Breaking Therapy Resistance: An Update on Oncolytic Newcastle Disease Virus for Improvements of Cancer Therapy



Abstract
1. Introduction
2. Basic Information
2.1. Evolution and Taxonomy of NDV
2.2. Molecular Biology of NDV
2.3. The Type I Interferon Response in Birds and Its Inhibition
2.4. The Type I Interferon Response in Mammalian Cells and Its Inhibition
3. Anti-Neoplastic and Immune-Stimulating Effects
3.1. Virus Infection of Tumor Cells from Non-Permissive Hosts: Tumor-Selective Virus Replication
3.2. Mechanism of Tumor-Selective NDV-Mediated Oncolysis and Immunogenic Cell Death
3.3. Post-Oncolytic Immunity and Direct Activation of Immune Cells by NDV
- NK cells: The viral surface protein HN of NDV is recognized by activatory NK cell receptors (NCR1) NKp46 and NKp44 of murine NK cells which transmit cytotoxicity inducing signals [64]. These two receptors are associated with the signaling chains CD3ζ (to NKp46) and DAP12 (to NKp44), respectively, which contain immunoreceptor tyrosine-based activation motif (ITAM) domains that facilitate signal transduction.
- Monocytes and macrophages: Upon contact with NDV, monocytes become activated to produce tumoricidal activity through TRAIL [71]. NDV activated macrophages exert anti-tumor activity not only in vitro [71] but also in vivo [72]. NDV-infected macrophages produce nitric oxide (NO) via activation of NFκB [73]. Caspase activation was required for NO-mediated, CD95-dependent and independent apoptosis in human neoplastic lymphoid cells [74]. The mechanism of NO-induced apoptosis in tumor cells has been reviewed [75].
- 4.
- T cells: HN molecules at the surface of infected tumor cells introduce new cell adhesive strength for interaction with lymphocytes [82,83]. Modification of tumor cells by a low dose of NDV introduced T cell costimulatory activity and augmented in vivo tumor-specific T cell response as a result of CD4+ and CD8+ immune T cell cooperation [84]. HN was demonstrated to augment peptide-specific CD8+ T cell responses [85]. Secondary tumor-specific cytotoxic T cell (CTL) responses in vitro were augmented when stimulator tumor cells were infected by NDV [86]. Such potentiation of CTL activity in vitro was mediated via induction of IFN-α, β (IFN-I) [87]. IFN-I plays an important role for the generation of CTLs not only in vitro but also in vivo. It was hardly possible to induce a CTL response in mice pretreated with an IFN-I neutralizing antiserum [87]. NDV infection of human melanoma cells was reported to break tolerance of a melanoma-specific CD4+ T helper cell line. This T-cell costimulatory activity was independent of B7-1/B7-2 [88].
3.4. Roles of Virus Infection and IFN-I for Interactions between NK Cells, DCs, and T Cells
4. Potential of NDV to Break Therapy Resistance and Anti-Viral Immunity
- Breaking resistance to chemotherapy (CT) or radiotherapy (RT): These therapies require cells to be in a proliferating state. Non-proliferating cells such as cancer stem cells or dormant tumor cells are not affected by CT or conventional photon radiotherapy using X-rays and gamma rays. In contrast to cytostatic drugs and RT, oncolysis by NDV does not depend on cell proliferation. Since NDV replicates in the cytoplasm of cells, it is independent from DNA replication. The virus is capable to replicate in X-irradiated cells such as ATV-NDV vaccine cells [103]. There is thus a potential of oncolytic NDV to target cancer stem cells and dormant tumor cells. In addition, NDV has the potential to break drug-resistance of cancer cells: (i) NDV was reported to induce apoptosis in cisplatin resistant human lung adenocarcinoma cells in vitro and in vivo [104]. Apoptosis was induced via the caspase pathway, particularly involving caspase-9. (ii) In another study, treatment with autophagy modulators was found an effective strategy to augment the therapeutic activity of oncolytic NDV against drug-resistant lung cancers [105]. (iii) Multidrug resistance, particularly resistance to temozolomide (TMZ), is a challenge in the treatment of GBM. NDV was found to enhance the growth-inhibiting and proapoptotic effects of TMZ on GBM cells in vitro and in vivo [106].
- Breaking resistance to apoptosis: Oncolytic NDV was demonstrated to be capable to break resistance to apoptosis. It was reported to even have selectivity for apoptosis-resistant cells [107]. Utilizing a human non-small-cell lung cancer cell line (A549) overexpressing the antiapoptotic protein Bcl-xL, the authors showed significant enhancement of oncolytic activity and NDV replication in comparison to A549 cells not resistant to apoptosis. The increased oncolytic activity seen was secondary to enhanced viral replication and syncytium formation [107]. Another study was performed with apoptosis-resistant primary melanoma cells that overexpress the inhibitor of apoptosis protein Livin [108]. In that study, NDV-HUJ, a one-cycle replicating virus, was capable to overcome the resistance of this cell line to apoptosis. Under the robust apoptotic stimulation of NDV-HUJ, caspases could cleave Livin to create a truncated protein with proapoptotic activity. Both studies reported that the interferon system was not involved in this type of NDV-induced oncolysis [107,108].
- Breaking resistance to hypoxia: Solid tumor microenvironments contain regions of hypoxia, in which a transcription factor, hypoxia inducible factor (HIF) is active. It influences gene expression and contributes to the tumors radio-, and chemo-resistance. A velogenic NDV strain was applied to compare the oncolytic effect against a clear cell renal carcinoma line under normoxic and hypoxic conditions. It was found that hypoxia augmented oncolytic activity regardless of the cells HIF levels [109].
- Breaking resistance to TRAIL: Trail-resistant hepatocellular carcinoma-derived cell lines were found to be more susceptible to NDV-mediated oncolysis than TRAIL-sensitive cells. IFN-stimulated gene (ISG)-12a over-expression or silencing enhanced or reduced the cells TRAIL sensitivities [110].
- Breaking resistance to immune checkpoint blockade: Zamarin et al. reported in 2014 that localized (i.t.) oncolytic virotherapy with NDV in B16 mouse melanoma could break systemic tumor resistance to immune checkpoint blockade immunotherapy [111]. The therapeutic effect was associated with marked distant tumor infiltration with activated CD8+ and CD4+ effector but not with regulatory T cells, and was dependent on CD8+ T cells, NK cells, and type I interferon. In 2018, the same group explored the immunogenic potential of NDV in bladder cancer. Immunotherapy in bladder cancer with checkpoint inhibitory antibodies revealed only suboptimal response rates. NDV was able to infect human and mouse bladder cancer cells and induced ICD. This was associated with upregulation of MHC and PD-L1 and occured even in cell lines that had been resistant to NDV oncolysis. In vivo, in a bilateral flank test model, localized NDV therapy was able to potentiate checkpoint blockade therapy in both treated and distant tumors and even when employing lysis-resistant tumor cells [112]. The latter study is reminiscent of a study from 2007, in which a host mediated anti-tumor effect of oncolytic NDV was described after intra-hepatic locoregional virus application when using a luciferase-transfected NDV-lysis resistant murine colon carcinoma cell line [113].
- Breaking resistance to anti-viral immunity: Anti-viral immunity is considered as a major hurdle for effective therapeutic activity of oncolytic viruses [1]. Surprisingly, in a recent study from Ricca et al. it was reported that pre-existing immunity to oncolytic NDV potentiates rather than inhibits its immunotherapeutic efficacy [114]. This suggests for NDV a potential to break resistance to anti-viral immunity.
5. Comparison of NDV to other OVs
6. Over 50 Years of Clinical NDV Application
6.1. 1960s to 1970s: Post-Operative Treatment with Oncolysate Vaccines
6.2. 1990s to 2000s: Post-Operative Treatment with Autologous Live NDV-Modified Tumor Cell Vaccine (ATV-NDV)
6.3. 2000s: Systemic Application Studies with Oncolytic NDV
6.4. 2010s: Combining Oncolytic Virus Modified Vaccines (ATV-NDV) with Costimulatory Bispecific Antibodies
7. A New Concept of Multimodal Cancer Immunotherapy
7.1. Treatment of Cancer Patients with Autologous Viral Oncolysate Pulsed DC Vaccine (IO-VACR)
7.2. The Two Steps of Multimodal Immunotherapy at IOZK
- Immune triggering of the cancer patient’s immune system. This is done by combining systemic (i.v.) application of NDV with a physical modality inducing ICD: Local moderate electrohyperthermia (mEHT). mEHT is a type of hyperthermia. This is only one of several physical modalities inducing immunogenic cell death (ICD). Others are ionizing irradiation, ultraviolet C light, photodynamic therapy with hypericin, and high hydrostatic pressure [157]. This pre-treatment by mEHT and NDV, which takes about one hour per day, is repeated 5 times over a period of 5 days. Since both, mEHT and NDV, induce in tumor cells ICD, this first step of treatment can be considered as “in situ” vaccination [158].
- Active-specific vaccination is then performed at the eigth day of the treatment by intradermal application of the “ex vivo” produced vaccine IO-VACR (see above). This is done in combination with a sixth dose of NDV and sixth session of mEHT. The patient-derived DCs for the preparation of IO-VACR are obtained by a differentiation process from adherent monocytes (CD14++,CD86+,CD209−,CD83−) via semiadherent immature DCs (CD14+,CD86+,CD209++,CD83−) to floating mature DCs (CD14−,CD86++,CD209+,CD83++). This process is followed regularly by flow cytometry.
7.3. Side Effects
7.4. Limitations and Challenges
8. Future Perspective: OV-mediated Gene Therapy and Combinatorial Approaches with Bispecific Antibodies and Checkpoint Inhibitors
8.1. OV-Mediated Gene Therapy
8.2. Combining NDV with Carrier Cells for Improving Tumor Targeting
8.3. Combinatorial Approaches of OVs with Bi- or Tri-specific Antibodies
8.4. Combinatorial Approaches with Checkpoint Inhibitors
8.5. Modulation of OV Therapy by Pharmacologicals
- Combining OVs with DNA alkylating drugs. Two examples are cyclophosphamide (CPA) and Temozolomide (TMZ). CPA has been used successfully in combination with OVs including HSV-1, adenovirus, vaccinia virus, reovirus, measles, and vesicular stomatitis virus. The best progression-free survival and OS rates were seen with a combination of low-dose metronomic CPA and intratumoral infection by gene-modified adenovirus Ad-GM-CSF [216]. In another study, using Ad5/3-D24-GM-CSF with or without low-dose CPA to reduce Tregs, co-treatment with TMZ increased tumor cell autophagy, anti-tumor immunity, and reduced tumor burden [219].
- Combining OVs with inhibition of DNA replication. Gemcitabine, a fluorinated deoxycytidine nucleoside analog, was shown to increase the anti-tumor activity of adenovirus, parvovirus, reovirus, vesicular stomatitis virus, HSV, vaccinia, and myxoma virus. A Phase I study was performed in 16 patients with advanced cancers by combining gemcitabine with intravenous reovirus [220].
- Combining OVs with epigenetic modulators. Dysfunctional IFN pathways in cancers are often due to epigenetic silencing including DNA promoter hypermethylation and transcriptionally suppressive histone modifications. Transcriptional activation of ISGs, which are often epigenetically silenced, requires HDAC activity [221]. HDAC inhibitors include valproic acid (VPA) and trichostatin A (TSA). These have been used in combination with OVs to effectively “reprogram” IFN-responsive tumors to become permissive to OV infection. Epigenetic modifiers have also been recommended to boost cancer immunotherapies [222,223]. One example will be given: 5-AZA-2′-deoxycytidine (5-AZA) is a methyltransferase inhibitor that prevents DNA methylation and allows silenced DNA to regain accessibility to transcription factors. An increase in survival was observed in glioma bearing mice treated with oncolytic HSV rQNestin34.5 and 5-AZA compared to either treatment alone [223]. 5-AZA had been found much earlier to be capable in vitro to de-repress on immunoresistant tumor variants expression of a TA which could be recognized by specific CTLs [224]. Recent advances in epigenomics have illuminated epigenetics in cancer stem cells (CSC) [225]. CSCs have been identified in a number of solid tumors, including breast cancer, brain tumors, lung cancer, colon cancer, and melanoma. Lung cancer stem cells (CSC) were recently reported to be responsible for tumor initiation, treatment resistance and tumor recurrence [225].
- Combining OVs with pharmacological modulation of autophagy. Chloroquine is an autophagy inhibitor and rapamycin an inducer of autophagy. Oncolytic effects of NDV (strain FMW) on cisplatin or paclitaxel resistant lung carcinoma cell lines could be significantly potentiated in vitro and in vivo by combination treatment with these autophagy modulators [105]. Oncolytic NDV was demonstrated to replicate in CSC-enriched lung cancer spheroids, to lyse them and to inhibit their 3D growth potential. Treatment of such spheroids with chloroquine increased the NDV-induced cytotoxicity [226].
9. Summary
10. Conclusions
Acknowledgments
Conflicts of Interest
References
- Fournier, P.; Schirrmacher, V. Harnessing oncolytic virus-mediated antitumor immunity. Front. Oncol. 2014, 4, 1–109. [Google Scholar]
- Heidbuechel, J.P.W.; Engeland, C.E. Paramyxoviruses for tumor-targeted immuno-modulation: Design and evalution ex vivo. J. Vis. Exp. 2019, 143, e58651. [Google Scholar]
- Pol, J.G.; Lévesque, S.; Workenhe, S.T.; Gujar, S.; Le Boeuf, F.; Clements, D.; Fahmer, J.E.; Fend, L.; Bell, C.J.; Mossman, K.L.; et al. Trial Watch: Oncolytic viro-immunotherapy of hematologic and solid tumors. Oncoimmunology 2018, 7, e1503032. [Google Scholar] [CrossRef] [PubMed]
- Schirrmacher, V. Immunobiology of Newcastle Disease Virus and its use for prophylactic vaccination in poultry and as adjuvant for therapeutic vaccination in cancer patients. Int. J. Mol. Sci. 2017, 18, 1103. [Google Scholar] [CrossRef] [PubMed]
- Schirrmacher, V. Fifty years of clinical application of Newcastle disease virus: Time to celebrate! Biomedicines 2016, 4, 16. [Google Scholar] [CrossRef] [PubMed]
- Cassel, W.A.; Garrett, R.E. Newcastle disease virus as an antineoplastic agent. Cancer 1965, 18, 863–868. [Google Scholar] [CrossRef]
- Jarvis, E.D.; Mirarab, S.; Aberer, A.J.; Li, B.; Houde, P.; Li, C.; Ho, S.Y.; Faircloth, B.C.; Nabholz, B.; Howard, J.T.; et al. Whole-genome analyses resolve early branches in the tree of life of modern birds. Science 2014, 346, 1320–1331. [Google Scholar] [PubMed]
- Dimitrov, K.M.; Abolnik, C.; Afonso, C.L.; Albina, E.; Bahl, J.; Berg, M.; Briand, F.X.; Brown, I.H.; Choi, K.S.; Chvala, I.; et al. Updated unified phylogenetic classification system and revised nomenclature for Newcastle disease virus. Infect. Genet. Evol. 2019. [Google Scholar] [CrossRef]
- Miller, P.J.; Koch, G. Newcastle disease. In Diseases of Poultry, 13th ed.; Swayne, D.E., Ed.; Wiley-Blackwell: Hoboken NJ, USA, 2013; pp. 89–138. [Google Scholar]
- Fournier, P.; Zeng, J.; Von der Lieth, C.W.; Washburn, B.; Ahlert, T.; Schirrmacher, V. Importance of serine 200 for functional activities of the hemagglutinin-neuraminidase protein of Newcasle disease virus. Int. J. Oncol. 2004, 24, 623–634. [Google Scholar]
- Li, Q.; Wei, D.; Feng, F.; Wang, X.L.; Li, C.; Chen, Z.N.; Bian, H. α2,6-linked sialic acid serves as a high-affinity receptor for cancer Oncolytic virotherapy with Newcastle disease virus. J. Cancer Res. Clin. Oncol. 2017, 143, 2171–2181. [Google Scholar] [CrossRef]
- Tan, L.; Zhang, Y.; Zhan, Y.; Yuan, Y.; Sun, Y.; Qiu, X.; Meng, C.; Song, C.; Liao, Y.; Ding, C. Newcastle disease virus employs macropinocytosis and Rab5a-dependent intracellular trafficking to infect DF-1 cells. Oncotarget 2016, 7, 86117. [Google Scholar] [CrossRef] [PubMed]
- Samal, K.S. Newcastle disease and related avian paramyxoviruses. In The Biology of Paramyxoviruses, 1st ed.; Samal, S.K., Ed.; Caister Academic Press: Norfolk, UK, 2011; pp. 69–114. [Google Scholar]
- Fiola, C.; Peeters, B.; Fournier, P.; Arnold, A.; Bucur, M.; Schirrmacher, V. Tumor selective replication of Newcastle disease virus: Association with defects of tumor cells in antiviral defence. Int. J. Cancer 2006, 119, 328–338. [Google Scholar] [CrossRef] [PubMed]
- Rangaswamy, U.S.; Wang, W.; Cheng, X.; Mc Tamney, P.; Carroll, D.; Jin, H. Newcastle disease virus establishes persistent infection in tumor cells in vitro: Contribution of the cleavage site of fusion protein and second sialic acid binding site of hemagglutinin-neuraminidase. J. Virol. 2017, 91, e00770-17. [Google Scholar] [CrossRef] [PubMed]
- Lech, P.J.; Russel, S.J. Use of attenuated paramyxoviruses for cancer therapy. Expert. Rev. Vaccines 2010, 9, 1275–1302. [Google Scholar]
- Feng, H.; Wei, D.; Nan, G.; Cui, S.J.; Chen, Z.N.; Bian, H. Construction of a minigenome rescue system for Newcastle disease virus strain Italien. Arch. Virol. 2011, 156, 611–616. [Google Scholar] [CrossRef] [PubMed]
- Ayllon, J.; Garcia-Sastre, A.; Martinez-Sobrido, L. Rescue of recombinant Newcastle disease virus from cDNA. J. Vis. Exp. 2013, 80, e50830. [Google Scholar] [CrossRef] [PubMed]
- Cardenas-Garcia, S.; Afonso, C.L. Reverse genetics of Newcastle disease virus. Methods Mol. Biol. 2017, 1602, 141–158. [Google Scholar]
- Hervas-Stubbs, S.; Perez-Gracia, J.L.; Rouzaut, A.; Sanmamed, M.F.; Le Bon, A.; Melero, I. Direct effects of type I interferons on cells of the immune system. Clin. Cancer Res. 2011, 17, 2619–2627. [Google Scholar] [CrossRef]
- Xiang, B.; Zhu, W.; Li, Y.; Gao, P.; Liang, J.; Liu, D.; Ding, C.; Liao, M.; Kang, Y.; Ren, T. Immune responses of mature chicken bone-marrow-derived dendritic cells infected with Newcastle disease virus strains with differing pathogenicity. Arch. Virol. 2018, 163, 1407–1417. [Google Scholar] [CrossRef]
- Wang, X.; Dang, R.; Yang, Z. The interferon antagonistic activities of the V proteins of NDV correlated with their virulence. Virus Genes 2019, 55, 233–237. [Google Scholar] [CrossRef]
- Alamares, J.G.; Elankumaran, S.; Samal, S.K.; Iorio, R.M. The interferon antagonistic activities of the V protein from two strains of Newcastle disease virus correlate with their known virulence properties. Virus Res. 2010, 147, 153–157. [Google Scholar] [CrossRef] [PubMed]
- Hornung, V.; Ellegast, J.; Kim, S.; Brzózka, K.; Jung, A.; Kato, H.; Poeck, H.; Akira, S.; Conzelmann, K.K.; Schlee, M.; et al. 5′-Triphosphate RNA is the ligand for RIG-I. Science 2005, 314, 994–997. [Google Scholar] [CrossRef] [PubMed]
- Fournier, P.; Wilden, H.; Schirrmacher, V. Importance of retinoic acid-inducible gene I and of receptor for type I interferon for cellular resistance to infection by Newcastle disease virus. Int. J. Oncol. 2012, 40, 287–298. [Google Scholar] [PubMed]
- Sun, Y.; Mao, X.; Zheng, H.; Wu, W.; Rehman, Z.U.; Liao, Y.; Meng, C.; Qiu, X.; Tan, L.; Song, C.; et al. Goose MAVS functions in RIG-I-mediated IFN-ß signaling activation. Dev. Comp. Immunol. 2019, 93, 58–65. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Hanson, R.P. In vivo interference by Newcastle disease virus in chickens, the natural host of the virus. Arch. Virol. 1989, 108, 229–245. [Google Scholar] [CrossRef] [PubMed]
- Ng, C.S.; Kato, H.; Fujita, T. Fueling type I interferonopathies: Regulation and function of Type I interferon antiviral responses. J. Interferon Cytokine Res. 2019, 39. [Google Scholar] [CrossRef] [PubMed]
- Kumagai, Y.; Kumar, H.; Koyama, S.; Kawai, T.; Takeuchi, O.; Akira, S. Cutting edge: TLR-dependent viral recognition along with type I IFN positive feedback signaling masks the requirement of viral replication for IFN-(alpha) production in plasmacytoid dendritic cells. J. Immunol. 2009, 182, 3960–3964. [Google Scholar] [CrossRef] [PubMed]
- Tough, D.F. Type I interferon as a link between innate and adaptive immunity through dendritic cell stimulation. Leuk. Lymphoma 2004, 45, 257–264. [Google Scholar] [CrossRef]
- Ivashkiv, L.B.; Donlin, L.T. Regulation of type I interferon responses. Nat. Rev. Immunol. 2014, 14, 36–49. [Google Scholar]
- Lee, J.-H.; Chiang, C.; Gack, M.U. Endogenous nucleic acid recognition by RIG-I-like receptors and cGAS. J. Interferon Cytokine Res. 2019, 39, 450–458. [Google Scholar] [CrossRef]
- Waldmann, T.A.; Chen, J. Disorders of the JAK/STAT pathway in T cell lymphoma pathogenesis: Implications for immunotherapy. Annu. Rev. Immunol. 2017, 35, 533–550. [Google Scholar] [CrossRef] [PubMed]
- Zaslavsky, E.; Hershberg, U.; Seto, J.; Pham, A.M.; Marques, S.; Duke, J.L.; Wetmur, J.G.; Tenoever, B.R.; Sealfon, S.C.; Kleinstein, S.H. Antiviral response dictated by choreographed cascade of transcription factors. J. Immunol. 2010, 184, 2908–2917. [Google Scholar] [CrossRef] [PubMed]
- Fournier, P.; Arnold, A.; Schirrmacher, V. Polarization of human monocyte-derived dendritic cells to DC1 by in vitro stimulation with Newcastle disease virus. J. BUON 2009, 14, S111–S122. [Google Scholar] [PubMed]
- Schirrmacher, V. Signaling through RIG-I and type I interferon receptor: Immune activation by Newcastle disease virus in man versus immune evasion by Ebola virus (Review). Int. J. Mol. Med. 2015, 36, 3–10. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, A.; Kim, S.H.; Manoharan, V.K.; Varghese, B.P.; Paldurai, A.; Samal, S.K. Novel avian paramyxovirus-based vaccine vectors expressing Ebola virus glycoprotein elicit mucosal and humoral immune responses in guinea pigs. Sci. Rep. 2019, 9, 5520. [Google Scholar] [CrossRef] [PubMed]
- Wilden, H.; Fournier, P.; Zawatzky, R.; Schirrmacher, V. Expression of RIG-I, IRF3, IFN-beta and IRF7 determines resistance or susceptibility of cells to infection by Newcastle disease virus. Int. J. Oncol. 2009, 34, 971–982. [Google Scholar] [PubMed]
- Reichard, K.W.; Lorence, R.M.; Cascino, C.L.; Peeples, M.E.; Walter, R.J.; Fernando, M.B.; Reyes, H.M.; Greager, J.A. Newcastle disease virus selectively kills human tumor cells. J. Surg. Res. 1992, 52, 448–453. [Google Scholar] [CrossRef]
- Phuangsab, A.; Lorence, R.M.; Reichard, K.W.; Peeples, M.E.; Walter, R.J. Newcastle disease virus therapy of human tumor xenografts: Antitumor effects of local or systemic administration. Cancer Lett. 2001, 172, 27–36. [Google Scholar] [CrossRef]
- Schnurr, M.; Duewell, P. Induction of immunogenic cell death by targeting RIG-I-like helicases in pancreatic cancer. Oncoimmunology 2014, 3, e955687. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Puhlmann, J.; Puehler, F.; Mumberg, D.; Boukamp, P.; Beier, R. Rac1 is required for oncolytic NDV replication in human cancer cells and establishes a link between tumorigenesis and sensitivity to oncolytic virus. Oncogene 2010, 29, 2205–2215. [Google Scholar] [CrossRef]
- Abdullah, J.M.; Mustafa, Z.; Ideris, A. Newcastle disease virus interaction in targeted therapy against proliferation and invasion pathways of glioblastoma multiforme. Biomed Res. Int. 2014, 2014, 386470. [Google Scholar] [CrossRef] [PubMed]
- Buijs, P.R.; van Eijck, C.H.; Hofland, L.J.; Fouchier, R.A.; van den Hoogen, B.G. Different responses of human pancreatic adenocarcinoma cell lines to oncolytic Newcastle disease virus infection. Cancer Gene Ther. 2014, 21, 24–30. [Google Scholar] [CrossRef]
- Buijs, P.; van Nieuwkoop, S.; Vaes, V.; Fouchier, R.; van Eijck, C.; van den Hoogen, B. Recombinant immunomodulating lentogenic or mesogenic oncolytic Newcastle disease virus for treatment of pancreatic adenocarcinoma. Viruses 2015, 7, 2980–2998. [Google Scholar] [CrossRef] [PubMed]
- Schwaiger, T.; Knitter, M.R.; Grund, C.; Roemer-Oberdoerfer, A.; Kapp, J.F.; Lerch, M.M.; Mettenleiter, T.C.; Mayerle, J.; Blohm, U. Newcastle disease virus mediates pancreatic tumor rejection via NK cell activation and prevents cancer relapse by prompting adaptive immunity. Int. J. Cancer 2017, 141, 2505–2516. [Google Scholar] [CrossRef] [PubMed]
- Duan, Z.; Xu, H.; Ji, X.; Zhao, J.; Xu, H.; Hu, Y.; Deng, S.; Hu, S.; Liu, X. Importin α5 negatively regulates importin ß1-mediated nuclear import of Newcasle disease virus matrix protein and viral replication and pathogenicity in chicken fibroblasts. Virulence 2018, 9, 783–803. [Google Scholar] [CrossRef] [PubMed]
- Kumar, N.; Khandelwal, N.; Kumar, R.; Chander, Y.; Rawat, K.D.; Chaubey, K.K.; Sharma, S.; Singh, S.V.; Riyesh, T.; Tripathi, B.N.; et al. Inhibitor of Sarco/Endoplasmatic reticulum calcium- ATPase impairs multiple steps of paramyxovirus replication. Front. Microbiol. 2019, 10, 209. [Google Scholar] [CrossRef] [PubMed]
- Ren, S.; Rehman, Z.U.; Shi, M.; Yang, B.; Qu, Y.; Yang, X.F.; Shao, Q.; Meng, C.; Yang, Z.; Gao, X.; et al. Syncytia generated by hemagglutinin-neuraminidase and fusion proteins of virulent Newcastle disease virus induce complete autophagy by activating AMPK-mTORC1-ULK1 signaling. Vet. Microbiol. 2019, 230, 283–290. [Google Scholar] [CrossRef] [PubMed]
- Szymanska, P.; Martin, K.R.; MacKeigan, J.P. Computational analysis of an autophagy/translation switch on mutual inhibition of MTORC1 and ULK. PLoS ONE 2015, 10. [Google Scholar] [CrossRef] [PubMed]
- Bai, L.; Koopmann, J.; Fiola, C.; Fournier, P.; Schirrmacher, V. Dendritic cells pulsed with viral oncolysaes potently stimulate autologous T cells from cancer patients. Int. J. Oncol. 2002, 21, 685–694. [Google Scholar]
- Alderson, T.R.; Roche, J.; Gastall, H.Y. Local unfolding of the HSP27 monomer regulates chaperon activity. Nat. Commun. 2019, 10, 1068. [Google Scholar] [CrossRef] [PubMed]
- Xu, T.; Jiang, L.; Wang, Z. The progression of HMGB1-induced autophagy in cancer biology. Oncotargets Ther. 2018, 12, 365–377. [Google Scholar] [CrossRef] [PubMed]
- Chun, Y.; Kim, J. Autophagy: An essential degradation program for cellular homeostasis and life. Cells 2018, 7, 278. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Qian, J.; Ding, J.; Li, J.; Nan, F.; Wang, W.; Qin, Q.; Fei, Y.; Xue, C.; Wang, J.; et al. Detection of viral components in exosomes derived from NDV-infected DF-1 cells and their promoting ability in virus replication. Microb. Pathog. 2019, 128, 414–422. [Google Scholar] [CrossRef] [PubMed]
- Zhou, C.; Tan, L.; Sun, Y.; Qiu, X.; Liao, Y.; Song, C.; Liu, W.; Nair, V.; Ding, C. Exosomes carry microRNAs into neighboring cells to promote diffusive infection of Newcastle disease virus. Viruses 2019, 11, 527. [Google Scholar] [CrossRef] [PubMed]
- Elankumaran, S.; Rockemann, D.; Samal, S.K. Newcastle disease virus exerts oncolysis by both intrinsic and extrinsic caspase-dependent pathways of cell death. J. Virol. 2006, 80, 7522–7534. [Google Scholar] [CrossRef] [PubMed]
- Liang, Y.; Song, D.Z.; Liang, S.; Zhang, Z.F.; Gao, L.X.; Fan, X.H. The hemagglutinin-neuraminidase protein of Newcastle disease virus upregulates expression of the TRAIL gene in murine natural killer cells through the activation of Syk and NF-κB. PLoS ONE 2017, 12, e0178746. [Google Scholar] [CrossRef] [PubMed]
- Ushmorov, A.; Ratter, F.; Lehmann, V.; Dröge, W.; Schirrmacher, V.; Umansky, V. Nitric-oxide-induced apoptosis in human leukemic lines requires mitochondrial lipid degradation and cytochrome C release. Blood 1999, 93, 2342–2352. [Google Scholar] [PubMed]
- Ren, S.; Rehman, Z.U.; Shi, M.; Yang, B.; Liu, P.; Yin, Y.; Qu, Y.; Meng, C.; Yang, Z.; Gao, X.; et al. Hemagglutinin-neuraminidase and fusion proteins of virulent Newcastle disease virus cooperately disturb fusion-fission homeostasis to enhance mitochondrial function by activating the unfolded protein response of endoplasmatic and reticulum stress. Vet. Res. 2019, 50, 37. [Google Scholar] [CrossRef]
- Bian, J.; Wang, K.; Kong, X.; Liu, H.; Chen, F.; Hu, M.; Zhang, X.; Jiao, X.; Ge, B.; Wu, Y.; et al. Caspase- and p38-MAPK-dependent induction of apoptosis in A549 lung cancer cells by Newcastle disease virus. Arch. Virol. 2011, 156, 1335–1344. [Google Scholar] [CrossRef]
- Samal, SK The Biology of Paramyxoviruses; Caister Academic Press: Wymondham, UK, 2011; pp. 1–469. ISBN 978-1-904455-85-1.
- Washburn, B.; Schirrmacher, V. Human tumor cell infection by Newcastle disease virus leads to upregulation of HLA and cell adhesion molecules and to induction of interferons, chemokines and finally apoptosis. Int. J. Oncol. 2002, 21, 85–93. [Google Scholar] [CrossRef]
- Jarahian, M.; Watzl, C.; Fournier, P.; Arnold, A.; Djandji, D.; Zahedi, S.; Cerwenka, A.; Paschen, A.; Schirrmacher, V.; Momburg, F. Activation of natural killer cells by Newcastle disease virus hemagglutinin-neuraminidase. J. Virol. 2009, 83, 8108–8121. [Google Scholar] [CrossRef] [PubMed]
- Koks, C.A.; Garg, A.D.; Ehrhardt, M.; Riva, M.; Vandenberk, L.; Boon, L.; De Vleeschouwer, S.; Agostinis, P.; Graf, N.; Van Gool, S.W. Newcastle disease virotherapy induces long-term survival and tumor-specific immune memory in orthotopic glioma through the induction of immunogenic cell death. Int. J. Cancer 2015, 136, E313–E325. [Google Scholar] [CrossRef] [PubMed]
- Liao, Y.; Wang, H.X.; Mao, X.; Fang, H.; Wang, H.; Li, Y.; Sun, Y.; Meng, C.; Tan, L.; Song, C.; et al. RIP1 is a central signaling protein in regulation of TNF-α/TRAIL mediated apoptosis and necroptosis during Newcastle disease virus infection. Oncotarget 2017, 8, 43201–43217. [Google Scholar] [CrossRef] [PubMed]
- Sharma, K.K.; Kalyani, I.H.; Mohapatra, J.; Patel, S.D.; Patel, D.R.; Vihol, P.D.; Chatterjee, A.; Patel, D.R.; Vyas, B. Evaluation of the oncolytic potential of R2B Mukteshwar vaccine strain of Newcastle disease virus (NDV) in a colon cancer cell line (SW-620). Arch. Virol. 2017, 162, 2705–2713. [Google Scholar] [CrossRef] [PubMed]
- Kalyanasundram, J.; Hamid, A.; Yusoff, K.; Chia, S.L. Newcastle disease virus strain AF2240 as an oncolytic virus: A review. Acta Trop. 2018, 183, 126–133. [Google Scholar] [CrossRef] [PubMed]
- Raihan, J.; Ahmad, U.; Yong, Y.K.; Eshak, Z.; Othman, F.; Ideris, A. Regression of solid breast tumors in mice by Newcastle disease virus is associated with production of apoptosis-related cytokines. BMC Cancer 2019, 19, 315. [Google Scholar]
- Ali-Saeed, R.; Alabsi, A.M.; Ideris, A.; Omar, A.R.; Yusoff, K.; Ali, A.M. Evaluation of ultra-microscopic changes and proliferation of apoptotic glioblastoma multiforme cells induced by velogenic strain of Newcastle disease virus AF2240. Asian Pac. J. Cancer Prev. 2019, 20, 757–765. [Google Scholar] [CrossRef] [PubMed]
- Washburn, B.; Weigand, M.A.; Grosse-Wilde, A.; Janke, M.; Stahl, H.; Rieser, E.; Sprick, M.R.; Schirrmacher, V.; Walczak, H. TNF-related apoptosis-inducing ligand mediates tumoricidal activity of human monocytes stimulated Newcastle disease virus. J. Immunol. 2003, 170, 1814–1821. [Google Scholar] [CrossRef] [PubMed]
- Schirrmacher, V.; Bai, L.; Umansky, V.; Yu, L.; Xing, Y.; Qian, Z. Newcastle disease virus activates macrophages for anti-tumor activity. Int. J. Oncol. 2000, 16, 363–373. [Google Scholar] [CrossRef] [PubMed]
- Umansky, V.; Shatrov, V.A.; Lehmann, V.; Schirrmacher, V. Induction of NO synthesis in macrophages by Newcastle disease virus is associated with activation of nuclear factor-kappa B. Int. Immunol. 1996, 8, 491–498. [Google Scholar] [CrossRef]
- Schirrmacher, V.; Umansky, V. Caspase activation is required for nitric oxide-mediated, CD95(APO-1/Fas)-dependent and independent apoptosis in human neoplastic lymphoid cells. Blood 1998, 91, 4311–4320. [Google Scholar]
- Umansky, V.; Schirrmacher, V. Nitric oxide-induced apoptosis in tumor cells. Adv. Cancer Res. 2001, 82, 107–131. [Google Scholar] [PubMed]
- Vander Lugt, B.; Riddel, J.; Khan, A.A.; Hackney, J.A.; Lesch, J.; De Voss, J.; Weirauch, M.T.; Singh, H.; Mellman, I. Transcriptional determinants of tolerogenic and immunogenic states during dendritic cell maturation. J. Cell Biol. 2017, 216, 779–792. [Google Scholar] [CrossRef] [PubMed]
- Lattanzi, L.; Rozera, C.; Marescotti, D.; D’Agostino, G.; Santodonato, L.; Cellini, S.; Belardelli, F.; Gavioli, R.; Ferrantini, M. IFN-α boosts epitope cross-presentation by dendritic cells via modulation of proteasome activity. Immunobiology 2011, 216, 537–547. [Google Scholar] [CrossRef] [PubMed]
- Lorenzi, S.; Mattei, F.; Sistigu, A.; Bracci, L.; Spadaro, F.; Sanchez, M.; Spada, M.; Belardelli, F.; Gabriele, L.; Schiavoni, G. Type I IFNs control antigen retention and survival of CD8 (+) dendritic cells after uptake of tumor apoptotic cells leading to cross-priming. J. Immunol. 2011, 186, 5142–5150. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Niu, C.; Shi, X.; Xu, D.; Li, M.; Cui, J.; Li, W.; Xu, J.; Jin, H. Dendritic cells loaded with the lysate of tumor cells infected with Newcastle diasease virus trigger potent anti-tumor immunity by promoting the secretion of IFN-γ and IL-2 from T cells. Oncol. Lett. 2018, 16, 1180–1188. [Google Scholar] [PubMed]
- Schirrmacher, V.; Lorenzen, D.; Van Gool, S.W.; Stuecker, W. A new strategy of cancer immunotherapy combining hyperthermia/oncolytic virus pretreatment with specific autologous anti-tumor vaccination—A Review. Austin Oncol. Case Rep. 2017, 2, 1006. [Google Scholar]
- Garg, A.D.; Coulie, P.G.; Van den Eynde, B.J.; Agostinis, P. Integrating next-generation dendritic cell vaccines into the current cancer immunotherapy landscape. Trends Immunol. 2017, 1392, 1–17. [Google Scholar] [CrossRef]
- Haas, C.; Ertel, C.; Gerhards, R.; Schirrmacher, V. Introduction of adhesive and costimulatory immune functions into tumor cells by infection with Newcastle disease virus. Int. J. Oncol. 1998, 13, 1105–1115. [Google Scholar] [CrossRef]
- Schirrmacher, V.; Haas, C.; Bonifer, R.; Ertel, C. Virus potentiation of tumor vaccine T-cell stimulatory capacity requires cell surface binding but not infection. Clin. Cancer Res. 1997, 3, 1135–1148. [Google Scholar]
- Schild, H.; von Hoegen, P.; Schirrmacher, V. Modification of tumor cells by a low dose of Newcastle disease virus. II. Augmented tumor-specific T cell response as a result of CD4+ and CD8+ immune T cell cooperation. Cancer Immunol. Immunother. 1989, 28, 22–28. [Google Scholar] [CrossRef] [PubMed]
- Ertel, C.; Millar, N.S.; Emmerson, P.T.; Schirrmacher, V.; von Hoegen, P. Viral hemagglutinin augments peptide-specific cytotoxic T cell responses. Eur. J. Immunol. 1993, 23, 2592–2596. [Google Scholar] [CrossRef] [PubMed]
- Von Hoegen, P.; Weber, E.; Schirrmacher, V. Modification of tumor cells by a low dose of Newcastle disease virus. I. Augmentation of the tumor-specific T cell response in the absence of an anti-viral response. Eur. J. Immunol. 1988, 18, 1159–1166. [Google Scholar] [CrossRef] [PubMed]
- Von Hoegen, P.; Zawatzky, R.; Schirrmacher, V. Modification of tumor cells by a low dose of Newcastle disease virus. III. Potentiation of tumor-specific cytolytic activity via induction of interferon-alpha/beta. Cell. Immunol. 1990, 126, 80–90. [Google Scholar] [CrossRef]
- Termeer, C.C.; Schirrmacher, V.; Bröcker, E.B.; Becker, J.C. Newcastle disease virus infection induces B7-1/B7-2-independent T-cell costimulatory activity in human melanoma cells. Cancer Gene Ther. 2000, 7, 316–323, 2000. [Google Scholar] [CrossRef] [PubMed]
- Zeng, J.; Fournier, P.; Schirrmacher, V. Induction of interferon-alpha and tumor necrosis factor-related apoptosis-inducing ligand in human blood mononuclear cells by hemagglutinin-neuraminidase but not F protein of Newcastle disease virus. Virology 2002, 297, 19–30. [Google Scholar] [CrossRef] [PubMed]
- Zeng, J.; Fournier, P.; Schirrmacher, V. Stimulation of human natural interferon-alpha response via paramyxovirus hemagglutinin lectin–cell interaction. J. Mol. Med. 2002, 80, 443–451. [Google Scholar] [CrossRef]
- Fournier, P.; Zeng, J.; Schirrmacher, V. Two ways to induce innate immune responses in human PBMCs: Paracrine stimulation of IFN-alpha responses by viral protein or dsRNA. Int. J. Oncol. 2003, 23, 673–680. [Google Scholar] [CrossRef]
- Ni, J.; Schirrmacher, V.; Fournier, P. The hemagglutinin-neuraminidase gene of Newcastle disease virus: A powerful molecular adjuvant for DNA anti-tumor vaccination. Vaccine 2010, 28, 6891–6900. [Google Scholar] [CrossRef]
- Sobo-Vujanovic, A.; Munich, S.; Vujanovic, N.L. Dendritic-cell exosomes cross-present Toll-like receptor-ligands and activate bystander dendritic cells. Cell. Immunol. 2014, 289, 119–127. [Google Scholar] [CrossRef]
- Kalinski, P.; Maillard, R.B.; Giermasz, A.; Zeh, H.J.; Basse, P.; Bartlett, D.L.; Kirkwood, J.M.; Lotze, M.T.; Herberman, R.B. Natural killer-dendritic cell cross-talk in cancer immunotherapy. Expert Opin. Ther. 2005, 5, 1303–1315. [Google Scholar] [CrossRef] [PubMed]
- Lucas, M.; Schachterle, W.; Oberle, K.; Aichele, P.; Diefenbach, A. Dendritic cells prime natural killer cells by trans-presenting interleukin 15. Immunity 2007, 26, 503–517. [Google Scholar] [CrossRef] [PubMed]
- Crouse, J.; Bedenikovic, G.; Wiesel, M.; Ibberson, M.; Xenarios, I.; Von Laer, D.; Kalinke, U. Type I interferons protect T cells against NK cell attack mediated by the activating receptor NCR1. Immunity 2014, 40, 961–973. [Google Scholar] [CrossRef] [PubMed]
- Graham, J.P.; Authie, P.; Karolina Palucka, A.; Zurawski, G. Targeting interferon-alpha to dendritic cells enhances a CD8+ T cell response to a human CD40-targeted cancer vaccine. Vaccine 2017, 35, 4532–4539. [Google Scholar] [CrossRef]
- Tiao, M.M.; Lu, L.; Huang, L.T.; Liang, C.D.; Chen, C.L.; Tao, R.; Fung, J.J.; Qian, S. Cross-tolerance of recipient-derived transforming growth factor-beta dendritic cells. Transplant. Proc. 2007, 39, 281–282. [Google Scholar] [CrossRef]
- Raftery, M.J.; Schwab, M.; Diesner, S.; Egerer, G.; Schönrich, G. Dendritic cells cross-presenting viral antigens derived from autologous cells as a sensitive tool for visualization of human cytomegalovirus-reactive CD8+ T cells. Transplantation 2002, 73, 998–1002. [Google Scholar] [CrossRef]
- Blachère, N.E.; Morris, H.K.; Braun, D.; Saklani, H.; Di Santo, J.P.; Darnell, R.B.; Albert, M.L. IL-2 is required for the activation of memory CD8+ T cells via antigen cross-presentation. J. Immunol. 2006, 176, 7288–7300. [Google Scholar] [CrossRef]
- Bai, L.; Beckhove, P.; Feuerer, M.; Umansky, V.; Choi, C.; Solomayer, F.S.; Diel, I.J.; Schirrmacher, V. Cognate interactions between memory T cells and tumor antigen-presenting dendritic cells from bone marrow of breast cancer patients: Bidirectional cell stimulation, survival and antitumor immunity in vivo. Int. J. Cancer. 2003, 103, 73–83. [Google Scholar] [CrossRef]
- Schirrmacher, V. Oncolytic Newcastle disease virus as a prospective anti-cancer therapy. A biological agent with potential to break therapy resistance. Exp. Opin. Biol. Ther. 2015, 15, 1–15. [Google Scholar] [CrossRef]
- Schirrmacher, V.; Jurianz, K.; Roth, C.; Griesbach, A.; Bonifer, R.; Zawatzky, R. Tumor stimulator cell modification by infection with Newcastle disease virus: Analysis of effects and mechanism in MLT-CML cultures. Int. J. Oncol. 1999, 14, 205–215. [Google Scholar] [CrossRef]
- Meng, S.; Zhou, Z.; Chen, F.; Kong, X.; Liu, H.; Jiang, K.; Liu, W.; Hu, M.; Zhang, X.; Ding, C.; et al. Newcastle disease virus induces apoptosis in cisplatin-resistant human lung adenocarcinoma A549 cells in vitro and in vivo. Cancer Lett. 2012, 317, 56–64. [Google Scholar] [CrossRef] [PubMed]
- Jiang, K.; Li, Y.; Xu, J.; Wang, Y.; Deng, W.; Liu, Q.; Zhang, G.; Meng, S. Pharmacological modulation of autophagy enhances Newcastle disease virus-mediated oncolysis in drug-resistant lung cancer cells. BMC Cancer 2014, 14, 551. [Google Scholar] [CrossRef] [PubMed]
- Bai, Y.; Chen, Y.; Hong, X.; Liu, X.; Su, X.; Li, S.; Dong, X.; Zhao, G.; Li, Y. Newcastle disease virus enhances the growth-inhibiting and proapoptotic effects of temozolomide on glioblastoma cells in vitro and in vivo. Sci. Rep. 2018, 8, 11470. [Google Scholar] [CrossRef] [PubMed]
- Mansour, M.; Palese, P.; Zamarin, D. Oncolytic specificity of Newcastle disease virus is mediated by selectivity for aopotosis-resistant cells. J. Virol. 2011, 85, 6015–6023. [Google Scholar] [CrossRef] [PubMed]
- Lazar, I.; Yaacov, B.; Shiloach, T. The oncolytic activity of Newcastle disease virus NDV-HUJ on chemoresistant primary melanoma cells is dependent on the proapoptotic activity of the inhibitor of apoptosis protein Livin. J. Virol. 2010, 84, 639–646. [Google Scholar] [CrossRef] [PubMed]
- Ch’ng, W.C.; Stanbridge, E.J.; Yusoff, K.; Shafee, N. The oncolytic activity of Newcastle disease virus in clear cell renal carcinoma cells in normoxic and hypoxic conditions: The interplay between von Hippel-Lindau and interferon-β signaling. J. Interferon Cytokine Res. 2013, 33, 346–354. [Google Scholar] [CrossRef] [PubMed]
- Liu, N.; Long, Y.; Liu, B.; Yang, D.; Li, C.; Chen, T.; Wang, X.; Liu, C.; Zhu, H. ISG12a mediates cell response to Newcastle disease viral infection. Virology 2014, 462–463, 283–294. [Google Scholar] [CrossRef]
- Zamarin, D.; Holmgaard, R.B.; Subudhi, S.K.; Park, J.S.; Mansour, M.; Palese, P.; Merghoub, T.; Wolcheok, J.D.; Allison, J.P. Localized oncolytic virotherapy overcomes systemic tumor resistance to immune checkpoint blockade immunotherapy. Sci. Transl. Med. 2014, 6, 226ra32. [Google Scholar] [CrossRef]
- Oseledchyk, A.; Ricca, J.M.; Gigoux, M.; Ko, B.; Redelman-Sidi, G.; Walther, T.; Liu, C.; Iyer, G.; Merghoub, T.; Wolchok, J.D.; et al. Lysis-independent potentiation of immune checkpoint blockade by oncolytic virus. Oncotarget 2018, 9, 28702–28716. [Google Scholar] [CrossRef][Green Version]
- Apostolidis, L.; Schirrmacher, V.; Fournier, P. Host mediated anti-tumor effect of oncolytic Newcastle disease virus after locoregional application. Int. J. Oncol. 2007, 31, 1009–1019. [Google Scholar]
- Ricca, J.M.; Oseledchyk, A.; Walther, T.; Liu, C.; Mangarin, L.; Merghoub, T.; Wolchok, J.D.; Zamarin, D. Pre-existing immunity to oncolytic virus potentiates its immunotherapeutic efficacy. Mol. Ther. 2018, 26, 1008–1019. [Google Scholar] [CrossRef] [PubMed]
- Harrington, K.J.; Vile, R.G.; Pandha, H.S. (Eds.) Viral Therapy of Cancer; Wiley & Sons: Hoboken, NJ, USA, 2008; pp. 1–404. [Google Scholar]
- Lundstrom, K. Viral vectors in gene therapy. Diseases 2018, 6, 42. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; He, B. Selective editing of herpes simplex virus 1 enables interferon induction and viral replication that destroy malignant cells. J. Virol. 2019, 93. [Google Scholar] [CrossRef] [PubMed]
- Schuster, P.; Lindner, G.; Thomann, S.; Haferkamp, S.; Schmidt, B. Prospect of plasmacytoid dendritic cells in enhancing anti-tumor immunity of oncolytic herpes viruses. Cancers 2019, 11, 651. [Google Scholar] [CrossRef]
- Thomas, S.; Kuncheria, L.; Roulstone, V.; Kyula, J.N.; Mansfield, D.; Bommareddy, P.K.; Smith, H.; Kaufman, H.L.; Harrington, K.J.; Coffin, R.S. Development of a new fusion-enhanced oncolytic immunotherapy platform based on herpes simplex virus type 1. J. Immunother. Cancer 2019, 7, 214. [Google Scholar] [CrossRef] [PubMed]
- Raman, S.S.; Hecht, J.R.; Chan, E. Talimogene laherparepvec: Review of its mechanism of action and clinical efficacy and safety. Immunotherapy 2019, 11, 705–723. [Google Scholar] [CrossRef] [PubMed]
- Stepanenko, A.A.; Chekhonin, V.P. A compendium of adenovirus genetic modifications for enhanced replication, oncolysis, and tumor immunosurveillance in cancer therapy. Gene 2018, 679, 11–18. [Google Scholar] [CrossRef]
- Tazawa, H.; Kuroda, S.; Hasei, J.; Kagawa, S.; Fujiwara, T. Impact of autophagy in oncolytic adenoviral therapy for cancer. Int. J. Mol. Sci. 2017, 18, 1479. [Google Scholar] [CrossRef]
- Diaconu, I.; Cerullo, V.; Hirvinen, M.L.; Escutenaire, S.; Ugolini, M.; Pesonen, S.K.; Bramante, S.; Parviainen, S.; Kanerva, A.; Loskog, A.S. Immune response is an important aspect of the antitumor effect produced by a CD40L-encoding oncolytic adenovirus. Cancer Res. 2012, 72, 2327–2338. [Google Scholar] [CrossRef]
- Havunen, R.; Santos, J.M.; Sorsa, S.; Rantapero, T.; Lumen, D.; Siurala, M.; Airaksiren, A.J.; Cervera-Carrascon, V.; Tähtinen, S.; Kanerva, A.; et al. Abscopal effect in non-injected tumors achieved with cytokine-armed oncolytic adenovirus. Mol. Ther. Oncolytics 2018, 11, 109–121. [Google Scholar] [CrossRef]
- Shoji, M.; Tachibana, M.; Katayama, K.; Tomita, K.; Tsuzuki, S.; Sakurai, F.; Kawabata, K.; Ishii, K.J.; Akira, S.; Mizuguchi, H. Type-I IFN signaling is required for the induction of antigen-specific CD8(+) T cell responses by adenovirus vector vaccine in the gut mucosa. Biochem. Biophys. Res. Commun. 2012, 425, 89–93. [Google Scholar] [CrossRef] [PubMed]
- Tähtinen, S.; Grönberg-Vähä-Koskela, S.; Lumen, D.; Merisalo-Soikkeli, M.; Siuraha, M.; Airaksinen, A.J.; Vähä-Koskela, M.; Hemminki, A. Adenovirus improves the efficacy of adoptive T-cell therapy by recruiting immune cells to and promoting their acticity at the tumor. Cancer Immunol. Res. 2015, 3, 915–925. [Google Scholar] [CrossRef] [PubMed]
- Sakhawat, A.; Ma, L.; Muhammad, T.; Khan, A.A.; Chen, X.; Huang, Y. A tumor targeting oncolytic adenovirus can improve therapeutic outcomes in chemotherapy resistant metastatic human breast carcinoma. Sci. Rep. 2019, 9, 7504. [Google Scholar] [CrossRef] [PubMed]
- Müller, L.M.E.; Holmes, M.; Michael, J.L.; Scott, G.B.; West, E.J.; Scott, K.J.; Parrish, C.; Hall, K.; Stäble, S.; Jennings, V.A. Plasmacytoid dendritic cells orchestrate innate and adaptive anti-tumor immunity induced by oncolytic coxsackievirus A21. J. Immunother. Cancer. 2019, 7, 164. [Google Scholar] [CrossRef] [PubMed]
- Annels, N.E.; Arif, M.; Simpson, G.R.; Denyer, M.; Moller-Levet, C.; Mansfield, D.; Butler, R.; Shafren, D.; Au, G.; Knowles, M. Oncolytic immunotherapy for bladder cancer using coxsackie A21 virus. Mol. Ther. Oncolytics 2018, 9, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Ogata, H.; Takishima, Y.; Miyamotos, S.; Inoue, H.; Kuroda, M.; Yamada, K.; Hijikata, Y.; Murahashi, M.; Shimizu, H.; et al. A novel combination therapy for human oxaliplatin-resistant colorectal cancer using oxaliplatin and coxsackievirus A11. Anticancer Res. 2018, 38, 6121–6126. [Google Scholar] [CrossRef]
- Delauny, T.; Violland, M.; Boisgerault, N.; Dutoit, S.; Vignard, V.; Münz, C.; Gannage, M.; Drèno, B.; Vaivode, K.; Pjanova, D. Oncolytic viruses sensitize human tumor cells for NY-ESO-1 tumor antigen recognition by CD4+ effector T cells. Oncoimmunology 2017, 7, e1407897. [Google Scholar] [CrossRef]
- Cassel, W.A.; Garrett, R.E. Tumor immunity after viral oncolysis. J. Bacteriol. 1966, 92, 792. [Google Scholar]
- Cassel, W.A.; Murray, D.R. A ten-year follow-up on stage II malignant melanoma patients treated postsurgically with Newcastle disease virus oncolysate. Med. Oncol. Tumor Pharmacother. 1992, 9, 169–171. [Google Scholar]
- Kirchner, H.H.; Anton, P.; Atzpodien, J. Adjuvant treatment of locally advanced renal cancer with autologous virus-modified tumor vaccines. World J. Urol. 1995, 13, 171–173. [Google Scholar] [CrossRef]
- Heicappell, R.; Schirrmacher, V.; von Hoegen, P.; Ahlert, T.; Appelhans, B. Prevention of metastatic spread by postoperative immunotherapy with virally modified autologous tumor cells. I. Parameters for optimal therapeutic effects. Int. J. Cancer 1986, 37, 569–577. [Google Scholar] [CrossRef] [PubMed]
- Schirrmacher, V.; Heicappell, R. Prevention of metastatic spread by postoperative immunotherapy with virally modified autologous tumor cells. II. Establishment of specific systemic immunity. Clin. Exp. Metastasis 1987, 5, 147–156. [Google Scholar] [CrossRef] [PubMed]
- Schirrmacher, V.; Haas, C.; Bonifer, R.; Ahlert, T.; Gerhards, R.; Ertel, C. Human tumor cell modification by virus infection: An efficient and safe way to produce cancer vaccine with pleiotropic immune stimulatory properties when using Newcastle disease virus. Gene Ther. 1999, 6, 63–73. [Google Scholar] [CrossRef] [PubMed]
- Ockert, D.; Schirrmacher, V.; Beck, N.; Stoelben, E.; Ahlert, T.; Flechtenmacher, J.; Hagmüller, E.; Buchcik, R.; Nagel, M.; Saeger, H.D. Newcastle disease virus-infected intact autologous tumor cell vaccine for adjuvant active specific immunotherapy of resected colorectal carcinoma. Clin. Cancer Res. 1996, 2, 21–28. [Google Scholar] [PubMed]
- Ahlert, T.; Sauerbrei, W.; Bastert, G.; Ruhland, S.; Bartik, B.; Simiantonaki, N.; Schumacher, J.; Häcker, B.; Schumacher, M.; Schirrmacher, V. Tumor-cell number and viability as quality and efficacy parameters of autologous virus-modified cancer vaccines in patients with breast and ovarian cancer. J. Clin. Oncol. 1997, 15, 2763. [Google Scholar] [CrossRef]
- Steiner, H.H.; Bonsanto, M.M.; Beckhove, P.; Brysch, M.; Geletneky, K.; Ahmadi, R.; Schuele-Freyer, R.; Kremer, P.; Ranaie, G.; Matejic, D. Antitumor vaccination of patients with glioblastoma multiforme: A pilot study to assess feasibility, safety, and clinical benefit. J. Clin. Oncol. 2004, 22, 4272–4281. [Google Scholar] [CrossRef] [PubMed]
- Karcher, J.; Dyckhoff, G.; Beckhove, P.; Reisser, C.; Brysch, M.; Ziouta, Y.; Helmke, B.H.; Weidauer, H.; Schirrmacher, V.; Herold-Mende, C. Antitumor vaccination in patients with head and neck squamous cell carcinomas with autologous virus-modified tumor cells. Cancer Res. 2004, 64, 8057–8061. [Google Scholar] [CrossRef]
- Schulze, T.; Kemmner, W.; Weitz, J.; Wernecke, K.D.; Schirrmacher, V.; Schlag, P.M. Efficiency of adjuvant active specific immunization with Newcastle disease virus modified tumor cells in colorectal carcinoma patients following resection of liver metastases. Results of a prospective randomized trial. Cancer Immunol. Immunother. 2009, 58, 61–69. [Google Scholar] [CrossRef] [PubMed]
- Schirrmacher, V.; Fournier, P.; Schlag, P. Autologous tumor cell vaccines for post-operative active-specific immunotherapy of colorectal carcinoma: Long-term patient survival and mechanism of function. Expert Rev. Vaccines 2014, 13, 117–130. [Google Scholar] [CrossRef] [PubMed]
- Liang, W.; Wang, H.; Sun, T.M.; Yao, W.Q.; Chen, L.L.; Jin, Y.; Li, C.L.; Meng, F.J. Application of autologous tumor cell vaccine and NDV vaccine in treatment of tumors of the digestive tract. World J. Gastroenterol. 2003, 9, 495–498. [Google Scholar] [CrossRef] [PubMed]
- Csatary, L.K.; Gosztonyi, G.; Szeberenyi, J.; Fabian, Z.; Liszka, V.; Bodey, B.; Csatary, C.M. MTH-68/H oncolytic viral treatment in human high-grade gliomas. J. Neurooncol. 2004, 67, 83–93. [Google Scholar] [CrossRef] [PubMed]
- Csatary, L.K.; Eckhardt, S.; Bukosza, I.; Czegledi, F.; Fenyvesi, C.; Gergely, P.; Bodey, B.; Csatary, C.M. Attenuated veterinary virus vaccine for the treatment of cancer. Cancer Detect. Prev. 1993, 17, 619–627. [Google Scholar] [PubMed]
- Freeman, A.I.; Zakay-Rones, Z.; Gomori, J.M.; Linetsky, E.; Rasooly, L.; Greenbaum, E.; Rozenman-Yair, S.; Panet, A.; Libson, E.; Irving, C.S.; et al. Phase I/II trial of intravenous DNV-HUJ oncolytic virus in recurrent glioblastoma multiforme. Mol. Ther. 2006, 13, 221–228. [Google Scholar] [CrossRef]
- Pecora, A.L.; Rizvi, N.; Cohen, G.I.; Meropol, N.J.; Sterman, D.; Marshall, I.L.; Goldberg, S.; Gross, P.; O’Neil, J.D.; Groene, W.S. Phase I trial of intravenous administration of PV701, an oncolytic virus, in patients with advanced cancer. J. Clin. Oncol. 2002, 20, 2251–2266. [Google Scholar] [CrossRef] [PubMed]
- Lorence, R.M.; Pecora, A.L.; Major, P.P.; Hotte, S.J.; Laurie, S.A.; Roberts, M.S.; Groene, W.S.; Bamat, M.K. Overview of phase I studies of intravenous administration of PV701, an oncolytic virus. Curr. Opin. Mol. Ther. 2003, 5, 618–624. [Google Scholar]
- Li, Y.L.; Wu, J.; Wie, D.; Zhang, D.W.; Feng, H.; Chen, Z.N.; Bian, H. Newcastle disease virus represses the activation of human hepatic stellate cells and reverses the development of hepatic fibrosis in mice. Liver Int. 2009, 29, 593–602. [Google Scholar] [CrossRef] [PubMed]
- Haas, C.; Herold-Mende, C.; Gerhards, R.; Schirrmacher, V. An effective strategy of human tumor vaccine modification by coupling bispecific costimulatory molecules. Cancer Gene Ther. 1999, 6, 254–262. [Google Scholar] [CrossRef]
- Haas, C.; Lulei, M.; Fournier, P.; Arnold, A.; Schirrmacher, V. A tumor vaccine containing anti-CD3 and anti-CD28 bispecific antibodies triggers strong and durable antitumor activity in human lymphocytes. Int. J. Cancer 2006, 118, 658–667. [Google Scholar] [CrossRef] [PubMed]
- Aigner, M.; Janke, M.; Lulei, M.; Beckhove, P.; Fournier, P.; Schirrmacher, V. An effective tumor vaccine optimized for costimulation via bispecific and trispecific fusion proteins. Int. J. Oncol. 2008, 32, 777–789. [Google Scholar]
- Fournier, P.; Aigner, M.; Schirrmacher, V. Transcriptome analysis and cytokine profiling of naive T cells stimulated by a tumor vaccine via CD3 and CD25. Int. J. Oncol. 2010, 37, 1439–1452. [Google Scholar] [PubMed]
- Schirrmacher, V.; Schlude, C.; Weitz, J.; Beckhove, P. Strong T-cell costimulation can reactivate tumor antigen-specific T cells in late-stage metastasized colorectal carcinoma patients: Results from a phase I clinical study. Int. J. Oncol. 2015, 46, 71–77. [Google Scholar] [CrossRef]
- Fournier, P.; Schirrmacher, V. Bispecific antibodies and trispecific immunocytokines for targeting the immune system against cancer: Preparing for the future. Biodrugs 2013, 27, 35–53. [Google Scholar] [CrossRef] [PubMed]
- Adkins, I.; Fucikova, J.; Garg, A.D.; Agostinis, P.; Spisek, R. Physical modalities inducing immunogenic tumor cell death for cancer immunotherapy. Oncoimmunology 2015, 3, e968434. [Google Scholar] [CrossRef]
- Hammerich, L.; Binder, A.; Brody, J.D. In situ vaccination: Cancer immunotherapy both personalized and off-the-shelf. Mol. Oncol. 2015, 9, 1966–1981. [Google Scholar] [CrossRef]
- Nishiyama-Fujita, Y.; Kawana-Tachikawa, A.I.; Ono, T.; Tanaka, Y.; Kato, T.; Heslop, H.E.; Morio, T.; Takahashi, S. Generation of multivirus-specific T cells by a single stimulation of peripheral blood mononuclear cells with a peptide mixture using serum-free medium. Cytotherapy 2018, 20, 1182–1190. [Google Scholar] [CrossRef]
- Schirrmacher, V.; Bihari, A.S.; Stuecker, W.; Sprenger, T. Long-term remission of prostate cancer with extensive bone metastases upon immuno- and virotherapy: A case report. Oncol. Lett. 2014, 8, 2403–2406. [Google Scholar] [CrossRef] [PubMed]
- Schirrmacher, V.; Stuecker, W.; Lulei, M.; Bihari, A.S.; Sprenger, T. Long-term survival of a breast cancer patient with extensive liver metastases upon immune and virotherapy: A case report. Immunotherapy 2015, 7, 855–860. [Google Scholar] [CrossRef] [PubMed]
- Van Gool, S.W.; Makalowski, J.; Feyen, O.; Prix, L.; Schirrmacher, V.; Stuecker, W. The induction of immunonogenic cell death (ICD) during maintenance chemotherapy and subsequent multimodal immunotherapy for glioblastoma (GBM). Austin Oncol. Case Rep. 2018, 3, 1010. [Google Scholar]
- Van Gool, S.W.; Feyen, O.; Makalowski, J.; Neinhuis, A.; Schirrmacher, V.; Stuecker, W. How to monitor immunogenic cell death in patients with glioblastoma. Neuro Oncol. 2018, 20, v17–v18. [Google Scholar] [CrossRef][Green Version]
- Coy, J.F. EDIM-TKTL1/Apo10 blood test: An innate immune system based liquid biopsy for the early detection, characterization and targeted treatment of cancer. Int. J. Mol. Sci. 2017, 18, 878. [Google Scholar] [CrossRef]
- Schirrmacher, V. From chemotherapy to biological therapy: A review of novel concepts to reduce the side effects of systemic cancer treatment (Review). Int. J. Oncol. 2019, 54, 407–419. [Google Scholar] [PubMed]
- Schirrmacher, V. Quo Vadis Cancer Therapy? Fascinating Discoveries of the Last 60 Years; Lambert Academic Publishing: Saarbrücken, Germany, 2017; pp. 1–353. [Google Scholar]
- Walther, W.; Stein, U.S. Gene Therapy of Cancer: Methods and Protocols. In Methods in Molecular Biology 542, Springer Protocols; Humana Press: Totowa, NY, USA, 2009; pp. 1–731. [Google Scholar]
- Fournier, P.; Bian, H.; Szeberényi, J.; Schirrmacher, V. Analysis of three properties of Newcastle disease virus for fighting cancer: Tumor-selective replication, antitumor cytotoxicity, and immunostimulation. Methods Mol. Biol. 2012, 797, 177–204. [Google Scholar]
- Conry, R.M.; Westbrook, B.; McKee, S.; Norwood, T.G. Talimogene laherparepvec: First in class oncolytic virotherapy. Hum. Vaccin. Immunother. 2018, 14, 839–846. [Google Scholar] [CrossRef]
- Zhang, Z.; Zhao, W.; Li, D.; Yang, J.; Zsak, L.; Yu, Q. Development of a Newcastle disease virus vector expressing a foreign gene through an internal ribosomal entry site provides direct proof for a sequential transcription mechanism. J. Gen. Virol. 2015, 96, 2028–2035. [Google Scholar] [CrossRef]
- Vigil, A.; Martinez, O.; Chua, M.A.; Garcia-Sastre, A. Recombinant Newcastle disease virus as a vaccine vector for cancer therapy. Mol. Ther. 2008, 16, 1883–1890. [Google Scholar] [CrossRef]
- Shobana, R.; Samal, S.K.; Elankumaran, S. Prostate-specific antigen-retargeted recombinant newcastle disease virus for prostate cancer virotherapy. J. Virol. 2013, 87, 3792–3800. [Google Scholar] [CrossRef] [PubMed]
- Wei, D.; Li, Q.; Wang, X.L.; Wang, Y.; Xu, J.; Feng, F.; Nan, G.; Wang, B.; Li, C.; Guo, T.; et al. Oncolytic Newcastle disease virus expressing chimeric antibody enhanced anti-tumor efficacy in orthotopic hepatoma-bearing mice. J. Exp. Clin. Cancer Res. 2015, 34, 153. [Google Scholar] [CrossRef] [PubMed]
- Janke, M.; Peeters, B.; de Leeuw, O.; Moorman, R.; Arnold, R.; Fournier, P.; Schirrmacher, V. Recombinant Newcastle diasese virus (NDV) with inserted gene coding for GM-CSF as a new vector for cancer immunogene therapy. Gene Ther. 2007, 14, 1639–1649. [Google Scholar] [CrossRef]
- Janke, M.; Peeters, B.; Zhao, H.; de Leeuw, O.; Moorman, R.; Arnold, A.; Ziouta, Y.; Fournier, P.; Schirrmacher, V. Activation of human T cells by a tumor vaccine infected with recombinant Newcastle disease virus producing IL-2. Int. J. Oncol. 2008, 33, 823–832. [Google Scholar] [PubMed]
- Xu, X.; Sun, Q.; Yu, X.; Zhao, L. Rescue of nonlytic Newcastle disease virus (NDV) expressing IL-15 for cancer immunotherapy. Virus Res. 2017, 233, 35–41. [Google Scholar] [CrossRef]
- Zamarin, D.; Holmgaard, R.B.; Ricca, J.; Plitt, T.; Palese, P.; Sharma, P.; Merghoub, T.; Wolchok, J.D.; Allison, J.P. Intratumoral modulation of the inducible co-stimulator ICOS by recombinant oncolytic virus promotes systemic anti-tumor immunity. Nat. Commun. 2017, 8, 14340. [Google Scholar] [CrossRef] [PubMed]
- Bai, F.L.; Yu, Y.H.; Tian, H.; Ren, G.P.; Wang, H.; Zhou, B.; Han, X.H.; Yu, Q.Z.; Li, D.S. Genetically engineered Newcastle disease virus expressing interleukin-2 and TNF-related apoptosis-inducing ligand for cancer therapy. Cancer Biol. Ther. 2014, 15, 1226–1238. [Google Scholar] [CrossRef]
- Cuadrado-Castano, S.; Ayllon, J.; Mansour, M.; de la Iglesia-Vicente, J.; Jordan, S.; Tripathi, S.; Garcia-Sastre, A.; Villar, E. Enhancement of the proapoptotic properties of newcastle disease virus promotes remission in syngeneic murine cancer models. Mol. Cancer Ther. 2015, 14, 1247–1258. [Google Scholar] [CrossRef]
- Wu, Y.; Zhang, X.; Wang, X.; Wang, L.; Hu, S.; Liu, X.; Meng, S. Apoptin enhances the oncolytic properties of Newcastle disease virus. Intervirology 2012, 55, 276–286. [Google Scholar] [CrossRef] [PubMed]
- An, Y.; Liu, T.; He, J.; Wu, H.; Chen, R.; Liu, Y.; Wu, Y.; Bai, Y.; Guo, X.; Zheng, Q.; et al. Recombinant Newcastle disease virus expressing p53 demonstrates promising antitumor efficiency in hepatoma model. J. Biomed. Sci. 2016, 23, 55. [Google Scholar] [CrossRef]
- Altomonte, J.; Marozin, S.; Schmid, R.M.; Ebert, O. Engineered newcastle disease virus as an improved oncolytic agent against hepatocellular carcinoma. Mol. Ther. 2010, 18, 275–284. [Google Scholar] [CrossRef] [PubMed]
- Matuszewska, K.; Santry, L.A.; van Vloten, J.P.; AuYeung, A.W.K.; Major, P.P.; Lawler, J.; Wootton, S.K.; Bridle, B.W.; Petrik, J. Combining vascular normalization with an oncolytic virus enhances immunotherapy in a preclinical model of advanced-stage ovarian cancer. Clin. Cancer Res. 2019, 25, 1624–1638. [Google Scholar] [CrossRef] [PubMed]
- Pühler, F.; Willuda, J.; Puhlmann, J.; Mumberg, D.; Römer-Oberdörfer, A.; Beier, R. Generation of a recombinant oncolytic Newcastle disease virus and expression of a full IgG antibody from two transgenes. Gene Ther. 2008, 15, 371–383. [Google Scholar] [CrossRef]
- Scott, E.M.; Duffy, M.R.; Freedman, J.D.; Fisher, K.D.; Seymour, L.W. Solid tumor immunotherapy with T cell engager-armed oncolytic viruses. Macromol. Biosci. 2017, 18. [Google Scholar] [CrossRef]
- Yuan, M.; Webb, E.; Lemoine, N.R.; Wang, Y. CRISPR-Cas9 as a powerful tool for efficient creation of oncolytic viruses. Viruses 2016, 8, 72. [Google Scholar] [CrossRef]
- Mader, E.K.; Macyama, Y.; Lin, Y.; Butler, G.W.; Russell, H.M.; Galanis, E.; Russel, S.J.; Dietz, A.B.; Peng, K.W. Mesenchymal stem cell carriers protect oncolytic measles viruses from antibody neutralization in an orthotopic ovarian cancer therapy model. Clin. Cancer Res. 2009, 15, 7246–7255. [Google Scholar] [CrossRef]
- Castleton, A.; Dey, A.; Beaton, B.; Patel, B.; Aucher, A.; Davis, D.M.; Fielding, A.K. Human mesenchymal stromal cells deliver systemic oncolytic measles virus to treat acute lymphoblastic leukemia in the presence of humoral immunity. Blood 2014, 123, 1327–1335. [Google Scholar] [CrossRef] [PubMed]
- Kazimirsky, G.; Jiang, W.; Slavin, S.; Ziv-Av, A.; Brodie, C. Mesenchymal stem cells enhance the oncolytic effect of Newcastle disease virus in glioma cells and glioma stem cells via the secretion of TRAIL. Stem Cell Res. Ther. 2016, 7, 149. [Google Scholar] [CrossRef] [PubMed]
- Pfirschke, C.; Schirrmacher, V. Cross-infection of tumor cells by contact with T lymphocytes loaded with Newcastle disease virus. Int. J. Oncol. 2009, 34, 951–962. [Google Scholar] [PubMed]
- Bian, H.; Fournier, P.; Peeters, B.; Schirrmacher, V. Tumor-targeted gene transfer in vivo via recombinant Newcastle disease virus modified by a bispecific fusion protein. Int. J. Oncol. 2005, 27, 377–384. [Google Scholar] [CrossRef][Green Version]
- Bian, H.; Fournier, P.; Moormann, R.; Peeters, B.; Schirrmacher, V. Selective gene transfer to tumor cells by recombinant Newcastle disease virus via a bispecific fusion protein. Int. J. Oncol. 2005, 26, 431–439. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Bian, H.; Wilden, H.; Fournier, P.; Peeters, B.; Schirrmacher, V. In vivo efficacy of systemic tumor targeting of a viral RNA vector with oncolytic properties using a bispecific adapter protein. Int. J. Oncol. 2006, 29, 1359–1369. [Google Scholar] [CrossRef] [PubMed]
- Gujar, S.; Pol, J.G.; Kroemer, G. Heating it up: Oncolytic viruses make tumors ‘hot’ and suitable for checkpoint blockade immunotherapies. Oncoimmunology 2018, 7, e1442169. [Google Scholar] [CrossRef] [PubMed]
- Schirrmacher, V.; Fournier, P. Multimodal cancer therapy involving oncolytic Newcastle disease virus, autologous immune cells and bispecific antibodies. Front. Oncol. 2014, 4, 224. [Google Scholar] [CrossRef] [PubMed]
- Russel, S.J.; Barber, G.N. Oncolytic viruses as antigen-agnostic cancer vaccines. Cancer Cell. 2018, 33, 599–605. [Google Scholar] [CrossRef]
- Zamarin, D.; Ricca, J.M.; Sadekova, S.; Oseledchyk, A.; Yu, Y.; Blumenschein, W.M.; Wong, J.; Gigoux, M.; Merghoub, T.; Wolchok, J.D. PD-L1 in tumor microenvironment mediates resistance to oncolytic immunotherapy. J. Clin. Investig. 2018, 128, 1413–1428. [Google Scholar] [CrossRef] [PubMed]
- Engeland, C.E.; Grossardt, C.; Veinalde, R.; Bossow, S.; Lutz, D.; Kaufman, J.K.; Shevchenko, I.; Umansky, V.; Nettelbeck, D.M.; Weichert, W.; et al. CTLA-4 and PD-L1 checkpoint blockade enhances oncolytic measles virus therapy. Mol. Ther. 2014, 22, 1949–1959. [Google Scholar] [CrossRef] [PubMed]
- Fend, L.; Yamazaki, T.; Remy, C.; Fahrner, C.; Gantzer, M.; Nourtier, V.; Préville, X.; Quéméneur, E.; Kepp, O.; Adam, J. Immune checkpoint blockade, immunogenic chemotherapy or IFN-〈 blockade boost the local and abscopal effects of oncolytic virotherapy. Cancer Res. 2017, 77, 4146–4157. [Google Scholar] [CrossRef] [PubMed]
- Tober, R.; Banki, Z.; Egerer, L.; Muik, A.; Behmüller, S.; Kreppel, F.; Greczmiel, U.; Oxenius, A.; von Laer, D.; Kimpel, J. VSV-GP: A potent viral vaccine vector that boosts the immune response upon repeated applications. J. Virol. 2014, 88, 4897–4907. [Google Scholar] [CrossRef]
- Scherwitzl, I.; Hurtado, A.; Pierce, C.M.; Vogt, S.; Pampeno, C.; Meruelo, D. Systemically administered Sindbis virus in combination with immune checkpoint blockade induces curative anti-tumor immunity. Mol. Ther. Oncolytics 2018, 9, 51–63. [Google Scholar] [CrossRef]
- Ilett, E.; Koffke, T.; Thompson, J.; Rajanik, K.; Zaidi, S.; Evqin, L.; Coffey, M.; Ralph, C.; Diaz, R.; Pandha, H.; et al. Prime-boost using separate oncolytic viruses in combination with checkpoint blockade improves anti-tumor therapy. Gene Ther. 2017, 24, 21–30. [Google Scholar] [CrossRef]
- Becht, E.; Giraldo, N.A.; Germain, C.; de Reyniès, A.; Laurent-Puiq, P.; Zucman-Ross, J.; Dieu-Nosjean, M.J.; Sautès-Fridman, C.; Fridman, W.H. Immune contexture, immunoscore, and malignant cell subgroups for prognostic and theranostic classification of cancers. Adv. Immunol. 2016, 130, 95–190. [Google Scholar]
- Samson, A.; Scott, K.J.; Taggart, D.; West, E.J.; Wilson, E.; Nuovo, G.J.; Thomson, S.; Coms, R.; Mathew, R.K.; Fuller, M.J. Intravenous delivery of oncolytic reovirus to brain tumor patients immunologically primes for subsequent checkpoint blockade. Sci. Transl. Med. 2018, 10, eaam7577. [Google Scholar] [CrossRef] [PubMed]
- Bourgeois-Daigneault, M.C.; Roy, D.G.; Aitken, A.S.; El Sayes, N.; Martin, N.T.; Varette, O.; Falls, T.; St-Germain, L.E.; Pelin, A.; Lichty, B.D. Neoadjuvant oncolytic virotherapy before surgery sensitizes triple-negative breast cancer to immune checkpoint therapy. Sci. Transl. Med. 2018, 10, eaao1641. [Google Scholar] [CrossRef]
- Loi, S.; Drubay, D.; Adams, S.; Pruneri, G.; Francis, P.A.; Laccroix-Triki, M.; Joensuu, H.; Died, M.Y.; Badve, S.; Demaria, A.; et al. Tumor-infiltrating lymphocytes and prognosis: A pooled individual patient analysis of early-stage triple-negative breast cancers. J. Clin. Oncol. 2019, 37, 559–569. [Google Scholar] [CrossRef]
- Naijar, Y.G.; Menk, A.V.; Sander, C.; Rao, U.; Karunamurthy, A.; Bhatia, R.; Zhai, S.; Kirkwood, J.M.; Delgoffe, G.M. Tumor cell oxidative metabolism as a barrier to PD-1 blockade immunotherapy. JCI Insight 2019, 4, pii:124989. [Google Scholar] [CrossRef] [PubMed]
- Mariathasan, S.; Turley, S.J.; Nickles, D.; Castiglioni, A.; Yuen, K.; Wang, Y.; Kadel, E.E., III; Koeppen, H.; Astarita, J.L.; Cubas, R.; et al. TGFβ attenuates tumour response to PD-L1 blockade by contributing to exclusion of T cells. Nature 2018, 554, 544–548. [Google Scholar] [CrossRef] [PubMed]
- Garris, C.S.; Arlauckas, S.P.; Kohler, R.H.; Trefny, M.P.; Garren, S.; Piot, C.; Engblom, C.; Pfirschke, C.; Siwicki, M.; Gungabeesoon, J.; et al. Successful anti- PD-1 cancer immunotherapy requires T cell-Dendritic cell crosstalk involving the cytokines IFN-γ and IL-12. Immunity. 2018, 49, 1148–1161.e7. [Google Scholar] [CrossRef] [PubMed]
- Sade-Feldman, M.; Yizhak, K.; Bjorgaard, S.L.; Ray, J.P.; de Boer, C.G.; Jenkins, R.W.; Lieb, D.J.; Chen, J.H.; Frederick, D.T.; Barzily-Rokni, M.; et al. Defining T cell states associated with response to checkpoint immunotherapy in melanoma. Cell 2018, 175, 998–1013. [Google Scholar] [CrossRef] [PubMed]
- Marchini, A.; Scott, E.M.; Rommelaere, J. Overcoming barriers in oncolytic virotherapy with HDAC inhibitors and immune checkpoint blockade. Viruses 2016, 8, 9. [Google Scholar] [CrossRef] [PubMed]
- Vilgelm, A.E.; Johnson, D.B.; Richmond, A. Combinatorial approach to cancer immunotherapy: Strength in numbers. J. Leukoc. Biol. 2016, 100, 275–290. [Google Scholar] [CrossRef] [PubMed]
- Zamarin, D.; Postow, M.A. Immune ckeckpoint modulation: Rational design of combinatorial strategies. Pharmacol. Ther. 2015, 150, 23–32. [Google Scholar] [CrossRef] [PubMed]
- Ribas, A.; Dummer, R.; Puzanov, I.; VanderWalde, A.; Andtbacka, R.H.I.; Michielin, O.; Olszanski, A.J.; Malvehy, J.; Cebon, J.; Fernandez, E.; et al. Oncolytic virotherapy promotes intratumoral T cell infiltration and improves anti-PD-1 immunotherapy. Cell 2017, 170, 1109–1119. [Google Scholar] [CrossRef] [PubMed]
- Bommareddy, P.K.; Shettigar, M.; Kaufman, H.L. Integrating oncolytic viruses in combination cancer immunotherapy. Nat. Rev. Immunol. 2018, 18, 498–513. [Google Scholar] [CrossRef]
- Cerullo, V.; Dianconu, I.; Kangasniemi, L.; Rajecki, M.; Escutenaire, S.; Koski, A.; Romano, V.; Rouvinen, N.; Tuuminen, T.; Laasonen, L.; et al. Immunological effects of low-dose cyclophosphamide in cancer patients treated with oncolytic adenovirus. Mol. Ther. 2011, 19, 1737–1746. [Google Scholar] [CrossRef] [PubMed]
- Bommareddy, P.K.; Aspromonte, S.; Sloza, A.; Rabkin, S.D.; Kaufman, H.L. MEK inhibition enhances oncolytic virus immunotherapy through increased tumor cell killing and T cell activation. Sci. Transl. Med. 2018. [Google Scholar] [CrossRef]
- Forbes, N.E.; Krishnan, R.; Diallo, J.S. Pharmacological modulation of anti-tumor immunity induced by oncolytic viruses. Front. Oncol. 2014, 4, 89–100. [Google Scholar] [CrossRef] [PubMed]
- Liikanen, I.; Ahtiainen, L.; Hirvinen, M.L.; Bramante, S.; Cerullo, V.; Nokisalmi, P.; Hemminki, O.; Diaconu, I.; Pesonen, S.; Koski, A.; et al. Oncolytic adenovirus with temozolomide induces autophagy and antitumor immune responses in cancer patients. Mol. Ther. 2013, 21, 1212–1223. [Google Scholar] [CrossRef] [PubMed]
- Lolkema, M.P.; Arkenau, H.T.; Harrington, K.; Roxburgh, P.; Morrison, R.; Roulstone, V.; Twigger, K.; Coffey, M.; Mettinger, K.; Gill, G.; et al. A phase I study of the combination of intravenous reovirus type 3 Dearing and gemcitabine in patients with advanced cancer. Clin. Cancer Res. 2011, 17, 581–588. [Google Scholar] [CrossRef] [PubMed]
- Barrero, M.J. Epigenetic strategies to boost cancer immunotherapies. Int. J. Mol. Sci. 2017, 18, 8. [Google Scholar] [CrossRef]
- Gallagher, S.J.; Shklovskaya, E.; Hersey, P. Epigenetic modulation of cancer immunotherapy. Curr. Opin. Pharmacol. 2017, 35, 48–56. [Google Scholar] [CrossRef] [PubMed]
- Okemoto, K.; Kasai, K.; Wagner, B.; Haseley, A.; Meisen, H.; Bolyard, C.; Mo, X.; Wehr, A.; Lehman, A.; Fernandez, S.; et al. DNA demethylating agents synergize with oncolytic HSV1 against malignant gliomas. Clin. Cancer Res. 2013, 19, 5952–5959. [Google Scholar] [CrossRef] [PubMed]
- Altevogt, P.; von Hoegen, P.; Schirrmacher, V. Immunoresistant metastatic tumor variants can re-express their tumor antigen after treatment with DNA methylation-inhibiting agents. Int. J. Cancer 1986, 38, 707–711. [Google Scholar] [CrossRef] [PubMed]
- Toh, T.B.; Lim, J.J.; Chow, E.K. Epigenetics in cancer stem cells. Mol. Cancer 2017, 16, 29. [Google Scholar] [CrossRef] [PubMed]
- Hu, L.; Sun, S.; Wang, T.; Li, Y.; Jiang, K.; Lin, G.; Ma, Y.; Barr, M.P.; Song, F.; Zhang, G.; et al. Oncolytic newcastle disease virus triggers cell death of lung cancer spheroids and is enhanced by pharmacological inhibition of autophagy. Am. J. Cancer Res. 2015, 5, 3612–3623. [Google Scholar]


{kind=link}
| Induction of post-oncolytic immunity |
| Activation of natural killer (NK) cells |
| Activation of monocytes and macrophages |
| Activation of dendritic cells |
| Polarization of dendritic cells (DCs) towards DC1 |
| Induction of a strong type I interferon response |
| IFN-α mediating epitope cross-presentation by DCs |
| IFN-α mediating a link between innate and adaptive immunity |
| IFN-α being essential for the generation of cytotoxic T cells (CTLs) |
| IFN-α protecting T cells against attack by NK cells |
| NDV exerting co-stimulatory effects on CD4+ and CD8+ T cells |
| Targeting the oncogenic protein Rac1 |
| Tumor-selective virus replication |
| Tumor-selective oncolysis |
| Tumor-selective induction of immunogenic cell death |
| Potential to break T cell tolerance towards tumor-associated antigen (TA) expressing tumor cells |
| Potential to break resistance to chemotherapy or radiotherapy |
| Potential to break resistance to apoptosis |
| Potential to break resistance to hypoxia |
| Potential to break resistance to TNF-related apoptosis-inducing ligand (TRAIL) |
| Potential to break resistance to immune checkpoint blockade |
| Potential to break resistance to anti-viral immunity |
| Promotion of virus propagation via syncytia, autophagy, and exosomes |
| 1. Post-operative application of NDV oncolysate vaccines in Stage II melanoma (n = 83) by WA Cassel and DR Murray (1977) (oncolytic strain 73 T) |
| 2. Systemic treatment of advanced chemorefractory cancers; Phase II placebo-controlled trial (n = 33 versus 26 controls) by LK Csatary and S Eckhardt (1993) (oncolytic strain MTH/68) |
| 3. Systemic treatment of glioblastoma multiforme (GBM) (n = 14); Intra-patient dose escalation followed by 3 cycles of 55 billion infectious units by AI Freeman and Z Zakay-Rones (2006) (lentogenic strain HUJ) |
| 4. Phase I dose-escalation trials of intravenous virus administration in patients with advanced solid cancers resistant to standard therapy (n = 113) by AL Pecora and RM Lorence (2002, 2003) (oncolytic strain PV71) |
5. Post-operative treatment with the irradiated live tumor cell vaccine Autologous Live NDV-Modified Tumor Cell Vaccine (ATV-NDV):
|
| 6. Prospectively randomized Phase II/III trial to investigate the efficiency of ATV-NDV vaccination after liver resection for hepatic metastases of CRC as a tertiary prevention method (n = 51) by T Schulze and PM Schlag (2009) (lentogenic strain Ulster) |
| Single-case Report of Prostate Cancer with Extensive Bone Metastases |
| Single-case report of breast cancer with extensive liver metastases |
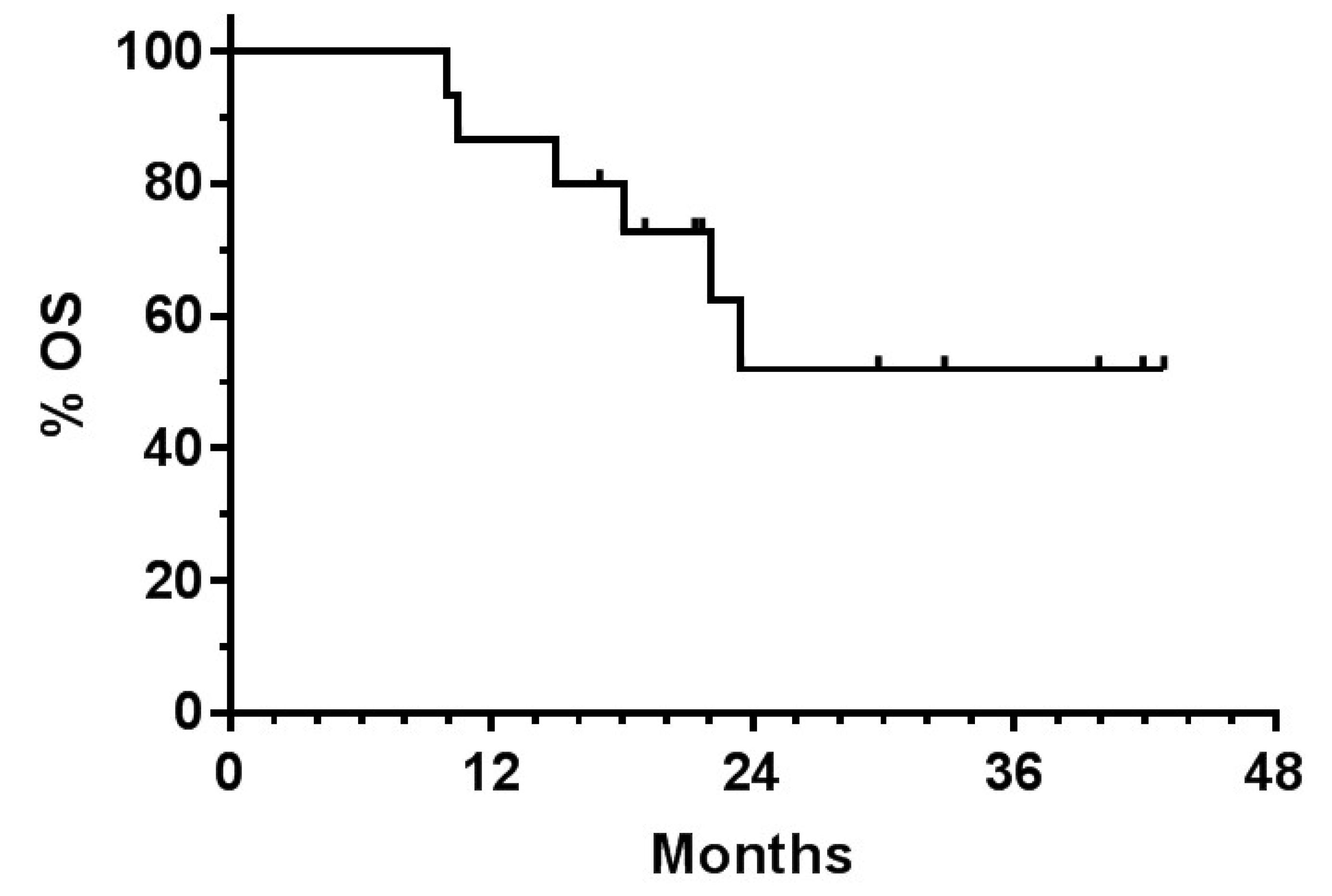
| Retrospective analysis of adults with GBM (median age 60) (n = 63) |
| Retrospective analysis of children (median age 6.5–9.5) (n = 76) with brain cancer; among them diffuse intrinsic pontine glioma (DIPG) (n = 28) 2015: IOZK succeeded in producing oncolytic NDV (derivative from strain Mukteshwar) from cell culture at high titers and with high purity according to GMP guidelines and received a permit for its use in cancer patients. This is used to produce the autologous NDV viral oncolysate-pulsed DC vaccine IO-VACR. |
| Melanoma (6, 88, 108, 132, 133) |
| Breast carcinoma (51, 63, 69, 101, 138, 163) |
| Ovarian carcinoma (119, 160, 164) |
| Colorectal carcinoma (67, 113, 139, 142, 143, 157) |
| Head and Neck squamous cell carcinoma (141) |
| Gastrointestinal carcinoma (144) |
| Pancreatic carcinoma (41, 44, 45, 46) |
| Renal cell carcinoma (109, 134) |
| Prostate carcinoma (162, 174) |
| Lung carcinoma (104, 105, 229) |
| Hepatocellular carcinoma (175, 183, 184) |
| Glioblastoma multiforme (65, 70, 106, 140, 145, 147, 164, 191) |
| Lymphoma (84, 86, 87, 135, 136, 193, 194, 195, 226) |
| OV-mediated gene therapy, incorporation of therapeutic genes |
| Improving tumor targeting of OVs by carrier cells |
| Improving T-cell costimulation by bi- or tri-specific antibodies |
| Improving OV targeting of immune cells by loading the cells with tri-specific antibodies |
| Combining OV therapy with checkpoint inhibitors |
| Combining OV therapy with physical modalities such as mEHT |
| Combining OV therapy with pharmacological modulation |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Schirrmacher, V.; van Gool, S.; Stuecker, W. Breaking Therapy Resistance: An Update on Oncolytic Newcastle Disease Virus for Improvements of Cancer Therapy. Biomedicines 2019, 7, 66. https://doi.org/10.3390/biomedicines7030066
Schirrmacher V, van Gool S, Stuecker W. Breaking Therapy Resistance: An Update on Oncolytic Newcastle Disease Virus for Improvements of Cancer Therapy. Biomedicines. 2019; 7(3):66. https://doi.org/10.3390/biomedicines7030066
Chicago/Turabian StyleSchirrmacher, Volker, Stefaan van Gool, and Wilfried Stuecker. 2019. "Breaking Therapy Resistance: An Update on Oncolytic Newcastle Disease Virus for Improvements of Cancer Therapy" Biomedicines 7, no. 3: 66. https://doi.org/10.3390/biomedicines7030066
APA StyleSchirrmacher, V., van Gool, S., & Stuecker, W. (2019). Breaking Therapy Resistance: An Update on Oncolytic Newcastle Disease Virus for Improvements of Cancer Therapy. Biomedicines, 7(3), 66. https://doi.org/10.3390/biomedicines7030066