- Ko, A.H.; LoConte, N.; Tempero, M.A.; Walker, E.J.; Kelley, R.K.; Lewis, S.; Chang, W.-C.; Kantoff, E.; Vannier, M.W.; Catenacci, D.V.; et al. A Phase I Study of FOLFIRINOX Plus IPI-926, a Hedgehog Pathway Inhibitor, for Advanced Pancreatic Adenocarcinoma. Pancreas 2016, 45, 370. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cohen, D.J.; Christos, P.J.; Kindler, H.L.; Catenacci, D.V.T.; Bekaii-Saab, T.B.; Tahiri, S.; Janjigian, Y.Y.; Gibson, M.K.; Chan, E.; Rajdev, L.; et al. Vismodegib (V), a Hedgehog (HH) Pathway Inhibitor, Combined with FOLFOX for First-Line Therapy of Patients (Pts) with Advanced Gastric and Gastroesophageal Junction (GEJ) Carcinoma: A New York Cancer Consortium Led Phase II Randomized Study. J. Clin. Oncol. 2013, 31, 4011. [Google Scholar] [CrossRef]
- Yoon, C.; Park, D.J.; Schmidt, B.; Thomas, N.J.; Lee, H.J.; Kim, T.S.; Janjigian, Y.Y.; Cohen, D.J.; Yoon, S.S. CD44 Expression Denotes a Subpopulation of Gastric Cancer Cells in Which Hedgehog Signaling Promotes Chemotherapy Resistance. Clin. Cancer Res. 2014, 20, 3974–3988. [Google Scholar] [CrossRef] [Green Version]
- Pietanza, M.C.; Litvak, A.M.; Varghese, A.M.; Krug, L.M.; Fleisher, M.; Teitcher, J.B.; Holodny, A.I.; Sima, C.S.; Woo, K.M.; Ng, K.K.; et al. A Phase I Trial of the Hedgehog Inhibitor, Sonidegib (LDE225), in Combination with Etoposide and Cisplatin for the Initial Treatment of Extensive Stage Small Cell Lung Cancer. Lung Cancer 2016, 99, 23. [Google Scholar] [CrossRef] [Green Version]
- Hanna, N.; Bunn, P.A.; Langer, C.; Einhorn, L.; Guthrie, T.; Beck, T.; Ansari, R.; Ellis, P.; Byrne, M.; Morrison, M.; et al. Randomized Phase III Trial Comparing Irinotecan/Cisplatin with Etoposide/Cisplatin in Patients with Previously Untreated Extensive-Stage Disease Small-Cell Lime Cancer. J. Clin. Oncol. 2006, 24, 2038–2043. [Google Scholar] [CrossRef] [PubMed]
- Lara, P.N., Jr.; Natale, R.; Crowley, J.; Lenz, H.J.; Redman, M.W.; Carleton, J.E.; Jett, J.; Langer, C.J.; Kuebler, J.P.; Dakhil, S.R.; et al. Phase III Trial of Irinotecan/Cisplatin Compared With Etoposide/Cisplatin in Extensive-Stage Small-Cell Lung Cancer: Clinical and Pharmacogenomic Results From SWOG S0124. J. Clin. Oncol. 2009, 27, 2530. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuehn, J.; Espinoza-Sanchez, N.A.; Teixeira, F.C.O.B.; Pavão, M.S.G.; Kiesel, L.; Győrffy, B.; Greve, B.; Götte, M. Prognostic Significance of Hedgehog Signaling Network-Related Gene Expression in Breast Cancer Patients. J. Cell. Biochem. 2021, 122, 577–597. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Yu, T.; Hu, Y.; Xiang, M.; Peng, H.; Lin, Y.; Han, L.; Zhang, L. Prognostic Role of Gli1 Expression in Breast Cancer: A Meta-Analysis. Oncotarget 2017, 8, 81088. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Im, S.; Choi, H.J.; Yoo, C.; Jung, J.-H.; Jeon, Y.-W.; Suh, Y.J.; Kang, C.S. Hedgehog Related Protein Expression in Breast Cancer: Gli-2 Is Associated with Poor Overall Survival. Korean J. Pathol. 2013, 47, 116. [Google Scholar] [CrossRef]
- Ten Haaf, A.; Bektas, N.; von Serenyi, S.; Losen, I.; Arweiler, E.C.; Hartmann, A.; Knüchel, R.; Dahl, E. Expression of the Glioma-Associated Oncogene Homolog (GLI) 1 in Human Breast Cancer Is Associated with Unfavourable Overall Survival. BMC Cancer 2009, 9, 298. [Google Scholar] [CrossRef] [Green Version]
- Cheng, J.; Gao, J.; Tao, K. Prognostic Role of Gli1 Expression in Solid Malignancies: A Meta-Analysis. Sci. Rep. 2016, 6, 22184. [Google Scholar] [CrossRef]
- Richtig, G.; Aigelsreiter, A.M.; Asslaber, M.; Weiland, T.; Pichler, M.; Eberhard, K.; Sygulla, S.; Schauer, S.; Hoefler, G.; Aigelsreiter, A. Hedgehog Pathway Proteins SMO and GLI Expression as Prognostic Markers in Head and Neck Squamous Cell Carcinoma. Histopathology 2019, 75, 118. [Google Scholar] [CrossRef] [Green Version]
- Li, R.; Liu, Z.; Chen, Y.; Hu, X.; Peng, X. GLI1 Expression in Pancreatic Ductal Adenocarcinoma Correlates the Clinical Significance and Prognosis: A Meta-Analysis. Medicine 2020, 99, e20950. [Google Scholar] [CrossRef]
- Liao, X.; Siu, M.K.Y.; Au, C.W.H.; Wong, E.S.Y.; Chan, H.Y.; Ip, P.P.C.; Ngan, H.Y.S.; Cheung, A.N.Y. Aberrant Activation of Hedgehog Signaling Pathway in Ovarian Cancers: Effect on Prognosis, Cell Invasion and Differentiation. Carcinogenesis 2009, 30, 131–140. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Li, N.; Pang, Y.; Zhao, J.; Wu, X. Gli Affects the Stemness and Prognosis of Epithelial Ovarian Cancer via Homeobox Protein NANOG. Mol. Med. Rep. 2021, 23, 128. [Google Scholar] [CrossRef] [PubMed]
- Ciucci, A.; De Stefano, I.; Vellone, V.G.; Lisi, L.; Bottoni, C.; Scambia, G.; Zannoni, G.F.; Gallo, D. Expression of the Glioma-Associated Oncogene Homolog 1 (Gli1) in Advanced Serous Ovarian Cancer Is Associated with Unfavorable Overall Survival. PLoS ONE 2013, 8, e60145. [Google Scholar]
- Rossi, M.; Magnoni, L.; Miracco, C.; Mori, E.; Tosi, P.; Pirtoli, L.; Tini, P.; Oliveri, G.; Cosci, E.; Bakker, A. β-Catenin and Gli1 Are Prognostic Markers in Glioblastoma. Cancer Biol. Ther. 2011, 753, 753–761. [Google Scholar] [CrossRef] [Green Version]
- Lv, L.; Yang, Z.; Ma, T.; Xuan, Y. Gli1, a Potential Cancer Stem Cell Marker, Is Strongly Associated with Prognosis in Prostate Cancer. Int. J. Clin. Exp. Pathol. 2018, 11, 4957. [Google Scholar]
- Xu, M.; Li, X.; Liu, T.; Leng, A.; Zhang, G. Prognostic Value of Hedgehog Signaling Pathway in Patients with Colon Cancer. Med. Oncol. 2011, 29, 1010–1016. [Google Scholar] [CrossRef]
- Ding, Y.L.; Zhou, Y.; Xiang, L.; Ji, Z.P.; Luo, Z. hong Expression of Glioma-Associated Oncogene Homolog 1 Is Associated with Invasion and Postoperative Liver Metastasis in Colon Cancer. Int. J. Med. Sci. 2012, 9, 334–338. [Google Scholar] [CrossRef] [Green Version]
- Lu, L.; Wu, M.; Zhao, F.; Fu, W.; Li, W.; Li, X.; Liu, T. Prognostic and Clinicopathological Value of Gli-1 Expression in Gastric Cancer: A Meta-Analysis. Oncotarget 2016, 7, 69087. [Google Scholar] [CrossRef] [Green Version]
- Qi, W.; Yang, Z.; Feng, Y.; Li, H.; Che, N.; Liu, L.; Xuan, Y. Gli1 Regulates Stemness Characteristics in Gastric Adenocarcinoma. Diagn. Pathol. 2020, 15, 60. [Google Scholar] [CrossRef]
- Shao, X.; Kuai, X.; Pang, Z.; Zhang, L.; Wu, L.; Xu, L.; Zhou, C. Correlation of Gli1 and HER2 Expression in Gastric Cancer: Identification of Novel Target. Sci. Rep. 2018, 8, 397. [Google Scholar] [CrossRef] [Green Version]
- Buczkowicz, P.; Ma, J.; Hawkins, C. GLI2 Is a Potential Therapeutic Target in Pediatric Medulloblastoma. J. Neuropathol. Exp. Neurol. 2011, 70, 430–437. [Google Scholar] [CrossRef] [Green Version]
- Ha, Y.-S.; Yun, S.-J.; Kim, Y.-J.; Lee, S.-C.; Kim, W.-J. Utility of Smo as a Prognostic Marker for Human Bladder Tumors. Korean J. Urol. 2007, 48, 997–1003. [Google Scholar] [CrossRef] [Green Version]
- Tu, Y.; Niu, M.; Xie, P.; Yue, C.; Liu, N.; Qi, Z.; Gao, S.; Liu, H.; Shi, Q.; Yu, R.; et al. Smoothened Is a Poor Prognosis Factor and a Potential Therapeutic Target in Glioma. Sci. Rep. 2017, 7, 42630. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tu, Y.; Chen, Z.; Zhao, P.; Sun, G.; Bao, Z.; Chao, H.; Fan, L.; Li, C.; You, Y.; Qu, Y.; et al. Smoothened Promotes Glioblastoma Radiation Resistance Via Activating USP3-Mediated Claspin Deubiquitination. Clin. Cancer Res. 2020, 26, 1749–1762. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Lui, N.; Cheng, T.; Tseng, H.-H.K.; Yue, D.; Giroux-Leprieur, E.; Do, H.T.; Sheng, Q.; Jin, J.Q.; Luh, T.W.; et al. Gli as a Novel Therapeutic Target in Malignant Pleural Mesothelioma. PLoS ONE 2013, 8, e57346. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boetto, J.; Bielle, F.; Sanson, M.; Peyre, M.; Kalamarides, M. SMO Mutation Status Defines a Distinct and Frequent Molecular Subgroup in Olfactory Groove Meningiomas. Neuro-Oncology 2017, 19, 345–351. [Google Scholar]
- Jin, M.Z.; Jin, W.L. The Updated Landscape of Tumor Microenvironment and Drug Repurposing. Signal Transduct. Target. Ther. 2020, 5, 166. [Google Scholar] [CrossRef]
- Gupta, S.; Takebe, N.; LoRusso, P. Targeting the Hedgehog Pathway in Cancer. Ther. Adv. Med. Oncol. 2010, 2, 237. [Google Scholar] [CrossRef] [Green Version]
- Tang, J.Y.; Mackay-Wiggan, J.M.; Aszterbaum, M.; Yauch, R.L.; Lindgren, J.; Chang, K.; Coppola, C.; Chanana, A.M.; Marji, J.; Bickers, D.R.; et al. Inhibiting the Hedgehog Pathway in Patients with the Basal-Cell Nevus Syndrome. N. Engl. J. Med. 2012, 366, 2180–2188. [Google Scholar] [CrossRef] [Green Version]
- Rodon, J.; Tawbi, H.A.; Thomas, A.L.; Stoller, R.G.; Turtschi, C.P.; Baselga, J.; Sarantopoulos, J.; Mahalingam, D.; Shou, Y.; Moles, M.A.; et al. A Phase I, Multicenter, Open-Label, First-in-Human, Dose-Escalation Study of the Oral Smoothened Inhibitor Sonidegib (LDE225) in Patients with Advanced Solid Tumors. Clin. Cancer Res. 2014, 20, 1900–1909. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Italiano, A.; Le Cesne, A.; Bellera, C.; Piperno-Neumann, S.; Duffaud, F.; Penel, N.; Cassier, P.; Domont, J.; Takebe, N.; Kind, M.; et al. GDC-0449 in Patients with Advanced Chondrosarcomas: A French Sarcoma Group/US and French National Cancer Institute Single-Arm Phase II Collaborative Study. Ann. Oncol. 2013, 24, 2922. [Google Scholar] [CrossRef] [PubMed]
- Ishii, T.; Shimizu, Y.; Nakashima, K.; Kondo, S.; Ogawa, K.; Sasaki, S.; Matsui, H. Inhibition Mechanism Exploration of Investigational Drug TAK-441 as Inhibitor against Vismodegib-Resistant Smoothened Mutant. Eur. J. Pharmacol. 2014, 723, 305–313. [Google Scholar] [CrossRef]
- Bender, M.H.; Hipskind, P.A.; Capen, A.R.; Cockman, M.; Credille, K.M.; Gao, H.; Bastian, J.A.; Clay, J.M.; Lobb, K.L.; Sall, D.J.; et al. Abstract 2819: Identification and Characterization of a Novel Smoothened Antagonist for the Treatment of Cancer with Deregulated Hedgehog Signaling. Cancer Res. 2011, 71, 2819. [Google Scholar]
- Tu, J.; Li, J.J.; Song, L.T.; Zhai, H.L.; Wang, J.; Zhang, X.Y. Molecular Modeling Study on Resistance of WT/D473H SMO to Antagonists LDE-225 and LEQ-506. Pharmacol. Res. 2017, 129, 491–499. [Google Scholar] [CrossRef]
- Jäger, T.; Ocker, M.; Kiesslich, T.; Neureiter, E.; Neureiter, D. Expert Opinion on Investigational Drugs Thoughts on Investigational Hedgehog Pathway Inhibitors for the Treatment of Cancer Thoughts on Investigational Hedgehog Pathway Inhibitors for the Treatment of Cancer. Expert Opin. Investig. Drugs 2016, 26, 133–136. [Google Scholar] [CrossRef] [Green Version]
- Li, Q.; Zhao, H.; Zhang, X.; Lang, H.; Yu, K. Novel-Smoothened Inhibitors for Therapeutic Targeting of Naïve and Drug-Resistant Hedgehog Pathway-Driven Cancers. Acta Pharmacol. Sin. 2019, 40, 257. [Google Scholar] [CrossRef]
- Hoch, L.; Faure, H.; Roudaut, H.; Schoenfelder, A.; Mann, A.; Girard, N.; Bihannic, L.; Ayrault, O.; Petricci, E.; Taddei, M.; et al. MRT-92 Inhibits Hedgehog Signaling by Blocking Overlapping Binding Sites in the Transmembrane Domain of the Smoothened Receptor. FASEB J. 2015, 29, 1817–1829. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, B.; Flaveny, C.A.; Giambelli, C.; Fei, D.L.; Han, L.; Hang, B.I.; Bai, F.; Pei, X.-H.; Nose, V.; Burlingame, O.; et al. Repurposing the FDA-Approved Pinworm Drug Pyrvinium as a Novel Chemotherapeutic Agent for Intestinal Polyposis. PLoS ONE 2014, 9, e101969. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carpenter, R.L.; Lo, H.W. Identification, Functional Characterization, and Pathobiological Significance of GLI1 Isoforms in Human Cancers. Vitam. Horm. 2012, 88, 115–140. [Google Scholar] [PubMed] [Green Version]
- Calcaterra, A.; Iovine, V.; Botta, B.; Quaglio, D.; D’Acquarica, I.; Ciogli, A.; Iazzetti, A.; Alfonsi, R.; Severini, L.L.; Infante, P.; et al. Chemical, Computational and Functional Insights into the Chemical Stability of the Hedgehog Pathway Inhibitor GANT61. J. Enzym. Inhib. Med. Chem. 2018, 33, 349. [Google Scholar] [CrossRef] [Green Version]
- Zhang, R.; Ma, J.; Avery, J.T.; Sambandam, V.; Nguyen, T.H.; Xu, B.; Suto, M.J.; Boohaker, R.J. GLI1 Inhibitor SRI-38832 Attenuates Chemotherapeutic Resistance by Downregulating NBS1 Transcription in BRAFV600E Colorectal Cancer. Front. Oncol. 2020, 10, 241. [Google Scholar] [CrossRef]
- Hyman, J.M.; Firestone, A.J.; Heine, V.M.; Zhao, Y.; Ocasio, C.A.; Han, K.; Sun, M.; Rack, P.G.; Sinha, S.; Wu, J.J.; et al. Small-Molecule Inhibitors Reveal Multiple Strategies for Hedgehog Pathway Blockade. Proc. Natl. Acad. Sci. USA 2009, 106, 14132–14137. [Google Scholar] [CrossRef] [Green Version]
- Xu, Y.; Chenna, V.; Hu, C.; Sun, H.-X.; Khan, M.; Bai, H.; Yang, X.-R.; Zhu, Q.-F.; Sun, Y.-F.; Maitra, A.; et al. Polymeric Nanoparticle-Encapsulated Hedgehog Pathway Inhibitor HPI-1 (NanoHHI) Inhibits Systemic Metastases in an Orthotopic Model of Human Hepatocellular Carcinoma. Clin. Cancer Res. 2012, 18, 1291–1302. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Severini, L.L.; Ghirga, F.; Bufalieri, F.; Quaglio, D.; Infante, P.; Marcotullio, L. Di The SHH/GLI Signaling Pathway: A Therapeutic Target for Medulloblastoma. Expert Opin. Ther. Targets 2020, 24, 1159–1181. [Google Scholar]
- Bariwal, J.; Kumar, V.; Dong, Y.; Mahato, R.I. Design of Hedgehog Pathway Inhibitors for Cancer Treatment. Med. Res. Rev. 2019, 39, 1137. [Google Scholar] [CrossRef] [PubMed]
- Infante, P.; Mori, M.; Alfonsi, R.; Ghirga, F.; Aiello, F.; Toscano, S.; Ingallina, C.; Siler, M.; Cucchi, D.; Po, A.; et al. Gli1/DNA Interaction Is a Druggable Target for Hedgehog-Dependent Tumors. EMBO J. 2015, 34, 200. [Google Scholar] [CrossRef] [PubMed]
- Infante, P.; Alfonsi, R.; Botta, B.; Mori, M.; Di Marcotullio, L. Targeting GLI Factors to Inhibit the Hedgehog Pathway. Trends Pharmacol. Sci. 2015, 36, 547–558. [Google Scholar] [CrossRef] [Green Version]
- Justilien, V.; Walsh, M.P.; Ali, S.A.; Thompson, E.A.; Murray, N.R.; Fields, A.P. The PRKCI and SOX2 Oncogenes Are Co-Amplified and Cooperate to Activate Hedgehog Signaling in Lung Squamous Cell Carcinoma. Cancer Cell 2014, 25, 139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, J.Y.; Ally, M.S.; Chanana, A.M.; Mackay-Wiggan, J.M.; Aszterbaum, M.; Lindgren, J.A.; Ulerio, G.; Rezaee, M.R.; Gildengorin, G.; Marji, J.; et al. Inhibition of the Hedgehog Pathway in Patients with Basal-Cell Nevus Syndrome: Final Results from the Multicentre, Randomised, Double-Blind, Placebo-Controlled, Phase 2 Trial. Lancet Oncol. 2016, 17, 1720–1731. [Google Scholar] [CrossRef]
- Huang, X.B.; Shi, Y.; Wang, C.S.; Wang, X.D.; Cheng, J.; Che, F.F. Synergistic Inhibitory Effect of Arsenic Trioxide Combined with Itraconazole on Hedgehog Pathway of Multiple Myeloma NCI-H929 Cells. Zhongguo Shi Yan Xue Ye Xue Za Zhi 2016, 24, 1459–1465. [Google Scholar]
- Zhang, Z.; Zhang, R.; Hao, C.; Pei, X.; Li, J.; Wang, L. GANT61 and Valproic Acid Synergistically Inhibited Multiple Myeloma Cell Proliferation via Hedgehog Signaling Pathway. Med. Sci. Monit. 2020, 26, e920541-1. [Google Scholar] [CrossRef]
- Berardozzi, S.; Bernardi, F.; Infante, P.; Ingallina, C.; Toscano, S.; De Paolis, E.; Alfonsi, R.; Caimano, M.; Botta, B.; Mori, M.; et al. Synergistic Inhibition of the Hedgehog Pathway by Newly Designed Smo and Gli Antagonists Bearing the Isoflavone Scaffold. Eur. J. Med. Chem. 2018, 156, 554–562. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bureta, C.; Saitoh, Y.; Tokumoto, H.; Sasaki, H.; Maeda, S.; Nagano, S.; Komiya, S.; Taniguchi, N.; Setoguchi, T. Synergistic Effect of Arsenic Trioxide, Vismodegib and Temozolomide on Glioblastoma. Oncol. Rep. 2019, 41, 3404–3412. [Google Scholar] [CrossRef] [PubMed]
- Pan, D.; Li, Y.; Li, Z.; Wang, Y.; Wang, P.; Liang, Y. Gli Inhibitor GANT61 Causes Apoptosis in Myeloid Leukemia Cells and Acts in Synergy with Rapamycin. Leuk. Res. 2012, 36, 742–748. [Google Scholar] [CrossRef] [PubMed]
- Cai, X.; Yu, K.; Zhang, L.; Li, Y.; Li, Q.; Yang, Z.; Shen, T.; Duan, L.; Xiong, W.; Wang, W. Synergistic Inhibition of Colon Carcinoma Cell Growth by Hedgehog-Gli1 Inhibitor Arsenic Trioxide and Phosphoinositide 3-Kinase Inhibitor LY294002. Onco Targets Ther. 2015, 8, 877. [Google Scholar] [PubMed] [Green Version]
- Hou, X.; Chen, X.; Zhang, P.; Fan, Y.; Ma, A.; Pang, T.; Song, Z.; Jin, Y.; Hao, W.; Liu, F.; et al. Inhibition of Hedgehog Signaling by GANT58 Induces Apoptosis and Shows Synergistic Antitumor Activity with AKT Inhibitor in Acute T Cell Leukemia Cells. Biochimie 2014, 101, 50–59. [Google Scholar] [CrossRef]
- Eckerdt, F.; Clymer, J.; Bell, J.B.; Beauchamp, E.M.; Blyth, G.T.; Goldman, S.; Platanias, L.C. Pharmacological MTOR Targeting Enhances the Antineoplastic Effects of Selective PI3Kα Inhibition in Medulloblastoma. Sci. Rep. 2019, 9, 12822. [Google Scholar] [CrossRef] [Green Version]
- Bai, X.Y.; Zhang, X.C.; Yang, S.Q.; An, S.J.; Chen, Z.H.; Su, J.; Xie, Z.; Gou, L.Y.; Wu, Y.L. Blockade of Hedgehog Signaling Synergistically Increases Sensitivity to Epidermal Growth Factor Receptor Tyrosine Kinase Inhibitors in Non-Small-Cell Lung Cancer Cell Lines. PLoS ONE 2016, 11, e0149370. [Google Scholar]
- Sternberg, C.; Gruber, W.; Eberl, M.; Tesanovic, S.; Stadler, M.; Elmer, D.P.; Schlederer, M.; Grund, S.; Roos, S.; Wolff, F.; et al. Synergistic Cross-Talk of Hedgehog and Interleukin-6 Signaling Drives Growth of Basal Cell Carcinoma. Int. J. Cancer 2018, 143, 2943–2954. [Google Scholar] [CrossRef] [Green Version]


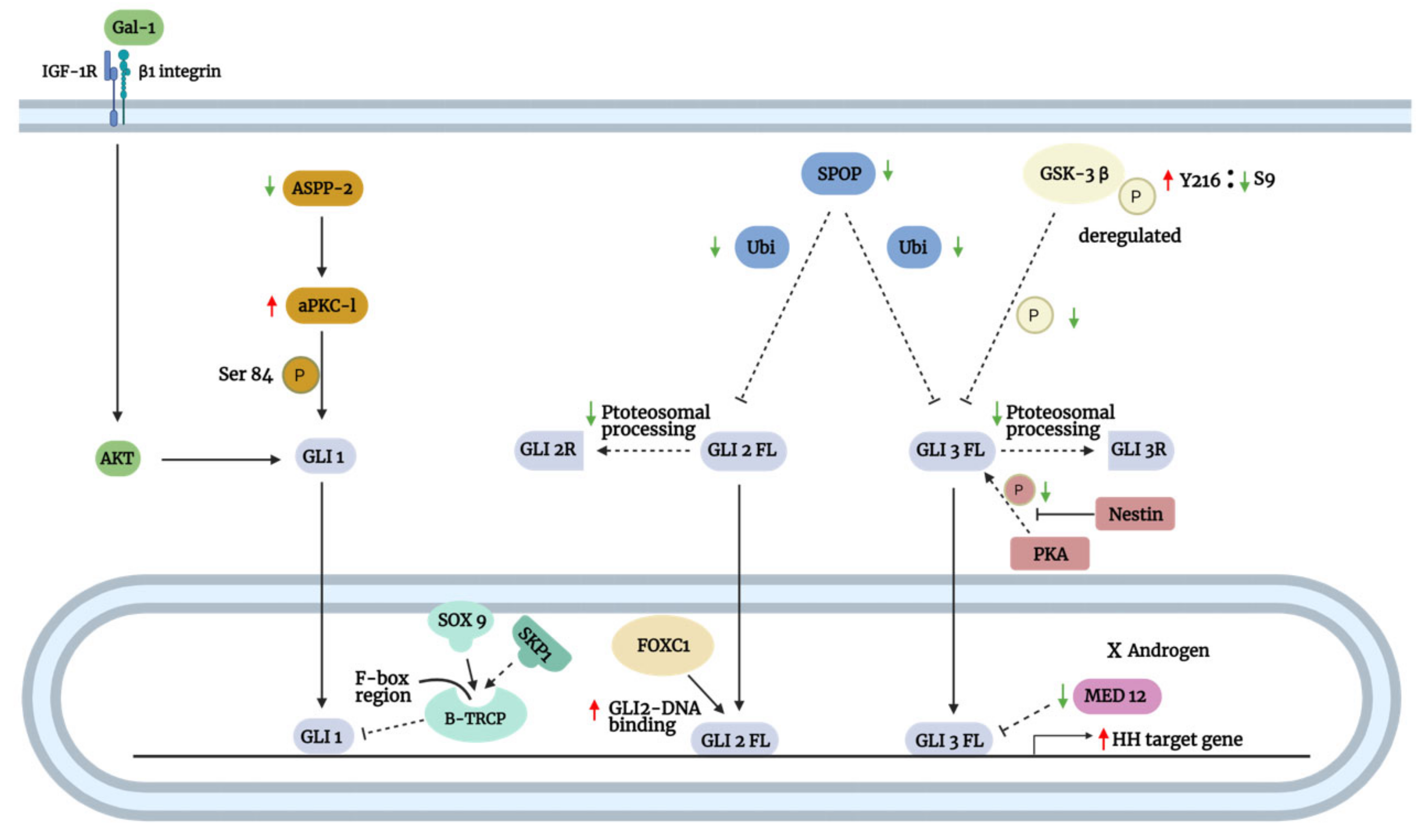

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| GLI Activation | Dysregulation | Regulators | Mechanism of Action | Cancer/Cell Type | Cancer Hallmarks | References |
|---|---|---|---|---|---|---|
| SMO-dependent | Mutations | PTCH1 | Inactivating PTCH1 mutation leads to SMO derepression and GLI1/2 activation | Basal cell carcinoma | Proliferation, resisting cell death, angiogenesis, genomic instability, invasion, metastasis, evading growth suppressor | [44,45,49,50,51,52] |
| Medulloblastoma | Proliferation | [53,54] | ||||
| Odontogenic keratocystic tumors | Proliferation, tumor-promoting inflammation | [55] | ||||
| T-cell acute lymphoblastic leukemia | Proliferation, resisting cell death | [56] | ||||
| Breast cancer | Stemness, resisting cell death | [57,58] | ||||
| Cervical carcinoma | Resisting cell death, invasion and metastasis | [59,60,61] | ||||
| SMO | SMO mutants constitutively activate GLI1/2 in the presence of vismodegib and are resistant to PTCH catalytic inhibition | Basal cell carcinoma | Proliferation, resisting cell death, angiogenesis, genomic instability, invasion and metastasis, and evading growth suppressor | [44,49,50,51,52,62,63,64,65,66] | ||
| SMO mutant constitutively activate GLI1 in the presence of vismodegib | Medulloblastoma | Resisting cell death | [67] | |||
| SMO mutant leads to enhance GLI1 expression and is more resistant to cyclopamine | Hepatocellular carcinoma | Proliferation | [68] | |||
| Transcriptional | NFκB | Transcriptionally upregulates Shh at the promoter level, leading to canonical Shh-GLI activation | Pancreatic cancer | Proliferation, resisting cell death, tumor-promoting inflammation | [69,70,71,72,73] | |
| Breast cancer | Stemness, activating migration | [74,75] | ||||
| CREB, AP1, AP2α, and SP1 | Transcriptionally upregulates SMO at the promoter level, leading to GLI activation | Prostate and breast cancer | NS | [76] | ||
| β-catenin/TCF-4 | Transcriptionally upregulates both SMO and GLI at the promoter level | Foreskin fibroblast | Proliferation | [77] | ||
| Epigenetic | DNA methyltransferase | Hypomethylation of Shh promoter leads to improve NFκB-induced Shh transcription and GLI1 expression | Breast cancer | Stemness, migration | [74] | |
| Hypomethylation of SMO promoter leads to improve SMO transcription and subsequent GLI3 activation | Colorectal cancer | Stemness, proliferation, invasion, deregulated cellular energetic | [78,79,80,81,82] | |||
| Hypomethylation of SMO promoter leads to improve SMO transcription and subsequent GLI2 expression | prostate, kidney, glioblastoma, and ovarian cancer | NS | [76] | |||
| Hypermethylation of PTCH1 promoter leads to decrease PTCH1 expression, causing enhance SMO-GLI1/2 activation and GLI1/2 nuclear translocation | Leiomyosarcoma | Proliferation, activating migration, resisting dell death | [83] | |||
| Hypermethylation of PTCH1 and HHIP lead to increase Hh-GLI signaling | Gastric cancer | Resisting cell death, proliferation, invasion and metastasis | [84,85,86,87,88,89,90] | |||
| SMO-independent | Oncogenic pathways | MAPK/ERK | Stimulation of NRP2 by VEGFa activates ERK, which phosphorylates GLI1 to promote its activation | Lung adenocarcinoma | Stemness, resisting cell death, angiogenesis | [91,92] |
| Stimulation of NRP2 by VEGF induced α6β1 integrin-mediated activation of RAS/MEK signaling through focal adhesion kinase FAK activation and consequently GLI1 expression | Breast cancer | Stemness, resisting cell death | [58,93] | |||
| Oncogenic mutant KRAS enhances GLI1 expression via RAF-MEK1-ERK | Pancreatic ductal adenocarcinoma | Resisting cell death, proliferation, invasion and metastasis | [94,95] | |||
| TGF-β/SMAD | TGF-β enhances GLI1 expression | Hepatocellular carcinoma | Stemness, proliferation, migration, and invasion | [96] | ||
| Pancreatic ductal adenocarcinoma | Resisting cell death, proliferation | [94] | ||||
| TGF-β/SMAD3 enhances GLI1 and GLI2 expression | Melanoma | Resisting apoptosis, proliferation, and invasion | [97] | |||
| Cooperative integration of TGF-β/SMAD and Wnt/β–catenin | β–catenin/TCF-4 and SMAD cooperatively bind to the GLI2 promoter and enhance its transcription | Breast cancer | Invasion and metastasis | [98] | ||
| Oral squamous cell carcinoma | [99] | |||||
| Wnt/β–catenin | β–catenin/TCF-4 upregulates CRD-BP, which binds and stabilizes GLI1 transcripts | Colorectal cancer | Proliferation | [100,101] | ||
| PI3K/AKT | p-AKT enhances GLI1 expression | Gastric cancer | Proliferation, migration, invasion, metastasis, resisting cell death, avoiding immune destruction | [102,103,104] | ||
| PI3K/mTOR regulates GLI1 expression | Lung squamous cell carcinoma | Proliferation | [105] | |||
| ErbB2 enhances GLI1 expression via PI3K/AKT/mTOR activation | Esophageal adenocarcinoma | Proliferation, resisting cell death | [106] | |||
| PI3K/AKT regulates the nuclear translocation of GLI1 | Osteosarcoma | Resisting cell death | [107] | |||
| DYRK1B activates PI3K/AKT/mTOR pathway to promote GLI1 stabilization | Pancreatic and ovarian cancer | Proliferation | [108] | |||
| PI3K/AKT enhances GLI1/2 expression and nuclear translocation | Renal cell carcinoma | Proliferation, resisting cell death | [109] | |||
| TNFα induces S6K1 phosphorylation and GLI1 expression | Prostate cancer | Proliferation | [110] | |||
| TNFα/mTOR activation of S6K1 induces phosphorylation of GLI1, promoting its stability | Esophageal adenocarcinoma | Proliferation and invasion | [111] | |||
| p70S6K2 phosphorylates and inhibits GSK3β function, promoting GLI1 stability | Non-small cell lung cancer | Proliferation, resisting cell death | [112] | |||
| RAS-MEK/AKT | Endogenous RAS-MEK and AKT signaling regulate GLI1 transcription and nuclear localization | Melanoma | Proliferation, resisting cell death, metastasis | [113] | ||
| NFκB | P65 transcriptionally upregulates GLI1 expression by binding to GLI1 promoter | Breast cancer | Cell proliferation, stemness, migration | [114] | ||
| Oncogenic proteins | SOX-9 | SOX9 binds and inhibits β-TrCP, promoting GLI1 stability | Pancreatic ducal adenocarcinoma | Stemness, proliferation, and resisting cell death | [115] | |
| FOXC1 | FOXC1 binds to GLI2 and enhance its DNA binding | Breast cancer | Stemness, proliferation, resisting cell death | [116] | ||
| Nestin | Nestin binds to GLI3 to prevent phosphorylation by PKA, thereby enhancing GLI3 stability | Medulloblastoma | Proliferation | [117] | ||
| Gal-1 | Gal-1 enhance GLI1 expression by binding and activating β1 integrin | Gastric cancer | Invasion, metastasis, vasculogenic mimicry, avoiding immune destruction | [118,119,120,121] | ||
| Gal-1 enhances GLI1 expression | Pancreatic ductal adenocarcinoma | Proliferation, angiogenesis | [122] | |||
| Tumor suppressors | SPOP | Downregulation of SPOP enhance GLI1/2 expression | Ovarian cancer | Proliferation, resisting cell death | [123] | |
| Downregulation of SPOP prevents proteasomal-dependent degradation of GLI2, thus promoting its stability | Gastric cancer | Proliferation, migration, and resisting cell death | [124] | |||
| C3H10T1/2 | NS | [125] | ||||
| ASPP2 | Downregulation of ASPP2 enhances aPKC-ι-mediated phosphorylation of GLI1 to promote its nuclear translocation | Gallbladder cancer | Invasion, metastasis, and tumor-promoting inflammation | [126] | ||
| GSK3β | Deregulated GSK3β function as a result of imbalance activating and inactivating phosphorylation impaired its ability to phosphorylate GLI3, promoting GLI3 stabilization | Colon cancer | Proliferation and resisting cell death | [127] | ||
| MED12 | Downregulation of MED12 relieve its constraint on GLI3, promoting its hyperactivation | Prostate cancer | Proliferation | [128] |
| Hh Inhibitor | Clinical Trial Phase | Cancer Type (Patients Enrolled) | Treatment Interventions | Efficacy | Clinical Trial Number (Recruitment Status) |
|---|---|---|---|---|---|
| Vismodegib | Phase Ib [155] | Intermediate or highrisk MF (n = 10) | Vismodegib 150 mg daily with ruxolitinib 15 or 20 mg twice daily | Only symptom response (n = 5) | NCT02593760 (Completed) |
| Phase Ib [156] | Rel/ref AML (n = 38) | Vismodegib 150 mg once daily | ORR 6.1% | NCT01880437 (Terminated) | |
| Phase I [157] | Metastatic pancreatic cancer (n = 69) | Erlotinib 150 mg and vismodegib 150 mg once daily | No tumor response observed; SD (n = 13); paired biopsies analysis showed reduced GLI1 mRNA, phospho-GLI, and Hh target genes; | NS | |
| Phase I [158] | Pancreatic cancer or other solid tumors (n = 31) | Vismodegib 150 mg daily plus sirolimus at an increasing dose from 3 to 6 mg daily | SD (n = 6); No PR or CR observed, reduced GLI1 expression before and after the first cycle | NCT01537107 (Completed) | |
| Phase II [159] | Untreated PDA (n = 71) | Gemcitabine 1000 mg/m2 and nab-paclitaxel 125 mg/m2 × days 1-8-15, followed by the same regimen with oral vismodegib 150 mg daily × 28 days | ORR 27%, median OS 9.79 months, and median PFS 5.42 months | NCT01088815 (Completed) | |
| Phase II [160] | Multiple BCC (n = 229) | Group A: vismodegib 150 mg daily x 12 weeks, then placebo × 8 weeks, followed by vismodegib 150 mg daily × 12 weeks; Group B: vismodegib 150 mg daily x 24 weeks, then placebo × 8 weeks, followed by vismodegib 150 mg daily × 8 weeks | ORR 62.7% (Group A) and 54.0% (Group B) | NCT01815840 (Completed) | |
| Phase II [161] | laBCC (n = 55) | Vismodegib 150 mg once daily | CR 61.4% | NCT02667574 (Ongoing) | |
| Phase I [162] | laBCC (n = 71) and mBCC (n = 33) | Vismodegib 150 mg once daily until disease progression, intolerable toxicity, or study withdrawal | ORR 60.3% (laBCC) and 48.5% (mBCC); median OS 33.4 months (mBCC), not estimable in laBCC cohort | NCT00833417 (Completed) | |
| Phase II [163] | Infiltrative, Nodular, and Superficial BCC (n = 27) | Vismodegib 150 mg daily | CR 20%, PR 41.5%, and SD 36.9% | NCT01700049 (Completed) | |
| Phase II [164,165,166,167] | Italian cohort: laBCC (n = 159) and mBCC (n = 23) | Vismodegib 150 mg daily until progressive disease, unacceptable toxicity, or withdrawal | ORR 61.7% (laBCC) and 20% (mBCC) | NCT01367665 (Completed) | |
| Ocular or Periocular laBCC (n = 244) | CR 28.7% and PR 38.5% | ||||
| laBCC and mBCC (n = 1232) | ORR 68.5% (laBCC) and 36.9% (mBCC); SD 25.1% (laBCC) and 46.4% (mBCC) | ||||
| laBCC and mBCC (n = 1227) | ORR 64.9% | ||||
| Sonidegib/Erismodegib | Phase I [168] | Untreated AML (n = 15), rel/ref AML (n = 23), MDS (n = 18), CMML (n = 4), and MF (n = 2) | Sonidegib 200–400 mg daily with azacitidine 75 mg/m2 | AML: ORR 23.1%; rel/ref AML: ORR 7.1%, SD 76%, and OS 7.6 months | NCT02129101 (Completed) |
| Phase II [169] | Multiple myeloma (n = 28) | Sonidegib 400 mg daily with Lenalidomide 10 mg daily | CR 46%, VGPR 85%, and 24 month PFS 73% | NCT02086552 (Ongoing) | |
| Phase Ib/II [170] | MF without prior therapy with JAKi (n = 50) | Sonidegib 400 mg daily with ruxolitinib 20 mg twice daily | 29.6% patients achieved > 35% reduction in spleen volume; 26% patients achieved 50% reduction in MFSAF and TSS; minimal change in GLI1 expression | NCT01787552 (Completed) | |
| Phase II [171] | Hypomethylating agent failure: MDS (n = 26), CMML (n = 5), and AML (n = 4) | Oral glasdegib 100 mg daily | ORR 6%, SD 56%, median OS 10.4 months, and EFS 6.4 months | NCT01842646 (Completed) | |
| Phase I [172] | AML (n = 7), MDS (n = 4), CMML (n = 1), and MF (n = 1) | Glasdegib 25/50/100 mg once daily | >80% suppression of GLI1 expression; AML: CR 8% and SD 31%; MDS: CR 8% and SD 16% | NCT02038777 (Ongoing) | |
| Phase Ib [173] | CML-CP (n = 11) | Erismodegib 200 mg once daily with nilotinib 400 mg twice daily | No clear clinical benefits were observed in terms of MMR and CCyR | NCT01456676 (Completed) | |
| I/II [174] | MB (n = 55) and others (n = 21) | Sonidegib 800 mg (adult) or 680 mg/m2 (pediatric) once daily | ORR 6.58% (50% responses were in Hh-positive MB patients); SD (n = 11; 27.7% responses were in Hh-positive MB patients) | NCT01125800 (Completed) | |
| Phase II [175] | mBCC (n = 36), laBCC: aggressive (n = 112) and nonaggressive (n = 82) | Sonidegib 200 or 800 mg once daily | ORR 51.1% (laBCC) and 12.6% (mBCC) | NCT01327053 (Completed) | |
| Phase II [176] | NBCCS (n = 10) | Sonidegib 400 mg or placebo | Total BCC reduced by 40% and 45% at weeks 12 and 16, respectively, vs. zero reduction for placebo | NCT01350115 (Completed) | |
| Phase Ib [177] | Metastatic pancreatic cancer: Chemo-naïve (n = 17) and prior-chemo (n = 9) | Escalated dose of sonidegib (800 mg and 200 mg for chemo-naïve and prior-chemo group, respectively) with gemcitabine 1000 mg/m2 and nab-paclitaxel 125 mg/m2 | SD 8%, 35% PR, and 4% CR. | NCT02358161 (Completed) | |
| Phase I/II [178] | Metastatic pancreatic cancer (n = 25) | Sonidegib 200 mg once daily with gemcitabine 1000 mg/m2 and nab-paclitaxel 125 mg/m2 | PR 10%, SD 53%, and OS 6 months | ||
| Phase Ib [179] | Triple-negative advanced breast cancer (n = 12) | Sonidegib 400/600/800 mg with docetaxel 75 mg/m2 | ORR 30% | NCT02027376 (Completed) | |
| Phase I [180] | High-risk localized prostate cancer (n = 14) | Sonidegib 800 mg once daily or no treatment × 4 weeks before prostatectomy | 86% in the Sonidegib arm achieved at least two-fold GLI1 suppression; no significant difference in DFS between sonidegib and observation arms | NCT02111187 (Completed) | |
| Glasdegib | Phase Ib/II [181] | Primary or secondary MF treated previously with ruxolitinib (n = 21) | Glasdegib or placebo 100 mg once daily | 9.5% and 40% patients had 50% and more than 20% reduced in TSS at week 12, respectively; SVR 4.8% | NCT02226172 (Terminated) |
| Phase Ib [182] | AML or high risk MDS (n = 52) | Glasdegib 100 or 200 mg once daily, either with LDAC 20 mg twice daily, decitabine 20 mg/m2, or cytarabine 100 mg/m2 and daunorubicin 60 mg/m2 | CR 31% | NCT01546038 (Completed) | |
| Phase II [183,184] | AML ineligible for intensive chemotherapy: de novo (n = 56) and secondary (n = 60) | Glasdegib 100 mg daily with LDAC 20 mg twice daily or LDAC 20 mg alone | Median OS: de novo (6.6 vs. 4.3 months) and secondary (9.1 vs. 4.1 months) | ||
| AML ineligible for intensive chemotherapy (n = 116) | Median OS 8.3 vs. 4.3 months | ||||
| Phase II [185] | AML (n = 66) and MDS (n = 5) | Glasdegib 100 mg once daily with cytarabine 100 mg/m2 and daunorubicin 60 mg/m2 | CR 46.4% (≥ 55 years old 40% CR), median OS 14.9 months | ||
| Saridegib/Patadegib | Phase Ib [186] | Metastatic pancreatic cancer (n = 16) | Saridegib (110, 130, or 160 mg) once daily with gemcitabine 1000 mg/m2 | Radiological PR 31% and median PFS > 7 month | NCT01130142 (Completed) |
| Phase II [187] | Gorlin syndrome BCC (n = 17) | Vehicle or 2/4% patidegib twice daily | ORR 25% (patidegib) vs. 0% (vehicle); shrinkage of SEBs was observed only in patients with successful reduction in Hh pathway activity. | NCT02762084 (Completed) | |
| Taladegib | Phase I [188] | Advanced solid tumors (n = 19) | Taladegib 100/200/300 mg once daily | All dose levels significantly inhibit GLI1 transcript levels; PR 5.3% and SD 21.1% | NCT01919398 (Completed) |
| Phase I/Ib [189] | Advanced solid tumors (n = 16) | Taladegib 50/100 mg once or 400 mg twice daily with paclitaxel 80 mg/m2 | PR (n = 3) | ISRCTN15903698 (NS) | |
| Phase I [190] | Treatment-naïve and previously treated BCC (n = 84) | Taladegib 400 mg once daily | Unaffected skin biopsies GLI1 expression showed an inhibition median of 92.3%; ORR 46.8% | NCT01226485 (Completed) | |
| Itraconazole | Phase II [191] | stomach cancer and gastroesophageal junction cancer (n = 23) | 160 mg/m2 nab-paclitaxel and 100 mg/m2 oxaliplatin x 1 day, S-1 60 mg/m2 × 3 days, and itraconazole 400 mg × days 3 days | ORR 70%, median OS 24 months, and 1-year OS rate 95%, | UMIN000021340 (Preinitiation) |
| Phase II [192] | Biochemically recurrent prostate cancer (n = 21) | Oral itraconazole 300 mg twice daily | >50% PSA decline 5%; any PSA decline 47%; among 10 patients without a PSA decline, no significant difference between on-treatment and pretreatment PSADT | NCT01787331 (Completed) | |
| Phase I/II [193] | Platinum-resistant ovarian cancer (n = 11) | Itraconazole 300 mg twice daily with hydroxychloroquine (dose escalation 200–600 mg twice daily) | ORR none, SD (n = 1), and median PFS 1.6 months | NCT03081702 (Completed) | |
| Arsenic trioxide | Phase II [153] | Refractory mBCC (n = 5) | Arsenic trioxide 0.3 mg/kg × 5 days followed by itraconazole 400 mg × 23 days | SD 60%; 75% reduction in GLI1 expression | NCT01791894 (Completed) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chai, J.Y.; Sugumar, V.; Alshawsh, M.A.; Wong, W.F.; Arya, A.; Chong, P.P.; Looi, C.Y. The Role of Smoothened-Dependent and -Independent Hedgehog Signaling Pathway in Tumorigenesis. Biomedicines 2021, 9, 1188. https://doi.org/10.3390/biomedicines9091188
Chai JY, Sugumar V, Alshawsh MA, Wong WF, Arya A, Chong PP, Looi CY. The Role of Smoothened-Dependent and -Independent Hedgehog Signaling Pathway in Tumorigenesis. Biomedicines. 2021; 9(9):1188. https://doi.org/10.3390/biomedicines9091188
Chicago/Turabian StyleChai, Jian Yi, Vaisnevee Sugumar, Mohammed Abdullah Alshawsh, Won Fen Wong, Aditya Arya, Pei Pei Chong, and Chung Yeng Looi. 2021. "The Role of Smoothened-Dependent and -Independent Hedgehog Signaling Pathway in Tumorigenesis" Biomedicines 9, no. 9: 1188. https://doi.org/10.3390/biomedicines9091188