Unraveling the Role of Inwardly Rectifying Potassium Channels in the Hippocampus of an Aβ(1–42)-Infused Rat Model of Alzheimer’s Disease




Abstract
:1. Introduction
2. Materials and Methods
2.1. Animals
2.2. Aβ(1–42)-Infused Rat Model
2.3. Experimental Design
2.4. Histological Congo Red Staining
2.5. Immunofluorescence Analysis
2.6. cDNA Synthesis and Real-Time PCR
2.7. Western blotting (WB)
2.8. Statistical Analysis
3. Results
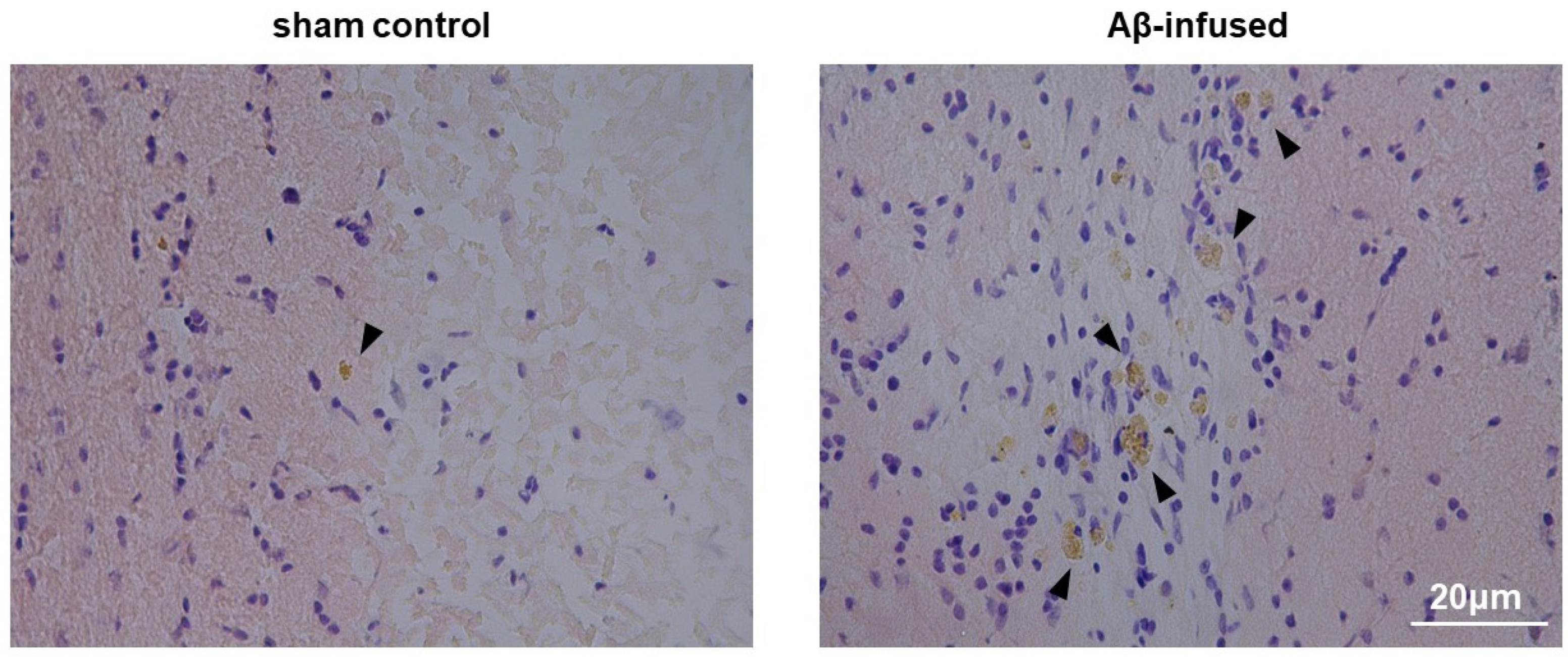
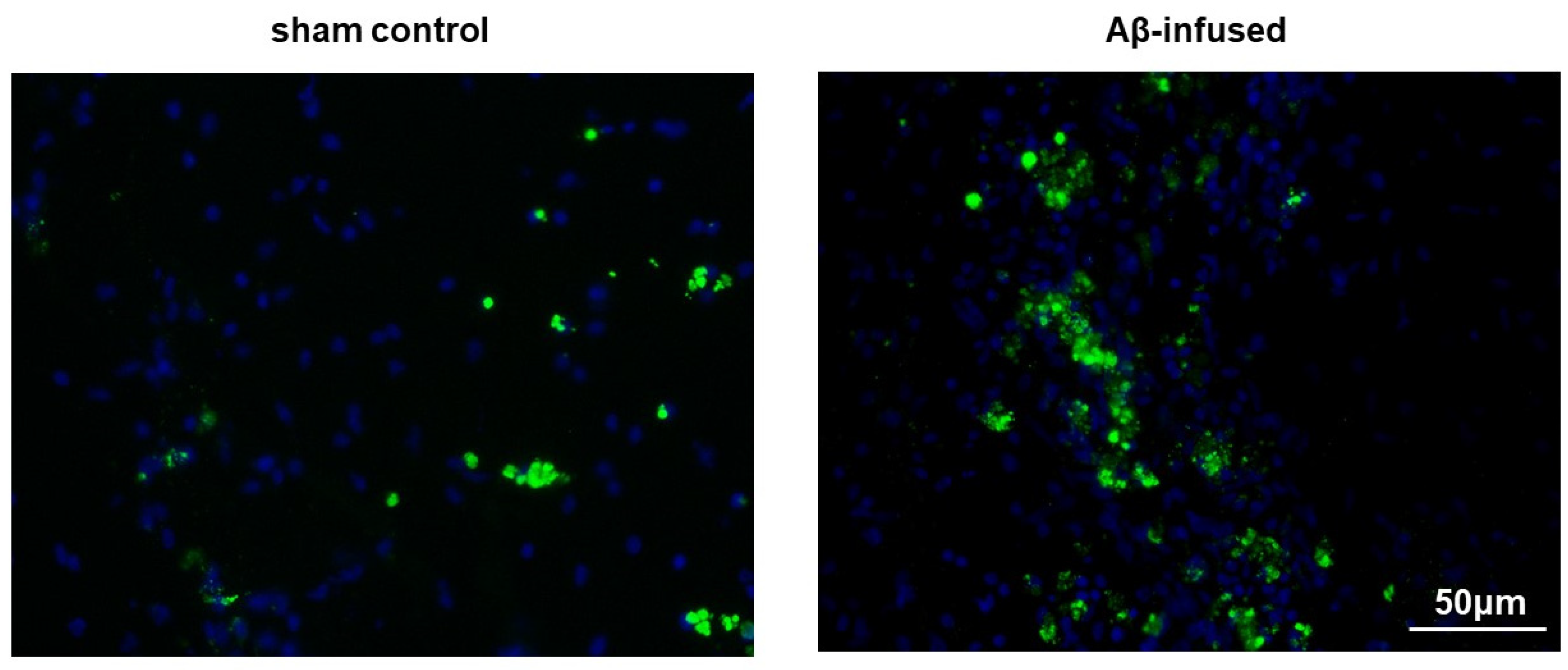
3.1. Injection of Aβ(1–42) Oligomers Mimicked Alzheimer’s Disease in Rats
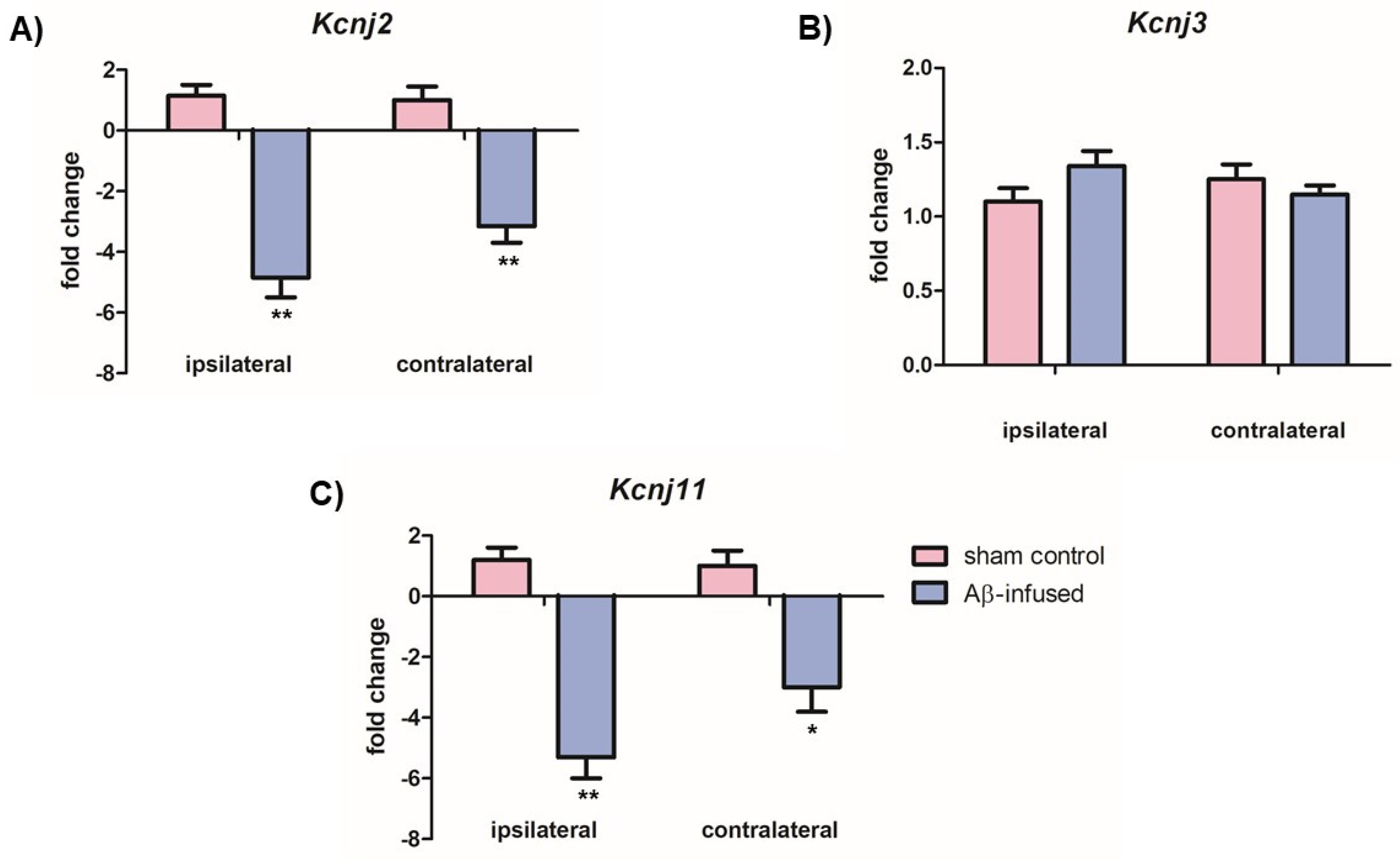
3.2. Aβ(1–42)-Infused Rats Exhibited Low mRNA Levels of Neuronal Kir2.1 and Kir6.2 Channels
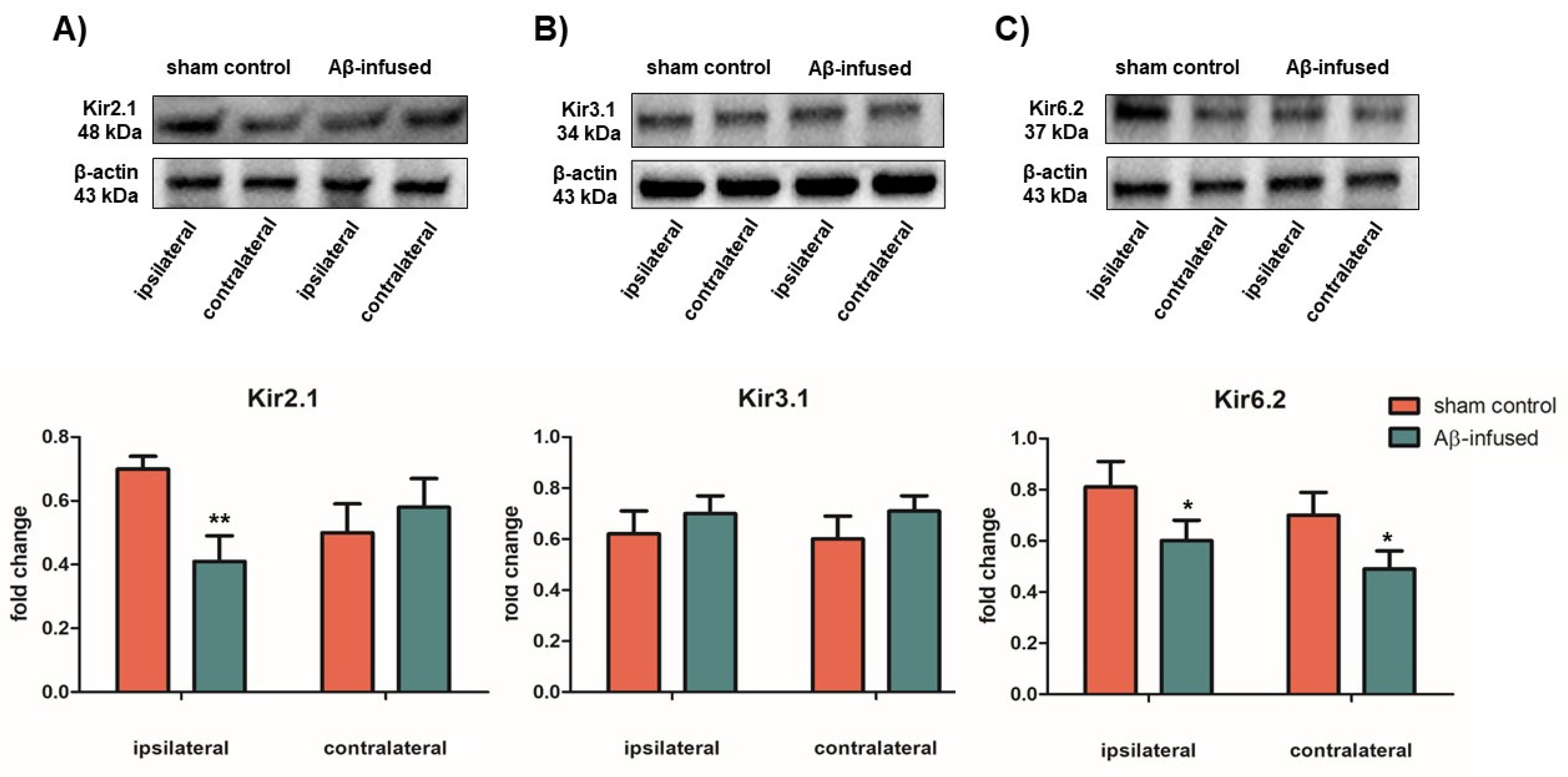
3.3. Low mRNA Levels of Kir2.1 and Kir6.2 Channels Correlate with Decreased Protein Levels in Aβ(1–42)-Infused Rat Model
4. Discussion
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Crous-Bou, M.; Minguillón, C.; Gramunt, N.; Molinuevo, J.L. Alzheimer’s disease prevention: From risk factors to early intervention. Alzheimers Res. Ther. 2017, 9, 71. [Google Scholar] [CrossRef]
- Anand, R.; Gill, K.D.; Mahdi, A.A. Therapeutics of Alzheimer’s disease: Past, present and future. Neuropharmacology 2014, 76 (Pt. A), 27–50. [Google Scholar] [CrossRef]
- Sanabria-Castro, A.; Alvarado-Echeverria, I.; Monge-Bonilla, C. Molecular Pathogenesis of Alzheimer’s Disease: An Update. Ann. Neurosci. 2017, 24, 46–54. [Google Scholar] [CrossRef] [PubMed]
- Haass, C.; Selkoe, D.J. Soluble protein oligomers in neurodegeneration: Lessons from the Alzheimer’s amyloid beta-peptide. Nat. Rev. Mol. Cell Biol. 2007, 8, 101–112. [Google Scholar] [CrossRef] [PubMed]
- Tönnies, E.; Trushina, E. Oxidative stress, synaptic dysfunction, and alzheimer’s disease. J. Alzheimers Dis. 2017, 57, 1105–1121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Castro, A.; Martinez, A. Targeting beta-amyloid pathogenesis through acetylcholinesterase inhibitors. Curr. Pharm. Des. 2006, 12, 4377–4387. [Google Scholar] [CrossRef] [PubMed]
- Mangialasche, F.; Solomon, A.; Winblad, B.; Mecocci, P.; Kivipelto, M. Alzheimer’s disease: Clinical trials and drug development. Lancet Neurol. 2010, 9, 702–716. [Google Scholar] [CrossRef]
- Akyuz, E.; Mega Tiber, P.; Beker, M.; Akbas, F. Expression of cardiac inwardly rectifying potassium channels in pentylenetetrazole kindling model of epilepsy in rats. Cell. Mol. Biol. (Noisy-le-grand) 2018, 64, 47–54. [Google Scholar] [CrossRef]
- Villa, C.; Combi, R. Potassium Channels and Human Epileptic Phenotypes: An Updated Overview. Front. Cell. Neurosci. 2016, 10, 81. [Google Scholar] [CrossRef] [Green Version]
- Binda, A.; Rivolta, I.; Villa, C.; Chisci, E.; Beghi, M.; Cornaggia, C.M.; Giovannoni, R.; Combi, R. A Novel KCNJ2 Mutation Identified in an Autistic Proband Affects the Single Channel Properties of Kir2.1. Front. Cell. Neurosci. 2018, 12, 76. [Google Scholar] [CrossRef] [Green Version]
- Akyuz, E.; Villa, C. A novel role of cardiac inwardly rectifying potassium channels explaining autonomic cardiovascular dysfunctions in a cuprizone-induced mouse model of multiple sclerosis. Auton. Neurosci. 2020, 225, 102647. [Google Scholar] [CrossRef] [PubMed]
- Villa, C.; Suphesiz, H.; Combi, R.; Akyuz, E. Potassium channels in the neuronal homeostasis and neurodegenerative pathways underlying Alzheimer’s Disease: An update. Mech. Ageing Dev. 2019, 185, 111197. [Google Scholar] [CrossRef] [PubMed]
- Hibino, H.; Inanobe, A.; Furutani, K.; Murakami, S.; Findlay, I.; Kurachi, Y. Inwardly rectifying potassium channels: Their structure, function, and physiological roles. Physiol. Rev. 2010, 90, 291–366. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mayordomo-Cava, J.; Yajeya, J.; Navarro-Lopez, D.J.; Jimenez-Diaz, L. Amyloidbeta(25-35) modulates the expression of GirK and KCNQ channel genes in the Hippocampus. PLoS ONE 2015, 10, e0134385. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, X.H.; Pan, Y.P.; Wang, X.L. mRNA expression alterations of inward rectifier potassium channels in rat brain with cholinergic impairment. Neurosci. Lett. 2002, 322, 25–28. [Google Scholar] [CrossRef]
- Serpente, M.; Fenoglio, C.; Villa, C.; Cortini, F.; Cantoni, C.; Ridolfi, E.; Clerici, F.; Marcone, A.; Benussi, L.; Ghidoni, R.; et al. Role of OLR1 and its regulating hsa-miR369-3 p in Alzheimer’s disease: Genetics and expression analysis. J. Alzheimers Dis. 2011, 26, 787–793. [Google Scholar] [CrossRef]
- Zangerl-Plessl, E.M.; Qile, M.; Bloothooft, M.; Stary-Weinzinger, A.; van der Heyden, M.A.G. Disease Associated Mutations in KIR Proteins Linked to Aberrant Inward Rectifier Channel Trafficking. Biomolecules. 2019, 9, 650. [Google Scholar] [CrossRef] [Green Version]
- Vitvitsky, V.M.; Garg, S.K.; Keep, R.F.; Albin, R.L.; Banerjee, R. Na+ and K+ ion imbalances in Alzheimer’s disease. Biochim. Biophys. Acta. 2012, 1822, 1671–1681. [Google Scholar] [CrossRef] [Green Version]
- Anumonwo, J.M.; Lopatin, A.N. Cardiac strong inward rectifier potassium channels. J. Mol. Cell. Cardiol. 2010, 48, 45–54. [Google Scholar] [CrossRef] [Green Version]
- D’Adamo, M.C.; Catacuzzeno, L.; Di Giovanni, G.; Franciolini, F.; Pessia, M. K(+) channelepsy: Progress in the neurobiology of potassium channels and epilepsy. Front. Cell. Neurosci. 2013, 7, 134. [Google Scholar] [CrossRef] [Green Version]
- Haider, S.; Antcliff, J.F.; Proks, P.; Sansom, M.S.; Ashcroft, F.M. Focus on Kir6.2: A key component of the ATP-sensitive potassium channel. J. Mol. Cell Cardiol. 2005, 38, 927–936. [Google Scholar] [CrossRef] [PubMed]
- Griffith, C.M.; Xie, M.X.; Qiu, W.Y.; Sharp, A.A.; Ma, C.; Pan, A.; Yan, X.X.; Patrylo, P.R. Aberrant expression of the pore-forming KATP channel subunit Kir6.2 in hippocampal reactive astrocytes in the 3 xTg-AD mouse model and human Alzheimer’s disease. Neuroscience 2016, 336, 81–101. [Google Scholar] [CrossRef] [PubMed]
- Héron-Milhavet, L.; Xue-Jun, Y.; Vannucci, S.; Wood, T.L.; Willing, L.B.; Stannard, B.; Hernandez-Sanchez, C.; Mobbs, C.; Virsolvy, A.; LeRoith, D. Protection against hypoxic-ischemic injury in transgenic mice overexpressing Kir6.2 channel pore in forebrain. Mol. Cell Neurosci. 2004, 25, 585–593. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choeiri, C.; Staines, W.A.; Miki, T.; Seino, S.; Renaud, J.M.; Teutenberg, K.; Messier, C. Cerebral glucose transporters expression and spatial learning in the K-ATP Kir6.2(-/-) knockout mice. Behav. Brain. Res. 2006, 172, 233–239. [Google Scholar] [CrossRef] [PubMed]
- Betourne, A.; Bertholet, A.M.; Labroue, E.; Halley, H.; Sun, H.S.; Lorsignol, A.; Feng, Z.P.; French, R.J.; Penicaud, L.; Lassalle, J.M.; et al. Involvement of hippocampal CA3 K(ATP) channels in contextual memory. Neuropharmacology 2009, 56, 615–625. [Google Scholar] [CrossRef]
- May, L.M.; Anggono, V.; Gooch, H.M.; Jang, S.E.; Matusica, D.; Kerbler, G.M.; Meunier, F.A.; Sah, S.; Coulson, E.J. G-protein-Coupled inwardly rectifying potassium (GIRK) channel activation by the p75 neurotrophin receptor is required for amyloid beta toxicity. Front. Neurosci. 2017, 11, 455. [Google Scholar] [CrossRef] [Green Version]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Genes | Forward Primer (5′–3′) | Reverse Primer (5′–3′) |
|---|---|---|
| Kcnj2 | GCAAACTCTGCTTGATGTGG | TCATACAAAGGGCTGTCTTCG |
| Kcnj3 | CTGACCGCTTCACATAGC | CTCCAGACTGGGATAGAC |
| Kcnj11 | CCTACACCAGGTGGACATCC | CAGGCTGCGGTCCTCATCAA |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Akyuz, E.; Villa, C.; Beker, M.; Elibol, B. Unraveling the Role of Inwardly Rectifying Potassium Channels in the Hippocampus of an Aβ(1–42)-Infused Rat Model of Alzheimer’s Disease. Biomedicines 2020, 8, 58. https://doi.org/10.3390/biomedicines8030058
Akyuz E, Villa C, Beker M, Elibol B. Unraveling the Role of Inwardly Rectifying Potassium Channels in the Hippocampus of an Aβ(1–42)-Infused Rat Model of Alzheimer’s Disease. Biomedicines. 2020; 8(3):58. https://doi.org/10.3390/biomedicines8030058
Chicago/Turabian StyleAkyuz, Enes, Chiara Villa, Merve Beker, and Birsen Elibol. 2020. "Unraveling the Role of Inwardly Rectifying Potassium Channels in the Hippocampus of an Aβ(1–42)-Infused Rat Model of Alzheimer’s Disease" Biomedicines 8, no. 3: 58. https://doi.org/10.3390/biomedicines8030058