Endoplasmic Reticulum Stress in Macrophages: The Vicious Circle of Lipid Accumulation and Pro-Inflammatory Response

 ,
,  ,
,  , and
, and
Abstract
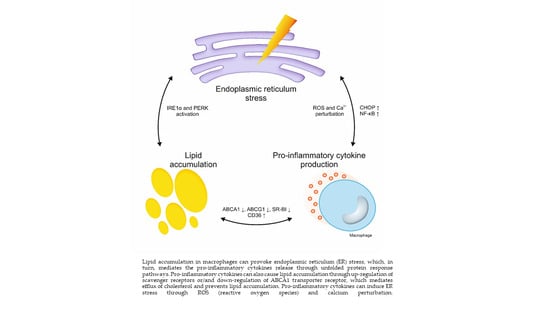
:
1. Introduction
2. The Role of ER Stress in the Formation of Foam Cells
3. The Role of ER Stress in Apoptosis
4. ER Stress and Inflammation
5. Relationship between Cholesterol Accumulation, ER Stress, and Pro-Inflammatory Response
6. Conclusions
Funding
Conflicts of Interest
References
- Kobiyama, K.; Ley, K. Atherosclerosis. Circ. Res. 2018, 123, 1118–1120. [Google Scholar] [CrossRef] [PubMed]
- Koelwyn, G.J.; Corr, E.M.; Erbay, E.; Moore, K.J. Regulation of macrophage immunometabolism in atherosclerosis. Nat. Immunol. 2018, 19, 526–537. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hotamisligil, G.S. Endoplasmic reticulum stress and atherosclerosis. Nat. Med. 2010, 16, 396–399. [Google Scholar] [CrossRef] [Green Version]
- Scull, C.M.; Tabas, I. Mechanisms of ER Stress-Induced Apoptosis in Atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 2792–2797. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Linton, M.F.; Babaev, V.R.; Huang, J.; Linton, E.F.; Tao, H.; Yancey, P.G. Macrophage Apoptosis and Efferocytosis in the Pathogenesis of Atherosclerosis. Circ. J. 2016, 80, 2259–2268. [Google Scholar] [CrossRef] [Green Version]
- Han, J.; Kaufman, R.J. The role of ER stress in lipid metabolism and lipotoxicity. J. Lipid Res. 2016, 57, 1329–1338. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Guo, X.; Ge, Q.; Zhao, Y.; Mu, H.; Zhang, J. ER Stress Activates the NLRP3 Inflammasome: A Novel Mechanism of Atherosclerosis. Oxidative Med. Cell. Longev. 2019, 2019. [Google Scholar] [CrossRef]
- Wu, H.; Carvalho, P.; Voeltz, G.K. Here, there, and everywhere: The importance of ER membrane contact sites. Science 2018, 361, eaan5835. [Google Scholar] [CrossRef] [Green Version]
- Oakes, S.A.; Papa, F.R. The Role of Endoplasmic Reticulum Stress in Human Pathology. Annu. Rev. Pathol. Mech. Dis. 2015, 10, 173–194. [Google Scholar] [CrossRef] [Green Version]
- Cominacini, L.; Garbin, U.; Mozzini, C.; Stranieri, C.; Pasini, A.; Solani, E.; Tinelli, I.; Pasini, A. The Atherosclerotic Plaque Vulnerability: Focus on the Oxidative and Endoplasmic Reticulum Stress in Orchestrating the Macrophage Apoptosis in the Formation of the Necrotic Core. Curr. Med. Chem. 2015, 22, 1565–1572. [Google Scholar] [CrossRef]
- Ivanova, E.; Orekhov, A.N. The Role of Endoplasmic Reticulum Stress and Unfolded Protein Response in Atherosclerosis. Int. J. Mol. Sci. 2016, 17, 193. [Google Scholar] [CrossRef] [PubMed]
- Tabas, I.; Ron, D. Integrating the mechanisms of apoptosis induced by endoplasmic reticulum stress. Nat. Cell Biol. 2011, 13, 184–190. [Google Scholar] [CrossRef] [PubMed]
- Hetz, C. The unfolded protein response: Controlling cell fate decisions under ER stress and beyond. Nat. Rev. Mol. Cell Biol. 2012, 13, 89–102. [Google Scholar] [CrossRef] [PubMed]
- Hetz, C.; Papa, F.R. The Unfolded Protein Response and Cell Fate Control. Mol. Cell 2018, 69, 169–181. [Google Scholar] [CrossRef] [Green Version]
- Nishitoh, H. CHOP is a multifunctional transcription factor in the ER stress response. J. Biochem. 2012, 151, 217–219. [Google Scholar] [CrossRef] [Green Version]
- Tabas, I. The Role of Endoplasmic Reticulum Stress in the Progression of Atherosclerosis. Circ. Res. 2010, 107, 839–850. [Google Scholar] [CrossRef]
- Chen, F.; Jin, J.; Hu, J.; Wang, Y.; Ma, Z.; Zhang, J. Endoplasmic Reticulum Stress Cooperates in Silica Nanoparticles-Induced Macrophage Apoptosis via Activation of CHOP-Mediated Apoptotic Signaling Pathway. Int. J. Mol. Sci. 2019, 20, 5846. [Google Scholar] [CrossRef] [Green Version]
- Hu, H.; Tian, M.; Ding, C.; Yu, S. The C/EBP Homologous Protein (CHOP) Transcription Factor Functions in Endoplasmic Reticulum Stress-Induced Apoptosis and Microbial Infection. Front. Immunol. 2019, 9. [Google Scholar] [CrossRef] [Green Version]
- Lee, A.-H.; Iwakoshi, N.N.; Glimcher, L.H. XBP-1 Regulates a Subset of Endoplasmic Reticulum Resident Chaperone Genes in the Unfolded Protein Response. Mol. Cell. Biol. 2003, 23, 7448–7459. [Google Scholar] [CrossRef] [Green Version]
- Ahmadiany, M.; Alavi-Samani, M.; Hashemi, Z.; Moosavi, M.A.; Rahmati, M. The increased rnase activity of ire1α in PBMCs from patients with rheumatoid arthritis. Adv. Pharm. Bull. 2019, 9, 505–509. [Google Scholar] [CrossRef] [Green Version]
- Urano, F.; Wang, X.; Bertolotti, A.; Zhang, Y.; Chung, P.; Harding, H.P.; Ron, D. Coupling of Stress in the ER to Activation of JNK Protein Kinases by Transmembrane Protein Kinase IRE1. Science 2000, 287, 664–666. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, A.; Patel, S.; McAlpine, C.; Werstuck, G.H. The Role of Endoplasmic Reticulum Stress-Glycogen Synthase Kinase-3 Signaling in Atherogenesis. Int. J. Mol. Sci. 2018, 19, 1607. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Darling, N.J.; Cook, S.J. The role of MAPK signalling pathways in the response to endoplasmic reticulum stress. Biochim. Biophys. Acta (BBA) Bioenerg. 2014, 1843, 2150–2163. [Google Scholar] [CrossRef] [Green Version]
- Velho, R.V.; De Pace, R.; Klünder, S.; Di Lorenzo, G.; Schweizer, M.; Braulke, T.; Pohl, S. Site-1 protease and lysosomal homeostasis. Biochim. Biophys. Acta Mol. Cell Res. 2017, 1864, 2162–2168. [Google Scholar] [CrossRef] [PubMed]
- Walther, T.C.; Chung, J.; Farese, R.V. Lipid Droplet Biogenesis. Annu. Rev. Cell Dev. Biol. 2017, 33, 491–510. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.S.; Mendez, R.; Heng, H.; Yang, Z.Q.; Zhang, K. Pharmacological ER stress promotes hepatic lipogenesis and lipid droplet formation. Am. J. Transl. Res. 2012, 4, 102–113. [Google Scholar]
- Röhrl, C.; Eigner, K.; Winter, K.; Korbelius, M.; Obrowsky, S.; Kratky, D.; Kovacs, W.J.; Stangl, H. Endoplasmic reticulum stress impairs cholesterol efflux and synthesis in hepatic cells. J. Lipid Res. 2014, 55, 94–103. [Google Scholar] [CrossRef] [Green Version]
- Volmer, R.; Ron, D. Lipid-dependent regulation of the unfolded protein response. Curr. Opin. Cell Biol. 2015, 33, 67–73. [Google Scholar] [CrossRef] [Green Version]
- Zheng, G.; Li, H.; Zhang, T.; Yang, L.; Yao, S.; Chen, S.; Zheng, M.; Zhao, Q.; Tian, H. Irisin protects macrophages from oxidized low density lipoprotein-induced apoptosis by inhibiting the endoplasmic reticulum stress pathway. Saudi J. Biol. Sci. 2018, 25, 849–857. [Google Scholar] [CrossRef]
- Yao, S.; Tian, H.; Miao, C.; Zhang, D.-W.; Zhao, L.; Li, Y.; Yang, N.; Jiao, P.; Sang, H.; Guo, S.; et al. D4F alleviates macrophage-derived foam cell apoptosis by inhibiting CD36 expression and ER stress-CHOP pathway. J. Lipid Res. 2015, 56, 836–847. [Google Scholar] [CrossRef] [Green Version]
- Yao, S.; Miao, C.; Tian, H.; Sang, H.; Yang, N.; Jiao, P.; Han, J.; Zong, C.; Qin, S. Endoplasmic Reticulum Stress Promotes Macrophage-derived Foam Cell Formation by Up-regulating Cluster of Differentiation 36 (CD36) Expression. J. Biol. Chem. 2014, 289, 4032–4042. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kattoor, A.J.; Goel, A.; Mehta, J.L. LOX-1: Regulation, Signaling and Its Role in Atherosclerosis. Antioxidants 2019, 8, 218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choromańska, B.; Myśliwiec, P.; Choromańska, K.; Dadan, J.; Chabowski, A. The role of CD36 receptor in the pathogenesis of atherosclerosis. Adv. Clin. Exp. Med. 2017, 26, 717–722. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Silverstein, R.L.; Li, W.; Park, Y.M.; Rahaman, S.O. Mechanisms of cell signaling by the scavenger receptor CD36: Implications in atherosclerosis and thrombosis. Trans. Am. Clin. Climatol. Assoc. 2010, 121, 206–220. [Google Scholar] [PubMed]
- Febbraio, M.; Guy, E.; Silverstein, R.L. Stem Cell Transplantation Reveals That Absence of Macrophage CD36 Is Protective Against Atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2004, 24, 2333–2338. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yao, S.; Yang, N.; Song, G.; Sang, H.; Tian, H.; Miao, C.; Zhang, Y.; Qin, S. Minimally modified low-density lipoprotein induces macrophage endoplasmic reticulum stress via toll-like receptor 4. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2012, 1821, 954–963. [Google Scholar] [CrossRef]
- Guo, C.; Ma, R.; Liu, X.; Chen, T.; Li, Y.; Yu, Y.; Duan, J.; Zhou, X.; Li, Y.; Sun, Z. Silica nanoparticles promote oxLDL-induced macrophage lipid accumulation and apoptosis via endoplasmic reticulum stress signaling. Sci. Total Environ. 2018, 631–632, 570–579. [Google Scholar] [CrossRef]
- Schmitz, G.; Grandl, M. The Molecular Mechanisms of HDL and Associated Vesicular Trafficking Mechanisms to Mediate Cellular Lipid Homeostasis. Arterioscler. Thromb. Vasc. Biol. 2009, 29, 1718–1722. [Google Scholar] [CrossRef] [Green Version]
- Tavoosi, Z.; Moradi-Sardareh, H.; Saidijam, M.; Yadegarazari, R.; Borzuei, S.; Soltanian, A.; Goodarzi, M.T. Cholesterol Transporters ABCA1 and ABCG1 Gene Expression in Peripheral Blood Mononuclear Cells in Patients with Metabolic Syndrome. Cholesterol 2015, 2015, 1–6. [Google Scholar] [CrossRef] [Green Version]
- Zaiou, M.; Bakillah, A. Epigenetic Regulation of ATP-Binding Cassette Protein A1 (ABCA1) Gene Expression: A New Era to Alleviate Atherosclerotic Cardiovascular Disease. Diseases 2018, 6, 34. [Google Scholar] [CrossRef] [Green Version]
- Ananth, S.; Gnana-Prakasam, J.P.; Bhutia, Y.D.; Veeranan-Karmegam, R.; Martin, P.M.; Smith, S.B.; Ganapathy, V. Regulation of the cholesterol efflux transporters ABCA1 and ABCG1 in retina in hemochromatosis and by the endogenous siderophore 2,5-dihydroxybenzoic acid. Biochim. Biophys. Acta Mol. Basis Dis. 2014, 1842, 603–612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, Y.; Zhang, D.; Liu, X.; Li, X.; Liu, F.; Yu, Y.; Jia, S.; Zhou, Y.; Zhao, Y. Endoplasmic Reticulum Stress Affects Lipid Metabolism in Atherosclerosis Via CHOP Activation and Over-Expression of miR-33. Cell. Physiol. Biochem. 2018, 48, 1995–2010. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Lei, T.; Li, W.; Ou, H. Enhanced cellular cholesterol efflux by naringenin is mediated through inhibiting endoplasmic reticulum stress-ATF6 activity in macrophages. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2019, 1864, 1472–1482. [Google Scholar] [CrossRef] [PubMed]
- Fusakio, M.E.; Willy, J.A.; Wang, Y.; Mirek, E.T.; Al Baghdadi, R.J.T.; Adams, C.M.; Anthony, T.G.; Wek, R.C. Transcription factor ATF4 directs basal and stress-induced gene expression in the unfolded protein response and cholesterol metabolism in the liver. Mol. Biol. Cell 2016, 27, 1536–1551. [Google Scholar] [CrossRef] [PubMed]
- Sahebkar, A.; Watts, G.F. Developing role of microRNA-33 in lipid metabolism and atherosclerosis. Curr. Opin. Lipidol. 2016, 27, 197–199. [Google Scholar] [CrossRef] [PubMed]
- McAlpine, C.S.; Werstuck, G.H. Protein kinase R-like endoplasmic reticulum kinase and glycogen synthase kinase-3α/β regulate foam cell formation. J. Lipid Res. 2014, 55, 2320–2333. [Google Scholar] [CrossRef] [Green Version]
- Maruyama, R.; Kamoshida, Y.; Shimizu, M.; Inoue, J.; Sato, R. ATF6α stimulates cholesterogenic gene expression and de novo cholesterol synthesis. Biosci. Biotechnol. Biochem. 2013, 77, 1734–1738. [Google Scholar] [CrossRef]
- Bobrovnikova-Marjon, E.; Hatzivassiliou, G.; Grigoriadou, C.; Romero, M.; Cavener, D.R.; Thompson, C.B.; Diehl, J.A. PERK-dependent regulation of lipogenesis during mouse mammary gland development and adipocyte differentiation. Proc. Natl. Acad. Sci. USA 2008, 105, 16314–16319. [Google Scholar] [CrossRef] [Green Version]
- Kara, M.; Oztas, E. Endoplasmic Reticulum Stress-Mediated Cell Death. In Programmed Cell Death; Hala, G.-M., Omar, N.R., Eds.; IntechOpen: London, UK, 2020; Available online: https://www.researchgate.net/publication/332418056_Endoplasmic_Reticulum_Stress-Mediated_Cell_Death (accessed on 20 April 2019). [CrossRef] [Green Version]
- Korbecki, J.; Bajdak-Rusinek, K. The effect of palmitic acid on inflammatory response in macrophages: An overview of molecular mechanisms. Inflamm. Res. 2019, 68, 915–932. [Google Scholar] [CrossRef] [Green Version]
- Sanson, M.; Augé, N.; Vindis, C.; Muller, C.; Bando, Y.; Thiers, J.-C.; Marachet, M.-A.; Zarkovic, K.; Sawa, Y.; Salvayre, R.; et al. Oxidized Low-Density Lipoproteins Trigger Endoplasmic Reticulum Stress in Vascular Cells. Circ. Res. 2009, 104, 328–336. [Google Scholar] [CrossRef] [Green Version]
- Yoshino, H.; Kumai, Y.; Kashiwakura, I. Effects of endoplasmic reticulum stress on apoptosis induction in radioresistant macrophages. Mol. Med. Rep. 2017, 15, 2867–2872. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nascimento Da Conceicao, V.; Sun, Y.; Zboril, E.K.; De la Chapa, J.J.; Singh, B.B. Loss of Ca2+ entry via Orai–TRPC1 induces ER stress, initiating immune activation in macrophages. J. Cell Sci. 2020, 133, jcs237610. [Google Scholar] [CrossRef] [Green Version]
- Yu, P.; Qian, A.S.; Chathely, K.M.; Trigatti, B.L. Data on leukocyte PDZK1 deficiency affecting macrophage apoptosis but not monocyte recruitment, cell proliferation, macrophage abundance or ER stress in atherosclerotic plaques of LDLR deficient mice. Data Br. 2018, 19, 1148–1161. [Google Scholar] [CrossRef] [PubMed]
- Yao, S.; Tian, H.; Zhao, L.; Li, J.; Yang, L.; Yue, F.; Li, Y.; Jiao, P.; Yang, N.; Wang, Y.; et al. Oxidized high density lipoprotein induces macrophage apoptosis via toll-like receptor 4-dependent CHOP pathway. J. Lipid Res. 2017, 58, 164–177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bryan, A.M.; Del Poeta, M. Sphingosine-1-phosphate receptors and innate immunity. Cell. Microbiol. 2018, 20, e12836. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Diarte-Añazco, E.M.G.; A Mendez-Lara, K.; Pérez, A.; Alonso, N.; Blanco-Vaca, F.; Julve, J. Novel Insights into the Role of HDL-Associated Sphingosine-1-Phosphate in Cardiometabolic Diseases. Int. J. Mol. Sci. 2019, 20, 6273. [Google Scholar] [CrossRef] [Green Version]
- Gonzalez, L.; Qian, A.S.; Tahir, U.; Yu, P.; Trigatti, B.L. Sphingosine-1-phosphate receptor 1, expressed in myeloid cells, slows diet-induced atherosclerosis and protects against macrophage apoptosis in ldlr KO mice. Int. J. Mol. Sci. 2017, 18, 2721. [Google Scholar] [CrossRef] [Green Version]
- Guo, S.; Li, L.; Yin, H. Cholesterol Homeostasis and Liver X Receptor (LXR) in Atherosclerosis. Cardiovasc. Hematol. Disord. Targets 2018, 18, 27–33. [Google Scholar] [CrossRef]
- Zhao, Q.; Zhou, D.; You, H.; Lou, B.; Zhang, Y.; Tian, Y.; Guo, N.; Chen, X.; Liu, Y.; Wu, Y.; et al. IFN-γ aggravates neointimal hyperplasia by inducing endoplasmic reticulum stress and apoptosis in macrophages by promoting ubiquitin-dependent liver X receptor-α degradation. FASEB J. 2017, 31, 5321–5331. [Google Scholar] [CrossRef] [Green Version]
- Oh, S.; Lim, Y.; Choi, J.; Lee, J.; Cho, S.; Go, D.; Kim, S.; Song, C. TNF-α-mediated ER stress causes elimination of Mycobacterium fortuitum reservoirs by macrophage apoptosis. FASEB J. 2018, 32, 3993–4003. [Google Scholar] [CrossRef] [Green Version]
- Zhang, C.; Syed, T.W.; Liu, R.; Yu, J. Role of Endoplasmic Reticulum Stress, Autophagy, and Inflammation in Cardiovascular Disease. Front. Cardiovasc. Med. 2017, 4. [Google Scholar] [CrossRef] [PubMed]
- Shao, B.; Han, B.; Zeng, Y.; Su, D.; Liu, C. The roles of macrophage autophagy in atherosclerosis. Acta Pharmacol. Sin. 2016, 37, 150–156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zahid, M.K.; Rogowski, M.; Ponce, C.; Choudhury, M.; Moustaid-Moussa, N.; Rahman, S.M. CCAAT/enhancer-binding protein beta (C/EBPβ) knockdown reduces inflammation, ER stress, and apoptosis, and promotes autophagy in oxLDL-treated RAW264.7 macrophage cells. Mol. Cell. Biochem. 2020, 463, 211–223. [Google Scholar] [CrossRef] [PubMed]
- Reverendo, M.; Mendes, A.; Argüello, R.J.; Gatti, E.; Pierre, P. At the crossway of ER-stress and proinflammatory responses. FEBS J. 2019, 286, 297–310. [Google Scholar] [CrossRef] [Green Version]
- Nakajima, S.; Kitamura, M. Bidirectional regulation of NF-κB by reactive oxygen species: A role of unfolded protein response. Free Radic. Biol. Med. 2013, 65, 162–174. [Google Scholar] [CrossRef]
- Samtleben, S.; Jaepel, J.; Fecher, C.; Andreska, T.; Rehberg, M.; Blum, R. Direct imaging of ER calcium with targeted-esterase induced dye loading (TED). J. Vis. Exp. 2013. [Google Scholar] [CrossRef]
- Bagur, R.; Rgy Hajnó Czky, G. Molecular Cell Review Intracellular Ca2+ Sensing: Its Role in Calcium Homeostasis and Signaling. Mol. Cell 2017, 66, 780–788. [Google Scholar] [CrossRef] [Green Version]
- Chauhan, A.; Sun, Y.; Sukumaran, P.; Quenum Zangbede, F.O.; Jondle, C.N.; Sharma, A.; Evans, D.L.; Chauhan, P.; Szlabick, R.E.; Aaland, M.O.; et al. M1 Macrophage Polarization Is Dependent on TRPC1-Mediated Calcium Entry. iScience 2018, 8, 85–102. [Google Scholar] [CrossRef] [Green Version]
- Bergmeier, W.; Weidinger, C.; Zee, I.; Feske, S. Emerging roles of store-operated Ca2+ entry through STIM and ORAI proteins in immunity, hemostasis and cancer. Channels 2013, 7, 379–391. [Google Scholar] [CrossRef] [Green Version]
- Son, S.M.; Byun, J.; Roh, S.E.; Kim, S.J.; Mook-Jung, I. Reduced IRE1 mediates apoptotic cell death by disrupting calcium homeostasis via the InsP3 receptor. Cell Death Dis. 2014, 5, e1188. [Google Scholar] [CrossRef] [Green Version]
- Raffaello, A.; Mammucari, C.; Gherardi, G.; Rizzuto, R. Calcium at the Center of Cell Signaling: Interplay between Endoplasmic Reticulum, Mitochondria, and Lysosomes. Trends Biochem. Sci. 2016, 41, 1035–1049. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Appenzeller-Herzog, C.; Bánhegyi, G.; Bogeski, I.; Davies, K.E.; Delaunay-Moisan, A.; Forman, H.J.; Görlach, A.; Kietzmann, T.; Laurindo, F.; Margittai, E.; et al. Transit of H2O2 across the endoplasmic reticulum membrane is not sluggish. Free. Radic. Boil. Med. 2016, 94, 157–160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoboue, E.D.; Sitia, R.; Simmen, T. Redox crosstalk at endoplasmic reticulum (ER) membrane contact sites (MCS) uses toxic waste to deliver messages. Cell Death Dis. 2018, 9, 1–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, K.; Kaufman, R.J. From endoplasmic-reticulum stress to the inflammatory response. Nature 2008, 454, 455–462. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heo, J.-N.; Kim, D.-Y.; Lim, S.-G.; Lee, K.; Suk, K.; Lee, W.-H. ER stress differentially affects pro-inflammatory changes induced by mitochondrial dysfunction in the human monocytic leukemia cell line, THP-1. Cell Biol. Int. 2019, 43, 313–322. [Google Scholar] [CrossRef]
- Wang, Y.; Zhang, Y.; Yi, P.; Dong, W.; Nalin, A.P.; Zhang, J.; Zhu, Z.; Chen, L.; Benson, D.M.; Mundy-Bosse, B.L.; et al. The IL-15–AKT–XBP1s signaling pathway contributes to effector functions and survival in human NK cells. Nat. Immunol. 2019, 20, 10–17. [Google Scholar] [CrossRef]
- Shu, S.; Zhang, Y.; Li, W.; Wang, L.; Wu, Y.; Yuan, Z.; Zhou, J. The role of monocyte chemotactic protein-induced protein 1 (MCPIP1) in angiotensin II-induced macrophage apoptosis and vulnerable plaque formation. Biochem. Biophys. Res. Commun. 2019, 515, 378–385. [Google Scholar] [CrossRef]
- Kim, S.R.; Lee, S.-G.; Kim, S.H.; Kim, J.H.; Choi, E.; Cho, W.; Rim, J.H.; Hwang, I.; Lee, C.J.; Lee, M.; et al. SGLT2 inhibition modulates NLRP3 inflammasome activity via ketones and insulin in diabetes with cardiovascular disease. Nat. Commun. 2020, 11, 2127. [Google Scholar] [CrossRef]
- Liu, Y.; Li, C.; Yin, H.; Zhang, X.; Li, Y. NLRP3 Inflammasome: A Potential Alternative Therapy Target for Atherosclerosis. Evid. Based Complement. Altern. Med. 2020, 2020. [Google Scholar] [CrossRef]
- Namgaladze, D.; Khodzhaeva, V.; Brüne, B. ER-Mitochondria Communication in Cells of the Innate Immune System. Cells 2019, 8, 1088. [Google Scholar] [CrossRef] [Green Version]
- Tao, L.; Lin, H.; Wen, J.; Sun, Q.; Gao, Y.; Xu, X.; Wang, J.; Zhang, J.; Weng, D. The kinase receptor-interacting protein 1 is required for inflammasome activation induced by endoplasmic reticulum stress. Cell Death Dis. 2018, 9. [Google Scholar] [CrossRef] [Green Version]
- Place, D.E.; Samir, P.; Karki, R.; Briard, B.; Vogel, P.; Kanneganti, T.D. ASK Family Kinases Are Required for Optimal NLRP3 Inflammasome Priming. Am. J. Pathol. 2018, 188, 1021–1030. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hang, L.; Peng, Y.; Xiang, R.; Li, X.; Li, Z. Ox-LDL Causes Endothelial Cell Injury Through ASK1/NLRP3-Mediated Inflammasome Activation via Endoplasmic Reticulum Stress. Drug Des. Dev. Ther. 2020, 14, 731–744. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Campden, R.I.; Zhang, Y. The role of lysosomal cysteine cathepsins in NLRP3 inflammasome activation. Arch. Biochem. Biophys. 2019, 670, 32–42. [Google Scholar] [CrossRef] [PubMed]
- Chevriaux, A.; Pilot, T.; Derangère, V.; Simonin, H.; Martine, P.; Chalmin, F.; Ghiringhelli, F.; Rébé, C. Cathepsin B Is Required for NLRP3 Inflammasome Activation in Macrophages, Through NLRP3 Interaction. Front. Cell Dev. Biol. 2020, 8. [Google Scholar] [CrossRef] [PubMed]
- Szpigel, A.; Hainault, I.; Carlier, A.; Venteclef, N.; Batto, A.-F.; Hajduch, E.; Bernard, C.; Ktorza, A.; Gautier, J.-F.; Ferré, P.; et al. Lipid environment induces ER stress, TXNIP expression and inflammation in immune cells of individuals with type 2 diabetes. Diabetologia 2018, 61, 399–412. [Google Scholar] [CrossRef] [PubMed]
- Shah, P.K.; Lecis, D. Inflammation in atherosclerotic cardiovascular disease. F1000Research 2019, 8, 1402. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Schwabe, R.F.; DeVries-Seimon, T.; Yao, P.M.; Gerbod-Giannone, M.-C.; Tall, A.R.; Davis, R.J.; Flavell, R.; Brenner, D.A.; Tabas, I. Free Cholesterol-loaded Macrophages Are an Abundant Source of Tumor Necrosis Factor-α and Interleukin-6. J. Biol. Chem. 2005, 280, 21763–21772. [Google Scholar] [CrossRef] [Green Version]
- Feng, B.; Yao, P.M.; Li, Y.; Devlin, C.M.; Zhang, D.; Harding, H.P.; Sweeney, M.; Rong, J.X.; Kuriakose, G.; Fisher, E.A.; et al. The endoplasmic reticulum is the site of cholesterol-induced cytotoxicity in macrophages. Nat. Cell Biol. 2003, 5, 781–792. [Google Scholar] [CrossRef]
- Wu, J.; He, G.-T.; Zhang, W.-J.; Xu, J.; Huang, Q.-B. IRE1α Signaling Pathways Involved in Mammalian Cell Fate Determination. Cell. Physiol. Biochem. 2016, 38, 847–858. [Google Scholar] [CrossRef]
- Huang, S.; Xing, Y.; Liu, Y. Emerging roles for the ER stress sensor IRE1α in metabolic regulation and disease. J. Biol. Chem. 2019, 294, 18726–18741. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meares, G.P.; Liu, Y.; Rajbhandari, R.; Qin, H.; Nozell, S.E.; Mobley, J.A.; Corbett, J.A.; Benveniste, E.N. PERK-Dependent Activation of JAK1 and STAT3 Contributes to Endoplasmic Reticulum Stress-Induced Inflammation. Mol. Cell. Biol. 2014, 34, 3911–3925. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duvigneau, J.C.; Luís, A.; Gorman, A.M.; Samali, A.; Kaltenecker, D.; Moriggl, R.; Kozlov, A.V. Crosstalk between inflammatory mediators and endoplasmic reticulum stress in liver diseases. Cytokine 2019, 124, 154577. [Google Scholar] [CrossRef] [PubMed]
- Di Conza, G.; Ho, P.-C. ER Stress Responses: An Emerging Modulator for Innate Immunity. Cells 2020, 9, 695. [Google Scholar] [CrossRef] [Green Version]
- Grootjans, J.; Kaser, A.; Kaufman, R.J.; Blumberg, R.S. The unfolded protein response in immunity and inflammation. Nat. Rev. Immunol. 2016, 16, 469–484. [Google Scholar] [CrossRef] [Green Version]
- Sanchez, C.L.; Sims, S.G.; Nowery, J.D.; Meares, G.P. Endoplasmic reticulum stress differentially modulates the IL-6 family of cytokines in murine astrocytes and macrophages. Sci. Rep. 2019, 9. [Google Scholar] [CrossRef] [Green Version]
- Gotoh, T.; Mori, M. Nitric Oxide and Endoplasmic Reticulum Stress. Arterioscler. Thromb. Vasc. Biol. 2006, 26, 1439–1446. [Google Scholar] [CrossRef]
- Weidinger, A.; Müllebner, A.; Paier-Pourani, J.; Banerjee, A.; Miller, I.; Lauterböck, L.; Duvigneau, J.C.; Skulachev, V.P.; Redl, H.; Kozlov, A.V. Vicious Inducible Nitric Oxide Synthase-Mitochondrial Reactive Oxygen Species Cycle Accelerates Inflammatory Response and Causes Liver Injury in Rats. Antioxid. Redox Signal. 2015, 22, 572–586. [Google Scholar] [CrossRef]
- Reiss, A.B.; Siegart, N.M.; De Leon, J. Interleukin-6 in atherosclerosis: Atherogenic or atheroprotective? Clin. Lipidol. 2017, 12, 14–23. [Google Scholar] [CrossRef]
- Frisdal, E.; Lesnik, P.; Olivier, M.; Robillard, P.; Chapman, M.J.; Huby, T.; Guerin, M.; Le Goff, W. Interleukin-6 Protects Human Macrophages from Cellular Cholesterol Accumulation and Attenuates the Proinflammatory Response. J. Biol. Chem. 2011, 286, 30926–30936. [Google Scholar] [CrossRef] [Green Version]
- Liu, H.; Deng, Y.; Wu, L.; Li, Y.; Lin, N.; Li, W.; Dong, X.; Ma, L. Interleukin-1β Regulates Lipid Homeostasis in Human Glomerular Mesangial Cells. J. Nutr. Health Aging 2020, 24, 246–250. [Google Scholar] [CrossRef] [PubMed]
- Shiotsugu, S.; Okinaga, T.; Habu, M.; Yoshiga, D.; Yoshioka, I.; Nishihara, T.; Ariyoshi, W. The Biological Effects of Interleukin-17A on Adhesion Molecules Expression and Foam Cell Formation in Atherosclerotic Lesions. J. Interf. Cytokine Res. 2019, 39, 694–702. [Google Scholar] [CrossRef] [PubMed]
- Tang, X.-E.; Li, H.; Chen, L.-Y.; Xia, X.-D.; Zhao, Z.-W.; Zheng, X.-L.; Zhao, G.-J.; Tang, C.-K. IL-8 negatively regulates ABCA1 expression and cholesterol efflux via upregulating miR-183 in THP-1 macrophage-derived foam cells. Cytokine 2019, 122, 154385. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.; Dong, A.; Feng, Z.; Li, J. Interleukin-32 promotes lipid accumulation through inhibition of cholesterol efflux. Exp. Ther. Med. 2017, 14, 947–952. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhat, O.M.; Kumar, P.U.; Giridharan, N.V.; Kaul, D.; Kumar, M.J.M.; Dhawan, V. Interleukin-18-induced atherosclerosis involves CD36 and NF-κB crosstalk in Apo E−/− mice. J. Cardiol. 2015, 66, 28–35. [Google Scholar] [CrossRef] [Green Version]
- Liu, Q.; Fan, J.; Bai, J.; Peng, L.; Zhang, T.; Deng, L.; Wang, G.; Zhao, Y.; Nong, J.; Zhang, M.; et al. IL-34 promotes foam cell formation by enhancing CD36 expression through p38 MAPK pathway. Sci. Rep. 2018, 8, 17347. [Google Scholar] [CrossRef]
- Reiss, A.B.; Patel, C.A.; Rahman, M.M.; Chan, E.S.L.; Hasneen, K.; Montesinos, M.C.; Trachman, J.D.; Cronstein, B.N. Interferon-gamma impedes reverse cholesterol transport and promotes foam cell transformation in THP-1 human monocytes/macrophages. Med. Sci. Monit. 2004, 10, BR420-5. [Google Scholar]
- Hashimoto, R.; Kakigi, R.; Miyamoto, Y.; Nakamura, K.; Itoh, S.; Daida, H.; Okada, T.; Katoh, Y. JAK-STAT-dependent regulation of scavenger receptors in LPS-activated murine macrophages. Eur. J. Pharmacol. 2020, 871. [Google Scholar] [CrossRef]
- Hashizume, M.; Mihara, M. Atherogenic effects of TNF-α and IL-6 via up-regulation of scavenger receptors. Cytokine 2012, 58, 424–430. [Google Scholar] [CrossRef]
- Stewart, C.R.; Stuart, L.M.; Wilkinson, K.; van Gils, J.M.; Deng, J.; Halle, A.; Rayner, K.J.; Boyer, L.; Zhong, R.; Frazier, W.A.; et al. CD36 ligands promote sterile inflammation through assembly of a Toll-like receptor 4 and 6 heterodimer. Nat. Immunol. 2010, 11, 155–161. [Google Scholar] [CrossRef] [Green Version]
- Seimon, T.A.; Nadolski, M.J.; Liao, X.; Magallon, J.; Nguyen, M.; Feric, N.T.; Koschinsky, M.L.; Harkewicz, R.; Witztum, J.L.; Tsimikas, S.; et al. Atherogenic Lipids and Lipoproteins Trigger CD36-TLR2-Dependent Apoptosis in Macrophages Undergoing Endoplasmic Reticulum Stress. Cell Metab. 2010, 12, 467–482. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, L.; Varghese, Z.; Moorhead, J.F.; Chen, Y.; Ruan, X.Z. CD36 and lipid metabolism in the evolution of atherosclerosis. Br. Med. Bull. 2018, 126, 101–112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sheedy, F. Turning 21: Induction of miR-21 as a Key Switch in the Inflammatory Response. Front. Immunol. 2015, 6, 6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Canfrán-Duque, A.; Rotllan, N.; Zhang, X.; Fernández-Fuertes, M.; Ramírez-Hidalgo, C.; Araldi, E.; Daimiel, L.; Busto, R.; Fernández-Hernando, C.; Suárez, Y. Macrophage deficiency of miR-21 promotes apoptosis, plaque necrosis, and vascular inflammation during atherogenesis. EMBO Mol. Med. 2017, 9, 1244–1262. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
| Triggers of Foam Cells Formation | Effects |
|---|---|
| Modified Atherogenic LDL Particles | • oxLDL is bound and internalized by CD36 and TLR4; • oxLDL activates the ER stress through PERK, ATF6, and IRE1-mediated UPR pathways. |
| Reduced Cholesterol Efflux | • downregulation of miR-21 expression and subsequent activation of p38MAPK/CHOP and JNK signaling pathways inhibits expression of ABCG1 and SR-BI; • overexpression of miR-33, leading to activation of PI3K/Akt signaling pathway by ATF6, inhibits the ABCA1 and ABCG1 expression. • Activated PERK/eIF2α/ATF4 and IRE1α/XBP1 signaling pathways can suppress the ABCA1 expression. |
| Alterations in Lipid Biosynthesis | • ATF6 regulates SREBP2, that can lead to an increased expression of HMG-CoA reductase and HMG-CoA synthase; • PERK regulates the expression of SREBP1 and FAS through the eIF2-dependent manner. |
| Apoptosis Triggers | Effects |
|---|---|
| Excessive Lipid Accumulation | High concentrations of free cholesterol, oxysterols, and oxLDL cause prolonged ER-stress, which induces the macrophage apoptosis via activation of CHOP through IRE1α/JNK/MAPK, IRE1α/XBP1, ATF6/XBP1, and PERK/eIF2α/ATF4 pathways |
| Inflammatory Processes | IFN-γ accelerates STAT1-dependent degradation of the transcription factor LXRα and upregulate the PERK/eIIF2α/CHOP pathway, thus contributing to ER stress and enhancing apoptotic processes. |
| Oxidized HDL | Binding of oxHDL to TLR4 can induce macrophage apoptosis by activation an ER stress/CHOP pathway through enhanced oxidative stress. |
| Ca2+ | Increased calcium concentration in ER may lead to absorption of Ca2+ by mitochondria, which can lead to PTP opening, releasing of apoptotic factors and subsequent inducing of apoptosis. |
| The Cause of the Pro-Inflammatory Cell Response | Cytokines Involved |
|---|---|
| Change in the calcium influx | TNF-α, IL-6, and IL-1β |
| Excessive lipid accumulation | TNF-α and IL-6 |
| Activation of PERK/CHOP pathway, which is enhanced by mitochondrial dysfunction | IL-23 |
| Activation of PERK/STAT3 pathway | IL-6 and IL-8 |
| Activation of IRE1α/XBP1 pathway | IL-6, CCL2, and TNF-α |
| Activation of TLRs on the cell surface, leading to the formation of NLRP3 inflammasome | IL-1β and IL-18 |
| Stress Factors | Relationship |
|---|---|
| TLR activation | TLR activation leads to the IRE1α/XBP1 signaling pathway induction and subsequent production of the pro-inflammatory cytokines IL6, CCL2, TNF-α. Also, TLR activates the NF-κB that leads to NLRP3 inflammasome activation and production of IL-1β and IL-18. |
| CD36 | CD36/TLR2/TLR6 pathway activate NF-κB, resulting in apoptosis of endoplasmic reticulum-stressed cholesterol-overloaded foam cells, which promotes pro-inflammatory response. CD36 act as another regulator of NLRP3 inflammasome activation. |
| Lipid accumulation | Cholesterol accumulation triggers IRE1α and PERK signaling pathways, that can induce both apoptosis and secretion of TNF-α and IL-6. |
| Ca2+ and ROS | Ca2+ deficiency activates UPR, enhances macrophage differentiation by the pro-inflammatory M1 phenotype, induces cytokine production, and can lead to apoptosis. Elevated ROS levels and Ca2+ disruption can induce activation all UPR signaling pathways, as well as STAT3 and TRAF2-mediated pathways that aggravate the inflammatory response and ER stress. |
| Mitochondrial dysfunction during inflammation | Activation of the PERK/CHOP pathway promotes IL-23 expression, which is enhanced by mitochondrial dysfunction. |
| Proinflammatory agents | Cytokines can activate ER stress through perturbation in ROS and Ca2+ homeostasis; can promote lipid accumulation by upregulating expression of scavenger receptors (CD36, LOX-1, SR-AI) or by diminishing the ABCA1-depending cholesterol efflux. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sukhorukov, V.N.; Khotina, V.A.; Bagheri Ekta, M.; Ivanova, E.A.; Sobenin, I.A.; Orekhov, A.N. Endoplasmic Reticulum Stress in Macrophages: The Vicious Circle of Lipid Accumulation and Pro-Inflammatory Response. Biomedicines 2020, 8, 210. https://doi.org/10.3390/biomedicines8070210
Sukhorukov VN, Khotina VA, Bagheri Ekta M, Ivanova EA, Sobenin IA, Orekhov AN. Endoplasmic Reticulum Stress in Macrophages: The Vicious Circle of Lipid Accumulation and Pro-Inflammatory Response. Biomedicines. 2020; 8(7):210. https://doi.org/10.3390/biomedicines8070210
Chicago/Turabian StyleSukhorukov, Vasily N., Victoria A. Khotina, Mariam Bagheri Ekta, Ekaterina A. Ivanova, Igor A. Sobenin, and Alexander N. Orekhov. 2020. "Endoplasmic Reticulum Stress in Macrophages: The Vicious Circle of Lipid Accumulation and Pro-Inflammatory Response" Biomedicines 8, no. 7: 210. https://doi.org/10.3390/biomedicines8070210