
Regulation of Insulin Clearance by Non-Esterified Fatty Acids

 ,
,  and
and {kind=link}
{kind=link}
{kind=link}
{kind=link}
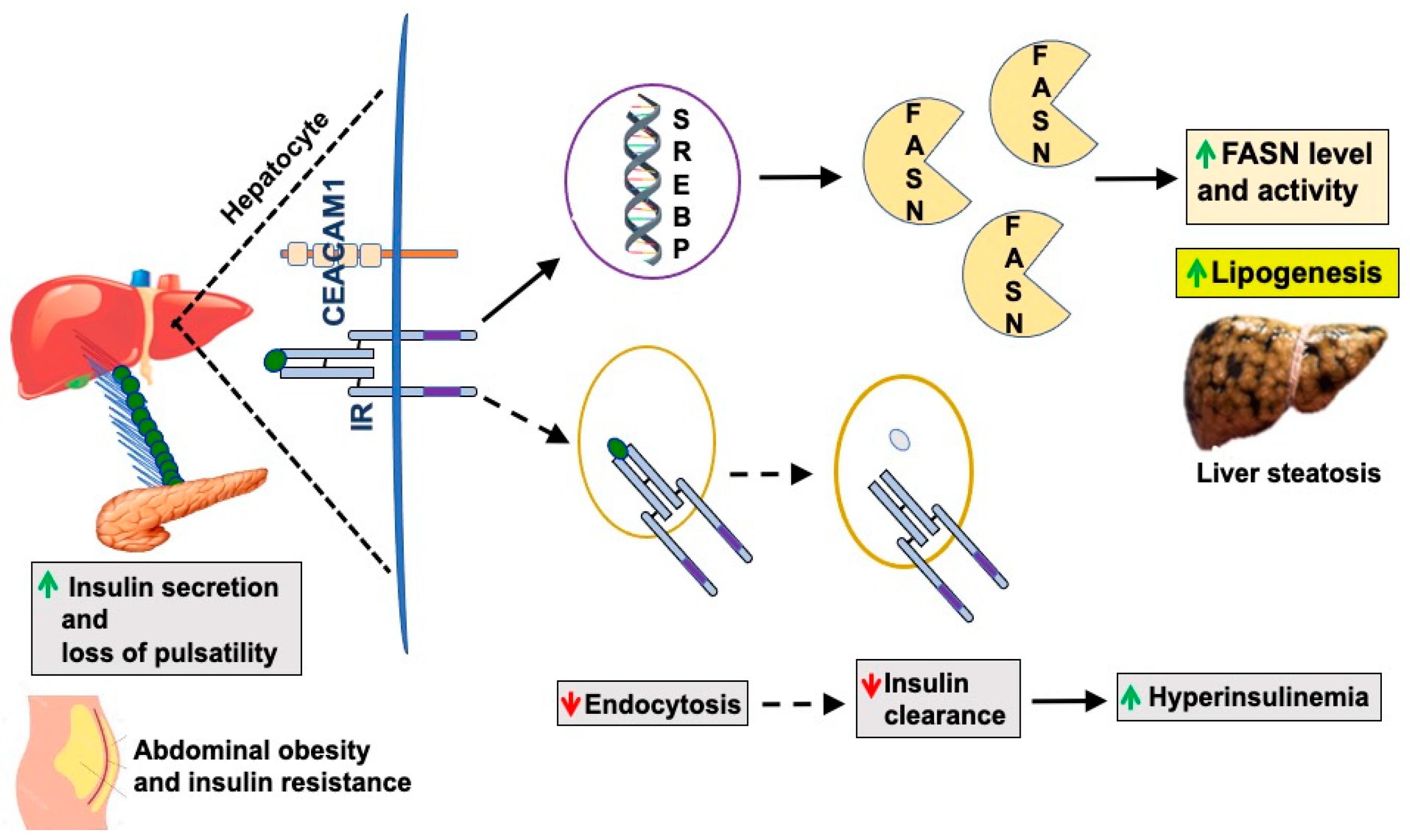
Abstract
:1. Mechanisms of Insulin Clearance
2. Reduced Insulin Clearance in Metabolic Disease
3. Elevated Plasma NEFA Play a Primary Role in Hepatic Insulin Resistance Followed by Reduced Insulin Clearance
4. Reduction in Hepatic Insulin Clearance Plays a Primary Role in Insulin Resistance Independently of NEFA Release
5. Concluding Remarks
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Najjar, S.M.; Perdomo, G. Hepatic Insulin Clearance: Mechanism and Physiology. Physiology 2019, 34, 198–215. [Google Scholar] [CrossRef] [PubMed]
- Matveyenko, A.V.; Liuwantara, D.; Gurlo, T.; Kirakossian, D.; Dalla Man, C.; Cobelli, C.; White, M.F.; Copps, K.D.; Volpi, E.; Fujita, S.; et al. Pulsatile portal vein insulin delivery enhances hepatic insulin action and signaling. Diabetes 2012, 61, 2269–2279. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haeusler, R.A.; McGraw, T.E.; Accili, D. Biochemical and cellular properties of insulin receptor signalling. Nat. Rev. Mol. Cell. Biol. 2018, 19, 31–44. [Google Scholar] [CrossRef] [PubMed]
- Saltiel, A.R. Insulin signaling in health and disease. J. Clin. Investig. 2021, 131, e142241. [Google Scholar] [CrossRef] [PubMed]
- Meijer, R.I.; Barrett, E.J. The Insulin Receptor Mediates Insulin’s Early Plasma Clearance by Liver, Muscle, and Kidney. Biomedicines 2021, 9, 37. [Google Scholar] [CrossRef]
- Najjar, S.M.; Yang, Y.; Fernstrom, M.A.; Lee, S.J.; Deangelis, A.M.; Rjaily, G.A.; Al-Share, Q.Y.; Dai, T.; Miller, T.A.; Ratnam, S.; et al. Insulin acutely decreases hepatic fatty acid synthase activity. Cell Metab. 2005, 2, 43–53. [Google Scholar] [CrossRef] [Green Version]
- Osborne, T.F. Sterol regulatory element-binding proteins (SREBPs): Key regulators of nutritional homeostasis and insulin action. J. Biol. Chem. 2000, 275, 32379–32382. [Google Scholar] [CrossRef] [Green Version]
- Satin, L.S.; Butler, P.C.; Ha, J.; Sherman, A.S. Pulsatile insulin secretion, impaired glucose tolerance and type 2 diabetes. Mol. Aspects Med. 2015, 42, 61–77. [Google Scholar] [CrossRef] [Green Version]
- Whitticar, N.B.; Nunemaker, C.S. Reducing Glucokinase Activity to Enhance Insulin Secretion: A Counterintuitive Theory to Preserve Cellular Function and Glucose Homeostasis. Front. Endocrinol. 2020, 11, 378. [Google Scholar] [CrossRef]
- Brown, M.S.; Goldstein, J.L. Selective versus total insulin resistance: A pathogenic paradox. Cell Metab. 2008, 7, 95–96. [Google Scholar] [CrossRef] [Green Version]
- Muturi, H.T.; Khuder, S.S.; Ghadieh, H.E.; Esakov, E.L.; Noh, H.; Kang, H.; McInerney, M.F.; Kim, J.K.; Lee, A.D.; Najjar, S.M. Insulin Sensitivity Is Retained in Mice with Endothelial Loss of Carcinoembryonic Antigen Cell Adhesion Molecule 1. Cells 2021, 10, 2093. [Google Scholar] [CrossRef]
- Tokarz, V.L.; MacDonald, P.E.; Klip, A. The cell biology of systemic insulin function. J. Cell Biol. 2018, 217, 2273–2289. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gonzalez-Casimiro, C.M.; Merino, B.; Casanueva-Alvarez, E.; Postigo-Casado, T.; Camara-Torres, P.; Fernandez-Diaz, C.M.; Leissring, M.A.; Cozar-Castellano, I.; Perdomo, G. Modulation of Insulin Sensitivity by Insulin-Degrading Enzyme. Biomedicines 2021, 9, 86. [Google Scholar] [CrossRef] [PubMed]
- Durham, T.B.; Toth, J.L.; Klimkowski, V.J.; Cao, J.X.; Siesky, A.M.; Alexander-Chacko, J.; Wu, G.Y.; Dixon, J.T.; McGee, J.E.; Wang, Y.; et al. Dual Exosite-binding Inhibitors of Insulin-degrading Enzyme Challenge Its Role as the Primary Mediator of Insulin Clearance in Vivo. J. Biol. Chem. 2015, 290, 20044–20059. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Villa-Perez, P.; Merino, B.; Fernandez-Diaz, C.M.; Cidad, P.; Lobaton, C.D.; Moreno, A.; Muturi, H.T.; Ghadieh, H.E.; Najjar, S.M.; Leissring, M.A.; et al. Liver-specific ablation of insulin-degrading enzyme causes hepatic insulin resistance and glucose intolerance, without affecting insulin clearance in mice. Metabolism 2018, 88, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Borges, D.O.; Patarrao, R.S.; Ribeiro, R.T.; de Oliveira, R.M.; Duarte, N.; Belew, G.D.; Martins, M.; Andrade, R.; Costa, J.; Correia, I.; et al. Loss of postprandial insulin clearance control by Insulin-degrading enzyme drives dysmetabolism traits. Metabolism 2021, 118, 154735. [Google Scholar] [CrossRef]
- Ader, M.; Stefanovski, D.; Kim, S.P.; Richey, J.M.; Ionut, V.; Catalano, K.J.; Hucking, K.; Ellmerer, M.; Van Citters, G.; Hsu, I.R.; et al. Hepatic insulin clearance is the primary determinant of insulin sensitivity in the normal dog. Obesity 2014, 22, 1238–1245. [Google Scholar] [CrossRef] [Green Version]
- Guo, X.; Cui, J.; Jones, M.R.; Haritunians, T.; Xiang, A.H.; Chen, Y.D.; Taylor, K.D.; Buchanan, T.A.; Davis, R.C.; Hsueh, W.A.; et al. Insulin clearance: Confirmation as a highly heritable trait, and genome-wide linkage analysis. Diabetologia 2012, 55, 2183–2192. [Google Scholar] [CrossRef] [Green Version]
- Lee, C.C.; Haffner, S.M.; Wagenknecht, L.E.; Lorenzo, C.; Norris, J.M.; Bergman, R.N.; Stefanovski, D.; Anderson, A.M.; Rotter, J.I.; Goodarzi, M.O.; et al. Insulin clearance and the incidence of type 2 diabetes in Hispanics and African Americans: The IRAS Family Study. Diabetes Care 2013, 36, 901–907. [Google Scholar] [CrossRef] [Green Version]
- Piccinini, F.; Polidori, D.C.; Gower, B.A.; Bergman, R.N. Hepatic but not extrahepatic insulin clearance is lower in African American than in European American women. Diabetes 2017, 66, 2564–2570. [Google Scholar] [CrossRef] [Green Version]
- Goff, L.M.; Ladwa, M.; Hakim, O.; Bello, O. Ethnic distinctions in the pathophysiology of type 2 diabetes: A focus on black African-Caribbean populations. Proc. Nutr. Soc. 2020, 79, 184–193. [Google Scholar] [CrossRef] [Green Version]
- Shah, M.H.; Piaggi, P.; Looker, H.C.; Paddock, E.; Krakoff, J.; Chang, D.C. Lower insulin clearance is associated with increased risk of type 2 diabetes in Native Americans. Diabetologia 2021, 64, 914–922. [Google Scholar] [CrossRef] [PubMed]
- Galderisi, A.; Polidori, D.; Weiss, R.; Giannini, C.; Pierpont, B.; Trico, D.; Caprio, S. Lower Insulin Clearance parallels a reduced insulin sensitivity in obese youths and is associated with a decline in beta-cell function over time. Diabetes 2019, 68, 2074–2084. [Google Scholar] [CrossRef] [PubMed]
- Trico, D.; Galderisi, A.; Mari, A.; Polidori, D.; Galuppo, B.; Pierpont, B.; Samuels, S.; Santoro, N.; Caprio, S. Intrahepatic fat, irrespective of ethnicity, is associated with reduced endogenous insulin clearance and hepatic insulin resistance in obese youths: A cross-sectional and longitudinal study from the Yale Pediatric NAFLD cohort. Diabetes Obes. Metab. 2020, 22, 1628–1638. [Google Scholar] [CrossRef] [PubMed]
- Al-Share, Q.Y.; DeAngelis, A.M.; Lester, S.G.; Bowman, T.A.; Ramakrishnan, S.K.; Abdallah, S.L.; Russo, L.; Patel, P.R.; Kaw, M.K.; Raphael, C.K.; et al. Forced Hepatic Overexpression of CEACAM1 Curtails Diet-Induced Insulin Resistance. Diabetes 2015, 64, 2780–2790. [Google Scholar] [CrossRef] [Green Version]
- Mittelman, S.D.; Van Citters, G.W.; Kim, S.P.; Davis, D.A.; Dea, M.K.; Hamilton-Wessler, M.; Bergman, R.N. Longitudinal compensation for fat-induced insulin resistance includes reduced insulin clearance and enhanced beta-cell response. Diabetes 2000, 49, 2116–2125. [Google Scholar] [CrossRef] [Green Version]
- Bakker, L.E.H.; van Schinkel, L.D.; Guigas, B.; Streefland, T.C.M.; Jonker, J.T.; van Klinken, J.B.; van der Zon, G.C.M.; Lamb, H.J.; Smit, J.W.A.; Pijl, H.; et al. A 5-Day High-Fat, High-Calorie Diet Impairs Insulin Sensitivity in Healthy, Young South Asian Men but Not in Caucasian Men. Diabetes 2014, 63, 248–258. [Google Scholar] [CrossRef] [Green Version]
- Peiris, A.N.; Mueller, R.A.; Smith, G.A.; Struve, M.F.; Kissebah, A.H. Splanchnic insulin metabolism in obesity. Influence of body fat distribution. J. Clin. Investig. 1986, 78, 1648–1657. [Google Scholar] [CrossRef] [Green Version]
- Polonsky, K.S.; Given, B.D.; Hirsch, L.; Shapiro, E.T.; Tillil, H.; Beebe, C.; Galloway, J.A.; Frank, B.H.; Karrison, T.; Van Cauter, E. Quantitative study of insulin secretion and clearance in normal and obese subjects. J. Clin. Investig. 1988, 81, 435–441. [Google Scholar] [CrossRef]
- Hansen, B.C.; Striffler, J.S.; Bodkin, N.L. Decreased hepatic insulin extraction precedes overt noninsulin dependent (Type II) diabetes in obese monkeys. Obes. Res. 1993, 1, 252–260. [Google Scholar] [CrossRef]
- Escobar, O.; Mizuma, H.; Sothern, M.S.; Blecker, U.; Udall, J.N., Jr.; Suskind, R.M.; Hilton, C.; Vargas, A. Hepatic insulin clearance increases after weight loss in obese children and adolescents. Am. J. Med. Sci. 1999, 317, 282–286. [Google Scholar] [CrossRef]
- Rossell, R.; Gomis, R.; Casamitjana, R.; Segura, R.; Vilardell, E.; Rivera, F. Reduced hepatic insulin extraction in obesity: Relationship with plasma insulin levels. J. Clin. Endocrinol. Metab. 1983, 56, 608–611. [Google Scholar] [CrossRef] [PubMed]
- Bonora, E.; Zavaroni, I.; Coscelli, C.; Butturini, U. Decreased hepatic insulin extraction in subjects with mild glucose intolerance. Metabolism 1983, 32, 438–446. [Google Scholar] [CrossRef]
- Giugliano, D.; Quatraro, A.; Minei, A.; De Rosa, N.; Coppola, L.; D’Onofrio, F. Hyperinsulinemia in hypertension: Increased secretion, reduced clearance or both? J. Endocrinol. Investig. 1993, 16, 315–321. [Google Scholar] [CrossRef]
- Kim, M.K.; Reaven, G.M.; Chen, Y.D.; Kim, E.; Kim, S.H. Hyperinsulinemia in individuals with obesity: Role of insulin clearance. Obesity 2015, 23, 2430–2434. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.P.; Ellmerer, M.; Kirkman, E.L.; Bergman, R.N. Beta-cell “rest” accompanies reduced first-pass hepatic insulin extraction in the insulin-resistant, fat-fed canine model. Am. J. Physiol. Endocrinol. Metab. 2007, 292, E1581–E1589. [Google Scholar] [CrossRef]
- Koh, H.E.; Cao, C.; Mittendorfer, B. Insulin Clearance in Obesity and Type 2 Diabetes. Int. J. Mol. Sci. 2022, 23, 596. [Google Scholar] [CrossRef]
- Zarghamravanbakhsh, P.; Frenkel, M.; Poretsky, L. Metabolic causes and consequences of nonalcoholic fatty liver disease (NAFLD). Metabol. Open 2021, 12, 100149. [Google Scholar] [CrossRef]
- Guerra, S.; Mocciaro, G.; Gastaldelli, A. Adipose tissue insulin resistance and lipidome alterations as the characterizing factors of non-alcoholic steatohepatitis. Eur. J. Clin. Investig. 2022, 52, e13695. [Google Scholar] [CrossRef]
- Bril, F.; Lomonaco, R.; Orsak, B.; Ortiz-Lopez, C.; Webb, A.; Tio, F.; Hecht, J.; Cusi, K. Relationship between disease severity, hyperinsulinemia, and impaired insulin clearance in patients with nonalcoholic steatohepatitis. Hepatology 2014, 59, 2178–2187. [Google Scholar] [CrossRef]
- Matsubayashi, Y.; Yoshida, A.; Suganami, H.; Ishiguro, H.; Yamamoto, M.; Fujihara, K.; Kodama, S.; Tanaka, S.; Kaku, K.; Sone, H. Role of fatty liver in the association between obesity and reduced hepatic insulin clearance. Diabetes Metab. 2018, 44, 135–142. [Google Scholar] [CrossRef] [PubMed]
- Lee, W. The CEACAM1 expression is decreased in the liver of severely obese patients with or without diabetes. Diagn. Pathol. 2011, 6, 40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heinrich, G.; Muturi, H.T.; Rezaei, K.; Al-Share, Q.Y.; DeAngelis, A.M.; Bowman, T.A.; Ghadieh, H.E.; Ghanem, S.S.; Zhang, D.; Garofalo, R.S.; et al. Reduced hepatic carcinoembryonic antigen-related cell adhesion molecule 1 level in obesity. Front. Endocrinol. 2017, 8, 54. [Google Scholar] [CrossRef] [PubMed]
- Kotronen, A.; Juurinen, L.; Tiikkainen, M.; Vehkavaara, S.; Yki-Jarvinen, H. Increased liver fat, impaired insulin clearance, and hepatic and adipose tissue insulin resistance in type 2 diabetes. Gastroenterology 2008, 135, 122–130. [Google Scholar] [CrossRef]
- Tiikkainen, M.; Hakkinen, A.M.; Korsheninnikova, E.; Nyman, T.; Makimattila, S.; Yki-Jarvinen, H. Effects of rosiglitazone and metformin on liver fat content, hepatic insulin resistance, insulin clearance, and gene expression in adipose tissue in patients with type 2 diabetes. Diabetes 2004, 53, 2169–2176. [Google Scholar] [CrossRef] [Green Version]
- Ghadieh, H.E.; Muturi, H.T.; Russo, L.; Marino, C.C.; Ghanem, S.S.; Khuder, S.S.; Hanna, J.C.; Jash, S.; Puri, V.; Heinrich, G.; et al. Exenatide induces carcinoembryonic antigen-related cell adhesion molecule 1 expression to prevent hepatic steatosis. Hepatol. Commun. 2018, 2, 35–47. [Google Scholar] [CrossRef]
- Boden, G. Fatty acid-induced inflammation and insulin resistance in skeletal muscle and liver. Curr. Diab. Rep. 2006, 6, 177–181. [Google Scholar] [CrossRef]
- Reaven, G.M. Pathophysiology of insulin resistance in human disease. Physiol. Rev. 1995, 75, 473–486. [Google Scholar] [CrossRef]
- Lewis, G.F.; Carpentier, A.; Adeli, K.; Giacca, A. Disordered fat storage and mobilization in the pathogenesis of insulin resistance and type 2 diabetes. Endocr. Rev. 2002, 23, 201–229. [Google Scholar] [CrossRef]
- Fabbrini, E.; Mohammed, B.S.; Magkos, F.; Korenblat, K.M.; Patterson, B.W.; Klein, S. Alterations in adipose tissue and hepatic lipid kinetics in obese men and women with nonalcoholic fatty liver disease. Gastroenterology 2008, 134, 424–431. [Google Scholar] [CrossRef] [Green Version]
- Gastaldelli, A.; Miyazaki, Y.; Pettiti, M.; Matsuda, M.; Mahankali, S.; Santini, E.; DeFronzo, R.A.; Ferrannini, E. Metabolic effects of visceral fat accumulation in type 2 diabetes. J. Clin. Endocrinol. Metab. 2002, 87, 5098–5103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, Z.; Hensrud, D.D.; Johnson, C.M.; Jensen, M.D. Regional postprandial fatty acid metabolism in different obesity phenotypes. Diabetes 1999, 48, 1586–1592. [Google Scholar] [CrossRef] [PubMed]
- Chiang, S.H.; Bazuine, M.; Lumeng, C.N.; Geletka, L.M.; Mowers, J.; White, N.M.; Ma, J.T.; Zhou, J.; Qi, N.; Westcott, D.; et al. The protein kinase IKKepsilon regulates energy balance in obese mice. Cell 2009, 138, 961–975. [Google Scholar] [CrossRef] [Green Version]
- Shi, H.; Kokoeva, M.V.; Inouye, K.; Tzameli, I.; Yin, H.; Flier, J.S. TLR4 links innate immunity and fatty acid-induced insulin resistance. J. Clin. Investig. 2006, 116, 3015–3025. [Google Scholar] [CrossRef] [PubMed]
- Saltiel, A.R.; Olefsky, J.M. Inflammatory mechanisms linking obesity and metabolic disease. J.Clin. Investig. 2017, 127, 1–4. [Google Scholar] [CrossRef] [Green Version]
- Perreault, M.; Marette, A. Targeted disruption of inducible nitric oxide synthase protects against obesity-linked insulin resistance in muscle. Nat. Med. 2001, 7, 1138–1143. [Google Scholar] [CrossRef]
- Hirosumi, J.; Tuncman, G.; Chang, L.; Gorgun, C.Z.; Uysal, K.T.; Maeda, K.; Karin, M.; Hotamisligil, G.S. A central role for JNK in obesity and insulin resistance. Nature 2002, 420, 333–336. [Google Scholar] [CrossRef]
- Cai, D.; Yuan, M.; Frantz, D.F.; Melendez, P.A.; Hansen, L.; Lee, J.; Shoelson, S.E. Local and systemic insulin resistance resulting from hepatic activation of IKK-beta and NF-kappaB. Nat. Med. 2005, 11, 183–190. [Google Scholar] [CrossRef]
- Berg, A.H.; Combs, T.P.; Du, X.; Brownlee, M.; Scherer, P.E. The adipocyte-secreted protein Acrp30 enhances hepatic insulin action. Nat. Med. 2001, 7, 947–953. [Google Scholar] [CrossRef]
- Shetty, A.; Hsu, J.W.; Manka, P.P.; Syn, W.K. Role of the Circadian Clock in the Metabolic Syndrome and Nonalcoholic Fatty Liver Disease. Dig. Dis. Sci. 2018, 63, 3187–3206. [Google Scholar] [CrossRef]
- Prasad, M.; Rajagopal, P.; Devarajan, N.; Veeraraghavan, V.P.; Palanisamy, C.P.; Cui, B.; Patil, S.; Jayaraman, S. A comprehensive review on high -fat diet-induced diabetes mellitus: An epigenetic view. J. Nutr. Biochem. 2022, 107, 109037. [Google Scholar] [CrossRef] [PubMed]
- Wesolowski, S.R.; Kasmi, K.C.; Jonscher, K.R.; Friedman, J.E. Developmental origins of NAFLD: A womb with a clue. Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 81–96. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baker, P.R., 2nd; Friedman, J.E. Mitochondrial role in the neonatal predisposition to developing nonalcoholic fatty liver disease. J. Clin. Investig. 2018, 128, 3692–3703. [Google Scholar] [CrossRef] [PubMed]
- Frayn, K.N. Adipose tissue as a buffer for daily lipid flux. Diabetologia 2002, 45, 1201–1210. [Google Scholar] [CrossRef] [Green Version]
- Boden, G.; She, P.; Mozzoli, M.; Cheung, P.; Gumireddy, K.; Reddy, P.; Xiang, X.; Luo, Z.; Ruderman, N. Free fatty acids produce insulin resistance and activate the proinflammatory nuclear factor-kappaB pathway in rat liver. Diabetes 2005, 54, 3458–3465. [Google Scholar] [CrossRef] [Green Version]
- Bajaj, M.; Suraamornkul, S.; Kashyap, S.; Cusi, K.; Mandarino, L.; DeFronzo, R.A. Sustained reduction in plasma free fatty acid concentration improves insulin action without altering plasma adipocytokine levels in subjects with strong family history of type 2 diabetes. J. Clin. Endocrinol. Metab. 2004, 89, 4649–4655. [Google Scholar] [CrossRef] [Green Version]
- Rosso, C.; Kazankov, K.; Younes, R.; Esmaili, S.; Marietti, M.; Sacco, M.; Carli, F.; Gaggini, M.; Salomone, F.; Moller, H.J.; et al. Crosstalk between adipose tissue insulin resistance and liver macrophages in non-alcoholic fatty liver disease. J. Hepatol. 2019, 71, 1012–1021. [Google Scholar] [CrossRef]
- Hennes, M.M.; Dua, A.; Kissebah, A.H. Effects of free fatty acids and glucose on splanchnic insulin dynamics. Diabetes 1997, 46, 57–62. [Google Scholar] [CrossRef]
- Wiesenthal, S.R.; Sandhu, H.; McCall, R.H.; Tchipashvili, V.; Yoshii, H.; Polonsky, K.; Shi, Z.Q.; Lewis, G.F.; Mari, A.; Giacca, A. Free fatty acids impair hepatic insulin extraction in vivo. Diabetes 1999, 48, 766–774. [Google Scholar] [CrossRef]
- Svedberg, J.; Stromblad, G.; Wirth, A.; Smith, U.; Bjorntorp, P. Fatty acids in the portal vein of the rat regulate hepatic insulin clearance. J. Clin. Investig. 1991, 88, 2054–2058. [Google Scholar] [CrossRef]
- Bosello, O.; Zamboni, M.; Armellini, F.; Zocca, I.; Bergamo Andreis, I.A.; Smacchia, C.; Milani, M.P.; Cominacini, L. Modifications of abdominal fat and hepatic insulin clearance during severe caloric restriction. Ann. Nutr. Metab. 1990, 34, 359–365. [Google Scholar] [CrossRef] [PubMed]
- Pereira, S.; Park, E.; Mori, Y.; Haber, C.A.; Han, P.; Uchida, T.; Stavar, L.; Oprescu, A.I.; Koulajian, K.; Ivovic, A.; et al. FFA-induced hepatic insulin resistance in vivo is mediated by PKCδ, NADPH oxidase, and oxidative stress. Am. J. Physiol. Endocrinol. Metab. 2014, 307, E34–E46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, M.; Vienberg, S.G.; Bezy, O.; O’Neill, B.T.; Kahn, C.R. Role of PKCdelta in Insulin Sensitivity and Skeletal Muscle Metabolism. Diabetes 2015, 64, 4023–4032. [Google Scholar] [CrossRef] [Green Version]
- Gassaway, B.M.; Petersen, M.C.; Surovtseva, Y.V.; Barber, K.W.; Sheetz, J.B.; Aerni, H.R.; Merkel, J.S.; Samuel, V.T.; Shulman, G.I.; Rinehart, J. PKCepsilon contributes to lipid-induced insulin resistance through cross talk with p70S6K and through previously unknown regulators of insulin signaling. Proc. Natl. Acad. Sci. USA 2018, 115, E8996–E9005. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, Y.B.; Kotani, K.; Ciaraldi, T.P.; Henry, R.R.; Kahn, B.B. Insulin-stimulated protein kinase C lambda/zeta activity is reduced in skeletal muscle of humans with obesity and type 2 diabetes: Reversal with weight reduction. Diabetes 2003, 52, 1935–1942. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, M.T.; Satoh, H.; Favelyukis, S.; Babendure, J.L.; Imamura, T.; Sbodio, J.I.; Zalevsky, J.; Dahiyat, B.I.; Chi, N.W.; Olefsky, J.M. JNK and tumor necrosis factor-alpha mediate free fatty acid-induced insulin resistance in 3T3-L1 adipocytes. J. Biol. Chem. 2005, 280, 35361–35371. [Google Scholar] [CrossRef] [Green Version]
- Svedberg, J.; Bjorntorp, P.; Smith, U.; Lonnroth, P. Free-fatty acid inhibition of insulin binding, degradation, and action in isolated rat hepatocytes. Diabetes 1990, 39, 570–574. [Google Scholar] [CrossRef]
- Hennes, M.M.; Shrago, E.; Kissebah, A.H. Receptor and postreceptor effects of free fatty acids (FFA) on hepatocyte insulin dynamics [see comments]. Int. J. Obes. 1990, 14, 831–841. [Google Scholar]
- Ferrante, A.W., Jr. Obesity-induced inflammation: A metabolic dialogue in the language of inflammation. J. Intern. Med. 2007, 262, 408–414. [Google Scholar] [CrossRef]
- Park, S.Y.; Cho, Y.R.; Kim, H.J.; Higashimori, T.; Danton, C.; Lee, M.K.; Dey, A.; Rothermel, B.; Kim, Y.B.; Kalinowski, A.; et al. Unraveling the temporal pattern of diet-induced insulin resistance in individual organs and cardiac dysfunction in C57BL/6 mice. Diabetes 2005, 54, 3530–3540. [Google Scholar] [CrossRef] [Green Version]
- Lee, Y.S.; Li, P.; Huh, J.Y.; Hwang, I.J.; Lu, M.; Kim, J.I.; Ham, M.; Talukdar, S.; Chen, A.; Lu, W.J.; et al. Inflammation is necessary for long-term but not short-term high-fat diet-induced insulin resistance. Diabetes 2011, 60, 2474–2483. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scherer, T.; Lindtner, C.; Zielinski, E.; O’Hare, J.; Filatova, N.; Buettner, C. Short term voluntary overfeeding disrupts brain insulin control of adipose tissue lipolysis. J. Biol. Chem. 2012, 287, 33061–33069. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kabir, M.; Catalano, K.J.; Ananthnarayan, S.; Kim, S.P.; Van Citters, G.W.; Dea, M.K.; Bergman, R.N. Molecular evidence supporting the portal theory: A causative link between visceral adiposity and hepatic insulin resistance. Am. J. Physiol. Endocrinol. Metab. 2005, 288, E454–E461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perry, R.J.; Camporez, J.P.; Kursawe, R.; Titchenell, P.M.; Zhang, D.; Perry, C.J.; Jurczak, M.J.; Abudukadier, A.; Han, M.S.; Zhang, X.M.; et al. Hepatic acetyl CoA links adipose tissue inflammation to hepatic insulin resistance and type 2 diabetes. Cell 2015, 160, 745–758. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Titchenell, P.M.; Quinn, W.J.; Lu, M.; Chu, Q.; Lu, W.; Li, C.; Chen, H.; Monks, B.R.; Chen, J.; Rabinowitz, J.D.; et al. Direct hepatocyte insulin signaling is required for lipogenesis but is dispensable for the suppression of glucose production. Cell Metab. 2016, 23, 1154–1166. [Google Scholar] [CrossRef] [Green Version]
- Divoux, A.; Tordjman, J.; Lacasa, D.; Veyrie, N.; Hugol, D.; Aissat, A.; Basdevant, A.; Guerre-Millo, M.; Poitou, C.; Zucker, J.D.; et al. Fibrosis in human adipose tissue: Composition, distribution, and link with lipid metabolism and fat mass loss. Diabetes 2010, 59, 2817–2825. [Google Scholar] [CrossRef] [Green Version]
- Yadav, H.; Quijano, C.; Kamaraju, A.K.; Gavrilova, O.; Malek, R.; Chen, W.; Zerfas, P.; Zhigang, D.; Wright, E.C.; Stuelten, C.; et al. Protection from obesity and diabetes by blockade of TGF-beta/Smad3 signaling. Cell Metab 2011, 14, 67–79. [Google Scholar] [CrossRef] [Green Version]
- Sun, K.; Park, J.; Gupta, O.T.; Holland, W.L.; Auerbach, P.; Zhang, N.; Goncalves Marangoni, R.; Nicoloro, S.M.; Czech, M.P.; Varga, J.; et al. Endotrophin triggers adipose tissue fibrosis and metabolic dysfunction. Nat. Commun. 2014, 5, 3485–3496. [Google Scholar] [CrossRef] [Green Version]
- Groop, L.C.; Saloranta, C.; Shank, M.; Bonadonna, R.C.; Ferrannini, E.; DeFronzo, R.A. The role of free fatty acid metabolism in the pathogenesis of insulin resistance in obesity and noninsulin-dependent diabetes mellitus. J. Clin. Endocrinol. Metab. 1991, 72, 96–107. [Google Scholar] [CrossRef]
- Groop, L.C.; Bonadonna, R.C.; Shank, M.; Petrides, A.S.; DeFronzo, R.A. Role of free fatty acids and insulin in determining free fatty acid and lipid oxidation in man. J. Clin. Investig. 1991, 87, 83–89. [Google Scholar] [CrossRef]
- Bays, H.; Mandarino, L.; DeFronzo, R.A. Role of the adipocyte, free fatty acids, and ectopic fat in pathogenesis of type 2 diabetes mellitus: Peroxisomal proliferator-activated receptor agonists provide a rational therapeutic approach. J. Clin. Endocrinol. Metab. 2004, 89, 463–478, Review. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramakrishnan, S.K.; Khuder, S.S.; Al-Share, Q.Y.; Russo, L.; Abdallah, S.L.; Patel, P.R.; Heinrich, G.; Muturi, H.T.; Mopidevi, B.R.; Oyarce, A.M.; et al. PPARalpha (peroxisome proliferator-activated receptor alpha) activation reduces hepatic CEACAM1 protein expression to regulate fatty acid oxidation during fasting-refeeding transition. J. Biol. Chem. 2016, 291, 8121–8129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gonzalez, E.; Flier, E.; Molle, D.; Accili, D.; McGraw, T.E. Hyperinsulinemia leads to uncoupled insulin regulation of the GLUT4 glucose transporter and the FoxO1 transcription factor. Proc. Natl. Acad. Sci. USA 2011, 108, 10162–10167. [Google Scholar] [CrossRef] [Green Version]
- Santoro, A.; McGraw, T.E.; Kahn, B.B. Insulin action in adipocytes, adipose remodeling, and systemic effects. Cell Metab 2021, 33, 748–757. [Google Scholar] [CrossRef]
- Russo, L.; Ghadieh, H.E.; Ghanem, S.S.; Al-Share, Q.Y.; Smiley, Z.N.; Gatto-Weis, C.; Esakov, E.L.; McInerney, M.F.; Heinrich, G.; Tong, X.; et al. Role for hepatic CEACAM1 in regulating fatty acid metabolism along the adipocyte-hepatocyte axis. J. Lipid Res. 2016, 57, 2163–2175. [Google Scholar] [CrossRef] [Green Version]
- Ben-Haroush Schyr, R.; Al-Kurd, A.; Moalem, B.; Permyakova, A.; Israeli, H.; Bardugo, A.; Arad, Y.; Hefetz, L.; Bergel, M.; Haran, A.; et al. Sleeve Gastrectomy Suppresses Hepatic Glucose Production and Increases Hepatic Insulin Clearance Independent of Weight Loss. Diabetes 2021, 70, 2289–2298. [Google Scholar] [CrossRef]
- Lester, S.G.; Russo, L.; Ghanem, S.S.; Khuder, S.S.; DeAngelis, A.M.; Esakov, E.L.; Bowman, T.A.; Heinrich, G.; Al-Share, Q.Y.; McInerney, M.F.; et al. Hepatic CEACAM1 over-expression protects against diet-induced fibrosis and inflammation in white adipose tissue. Front. Endocrinol. 2015, 6, 116–122. [Google Scholar] [CrossRef] [Green Version]
- Kabir, M.; Iyer, M.S.; Richey, J.M.; Woolcott, O.O.; Asare Bediako, I.; Wu, Q.; Kim, S.P.; Stefanovski, D.; Kolka, C.M.; Hsu, I.R.; et al. CB1R antagonist increases hepatic insulin clearance in fat-fed dogs likely via upregulation of liver adiponectin receptors. Am. J. Physiol. Endocrinol. Metab. 2015, 309, E747–E758. [Google Scholar] [CrossRef] [Green Version]
- Lundsgaard, A.M.; Sjoberg, K.A.; Hoeg, L.D.; Jeppesen, J.; Jordy, A.B.; Serup, A.K.; Fritzen, A.M.; Pilegaard, H.; Myrmel, L.S.; Madsen, L.; et al. Opposite Regulation of Insulin Sensitivity by Dietary Lipid Versus Carbohydrate Excess. Diabetes 2017, 66, 2583–2595. [Google Scholar] [CrossRef] [Green Version]
- Bojsen-Moller, K.N.; Lundsgaard, A.M.; Madsbad, S.; Kiens, B.; Holst, J.J. Hepatic insulin clearance in regulation of systemic insulin concentrations-role of carbohydrate and energy availability. Diabetes 2018, 67, 2129–2136. [Google Scholar] [CrossRef] [Green Version]
- Shah, P.; Vella, A.; Basu, A.; Basu, R.; Adkins, A.; Schwenk, W.F.; Johnson, C.M.; Nair, K.S.; Jensen, M.D.; Rizza, R.A. Effects of free fatty acids and glycerol on splanchnic glucose metabolism and insulin extraction in nondiabetic humans. Diabetes 2002, 51, 301–310. [Google Scholar] [CrossRef] [Green Version]
- Erdmann, J.; Kallabis, B.; Oppel, U.; Sypchenko, O.; Wagenpfeil, S.; Schusdziarra, V. Development of hyperinsulinemia and insulin resistance during the early stage of weight gain. Am. J. Physiol. Endocrinol. Metab. 2008, 294, E568–E575. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaga, H.; Tamura, Y.; Takeno, K.; Kakehi, S.; Funayama, T.; Furukawa, Y.; Nishitani-Yokoyama, M.; Shimada, K.; Daida, H.; Aoki, S.; et al. Correlates of insulin clearance in apparently healthy non-obese Japanese men. Sci. Rep. 2017, 7, 1462. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bergman, R.N.; Kabir, M.; Ader, M. The Physiology of Insulin Clearance. Int. J. Mol. Sci. 2022, 23, E568–E575. [Google Scholar] [CrossRef] [PubMed]
- Bergman, R.N.; Piccinini, F.; Kabir, M.; Kolka, C.M.; Ader, M. Hypothesis: Role of Reduced Hepatic Insulin Clearance in the Pathogenesis of Type 2 Diabetes. Diabetes 2019, 68, 1709–1716. [Google Scholar] [CrossRef]
- Ghadieh, H.E.; Russo, L.; Muturi, H.T.; Ghanem, S.S.; Manaserh, I.H.; Noh, H.L.; Suk, S.; Kim, J.K.; Hill, J.W.; Najjar, S.M. Hyperinsulinemia drives hepatic insulin resistance in male mice with liver-specific Ceacam1 deletion independently of lipolysis. Metabolism 2019, 93, 33–43. [Google Scholar] [CrossRef]
- Russo, L.; Muturi, H.T.; Ghadieh, H.E.; Ghanem, S.S.; Bowman, T.A.; Noh, H.L.; Dagdeviren, S.; Dogbey, G.Y.; Kim, J.K.; Heinrich, G.; et al. Liver-specific reconstitution of CEACAM1 reverses the metabolic abnormalities caused by its global deletion in male mice. Diabetologia 2017, 60, 2463–2474. [Google Scholar] [CrossRef] [PubMed]
- Helal, R.A.; Russo, L.; Ghadieh, H.E.; Muturi, H.T.; Asalla, S.; Lee, A.D.; Gatto-Weis, C.; Najjar, S.M. Regulation of hepatic fibrosis by carcinoembryonic antigen-related cell adhesion molecule 1. Metabolism 2021, 121, 154801. [Google Scholar] [CrossRef] [PubMed]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Najjar, S.M.; Abdolahipour, R.; Ghadieh, H.E.; Jahromi, M.S.; Najjar, J.A.; Abuamreh, B.A.M.; Zaidi, S.; Kumarasamy, S.; Muturi, H.T. Regulation of Insulin Clearance by Non-Esterified Fatty Acids. Biomedicines 2022, 10, 1899. https://doi.org/10.3390/biomedicines10081899
Najjar SM, Abdolahipour R, Ghadieh HE, Jahromi MS, Najjar JA, Abuamreh BAM, Zaidi S, Kumarasamy S, Muturi HT. Regulation of Insulin Clearance by Non-Esterified Fatty Acids. Biomedicines. 2022; 10(8):1899. https://doi.org/10.3390/biomedicines10081899
Chicago/Turabian StyleNajjar, Sonia M., Raziyeh Abdolahipour, Hilda E. Ghadieh, Marziyeh Salehi Jahromi, John A. Najjar, Basil A. M. Abuamreh, Sobia Zaidi, Sivarajan Kumarasamy, and Harrison T. Muturi. 2022. "Regulation of Insulin Clearance by Non-Esterified Fatty Acids" Biomedicines 10, no. 8: 1899. https://doi.org/10.3390/biomedicines10081899