
Absence of Cold-Inducible RNA-Binding Protein (CIRP) Promotes Angiogenesis and Regeneration of Ischemic Tissue by Inducing M2-Like Macrophage Polarization

 ,
,  ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Animals and Treatments
2.2. Femoral Artery Ligation and Tissue Processing
2.3. Histology and Immunohistology
2.4. Cell Culture
2.5. Quantitative Real-Time PCR (qRT-PCR)
2.6. Statistical Analyses
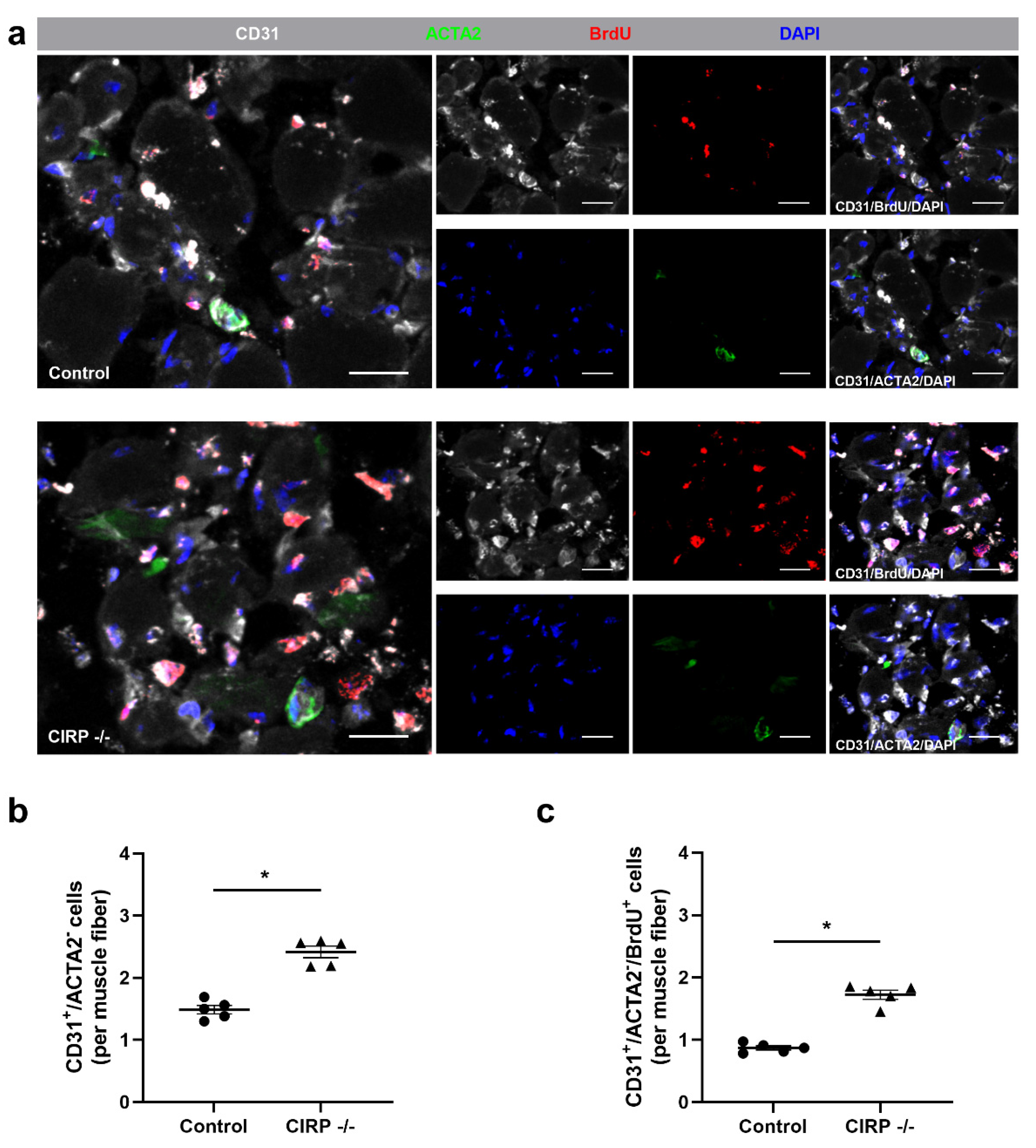
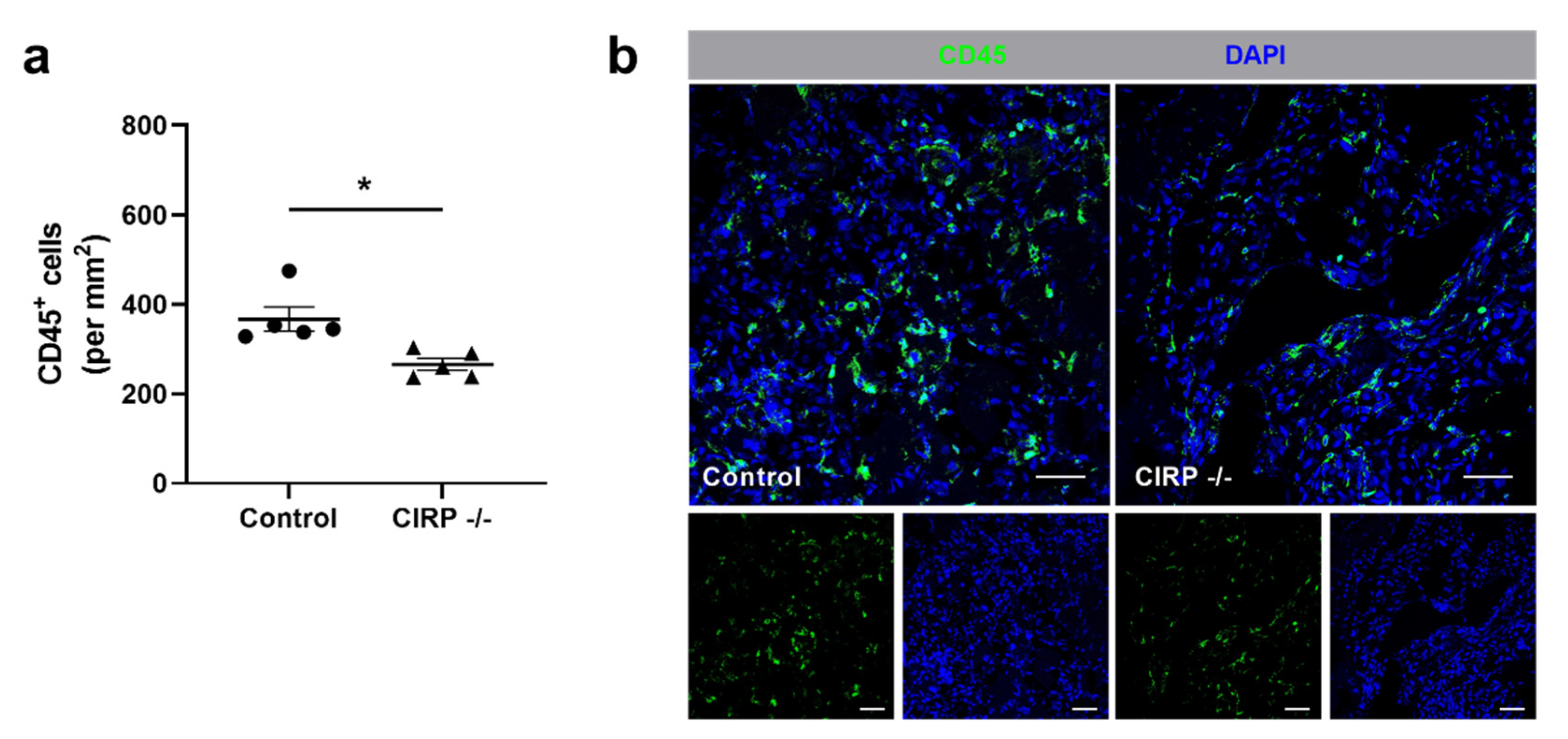
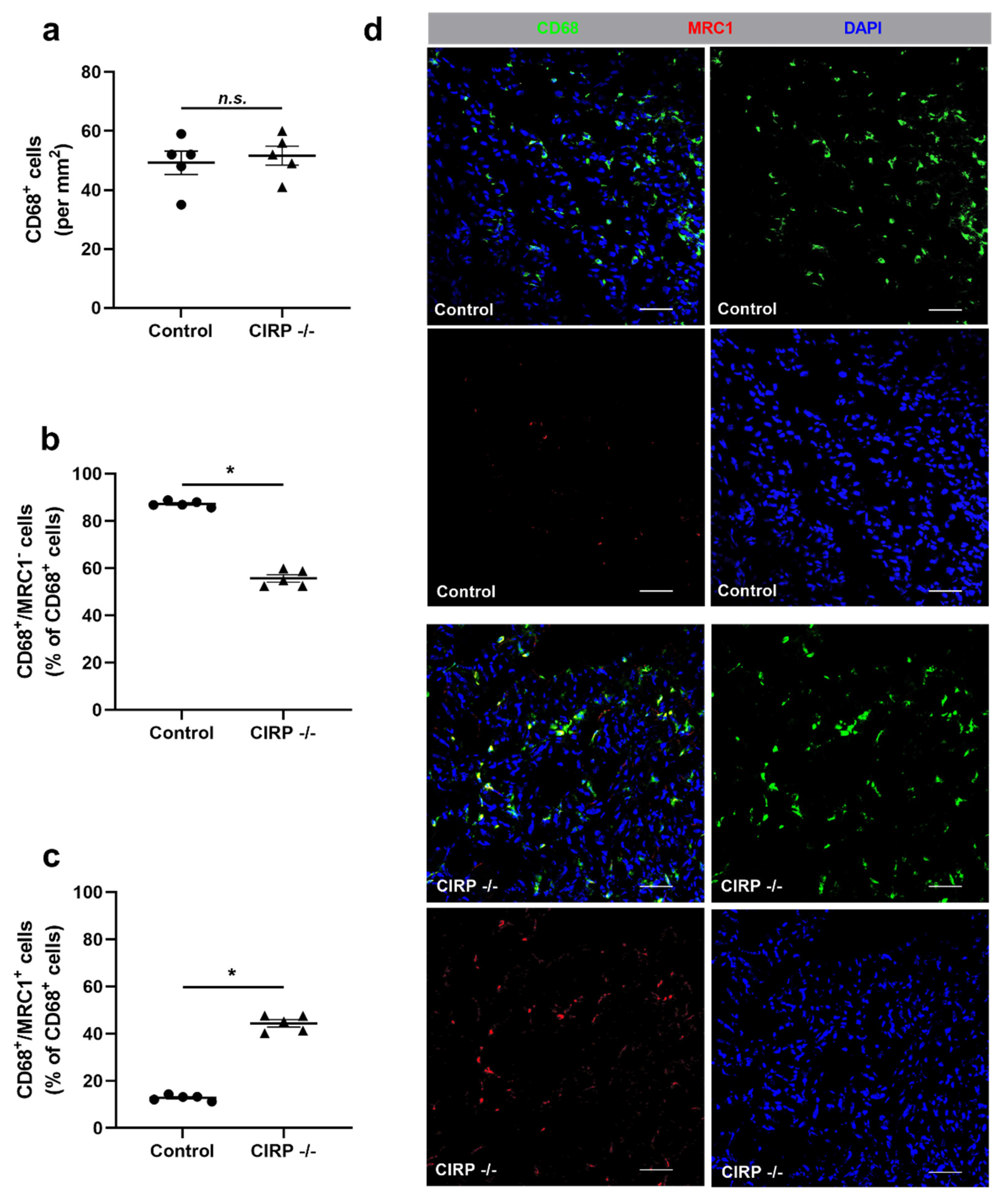
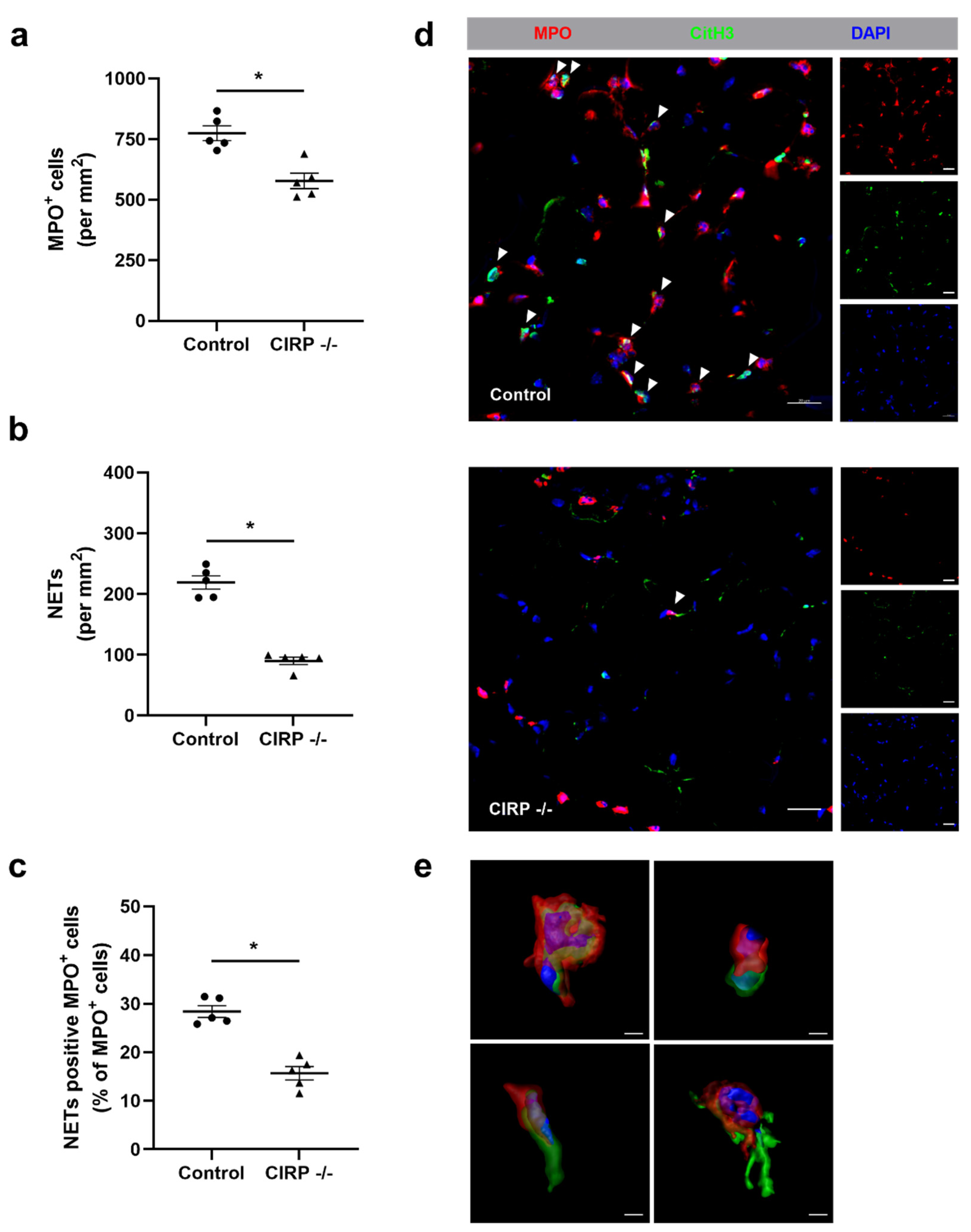
3. Results
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Carmeliet, P. Angiogenesis in health and disease. Nat. Med. 2003, 9, 653–660. [Google Scholar] [CrossRef]
- Jain, R.K. Molecular regulation of vessel maturation. Nat. Med. 2003, 9, 685–693. [Google Scholar] [CrossRef]
- Tonnesen, M.G.; Feng, X.; Clark, R.A. Angiogenesis in wound healing. J. Investig. Dermatol. Symp. Proc. 2000, 5, 40–46. [Google Scholar] [CrossRef] [Green Version]
- Folkman, J. Angiogenesis in cancer, vascular, rheumatoid and other disease. Nat. Med. 1995, 1, 27–30. [Google Scholar] [CrossRef]
- Priya, S.K.; Nagare, R.P.; Sneha, V.; Sidhanth, C.; Bindhya, S.; Manasa, P.; Ganesan, T. Tumour angiogenesis—Origin of blood vessels. Int. J. Cancer 2016, 139, 729–735. [Google Scholar] [CrossRef]
- Yu, J.; Ba, J.; Peng, R.-S.; Xu, D.; Li, Y.-H.; Shi, H.; Wang, Q. Intravitreal anti-VEGF injections for treating wet age-related macular degeneration: A systematic review and meta-analysis. Drug Des. Dev. Ther. 2015, 9, 5397–5405. [Google Scholar] [CrossRef] [Green Version]
- Garcia, J.; Hurwitz, H.I.; Sandler, A.B.; Miles, D.; Coleman, R.L.; Deurloo, R.; Chinot, O.L. Bevacizumab (Avastin®) in cancer treatment: A review of 15 years of clinical experience and future outlook. Cancer Treat. Rev. 2020, 86, 102017. [Google Scholar] [CrossRef] [PubMed]
- Elshabrawy, H.A.; Chen, Z.; Volin, M.V.; Ravella, S.; Virupannavar, S.; Shahrara, S. The pathogenic role of angiogenesis in rheumatoid arthritis. Angiogenesis 2015, 18, 433–448. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DiPietro, L.A. Angiogenesis and wound repair: When enough is enough. J. Leukoc. Biol. 2016, 100, 979–984. [Google Scholar] [CrossRef] [PubMed]
- Emanueli, C.; Madeddu, P. Angiogenesis gene therapy to rescue ischaemic tissues: Achievements and future directions. Br. J. Pharmacol. 2001, 133, 951–958. [Google Scholar] [CrossRef] [Green Version]
- Badimon, L.; Borrell, M. Microvasculature recovery by angiogenesis after myocardial infarction. Curr. Pharm. Des. 2018, 24, 2967–2973. [Google Scholar] [CrossRef]
- Inampudi, C.; Akintoye, E.; Ando, T.; Briasoulis, A. Angiogenesis in peripheral arterial disease. Curr. Opin. Pharmacol. 2018, 39, 60–67. [Google Scholar] [CrossRef] [PubMed]
- Ruan, L.; Wang, B.; ZhuGe, Q.; Jin, K. Coupling of neurogenesis and angiogenesis after ischemic stroke. Brain Res. 2015, 1623, 166–173. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deindl, E.; Schaper, W. The art of arteriogenesis. Cell Biophys. 2005, 43, 1–15. [Google Scholar] [CrossRef]
- Faber, J.E.; Chilian, W.M.; Deindl, E.; Van Royen, N.; Simons, M. A brief etymology of the collateral circulation. Arter. Thromb. Vasc. Biol. 2014, 34, 1854–1859. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heil, M.; Eitenmüller, I.; Schmitz-Rixen, T.; Schaper, W. Arteriogenesis versus angiogenesis: Similarities and differences. J. Cell. Mol. Med. 2006, 10, 45–55. [Google Scholar] [CrossRef] [Green Version]
- Rizzi, A.; Benagiano, V.; Ribatti, D. Angiogenesis versus arteriogenesis. Rom. J. Morphol. Embryol 2017, 58, 15–19. [Google Scholar] [PubMed]
- Adams, R.H.; Alitalo, K. Molecular regulation of angiogenesis and lymphangiogenesis. Nat. Rev. Mol. Cell Biol. 2007, 8, 464–478. [Google Scholar] [CrossRef]
- Egginton, S.; Zhou, A.-L.; Brown, M.D.; Hudlická, O. Unorthodox angiogenesis in skeletal muscle. Cardiovasc. Res. 2001, 49, 634–646. [Google Scholar] [CrossRef] [Green Version]
- Carmeliet, P. Mechanisms of angiogenesis and arteriogenesis. Nat. Med. 2000, 6, 389–395. [Google Scholar] [CrossRef] [PubMed]
- Mentzer, S.J.; Konerding, M.A. Intussusceptive angiogenesis: Expansion and remodeling of microvascular networks. Angiogenesis 2014, 17, 499–509. [Google Scholar] [CrossRef] [Green Version]
- Risau, W. Mechanisms of angiogenesis. Nat. Cell Biol. 1997, 386, 671–674. [Google Scholar] [CrossRef]
- Demir, R.; Yaba, A.; Huppertz, B. Vasculogenesis and angiogenesis in the endometrium during menstrual cycle and implantation. Acta Histochem. 2010, 112, 203–214. [Google Scholar] [CrossRef]
- Karizbodagh, M.P.; Rashidi, B.; Sahebkar, A.; Masoudifar, A.; Mirzaei, H. Implantation window and angiogenesis. J. Cell. Biochem. 2017, 118, 4141–4151. [Google Scholar] [CrossRef]
- Hudlicka, O.; Brown, M.; Egginton, S. Angiogenesis in skeletal and cardiac muscle. Physiol. Rev. 1992, 72, 369–417. [Google Scholar] [CrossRef]
- Weckbach, L.T.; Preissner, K.T.; Deindl, E. The role of midkine in arteriogenesis, involving mechanosensing, endothelial cell proliferation, and vasodilation. Int. J. Mol. Sci. 2018, 19, 2559. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gerhardt, H.; Golding, M.; Fruttiger, M.; Ruhrberg, C.; Lundkvist, A.; Abramsson, A.; Jeltsch, M.; Mitchell, C.; Alitalo, K.; Shima, D.; et al. VEGF guides angiogenic sprouting utilizing endothelial tip cell filopodia. J. Cell Biol. 2003, 161, 1163–1177. [Google Scholar] [CrossRef] [PubMed]
- Ferrara, N.; Henzel, W.J. Pituitary follicular cells secrete a novel heparin-binding growth factor specific for vascular endothelial cells. Biochem. Biophys. Res. Commun. 1989, 161, 851–858. [Google Scholar] [CrossRef]
- Ferrara, N.; Gerber, H.-P.; LeCouter, J. The biology of VEGF and its receptors. Nat. Med. 2003, 9, 669–676. [Google Scholar] [CrossRef] [PubMed]
- Schwarz, E.R.; Speakman, M.T.; Patterson, M.; Hale, S.S.; Isner, J.M.; Kedes, L.H.; A Kloner, R. Evaluation of the effects of intramyocardial injection of DNA expressing vascular endothelial growth factor (VEGF) in a myocardial infarction model in the rat—Angiogenesis and angioma formation. J. Am. Coll. Cardiol. 2000, 35, 1323–1330. [Google Scholar] [CrossRef] [Green Version]
- Scapini, P.; Morini, M.; Tecchio, C.; Minghelli, S.; Di Carlo, E.; Tanghetti, E.; Albini, A.; Lowell, C.; Berton, G.; Noonan, D.M.; et al. CXCL1/Macrophage inflammatory protein-2-induced angiogenesis in vivo is mediated by neutrophil-derived vascular endothelial growth factor-A. J. Immunol. 2004, 172, 5034–5040. [Google Scholar] [CrossRef] [Green Version]
- Gaudry, M.; Brégerie, O.; Andrieu, V.; El Benna, J.; A Pocidalo, M.; Hakim, J. Intracellular pool of vascular endothelial growth factor in human neutrophils. Blood 1997, 90, 4153–4161. [Google Scholar] [CrossRef]
- Scapini, P.; Calzetti, F.; A Cassatella, M. On the detection of neutrophil-derived vascular endothelial growth factor (VEGF). J. Immunol. Methods 1999, 232, 121–129. [Google Scholar] [CrossRef]
- Stockmann, C.; Kirmse, S.; Helfrich, I.; Weidemann, A.; Takeda, N.; Doedens, A.; Johnson, R.S. A Wound size–dependent effect of myeloid cell–derived vascular endothelial growth factor on wound healing. J. Investig. Dermatol. 2011, 131, 797–801. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nissen, N.N.; Polverini, P.J.; Koch, A.E.; Volin, M.V.; Gamelli, R.L.; DiPietro, L.A. Vascular endothelial growth factor mediates angiogenic activity during the proliferative phase of wound healing. Am. J. Pathol. 1998, 152, 1445–1452. [Google Scholar] [PubMed]
- Berse, B.; Brown, L.F.; Van De Water, L.; Dvorak, H.F.; Senger, D.R. Vascular permeability factor (vascular endothelial growth factor) gene is expressed differentially in normal tissues, macrophages, and tumors. Mol. Biol. Cell 1992, 3, 211–220. [Google Scholar] [CrossRef] [Green Version]
- Brinkmann, V.; Reichard, U.; Goosmann, C.; Fauler, B.; Uhlemann, Y.; Weiss, D.S.; Weinrauch, Y.; Zychlinsky, A. Neutrophil extracellular traps kill bacteria. Science 2004, 303, 1532–1535. [Google Scholar] [CrossRef]
- Rohrbach, A.S.; Slade, D.J.; Thompson, P.R.; Mowen, K.A. Activation of PAD4 in NET formation. Front. Immunol. 2012, 3, 360. [Google Scholar] [CrossRef] [Green Version]
- Zawrotniak, M.; Rapala-Kozik, M. Neutrophil extracellular traps (NETs) - formation and implications. Acta Biochim. Pol. 2013, 60, 277–284. [Google Scholar] [CrossRef] [Green Version]
- Aldabbous, L.; Abdul-Salam, V.; McKinnon, T.; Duluc, L.; Pepke-Zaba, J.; Southwood, M.; Ainscough, A.J.; Hadinnapola, C.; Wilkins, M.R.; Toshner, M.; et al. Neutrophil extracellular traps promote angiogenesis: Evidence from vascular pathology in pulmonary hypertension. Arterioscler. Thromb. Vasc. Biol. 2016, 36, 2078–2087. [Google Scholar] [CrossRef] [Green Version]
- Binet, F.; Cagnone, G.; Crespo-Garcia, S.; Hata, M.; Neault, M.; Dejda, A.; Wilson, A.M.; Buscarlet, M.; Mawambo, G.T.; Howard, J.P.; et al. Neutrophil extracellular traps target senescent vasculature for tissue remodeling in retinopathy. Science 2020, 369, 5356. [Google Scholar] [CrossRef] [PubMed]
- Lefrançais, E.; Mallavia, B.; Zhuo, H.; Calfee, C.S.; Looney, M.R. Maladaptive role of neutrophil extracellular traps in pathogen-induced lung injury. JCI Insight 2018, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wong, S.L.; Demers, M.; Martinod, K.; Gallant, M.; Wang, Y.; Goldfine, A.B.; Kahn, C.R.; Wagner, D.D. Diabetes primes neutrophils to undergo NETosis, which impairs wound healing. Nat. Med. 2015, 21, 815–819. [Google Scholar] [CrossRef] [Green Version]
- Saffarzadeh, M.; Juenemann, C.; Queisser, M.A.; Lochnit, G.; Barreto, G.; Galuska, S.P.; Lohmeyer, J.; Preissner, K.T. Neutrophil extracellular traps directly induce epithelial and endothelial cell death: A predominant role of histones. PLoS ONE 2012, 7, e32366. [Google Scholar] [CrossRef]
- Gerstberger, S.; Hafner, M.; Tuschl, T. A census of human RNA-binding proteins. Nat. Rev. Genet. 2014, 15, 829–845. [Google Scholar] [CrossRef]
- Lukong, K.E.; Chang, K.-W.; Khandjian, E.W.; Richard, S. RNA-binding proteins in human genetic disease. Trends Genet. 2008, 24, 416–425. [Google Scholar] [CrossRef] [PubMed]
- Chang, S.-H.; Hla, T. Gene regulation by RNA binding proteins and microRNAs in angiogenesis. Trends Mol. Med. 2011, 17, 650–658. [Google Scholar] [CrossRef] [Green Version]
- Nishiyama, H.; Itoh, K.; Kaneko, Y.; Kishishita, M.; Yoshida, O.; Fujita, J. A glycine-rich RNA-binding protein mediating cold-inducible suppression of mammalian cell growth. J. Cell Biol. 1997, 137, 899–908. [Google Scholar] [CrossRef]
- Zhong, P.; Huang, H. Recent progress in the research of cold-inducible RNA-binding protein. Future Sci. OA 2017, 3, FSO246. [Google Scholar] [CrossRef] [Green Version]
- De Leeuw, F.; Zhang, T.; Wauquier, C.; Huez, G.; Kruys, V.; Gueydan, C. The cold-inducible RNA-binding protein migrates from the nucleus to cytoplasmic stress granules by a methylation-dependent mechanism and acts as a translational repressor. Exp. Cell Res. 2007, 313, 4130–4144. [Google Scholar] [CrossRef]
- Yang, R.; Zhan, M.; Nalabothula, N.R.; Yang, Q.; Indig, F.E.; Carrier, F. Functional significance for a heterogenous ribonucleoprotein A18 signature RNA motif in the 3′-untranslated region of ataxia telangiectasia mutated and Rad3-related (ATR) transcript. J. Biol. Chem. 2010, 285, 8887–8893. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Liu, X.; Li, B.; Zhang, Q.; Wang, J.; Zhang, W.; Luo, W.; Chen, J. Cold inducible RNA binding protein is involved in chronic hypoxia induced neuron apoptosis by down-regulating HIF-1α expression and regulated by microRNA-23a. Int. J. Biol. Sci. 2017, 13, 518–531. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sumitomo, Y.; Higashitsuji, H.; Higashitsuji, H.; Liu, Y.; Fujita, T.; Sakurai, T.; Candeias, M.M.; Itoh, K.; Chiba, T.; Fujita, J. Identification of a novel enhancer that binds Sp1 and contributes to induction of cold-inducible RNA-binding protein (cirp) expression in mammalian cells. BMC Biotechnol. 2012, 12, 72. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, R.; Weber, D.J.; Carrier, F. Post-transcriptional regulation of thioredoxin by the stress inducible heterogenous ribonucleoprotein A18. Nucleic Acids Res. 2006, 34, 1224–1236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Wu, Y.; Mao, P.; Li, F.; Han, X.; Zhang, Y.; Jiang, S.; Chen, Y.; Huang, J.; Liu, D.; et al. Cold-inducible RNA-binding protein CIRP/hnRNP A18 regulates telomerase activity in a temperature-dependent manner. Nucleic Acids Res. 2016, 44, 761–775. [Google Scholar] [CrossRef]
- Na Lee, H.; Ahn, S.-M.; Jang, H.H. Cold-inducible RNA-binding protein, CIRP, inhibits DNA damage-induced apoptosis by regulating p53. Biochem. Biophys. Res. Commun. 2015, 464, 916–921. [Google Scholar] [CrossRef] [PubMed]
- Sakurai, T.; Itoh, K.; Higashitsuji, H.; Nonoguchi, K.; Liu, Y.; Watanabe, H.; Nakano, T.; Fukumoto, M.; Chiba, T.; Fujita, J. Cirp protects against tumor necrosis factor-α-induced apoptosis via activation of extracellular signal-regulated kinase. Biochim. Biophys. Acta 2006, 1763, 290–295. [Google Scholar] [CrossRef] [Green Version]
- Chang, E.T.; Parekh, P.R.; Yang, Q.; Nguyen, D.M.; Carrier, F. Heterogenous ribonucleoprotein A18 (hnRNP A18) promotes tumor growth by increasing protein translation of selected transcripts in cancer cells. Oncotarget 2016, 7, 10578–10593. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, D.; Lv, F.-L.; Wang, G.-H. Effects of HIF-1α on diabetic retinopathy angiogenesis and VEGF expression. Eur. Rev. Med. Pharmacol. Sci. 2018, 22, 5071–5076. [Google Scholar]
- Gerstberger, S.; Hafner, M.; Ascano, M.; Tuschl, T. Evolutionary conservation and expression of human RNA-binding proteins and their role in human genetic disease. Adv. Exp. Med. Biol. 2014, 825, 1–55. [Google Scholar]
- Xia, Z.; Zheng, X.; Zheng, H.; Liu, X.; Yang, Z.; Wang, X. Cold-inducible RNA-binding protein (CIRP) regulates target mRNA stabilization in the mouse testis. FEBS Lett. 2012, 586, 3299–3308. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coburn, K.; Melville, Z.; Aligholizadeh, E.; Roth, B.M.; Varney, K.M.; Carrier, F.; Pozharski, E.; Weber, D.J. Crystal structure of the human heterogeneous ribonucleoprotein A18 RNA-recognition motif. Acta Crystallogr. Sect. F Struct. Biol. Commun. 2017, 73, 209–214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, X.; Wu, Y.; Hartley, R.S. Cold-inducible RNA-binding protein contributes to human antigen R and cyclin E1 deregulation in breast cancer. Mol. Carcinog. 2009, 49, 130–140. [Google Scholar] [CrossRef] [Green Version]
- Lujan, D.A.; Ochoa, J.L.; Hartley, R.S. Cold-inducible RNA binding protein in cancer and inflammation. Wiley Interdiscip. Rev. RNA 2018, 9, e1462. [Google Scholar] [CrossRef] [PubMed]
- Aziz, M.; Brenner, M.; Wang, P. Extracellular CIRP (eCIRP) and inflammation. J. Leukoc. Biol. 2019, 106, 133–146. [Google Scholar] [CrossRef]
- Qiang, X.; Yang, W.-L.; Wu, R.; Zhou, M.; Jacob, A.; Dong, W.; Kuncewitch, M.; Ji, Y.; Yang, H.; Wang, H.; et al. Cold-inducible RNA-binding protein (CIRP) triggers inflammatory responses in hemorrhagic shock and sepsis. Nat. Med. 2013, 19, 1489–1495. [Google Scholar] [CrossRef] [Green Version]
- Takizawa, S.; Murao, A.; Ochani, M.; Aziz, M.; Wang, P. Frontline science: Extracellular CIRP generates a proinflammatory Ly6G + CD11b hi subset of low-density neutrophils in sepsis. J. Leukoc. Biol. 2020. [Google Scholar] [CrossRef]
- Ode, Y.; Aziz, M.; Jin, H.; Arif, A.; Nicastro, J.G.; Wang, P. Cold-inducible RNA-binding protein induces neutrophil extracellular traps in the lungs during sepsis. Sci. Rep. 2019, 9, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Ode, Y.; Aziz, M.; Wang, P. CIRP increases ICAM-1 + phenotype of neutrophils exhibiting elevated iNOS and NETs in sepsis. J. Leukoc. Biol. 2018, 103, 693–707. [Google Scholar] [CrossRef]
- Murao, A.; Arif, A.; Brenner, M.; Denning, N.-L.; Jin, H.; Takizawa, S.; Nicastro, B.; Wang, P.; Aziz, M. Extracellular CIRP and TREM-1 axis promotes ICAM-1-Rho-mediated NETosis in sepsis. FASEB J. 2020, 34, 9771–9786. [Google Scholar] [CrossRef]
- Welten, S.M.; Bastiaansen, A.J.; De Jong, R.C.; De Vries, M.R.; Peters, E.A.; Boonstra, M.C.; Sheikh, S.P.; La Monica, N.; Kandimalla, E.R.; Quax, P.H.; et al. Inhibition of 14q32 MicroRNAs miR-329, miR-487b, miR-494, and miR-495 increases neovascularization and blood flow recovery after ischemia. Circ. Res. 2014, 115, 696–708. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Velasco, A.D.R.; Welten, S.M.; Goossens, E.A.; Quax, P.H.; Rappsilber, J.; Michlewski, G.; Nossent, A.Y. Posttranscriptional regulation of 14q32 MicroRNAs by the CIRBP and HADHB during vascular regeneration after ischemia. Mol. Ther. Nucleic Acids 2019, 14, 329–338. [Google Scholar] [CrossRef] [Green Version]
- Wang, P.; Luo, Y.; Duan, H.; Xing, S.; Zhang, J.; Lu, D.; Feng, J.; Yang, N.; Song, L.; Yan, X. MicroRNA 329 suppresses angiogenesis by targeting CD146. Mol. Cell. Biol. 2013, 33, 3689–3699. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Idrovo, J.P.; Jacob, A.; Yang, W.L.; Wang, Z.; Yen, H.T.; Nicastro, J.; Coppa, G.F.; Wang, P. A deficiency in cold-inducible RNA-binding protein accelerates the inflammation phase and improves wound healing. Int. J. Mol. Med. 2016, 37, 423–428. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Limbourg, A.; Korff, T.; Napp, L.C.; Schaper, W.; Drexler, H.; Limbourg, F.P. Evaluation of postnatal arteriogenesis and angiogenesis in a mouse model of hind-limb ischemia. Nat. Protoc. 2009, 4, 1737–1748. [Google Scholar] [CrossRef] [PubMed]
- Olfert, I.M.; Baum, O.; Hellsten, Y.; Egginton, S. Advances and challenges in skeletal muscle angiogenesis. Am. J. Physiol. Circ. Physiol. 2016, 310, H326–H336. [Google Scholar] [CrossRef] [Green Version]
- Kumar, A.; D’Souza, S.S.; Moskvin, O.V.; Toh, H.; Wang, B.; Zhang, J.; Swanson, S.; Guo, L.-W.; Thomson, J.A.; Slukvin, I.I. Specification and diversification of pericytes and smooth muscle cells from mesenchymoangioblasts. Cell Rep. 2017, 19, 1902–1916. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Attwell, D.; Mishra, A.; Hall, C.N.; O’Farrell, F.M.; Dalkara, T. What is a pericyte? J. Cereb. Blood Flow Metab. 2016, 36, 451–455. [Google Scholar] [CrossRef] [Green Version]
- Shepro, D.; Morel, N.M.L. Pericyte physiology. FASEB J. 1993, 7, 1031–1038. [Google Scholar] [CrossRef] [PubMed]
- He, L.; Vanlandewijck, M.; Raschperger, E.; Mäe, M.A.; Jung, B.; Lebouvier, T.; Ando, K.; Hofmann, J.; Keller, A.; Betsholtz, C. Analysis of the brain mural cell transcriptome. Sci. Rep. 2016, 6, 35108. [Google Scholar] [CrossRef] [Green Version]
- Chillo, O.; Kleinert, E.C.; Lautz, T.; Lasch, M.; Pagel, J.-I.; Heun, Y.; Troidl, K.; Fischer, S.; Caballero-Martinez, A.; Mauer, A.; et al. Perivascular mast cells govern shear stress-induced arteriogenesis by orchestrating leukocyte function. Cell Rep. 2016, 16, 2197–2207. [Google Scholar] [CrossRef] [Green Version]
- Du Cheyne, C.; Tay, H.; De Spiegelaere, W. The complex TIE between macrophages and angiogenesis. Anat. Histol. Embryol. 2019, 49, 585–596. [Google Scholar] [CrossRef]
- Seignez, C.; Phillipson, M. The multitasking neutrophils and their involvement in angiogenesis. Curr. Opin. Hematol. 2017, 24, 3–8. [Google Scholar] [CrossRef]
- Wang, J. Neutrophils in tissue injury and repair. Cell Tissue Res. 2018, 371, 531–539. [Google Scholar] [CrossRef] [Green Version]
- Castanheira, F.V.S.; Kubes, P. Neutrophils and NETs in modulating acute and chronic inflammation. Blood 2019, 133, 2178–2185. [Google Scholar] [CrossRef] [PubMed]
- Pittman, K.; Kubes, P. Damage-associated molecular patterns control neutrophil recruitment. J. Innate Immun. 2013, 5, 315–323. [Google Scholar] [CrossRef] [PubMed]
- Denning, N.-L.; Aziz, M.; Murao, A.; Gurien, S.D.; Ochani, M.; Prince, J.M.; Wang, P. Extracellular CIRP as an endogenous TREM-1 ligand to fuel inflammation in sepsis. JCI Insight 2020, 5. [Google Scholar] [CrossRef]
- Yang, W.-L.; Sharma, A.; Wang, Z.; Li, Z.; Fan, J.; Wang, P. Cold-inducible RNA-binding protein causes endothelial dysfunction via activation of Nlrp3 inflammasome. Sci. Rep. 2016, 6, 26571. [Google Scholar] [CrossRef] [Green Version]
- Wellmann, S.; Bührer, C.; Moderegger, E.; Zelmer, A.; Kirschner, R.; Koehne, P.; Fujita, J.; Seeger, K. Oxygen-regulated expression of the RNA-binding proteins RBM3 and CIRP by a HIF-1-independent mechanism. J. Cell Sci. 2004, 117, 1785–1794. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Hossain, M.; Thanabalasuriar, A.; Gunzer, M.; Meininger, C.; Kubes, P. Visualizing the function and fate of neutrophils in sterile injury and repair. Science 2017, 358, 111–116. [Google Scholar] [CrossRef] [Green Version]
- Murray, P.J. Macrophage polarization. Annu. Rev. Physiol. 2017, 79, 541–566. [Google Scholar] [CrossRef] [PubMed]
- Duffield, J.S.; Forbes, S.J.; Constandinou, C.M.; Clay, S.; Partolina, M.; Vuthoori, S.; Wu, S.; Lang, R.; Iredale, J.P. Selective depletion of macrophages reveals distinct, opposing roles during liver injury and repair. J. Clin. Investig. 2005, 115, 56–65. [Google Scholar] [CrossRef] [Green Version]
- Lucas, T.; Waisman, A.; Ranjan, R.; Roes, J.; Krieg, T.; Müller, W.; Roers, A.; Eming, S.A. Differential roles of macrophages in diverse phases of skin repair. J. Immunol. 2010, 184, 3964–3977. [Google Scholar] [CrossRef] [Green Version]
- Hong, H.; Tian, X.Y. The role of macrophages in vascular repair and regeneration after ischemic injury. Int. J. Mol. Sci. 2020, 21, 6328. [Google Scholar] [CrossRef] [PubMed]
- Gurevich, D.B.; E Severn, C.; Twomey, C.; Greenhough, A.; Cash, J.; Toye, A.M.; Mellor, H.; Martin, P. Live imaging of wound angiogenesis reveals macrophage orchestrated vessel sprouting and regression. EMBO J. 2018, 37. [Google Scholar] [CrossRef] [PubMed]
- Goel, H.L.; Mercurio, A.M. VEGF targets the tumour cell. Nat. Rev. Cancer 2013, 13, 871–882. [Google Scholar] [CrossRef]
- Zajac, E.; Schweighofer, B.; Kupriyanova, T.A.; Juncker-Jensen, A.; Minder, P.; Quigley, J.P.; Deryugina, E.I. Angiogenic capacity of M1- and M2-polarized macrophages is determined by the levels of TIMP-1 complexed with their secreted proMMP-9. Blood 2013, 122, 4054–4067. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Muri, J.; Fitzgerald, G.; Gorski, T.; Gianni-Barrera, R.; Masschelein, E.; D’Hulst, G.; Gilardoni, P.; Turiel, G.; Fan, Z.; et al. Endothelial lactate controls muscle regeneration from ischemia by inducing M2-like macrophage polarization. Cell Metab. 2020, 31, 1136–1153.e7. [Google Scholar] [CrossRef]
- Willenborg, S.; Lucas, T.; Van Loo, G.; Knipper, J.A.; Krieg, T.; Haase, I.; Brachvogel, B.; Hammerschmidt, M.; Nagy, A.; Ferrara, N.; et al. CCR2 recruits an inflammatory macrophage subpopulation critical for angiogenesis in tissue repair. Blood 2012, 120, 613–625. [Google Scholar] [CrossRef] [Green Version]
- Dort, J.; Fabre, P.; Molina, T.; Dumont, N.A. Macrophages are key regulators of stem cells during skeletal muscle regeneration and diseases. Stem Cells Int. 2019, 2019, 4761427. [Google Scholar] [CrossRef]
- Gordon, S.; Martinez, F.O. Alternative activation of macrophages: Mechanism and functions. Immunity 2010, 32, 593–604. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moore, E.M.; West, J.L. Harnessing macrophages for vascularization in tissue engineering. Ann. Biomed. Eng. 2018, 47, 354–365. [Google Scholar] [CrossRef]
- Gordon, S.; Taylor, P.R. Monocyte and macrophage heterogeneity. Nat. Rev. Immunol. 2005, 5, 953–964. [Google Scholar] [CrossRef] [PubMed]
- Pollard, J.W. Trophic macrophages in development and disease. Nat. Rev. Immunol. 2009, 9, 259–270. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, G.; Yang, L.; Yuan, H.; Liu, Y.; He, Y.; Wu, X.; Jin, X. Cold-inducible RNA-binding protein plays a central role in the pathogenesis of abdominal aortic aneurysm in a murine experimental model. Surgery 2016, 159, 1654–1667. [Google Scholar] [CrossRef]
- Zhou, M.; Aziz, M.; Denning, N.-L.; Yen, H.-T.; Ma, G.; Wang, P. Extracellular CIRP induces macrophage endotoxin tolerance through IL-6R–mediated STAT3 activation. JCI Insight 2020, 5. [Google Scholar] [CrossRef]
- Ardi, V.C.; Kupriyanova, T.A.; Deryugina, E.I.; Quigley, J.P. Human neutrophils uniquely release TIMP-free MMP-9 to provide a potent catalytic stimulator of angiogenesis. Proc. Natl. Acad. Sci. USA 2007, 104, 20262–20267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gong, Y.; Koh, D.-R. Neutrophils promote inflammatory angiogenesis via release of preformed VEGF in an in vivo corneal model. Cell Tissue Res. 2009, 339, 437–448. [Google Scholar] [CrossRef]
- Christoffersson, G.; Vågesjö, E.; Vandooren, J.; Lidén, M.; Massena, S.; Reinert, R.B.; Brissova, M.; Powers, A.C.; Opdenakker, G.; Phillipson, M. VEGF-A recruits a proangiogenic MMP-9–delivering neutrophil subset that induces angiogenesis in transplanted hypoxic tissue. Blood 2012, 120, 4653–4662. [Google Scholar] [CrossRef]
- Kolaczkowska, E.; Kubes, P. Neutrophil recruitment and function in health and inflammation. Nat. Rev. Immunol. 2013, 13, 159–175. [Google Scholar] [CrossRef] [PubMed]
- Pizza, F.X.; Peterson, J.M.; Baas, J.H.; Koh, T.J. Neutrophils contribute to muscle injury and impair its resolution after lengthening contractions in mice. J. Physiol. 2005, 562, 899–913. [Google Scholar] [CrossRef]
- Toumi, H.; F’Guyer, S.; Best, T.M. The role of neutrophils in injury and repair following muscle stretch. J. Anat. 2006, 208, 459–470. [Google Scholar] [CrossRef]
- Teixeira, C.D.F.; Zamunér, S.R.; Zuliani, J.P.; Fernandes, C.M.; Höfling, M.A.C.; Fernandes, I.; Chaves, F.; Gutiérrez, J.M. Neutrophils do not contribute to local tissue damage, but play a key role in skeletal muscle regeneration, in mice injected with Bothrops aspersnake venom. Muscle Nerve 2003, 28, 449–459. [Google Scholar] [CrossRef]
- De Oliveira, S.; Rosowski, E.E.; Huttenlocher, A. Neutrophil migration in infection and wound repair: Going forward in reverse. Nat. Rev. Immunol. 2016, 16, 378–391. [Google Scholar] [CrossRef] [PubMed]
- Jin, H.; Aziz, M.; Ode, Y.; Wang, P. CIRP induces neutrophil reverse transendothelial migration in sepsis. Shock 2019, 51, 548–556. [Google Scholar] [CrossRef] [PubMed]
- Slaba, I.; Wang, J.; Kolaczkowska, E.; McDonald, B.; Lee, W.-Y.; Kubes, P. Imaging the dynamic platelet-neutrophil response in sterile liver injury and repair in mice. Hepatology 2015, 62, 1593–1605. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, K.; Murao, A.; Arif, A.; Takizawa, S.; Jin, H.; Jiang, J.; Aziz, M.; Wang, P. Inhibition of efferocytosis by extracellular CIRP–induced neutrophil extracellular traps. J. Immunol. 2021, 206, 797–806. [Google Scholar] [CrossRef] [PubMed]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kübler, M.; Beck, S.; Fischer, S.; Götz, P.; Kumaraswami, K.; Ishikawa-Ankerhold, H.; Lasch, M.; Deindl, E. Absence of Cold-Inducible RNA-Binding Protein (CIRP) Promotes Angiogenesis and Regeneration of Ischemic Tissue by Inducing M2-Like Macrophage Polarization. Biomedicines 2021, 9, 395. https://doi.org/10.3390/biomedicines9040395
Kübler M, Beck S, Fischer S, Götz P, Kumaraswami K, Ishikawa-Ankerhold H, Lasch M, Deindl E. Absence of Cold-Inducible RNA-Binding Protein (CIRP) Promotes Angiogenesis and Regeneration of Ischemic Tissue by Inducing M2-Like Macrophage Polarization. Biomedicines. 2021; 9(4):395. https://doi.org/10.3390/biomedicines9040395
Chicago/Turabian StyleKübler, Matthias, Sebastian Beck, Silvia Fischer, Philipp Götz, Konda Kumaraswami, Hellen Ishikawa-Ankerhold, Manuel Lasch, and Elisabeth Deindl. 2021. "Absence of Cold-Inducible RNA-Binding Protein (CIRP) Promotes Angiogenesis and Regeneration of Ischemic Tissue by Inducing M2-Like Macrophage Polarization" Biomedicines 9, no. 4: 395. https://doi.org/10.3390/biomedicines9040395