
Anomalous Angiogenesis in Retina



Abstract
1. Introduction
2. Materials and Methods
2.1. Cellular Potts Model
2.2. Continuum Fields at the Extracellular Scale
Durotaxis
2.3. Signaling Processes and Cell Dynamics
2.4. Retinal Configuration and Onset of Angiogenesis
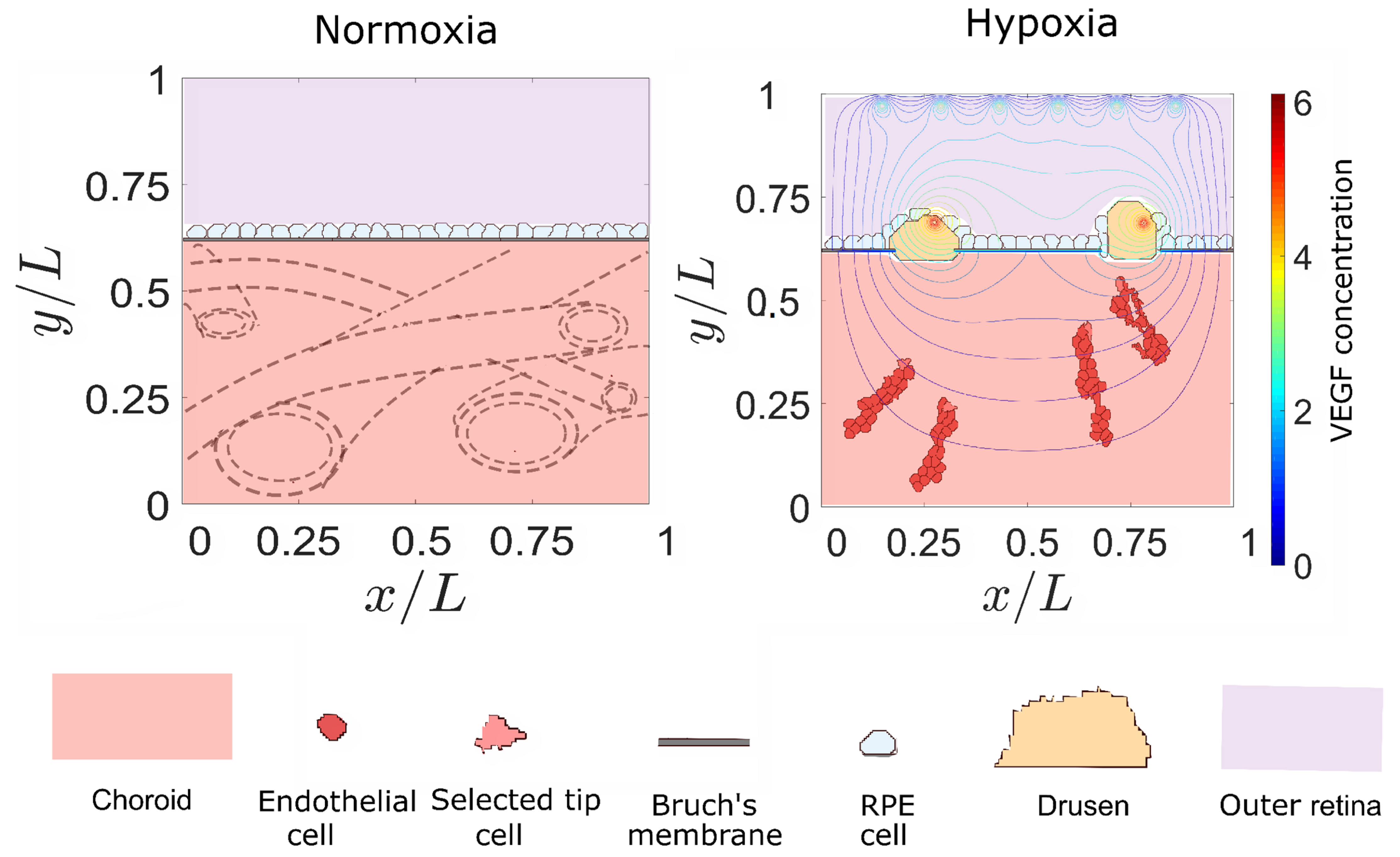
3. Results
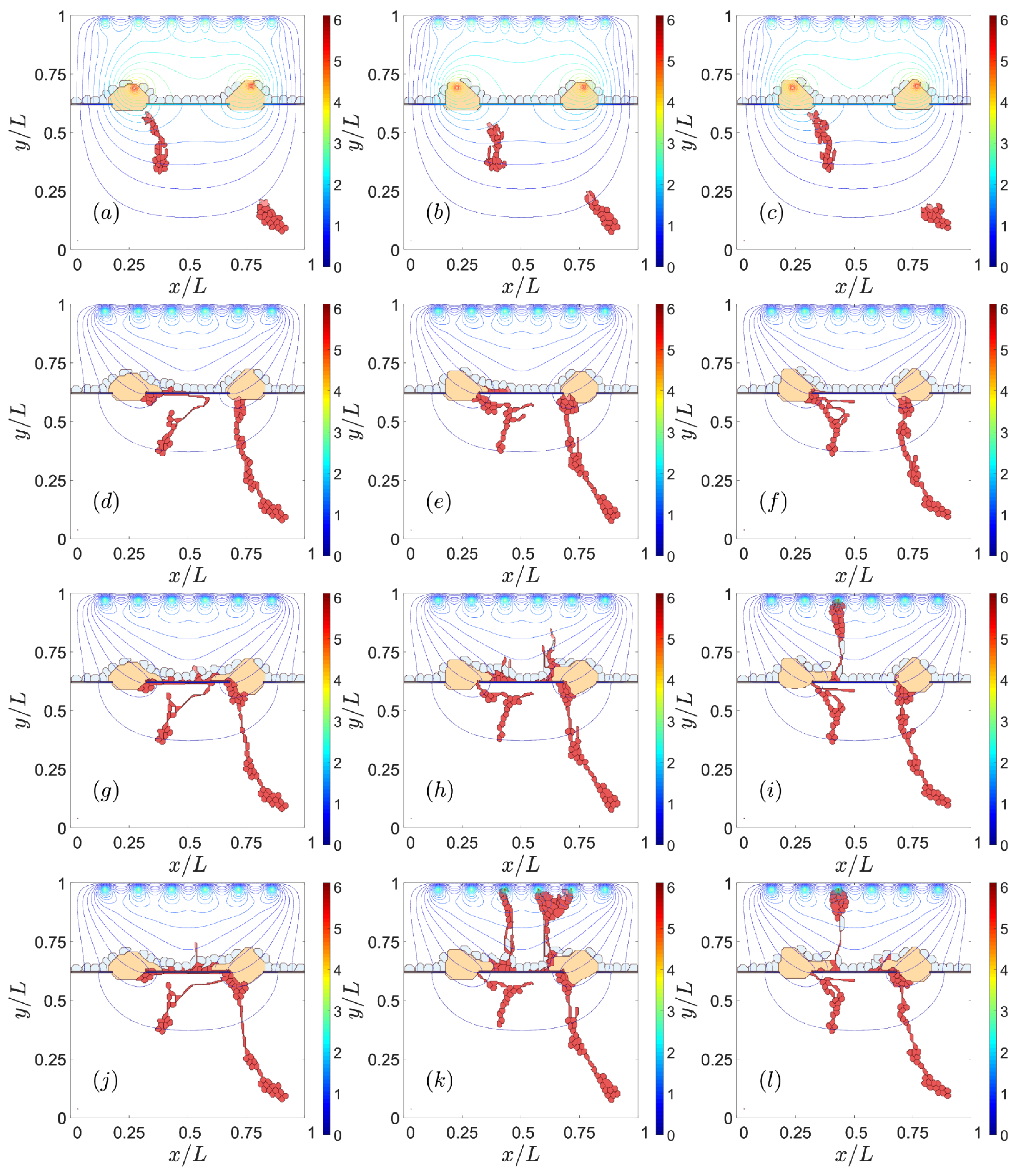
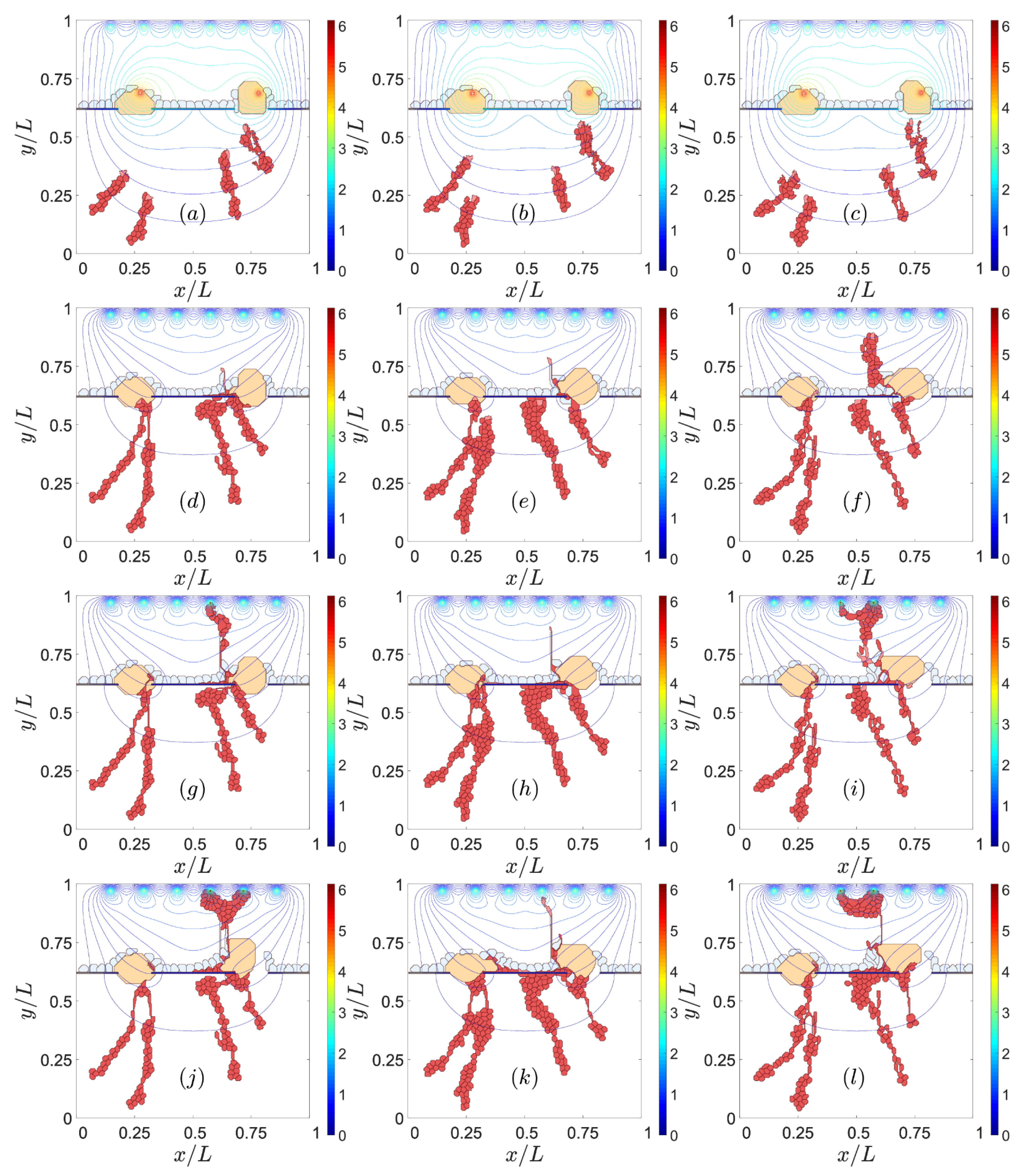
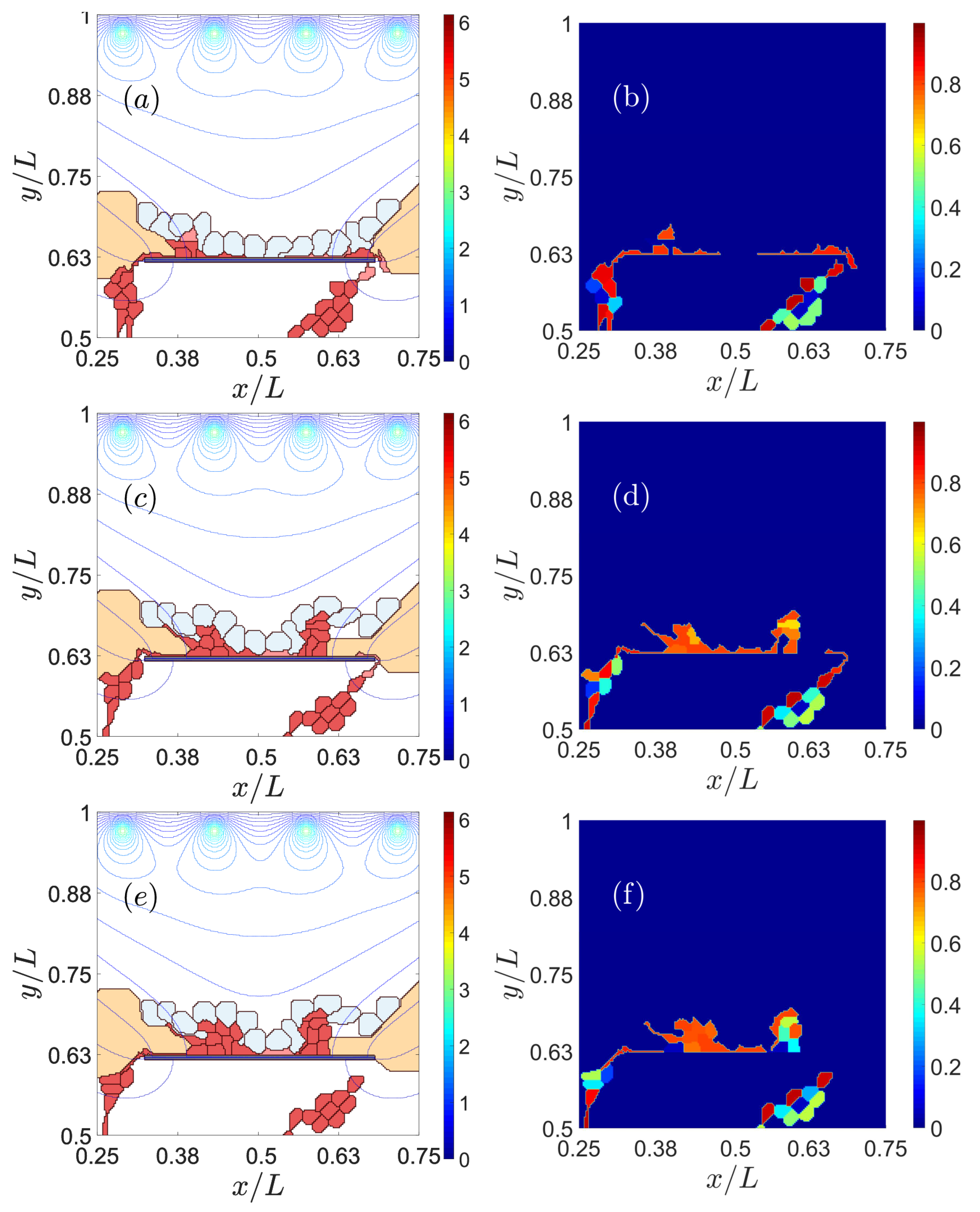
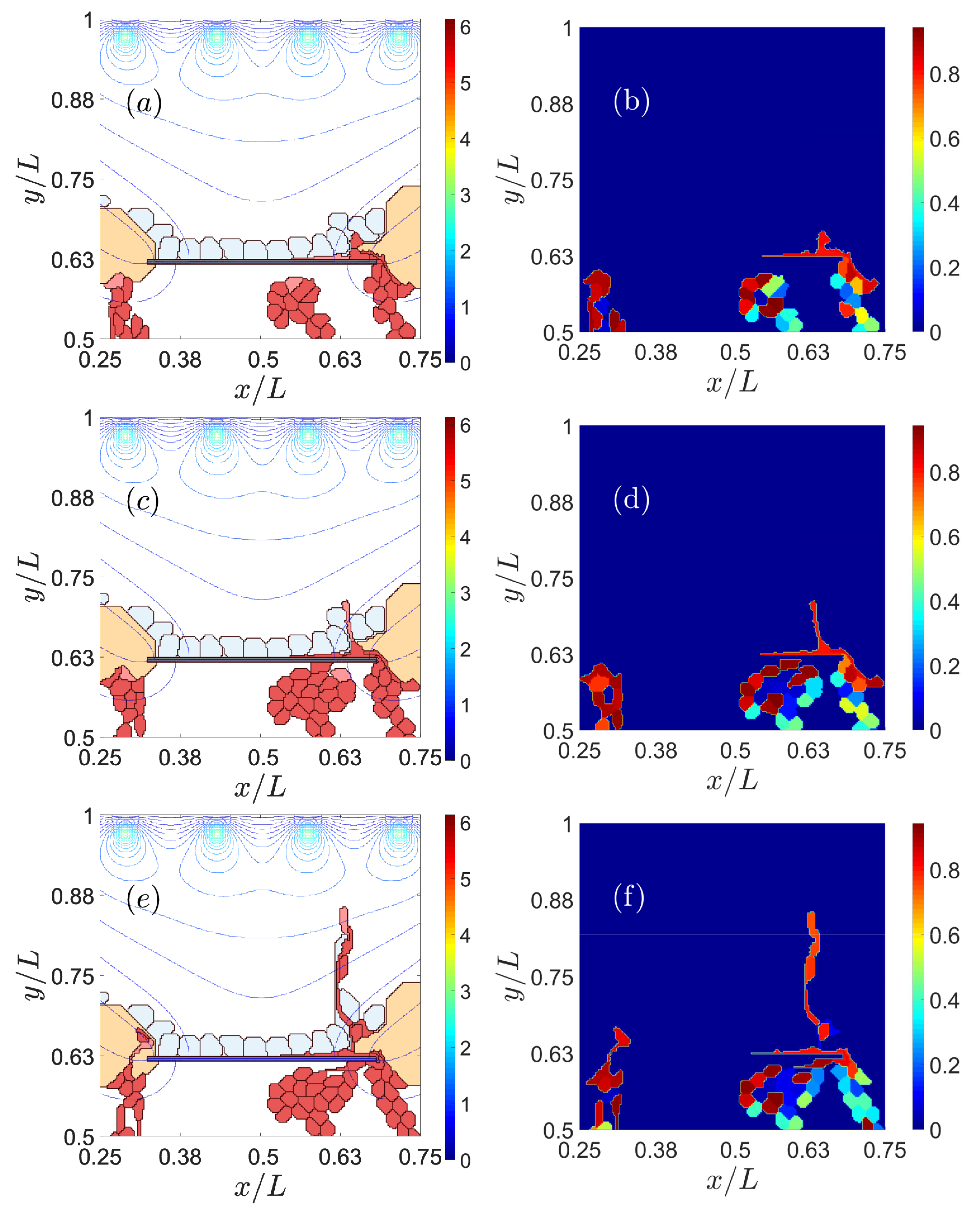
3.1. Impaired Adhesion
3.1.1. Adhesion between RPE and BM
3.1.2. RPE–RPE and EC–EC Adhesion
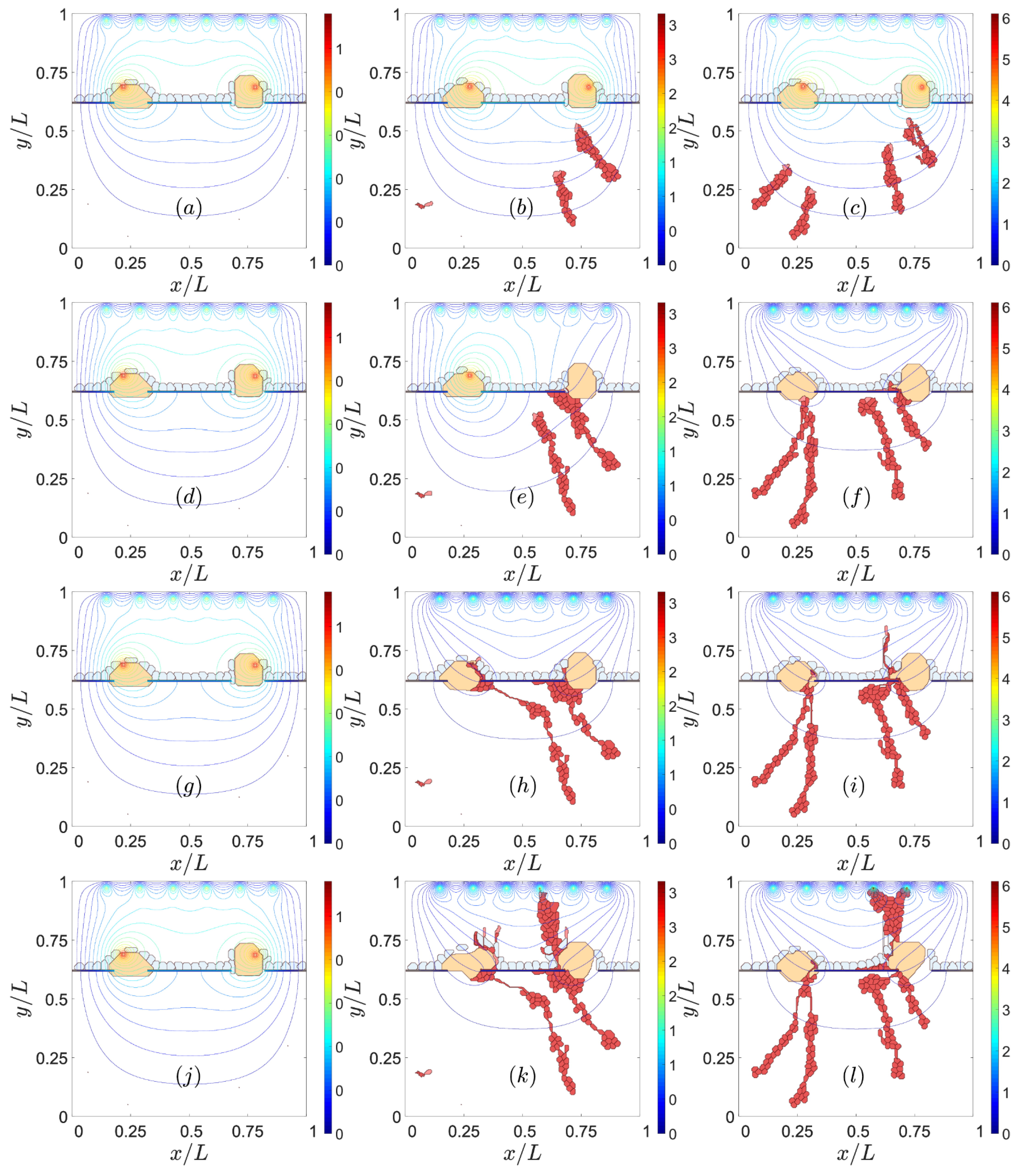
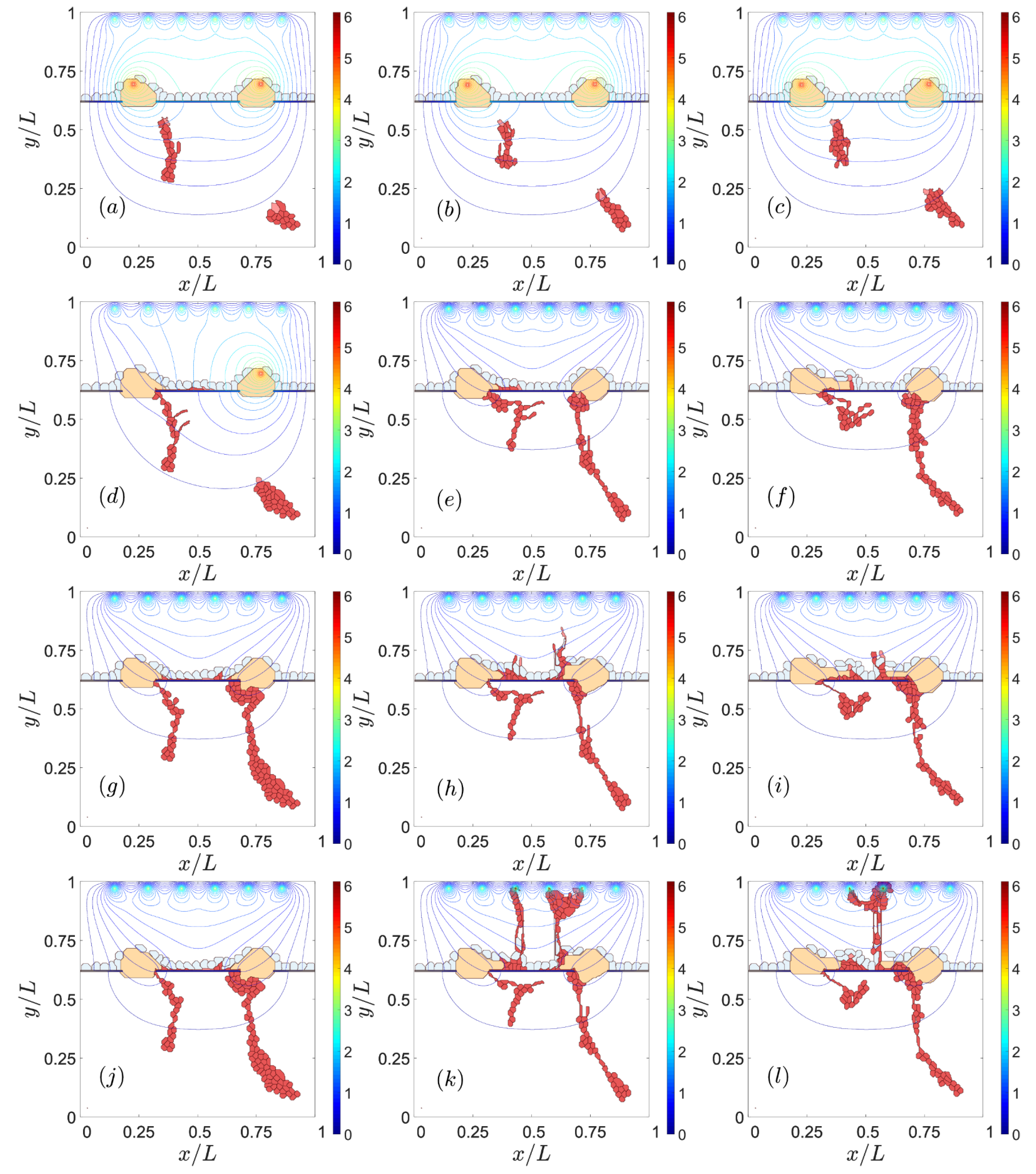
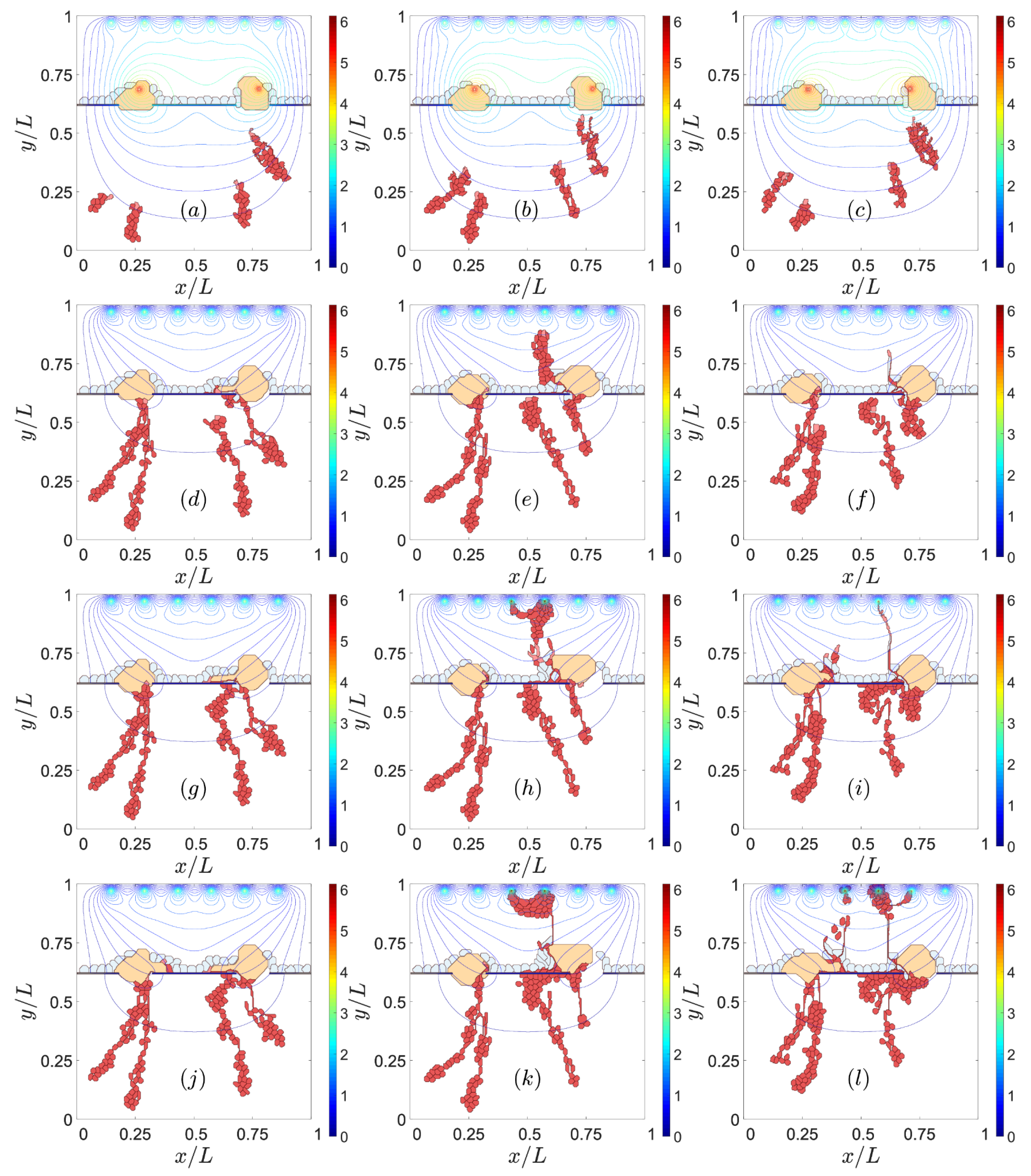
3.2. Sources of VEGF
3.3. Notch Signaling
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| 2D | Two Dimensional |
| AMD | Age-related Macular Degeneration |
| BM | Bruch’s membrane |
| CNV | Choroid Neo Vascularization |
| CPM | Cellular Potts Model |
| EC | Endothelial Cell |
| ECM | Extra Cellular Matrix |
| EMT | Epithelial to Mesenchymal Transition |
| MC | Monte Carlo |
| MCTS | Monte Carlo Time Step |
| RPE | Retinal Pigmentation Epithelium |
| VEGF | Vessel Endothelial Growth Factor |
References
- Wong, W.L.; Su, X.; Li, X.; Cheung, C.M.G.; Klein, R.; Cheng, C.-Y.; Wong, T.Y. Global prevalence of age-related macular degeneration and disease burden projection for 2020 and 2040: A systematic review and meta-analysis. Lancet Glob. Health 2014, 2, 106–116. [Google Scholar] [CrossRef]
- Jonas, J.B. Comment. Global prevalence of age-related macular degeneration. Lancet Glob. Health 2014, 2, e65–e66. [Google Scholar] [CrossRef]
- Jager, R.D.; Mieler, W.F.; Miller, J.W. Age-related macular degeneration. N. Engl. J. Med. 2008, 358, 2606–2617. [Google Scholar] [CrossRef]
- Nivison-Smith, L.; Milston, R.; Madigan, M.; Kalloniatis, M. Age-Related Macular Degeneration: Linking Clinical Presentation to Pathology. Optom. Vision Sci. 2014, 91, 832–848. [Google Scholar] [CrossRef] [PubMed]
- Manjunath, V.; Taha, M.; Fujimoto, J.G.; Duker, J.S. Choroidal Thickness in Normal Eyes Measured Using Cirrus-HD Optical Coherence Tomography. Am. J. Ophthalmol. 2010, 150, 325–329.e1. [Google Scholar] [CrossRef] [PubMed]
- Coscas, G.; Lupidi, M.; Coscas, F. Atlas of OCT: Angiography in AMD. Comparison with Multimodal Imaging; Societé Francaise de Retine, L’Europeenne d’Editions: Paris, France, 2015. [Google Scholar]
- Laforest, T.; Künzi, M.; Kowalczuk, L.; Carpentras, D.; Behar-Cohen, F.; Moser, C. Transscleral optical phase imaging of the human retina. Nat. Photonics 2020, 14, 439–445. [Google Scholar] [CrossRef]
- Gariano, R.F.; Gardner, T.W. Retinal angiogenesis in development and disease. Nature 2005, 438, 960–966. [Google Scholar] [CrossRef]
- Fruttiger, M. Development of the retinal vasculature. Angiogenesis 2007, 10, 77–88. [Google Scholar] [CrossRef] [PubMed]
- Scott, A.; Powner, M.B.; Gandhi, P.; Clarkin, C.; Gutmann, D.H.; Johnson, R.S.; Ferrara, N.; Fruttiger, M. Astrocyte-Derived Vascular Endothelial Growth Factor Stabilizes Vessels in the Developing Retinal Vasculature. PLoS ONE 2010, 5, e11863. [Google Scholar] [CrossRef]
- Selvam, S.; Kumar, T.; Fruttiger, M. Retinal vasculature development in health and disease. Prog. Retin. Eye Res. 2018, 63, 1–19. [Google Scholar] [CrossRef]
- Carmeliet, P.F. Angiogenesis in life, disease and medicine. Nature 2005, 438, 932–936. [Google Scholar] [CrossRef]
- Carmeliet, P.; Tessier-Lavigne, M. Common mechanisms of nerve and blood vessel wiring. Nature 2005, 436, 193–200. [Google Scholar] [CrossRef]
- Carmeliet, P.; Jain, R.K. Molecular mechanisms and clinical applications of angiogenesis. Nature 2011, 473, 298–307. [Google Scholar] [CrossRef] [PubMed]
- Tonnesen, M.G.; Feng, X.; Clark, R.A. Angiogenesis in wound healing. J. Investig. Dermatol. Symp. Proc. 2000, 5, 40–46. [Google Scholar] [CrossRef]
- Figg, W.D.; Folkman, J. Angiogenesis. An Integrative Approach from Science to Medicine; Springer: Berlin/Heidelberg, Germany, 2008. [Google Scholar]
- Gerhardt, H.; Golding, M.; Fruttiger, M.; Ruhrberg, C.; Lundkvist, A.; Abramsson, A.; Jeltsch, M.; Mitchell, C.; Alitalo, K.; Shima, D.; et al. VEGF guides angiogenic sprouting utilizing endothelial tip cell filopodia. J. Cell Biol. 2003, 161, 1163–1177. [Google Scholar] [CrossRef] [PubMed]
- Hellström, M.; Phng, L.K.; Hofmann, J.J.; Wallgard, E.; Coultas, L.; Lindblom, P.; Alva, J.; Nilsson, A.-K.; Karlsson, L.; Gaiano, N.; et al. Dll4 signalling through Notch1 regulates formation of tip cells during angiogenesis. Nature 2007, 445, 776–780. [Google Scholar] [CrossRef]
- Jolly, M.K.; Boareto, M.; Lu, M.; Onuchic, J.N.; Clementi, C.; Ben-Jacob, E. Operating principles of Notch-Delta-Jagged module of cell-cell communication. New J. Phys. 2015, 17, 055021. [Google Scholar] [CrossRef]
- Page, D.J.; Thuret, R.; Venkatraman, L.; Takahashi, T.; Bentley, K.; Herbert, S.P. Positive Feedback Defines the Timing, Magnitude, and Robustness of Angiogenesis. Cell Rep. 2019, 27, 3139–3151. [Google Scholar] [CrossRef] [PubMed]
- Vega, R.; Carretero, M.; Travasso, R.D.M.; Bonilla, L.L. Notch signaling and taxis mechanims regulate earlystage angiogenesis: A mathematical and computational model. PLoS Comput. Biol. 2020, 16, e1006919. [Google Scholar] [CrossRef] [PubMed]
- Gebala, V.; Collins, R.; Geudens, I.; Phng, L.-K.; Gerhardt, H. Blood flow drives lumen formation by inverse membrane blebbing during angiogenesis in vivo. Nat. Cell Biol. 2016, 18, 443–450. [Google Scholar] [CrossRef]
- Franco, C.A.; Jones, M.L.; Bernabeu, M.O.; Geudens, I.; Mathivet, T.; Rosa, A.; Lopes, F.M.; Lima, A.P.; Ragab, A.; Collins, R.T.; et al. Dynamic endothelial cell rearrangements drive developmental vessel regression. PLoS Biol. 2015, 13, e1002125. [Google Scholar]
- Szymborska, A.; Gerhardt, H. Hold me, but not too tight–endothelial cell-cell junctions in angiogenesis. Cold Spring Harb. Perspect. Biol. 2018, 10, a029223. [Google Scholar] [CrossRef]
- Dufraine, J.; Funahashi, Y.; Kitajewski, J. Notch signaling regulates tumor angiogenesis by diverse mechanisms. Oncogene 2008, 27, 5132–5137. [Google Scholar] [CrossRef]
- Booij, J.C.; Baas, D.C.; Beisekeeva, J.; Gorgels, T.G.M.F.; Bergen, A.A.B. The dynamic nature of Bruch’s membrane. Prog. Ret. Eye Res. 2010, 29, 1–18. [Google Scholar] [CrossRef]
- Mammadzada, P.; Corredoira, P.M.; André, H. The role of hypoxia-inducible factors in neovascular age-related macular degeneration: A gene therapy perspective. Cell. Mol. Life Sci. 2020, 77, 819–833. [Google Scholar] [CrossRef] [PubMed]
- Gehrs, K.M.; Anderson, D.H.; Johnson, L.V.; Hageman, G.S. Age-related macular degeneration–emerging pathogenetic and therapeutic concepts. Ann. Med. 2009, 38, 450–471. [Google Scholar] [CrossRef]
- Radeke, M.J.; Radeke, C.M.; Shih, Y.-H.; Hu, J.; Bok, D.; Johnson, L.V.; Coffey, P.J. Restoration of mesenchymal retinal pigmented epithelial cells by TGFβ pathway inhibitors: Implications for age-related macular degeneration. Genome Med. 2015, 7, 58. [Google Scholar] [CrossRef]
- Shu, D.Y.; Butcher, E.; Saint-Geniez, M. EMT and EndMT: Emerging Roles in Age-Related Macular Degeneration. Int. J. Mol. Sci. 2020, 21, 4271. [Google Scholar] [CrossRef]
- Tamiya, S.; Liu, L.; Kaplan, H.J. Epithelial-mesenchymal transition and proliferation of retinal pigment epithelial cells initiated upon loss of cell-cell contact. Investig. Ophthalmol. Vis. Sci. 2010, 51, 2755–2763. [Google Scholar] [CrossRef]
- Roberts, P.A.; Gaffney, E.A.; Luthert, P.J.; Foss, A.J.E.; Byrne, H.M. Mathematical and Computational Models of the Retina in Health, Development and Disease. Prog. Retin. Eye Res. 2016, 53, 48–69. [Google Scholar] [CrossRef]
- Flower, R.W.; von Kerczek, C.; Zhu, L.; Eggleton, E.C.; Topoleski, L.D.T. Theoretical Investigation of the Role of Choriocapillaris Blood Flow in Treatment of Subfoveal Choroidal Neovascularization Associated WithAge-related Macular Degeneration. Am. J. Opthtalmol. 2001, 132, 85–93. [Google Scholar] [CrossRef]
- Davies, A.E.; Williams, R.L.; Lugano, G.; Pop, S.R.; Kearns, V.R. In vitro and computational modelling of drug delivery across the outer blood-retinal barrier. Interface Focus 2020, 10, 20190132. [Google Scholar] [CrossRef]
- Shirinifard, A.; Glazier, J.A.; Swat, M.; Gens, J.S.; Family, F.; Jiang, Y.; Grossniklaus, H.E. Adhesion Failures Determine the Pattern of Choroidal Neovascularization in the Eye: A Computer Simulation Study. PLoS Comput. Biol. 2012, 8, e1002440. [Google Scholar] [CrossRef] [PubMed]
- Graner, F.; Glazier, J.A. Simulation of Biological Cell Sorting Using a Two-Dimensional Extended Potts Model. Phys. Rev. Lett. 1992, 69, 2013–2016. [Google Scholar] [CrossRef]
- Bauer, A.L.; Jackson, T.L.; Jiang, Y. A Cell-Based Model Exhibiting Branching and Anastomosis during Tumor-Induced Angiogenesis. Biophys. J. 2007, 92, 3105–3121. [Google Scholar] [CrossRef] [PubMed]
- Van Oers, R.F.M.; Rens, E.G.; La Valley, D.J.; Reinhart-King, C.A.; Merks, R.M.H. Mechanical cell-matrix feedback explains pairwise and collective endothelial cell behavior in vitro. PLoS Comput. Biol. 2014, 10, el003774. [Google Scholar] [CrossRef] [PubMed]
- Boareto, M.; Jolly, M.K. Ben-Jacob, E.; Onuchic, J.N. Jagged mediates differences in normal and tumor angiogenesis by affecting tip-stalk fate decision. Proc. Natl. Acad. Sci. USA 2015, 112, E3836–E3844. [Google Scholar] [CrossRef] [PubMed]
- Arima, S.; Nishiyama, K.; Ko, T.; Arima, Y.; Hakozaki, Y.; Sugihara, K.; Koseki, K.; Uchijima, H.; Kurihara, Y.; Kurihara, H. Angiogenic morphogenesis driven by dynamic and heterogeneous collective endothelial cell movement. Development 2011, 138, 4763–4776. [Google Scholar] [CrossRef]
- Sugihara, K.; Nishiyama, K.; Fukuhara, S.; Uemura, A.; Arima, S.; Kobayashi, R.; Köhn-Luque, A.; Mochizuki, N.; Suda, T.; Ogawa, H.; et al. Autonomy and Non-autonomy of Angiogenic Cell Movements Revealed by Experiment-Driven Mathematical Modeling. Cell Rep. 2015, 13, 1814–1827. [Google Scholar] [CrossRef]
- Mantzaris, N.V.; Webb, S.; Othmer, H.G. Mathematical modeling of tumor-induced angiogenesis. J. Math. Biol. 2004, 49, 111–187. [Google Scholar] [CrossRef]
- Bai, H.X.; Mao, Y.; Shen, L.; Xu, X.L.; Gao, F.; Zhang, Z.B.; Li, B.; Jonas, J.B. Bruch’s membrane thickness in relationship to axial length. PLoS ONE 2017, 12, e0182080. [Google Scholar] [CrossRef]
- Friberg, T.R.; Bilonick, R.A.; Peter Brennen, P. Is Drusen Area Really So Important? An Assessment of Risk of Conversion to Neovascular AMD Based on Computerized Measurements of Drusen. Investig. Opthalmology Vis. Sci. 2012, 53, 1742–1751. [Google Scholar] [CrossRef] [PubMed]
- Poh, S.; Tham, Y.; Chee, M.L.; Dai, W.; Majithia, S.; Soh, Z.D.; Fenwick, E.K.; Tao, Y.; Thakur, S.; Rim, T.H.; et al. Association between Macular Thickness Profiles and Visual Function in Healthy Eyes: The Singapore Epidemiology of Eye Diseases (SEED) Study. Sci. Rep. 2020, 10, 6142. [Google Scholar] [CrossRef] [PubMed]
- Gullapalli, V.K.; Sugino, I.K.; Van Patten, Y.; Shah, S.; Zarbin, M.A. Impaired RPE survival on aged submacular human Bruch’s membrane. Exp. Eye Res. 2005, 80, 235–248. [Google Scholar] [CrossRef] [PubMed]
- Imamura, Y.; Noda, S.; Hashizume, K.; Shinoda, K.; Yamaguchi, M.; Uchiyama, S.; Shimizu, T.; Mizushima, Y.; Shirasawa, T.; Tsubota, K. Drusen, choroidal neovascularization, and retinal pigment epithelium dysfunction in SOD1-deficient mice: A model of age-related macular degeneration. Procs. Natl. Acad. Sci. USA 2005, 103, 11282–11287. [Google Scholar] [CrossRef] [PubMed]
- Simó, R.; Villarroel, M.; Corraliza, L.; Hernández, C.; Garcia-Ramírez, M. The retinal pigment epithelium: Something more than a constituent of the blood-retinal barrier–implications for the pathogenesis of diabetic retinopathy. J. Biomed. Biotechnol. 2010, 2010, 190724. [Google Scholar] [CrossRef]
- Ramos, J.R.D.; Travasso, R. Carvalho, J. Capillary network formation from dispersed endothelial cells: Influence of cell traction, cell adhesion, and extracellular matrix rigidity. Phys. Rev. E 2018, 97, 012408. [Google Scholar] [CrossRef]
- Bhutto, I.A.; McLeod, D.S.; Hasegawa, T.; Kim, S.Y.; Merges, C.; Tong, P.; Lutty, G.A. Pigment epithelium-derived factor (PEDF) and vascular endothelial growth factor (VEGF) in aged human choroid and eyes with age-related macular degeneration. Exp. Eye Res. 2006, 82, 99–110. [Google Scholar] [CrossRef]
- Fogli, S.; Del Re, M.; Rofi, E.; Posarelli, C.; Figus, M.; Danesi, R. Clinical pharmacology of intravitreal anti-VEGF drugs. Eye 2018, 32, 1010–1020. [Google Scholar] [CrossRef]
- West, J.W.; Sagert, J.G.; Bessette, P.H.; Lowman, H.B.; Stagliano, N.E.; Vasiljeva, O.; Menendez, E.-E.M. Anti-Jagged 1/Jagged 2 Cross-Reactive Antibodies, Activatable Anti-Jagged Antibodies and Methods of Use Thereof. U.S. Patent 10,301,380, 28 May 2019. Available online: https://patents.google.com/patent/US9127053B2/en (accessed on 17 December 2020).
- Sierra, R.A.; Trillo-Tinoco, J.; Mohamed, E.; Yu, L.; Achyut, B.R.; Arbab, A.; Bradford, J.W.; Osborne, B.A.; Miele, L.; Rodriguez, P.C. Anti-Jagged immunotherapy inhibits MDSCs and overcomes tumor-induced tolerance. Cancer Res. 2017, 77, 5628–5638. [Google Scholar] [CrossRef]
- Zhou, M.; Yu, Q.; Huang, K.; Mahov, S.; Eslami, A.; Maier, M.; Lohmann, C.P.; Navab, N.; Zapp, D.; Knoll, A.; et al. Towards Robotic-assisted Subretinal Injection: A Hybrid Parallel-Serial Robot System Design and Preliminary Evaluation. IEEE Trans. Ind. Electron. 2020, 67, 6617–6628. [Google Scholar] [CrossRef]
- Parolini, B.; Nava, U.; Palmieri, M.; Lucente, A.; Finzi, A.; Frisina, R. RPE and Choroid Transplantation in Macular Degeneration. In Macular Surgery; Chang, A., Mieler, W.F., Ohji, M., Eds.; Springer Nature: Cham, Switzerland, 2020; pp. 401–422. [Google Scholar]
- Cheng, D.L.; Greenberg, P.B.; Borton, D.A. Advances in retinal prosthetic research: A systematic review of engineering and clinical characteristics of current prosthetic initiatives. Curr. Eye Res. 2017, 42, 334–347. [Google Scholar] [CrossRef]
- Maya-Vetencourt, J.F.; Ghezzi, D.; Antognazza, M.R.; Colombo, E.; Mete, M.; Feyen, P.; Desii, A.; Buschiazzo, A.; Di Paolo, M.; Di Marco, S.; et al. A fully organic retinal prosthesis restores vision in a rat model of degenerative blindness. Nat. Mater. 2017, 16, 681–689. [Google Scholar] [CrossRef] [PubMed]
- Tang, J.; Qin, N.; Chong, Y.; Diao, Y.; Yiliguma; Wang, Z.; Xue, T.; Jiang, M.; Zhang, J.; Zheng, G. Nanowire arrays restore vision in blind mice. Nat. Commun. 2018, 9, 786. [Google Scholar] [CrossRef] [PubMed]
- Gagliardi, G.; Ben M’Barek, K.; Goureau, O. Photoreceptor cell replacement in macular degeneration and retinitis pigmentosa: A pluripotent stem cell-based approach. Prog. Ret. Eye Res. 2019, 71, 1–25. [Google Scholar] [CrossRef]
- Singh, M.S.; Park, S.S.; Albini, T.A.; Canto-Soler, M.V.; Klassen, H.; MacLaren, R.E.; Takahashi, M.; Nagiel, A.; Schwartz, S.D.; Bharti, K. Retinal stem cell transplantation: Balancing safety and potential. Prog. Retin. Eye Res. 2020, 75, 100779. [Google Scholar] [CrossRef] [PubMed]
- Stieger, K.; Lorenz, B. RPE and gene therapy. In Retinal Pigment Epithelium in Health and Disease; Klettner, A.K., Dithmar, S., Eds.; Springer Nature: Cham, Switzerland, 2020; pp. 265–279. [Google Scholar]
- Jemni-Damer, N.; Guedan-Duran, A.; Cichy, J.; Lozano-Picazo, P.; Gonzalez-Nieto, D.; Perez-Rigueiro, J.; Rojo, F.; Guinea, G.V.; Virtuoso, A.; Cirillo, G.; et al. First steps for the development of silk fibroin-based 3D biohybrid retina for age-related macular degeneration (AMD). J. Neural Eng. 2020, 17, 055003. [Google Scholar] [CrossRef]
- Bonilla, L.L.; Carpio, A.; Trenado, C. Tracking collective cell motion by topological data analysis. PLoS Comput. Biol. 2020, 16, e1008407. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Param. | AEC | PEC | LEC | ARPE | PRPE | Adruse | Pdruse |
|---|---|---|---|---|---|---|---|
| Value | 78 µm2 | 50 µm | 60 µm | 169 µm2 | 52 µm | 2827 µm2 | 188 µm |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vega, R.; Carretero, M.; Bonilla, L.L. Anomalous Angiogenesis in Retina. Biomedicines 2021, 9, 224. https://doi.org/10.3390/biomedicines9020224
Vega R, Carretero M, Bonilla LL. Anomalous Angiogenesis in Retina. Biomedicines. 2021; 9(2):224. https://doi.org/10.3390/biomedicines9020224
Chicago/Turabian StyleVega, Rocío, Manuel Carretero, and Luis L Bonilla. 2021. "Anomalous Angiogenesis in Retina" Biomedicines 9, no. 2: 224. https://doi.org/10.3390/biomedicines9020224
APA StyleVega, R., Carretero, M., & Bonilla, L. L. (2021). Anomalous Angiogenesis in Retina. Biomedicines, 9(2), 224. https://doi.org/10.3390/biomedicines9020224