

Transposons Acting as Competitive Endogenous RNAs: In-Silico Evidence from Datasets Characterised by L1 Overexpression
 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Data Collection and Pre-Processing
2.2. Analysis of Locus-Specific TE Expression
2.3. Detection of L1 Autonomous Transcription
2.4. Gene Expression Analysis
2.5. Functional Enrichment Analysis
2.6. Overlap Analysis
2.7. Analysis of miRNA Target Sites Sharing
2.8. Identification of miRNAs Sequestered by L1s
2.9. miRNA-Gene Networks Identification
2.10. Analysis of miRNA-L1 Expression
2.11. Analysis of TE Expression at the Consensus Level
3. Results
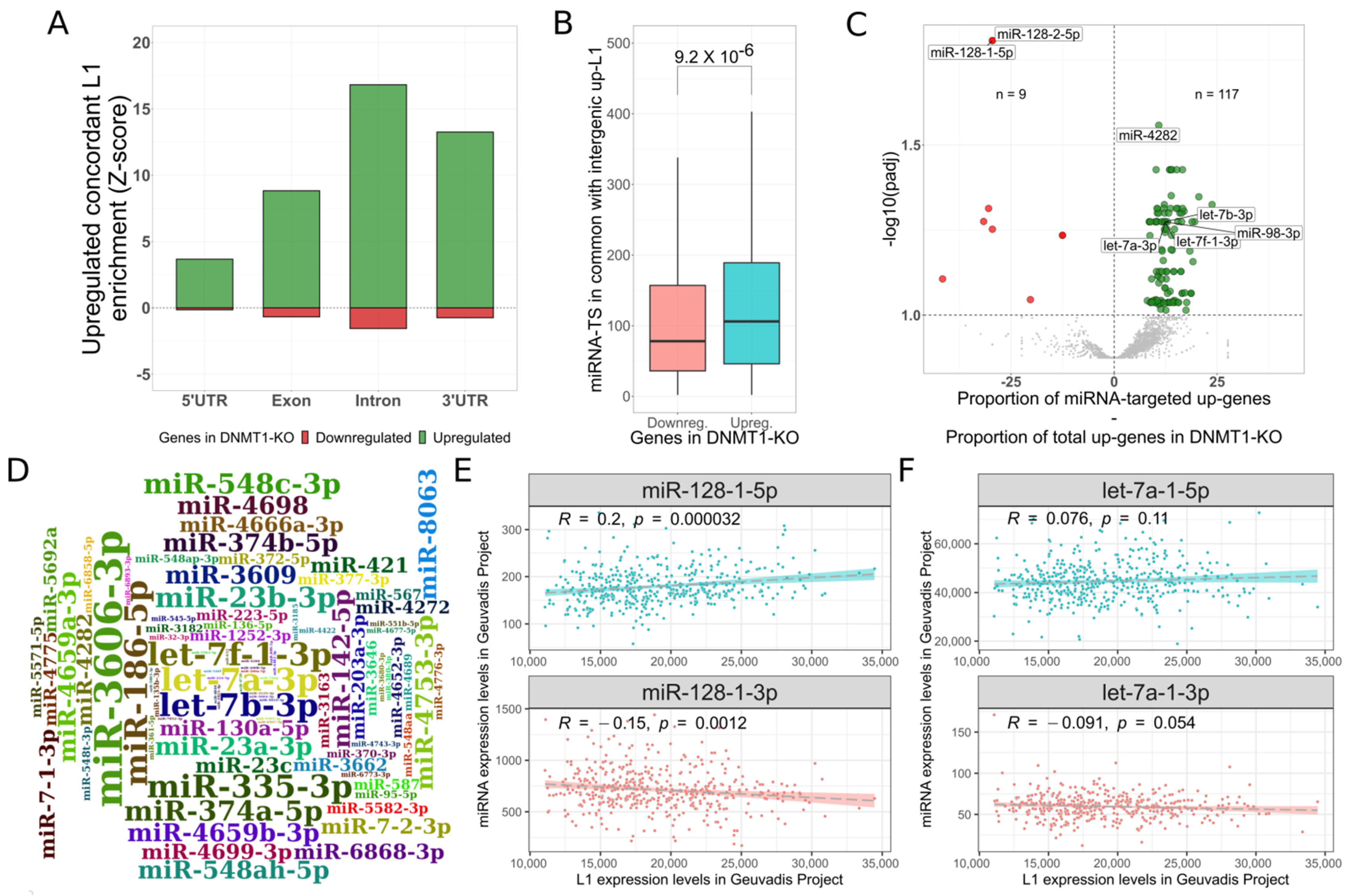
3.1. L1s Are Autonomously Transcribed upon DNMT1-KO
3.2. L1 Transcripts Could Act as ceRNA
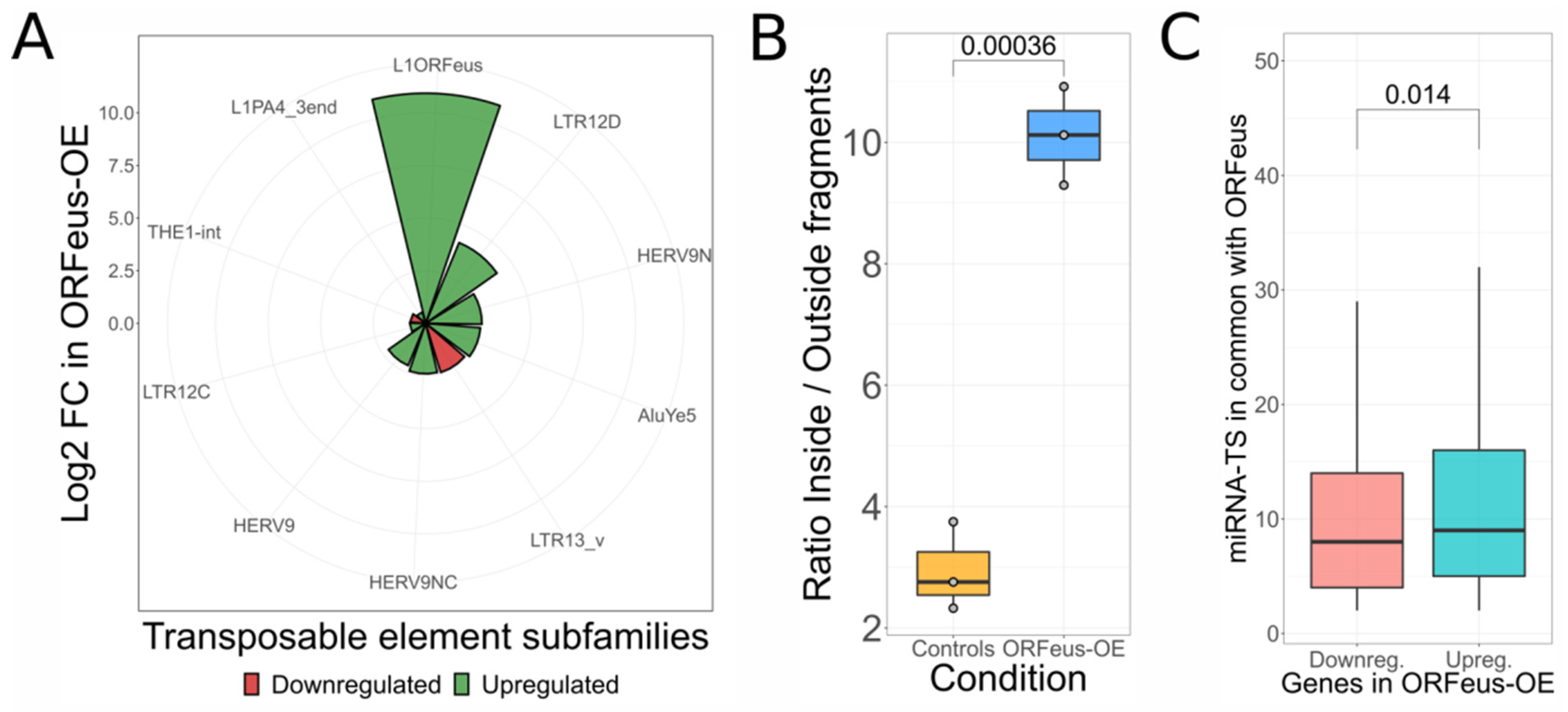
3.3. Support for L1 ceRNA Activity Using an Independent Experiment in Which a Specific L1 Is Artificially Overexpressed
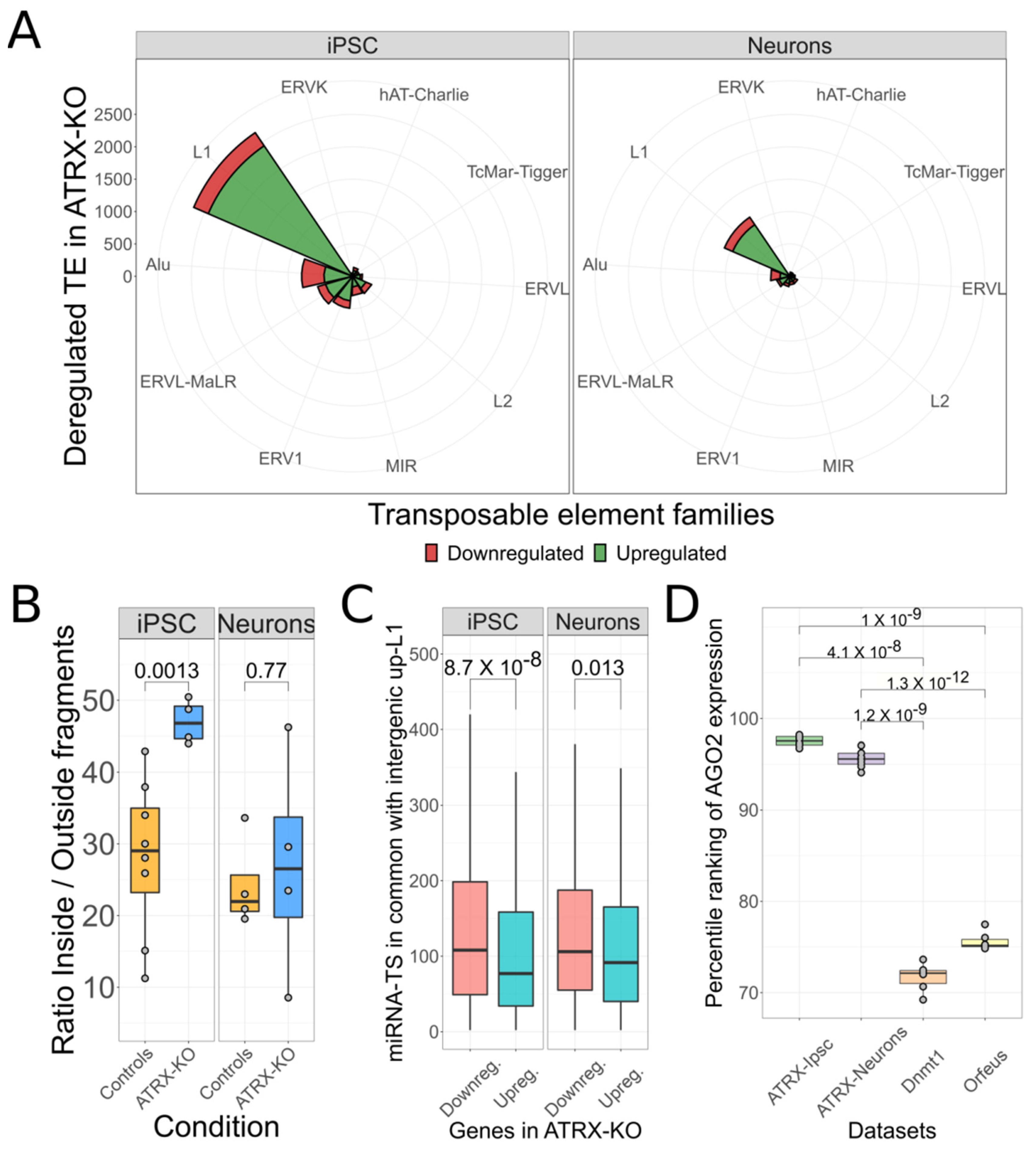
3.4. Putative L1 ceRNA Activity Might Depend on Autonomous L1 Transcription and AGO2
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Wicker, T.; Sabot, F.; Hua-Van, A.; Bennetzen, J.L.; Capy, P.; Chalhoub, B.; Flavell, A.; Leroy, P.; Morgante, M.; Panaud, O.; et al. A Unified Classification System for Eukaryotic Transposable Elements. Nat. Rev. Genet. 2007, 8, 973–982. [Google Scholar] [CrossRef] [PubMed]
- Lander, E.S.; Linton, L.M.; Birren, B.; Nusbaum, C.; Zody, M.C.; Baldwin, J.; Devon, K.; Dewar, K.; Doyle, M.; FitzHugh, W.; et al. Initial Sequencing and Analysis of the Human Genome. Nature 2001, 409, 860–921. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morrish, T.A.; Gilbert, N.; Myers, J.S.; Vincent, B.J.; Stamato, T.D.; Taccioli, G.E.; Batzer, M.A.; Moran, J.V. DNA Repair Mediated by Endonuclease-Independent LINE-1 Retrotransposition. Nat. Genet. 2002, 31, 159–165. [Google Scholar] [CrossRef] [PubMed]
- Mills, R.E.; Bennett, E.A.; Iskow, R.C.; Luttig, C.T.; Tsui, C.; Pittard, W.S.; Devine, S.E. Recently Mobilized Transposons in the Human and Chimpanzee Genomes. Am. J. Hum. Genet. 2006, 78, 671–679. [Google Scholar] [CrossRef] [Green Version]
- Sultana, T.; van Essen, D.; Siol, O.; Bailly-Bechet, M.; Philippe, C.; Zine El Aabidine, A.; Pioger, L.; Nigumann, P.; Saccani, S.; Andrau, J.-C.; et al. The Landscape of L1 Retrotransposons in the Human Genome Is Shaped by Pre-Insertion Sequence Biases and Post-Insertion Selection. Mol. Cell 2019, 74, 555–570.e7. [Google Scholar] [CrossRef]
- Kazazian, H.H. Mobile Elements: Drivers of Genome Evolution. Science 2004, 303, 1626–1632. [Google Scholar] [CrossRef] [Green Version]
- Jordan, I.K.; Rogozin, I.B.; Glazko, G.V.; Koonin, E.V. Origin of a Substantial Fraction of Human Regulatory Sequences from Transposable Elements. Trends Genet. TIG 2003, 19, 68–72. [Google Scholar] [CrossRef]
- Ni, J.Z.; Grate, L.; Donohue, J.P.; Preston, C.; Nobida, N.; O’Brien, G.; Shiue, L.; Clark, T.A.; Blume, J.E.; Ares, M. Ultraconserved Elements Are Associated with Homeostatic Control of Splicing Regulators by Alternative Splicing and Nonsense-Mediated Decay. Genes Dev. 2007, 21, 708–718. [Google Scholar] [CrossRef] [Green Version]
- Spengler, R.M.; Oakley, C.K.; Davidson, B.L. Functional MicroRNAs and Target Sites Are Created by Lineage-Specific Transposition. Hum. Mol. Genet. 2014, 23, 1783–1793. [Google Scholar] [CrossRef] [Green Version]
- Rangasamy, D.; Lenka, N.; Ohms, S.; Dahlstrom, J.E.; Blackburn, A.C.; Board, P.G. Activation of LINE-1 Retrotransposon Increases the Risk of Epithelial-Mesenchymal Transition and Metastasis in Epithelial Cancer. Curr. Mol. Med. 2015, 15, 588–597. [Google Scholar] [CrossRef]
- Sanchez-Luque, F.J.; Kempen, M.-J.H.C.; Gerdes, P.; Vargas-Landin, D.B.; Richardson, S.R.; Troskie, R.-L.; Jesuadian, J.S.; Cheetham, S.W.; Carreira, P.E.; Salvador-Palomeque, C.; et al. LINE-1 Evasion of Epigenetic Repression in Humans. Mol. Cell 2019, 75, 590–604.e12. [Google Scholar] [CrossRef] [PubMed]
- Hatanaka, Y.; Inoue, K.; Oikawa, M.; Kamimura, S.; Ogonuki, N.; Kodama, E.N.; Ohkawa, Y.; Tsukada, Y.; Ogura, A. Histone Chaperone CAF-1 Mediates Repressive Histone Modifications to Protect Preimplantation Mouse Embryos from Endogenous Retrotransposons. Proc. Natl. Acad. Sci. USA 2015, 112, 14641–14646. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Fazio, S.; Bartonicek, N.; Di Giacomo, M.; Abreu-Goodger, C.; Sankar, A.; Funaya, C.; Antony, C.; Moreira, P.N.; Enright, A.J.; O’Carroll, D. The Endonuclease Activity of Mili Fuels PiRNA Amplification That Silences LINE1 Elements. Nature 2011, 480, 259–263. [Google Scholar] [CrossRef] [PubMed]
- Servant, G.; Streva, V.A.; Derbes, R.S.; Wijetunge, M.I.; Neeland, M.; White, T.B.; Belancio, V.P.; Roy-Engel, A.M.; Deininger, P.L. The Nucleotide Excision Repair Pathway Limits L1 Retrotransposition. Genetics 2017, 205, 139–153. [Google Scholar] [CrossRef] [Green Version]
- Lee, R.C.; Ambros, V. An Extensive Class of Small RNAs in Caenorhabditis Elegans. Science 2001, 294, 862–864. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, T.A.; Jo, M.H.; Choi, Y.-G.; Park, J.; Kwon, S.C.; Hohng, S.; Kim, V.N.; Woo, J.-S. Functional Anatomy of the Human Microprocessor. Cell 2015, 161, 1374–1387. [Google Scholar] [CrossRef] [Green Version]
- Grishok, A.; Pasquinelli, A.E.; Conte, D.; Li, N.; Parrish, S.; Ha, I.; Baillie, D.L.; Fire, A.; Ruvkun, G.; Mello, C.C. Genes and Mechanisms Related to RNA Interference Regulate Expression of the Small Temporal RNAs That Control C. Elegans Developmental Timing. Cell 2001, 106, 23–34. [Google Scholar] [CrossRef] [Green Version]
- Rivas, F.V.; Tolia, N.H.; Song, J.-J.; Aragon, J.P.; Liu, J.; Hannon, G.J.; Joshua-Tor, L. Purified Argonaute2 and an SiRNA Form Recombinant Human RISC. Nat. Struct. Mol. Biol. 2005, 12, 340–349. [Google Scholar] [CrossRef]
- Bartel, D.P. MicroRNAs: Target Recognition and Regulatory Functions. Cell 2009, 136, 215–233. [Google Scholar] [CrossRef] [Green Version]
- Jo, M.H.; Shin, S.; Jung, S.-R.; Kim, E.; Song, J.-J.; Hohng, S. Human Argonaute 2 Has Diverse Reaction Pathways on Target RNAs. Mol. Cell 2015, 59, 117–124. [Google Scholar] [CrossRef]
- Behm-Ansmant, I.; Rehwinkel, J.; Doerks, T.; Stark, A.; Bork, P.; Izaurralde, E. MRNA Degradation by MiRNAs and GW182 Requires Both CCR4:NOT Deadenylase and DCP1:DCP2 Decapping Complexes. Genes Dev. 2006, 20, 1885–1898. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Braun, J.E.; Truffault, V.; Boland, A.; Huntzinger, E.; Chang, C.-T.; Haas, G.; Weichenrieder, O.; Coles, M.; Izaurralde, E. A Direct Interaction between DCP1 and XRN1 Couples MRNA Decapping to 5′ Exonucleolytic Degradation. Nat. Struct. Mol. Biol. 2012, 19, 1324–1331. [Google Scholar] [CrossRef] [PubMed]
- Hamdorf, M.; Idica, A.; Zisoulis, D.G.; Gamelin, L.; Martin, C.; Sanders, K.J.; Pedersen, I.M. MiR-128 Represses L1 Retrotransposition by Binding Directly to L1 RNA. Nat. Struct. Mol. Biol. 2015, 22, 824–831. [Google Scholar] [CrossRef] [PubMed]
- Tristán-Ramos, P.; Rubio-Roldan, A.; Peris, G.; Sánchez, L.; Amador-Cubero, S.; Viollet, S.; Cristofari, G.; Heras, S.R. The Tumor Suppressor MicroRNA Let-7 Inhibits Human LINE-1 Retrotransposition. Nat. Commun. 2020, 11, 5712. [Google Scholar] [CrossRef] [PubMed]
- Salmena, L.; Poliseno, L.; Tay, Y.; Kats, L.; Pandolfi, P.P. A CeRNA Hypothesis: The Rosetta Stone of a Hidden RNA Language? Cell 2011, 146, 353–358. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poliseno, L.; Salmena, L.; Zhang, J.; Carver, B.; Haveman, W.J.; Pandolfi, P.P. A Coding-Independent Function of Gene and Pseudogene MRNAs Regulates Tumour Biology. Nature 2010, 465, 1033–1038. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnson, R.; Guigó, R. The RIDL Hypothesis: Transposable Elements as Functional Domains of Long Noncoding RNAs. RNA 2014, 20, 959–976. [Google Scholar] [CrossRef] [Green Version]
- Carrieri, C.; Cimatti, L.; Biagioli, M.; Beugnet, A.; Zucchelli, S.; Fedele, S.; Pesce, E.; Ferrer, I.; Collavin, L.; Santoro, C.; et al. Long Non-Coding Antisense RNA Controls Uchl1 Translation through an Embedded SINEB2 Repeat. Nature 2012, 491, 454–457. [Google Scholar] [CrossRef] [Green Version]
- Fu, Z.; Li, G.; Li, Z.; Wang, Y.; Zhao, Y.; Zheng, S.; Ye, H.; Luo, Y.; Zhao, X.; Wei, L.; et al. Endogenous MiRNA Sponge LincRNA-ROR Promotes Proliferation, Invasion and Stem Cell-like Phenotype of Pancreatic Cancer Cells. Cell Death Discov. 2017, 3, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Jönsson, M.E.; Ludvik Brattås, P.; Gustafsson, C.; Petri, R.; Yudovich, D.; Pircs, K.; Verschuere, S.; Madsen, S.; Hansson, J.; Larsson, J.; et al. Activation of Neuronal Genes via LINE-1 Elements upon Global DNA Demethylation in Human Neural Progenitors. Nat. Commun. 2019, 10, 3182. [Google Scholar] [CrossRef]
- Marasca, F.; Sinha, S.; Vadalà, R.; Polimeni, B.; Ranzani, V.; Paraboschi, E.M.; Burattin, F.V.; Ghilotti, M.; Crosti, M.; Negri, M.L.; et al. LINE1 Are Spliced in Non-Canonical Transcript Variants to Regulate T Cell Quiescence and Exhaustion. Nat. Genet. 2022, 54, 180–193. [Google Scholar] [CrossRef]
- Ardeljan, D.; Steranka, J.P.; Liu, C.; Li, Z.; Taylor, M.S.; Payer, L.M.; Gorbounov, M.; Sarnecki, J.S.; Deshpande, V.; Hruban, R.H.; et al. Cell Fitness Screens Reveal a Conflict between LINE-1 Retrotransposition and DNA Replication. Nat. Struct. Mol. Biol. 2020, 27, 168–178. [Google Scholar] [CrossRef] [PubMed]
- Deneault, E.; White, S.H.; Rodrigues, D.C.; Ross, P.J.; Faheem, M.; Zaslavsky, K.; Wang, Z.; Alexandrova, R.; Pellecchia, G.; Wei, W.; et al. Complete Disruption of Autism-Susceptibility Genes by Gene Editing Predominantly Reduces Functional Connectivity of Isogenic Human Neurons. Stem Cell Rep. 2018, 11, 1211–1225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lappalainen, T.; Sammeth, M.; Friedländer, M.R.; ‘t Hoen, P.A.C.; Monlong, J.; Rivas, M.A.; Gonzàlez-Porta, M.; Kurbatova, N.; Griebel, T.; Ferreira, P.G.; et al. Transcriptome and Genome Sequencing Uncovers Functional Variation in Humans. Nature 2013, 501, 506–511. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Athar, A.; Füllgrabe, A.; George, N.; Iqbal, H.; Huerta, L.; Ali, A.; Snow, C.; Fonseca, N.A.; Petryszak, R.; Papatheodorou, I.; et al. ArrayExpress Update—From Bulk to Single-Cell Expression Data. Nucleic Acids Res. 2019, 47, D711–D715. [Google Scholar] [CrossRef] [PubMed]
- An, W.; Dai, L.; Niewiadomska, A.M.; Yetil, A.; O’Donnell, K.A.; Han, J.S.; Boeke, J.D. Characterization of a Synthetic Human LINE-1 Retrotransposon ORFeus-Hs. Mob. DNA 2011, 2, 2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andrews, S. FastQC: A Quality Control Tool for High Throughput Sequence Data [Online]. 2010. Available online: http://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (accessed on 20 November 2020).
- Yang, W.R.; Ardeljan, D.; Pacyna, C.N.; Payer, L.M.; Burns, K.H. SQuIRE Reveals Locus-Specific Regulation of Interspersed Repeat Expression. Nucleic Acids Res. 2019, 47, e27. [Google Scholar] [CrossRef] [Green Version]
- Love, M.I.; Huber, W.; Anders, S. Moderated Estimation of Fold Change and Dispersion for RNA-Seq Data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [Green Version]
- Dombroski, B.A.; Scott, A.F.; Kazazian, H.H. Two Additional Potential Retrotransposons Isolated from a Human L1 Subfamily That Contains an Active Retrotransposable Element. Proc. Natl. Acad. Sci. USA 1993, 90, 6513–6517. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Durbin, R. Fast and Accurate Short Read Alignment with Burrows-Wheeler Transform. Bioinforma. Oxf. Engl. 2009, 25, 1754–1760. [Google Scholar] [CrossRef]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. 1000 Genome Project Data Processing Subgroup The Sequence Alignment/Map Format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quinlan, A.R.; Hall, I.M. BEDTools: A Flexible Suite of Utilities for Comparing Genomic Features. Bioinformatics 2010, 26, 841–842. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tarailo-Graovac, M.; Chen, N. Using RepeatMasker to Identify Repetitive Elements in Genomic Sequences. Curr. Protoc. Bioinforma. 2009, 4, 4–10. [Google Scholar] [CrossRef] [PubMed]
- Karolchik, D.; Hinrichs, A.S.; Furey, T.S.; Roskin, K.M.; Sugnet, C.W.; Haussler, D.; Kent, W.J. The UCSC Table Browser Data Retrieval Tool. Nucleic Acids Res. 2004, 32, D493–D496. [Google Scholar] [CrossRef]
- Hubbard, T.; Barker, D.; Birney, E.; Cameron, G.; Chen, Y.; Clark, L.; Cox, T.; Cuff, J.; Curwen, V.; Down, T.; et al. The Ensembl Genome Database Project. Nucleic Acids Res. 2002, 30, 38–41. [Google Scholar] [CrossRef] [Green Version]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast Universal RNA-Seq Aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef]
- Anders, S.; Pyl, P.T.; Huber, W. HTSeq--a Python Framework to Work with High-Throughput Sequencing Data. Bioinforma. Oxf. Engl. 2015, 31, 166–169. [Google Scholar] [CrossRef] [Green Version]
- Kolberg, L.; Raudvere, U.; Kuzmin, I.; Vilo, J.; Peterson, H. Gprofiler2—An R Package for Gene List Functional Enrichment Analysis and Namespace Conversion Toolset g:Profiler. F1000Research 2020, 9, ELIXIR-709. [Google Scholar] [CrossRef]
- Griffiths-Jones, S.; Grocock, R.J.; van Dongen, S.; Bateman, A.; Enright, A.J. MiRBase: MicroRNA Sequences, Targets and Gene Nomenclature. Nucleic Acids Res. 2006, 34, D140–D144. [Google Scholar] [CrossRef]
- Licursi, V.; Conte, F.; Fiscon, G.; Paci, P. MIENTURNET: An Interactive Web Tool for MicroRNA-Target Enrichment and Network-Based Analysis. BMC Bioinformatics 2019, 20, 545. [Google Scholar] [CrossRef]
- Huang, H.-Y.; Lin, Y.-C.-D.; Li, J.; Huang, K.-Y.; Shrestha, S.; Hong, H.-C.; Tang, Y.; Chen, Y.-G.; Jin, C.-N.; Yu, Y.; et al. MiRTarBase 2020: Updates to the Experimentally Validated MicroRNA–Target Interaction Database. Nucleic Acids Res. 2020, 48, D148–D154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, E.Y.; Tan, C.M.; Kou, Y.; Duan, Q.; Wang, Z.; Meirelles, G.V.; Clark, N.R.; Ma’ayan, A. Enrichr: Interactive and Collaborative HTML5 Gene List Enrichment Analysis Tool. BMC Bioinform. 2013, 14, 128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ansaloni, F.; Gualandi, N.; Esposito, M.; Gustincich, S.; Sanges, R. TEspeX: Consensus-Specific Quantification of Transposable Element Expression Preventing Biases from Exonized Fragments. Bioinformatics 2022, 38, 4430–4433. [Google Scholar] [CrossRef] [PubMed]
- Hubley, R.; Finn, R.D.; Clements, J.; Eddy, S.R.; Jones, T.A.; Bao, W.; Smit, A.F.A.; Wheeler, T.J. The Dfam Database of Repetitive DNA Families. Nucleic Acids Res. 2016, 44, D81–D89. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hermann, A.; Goyal, R.; Jeltsch, A. The Dnmt1 DNA-(Cytosine-C5)-Methyltransferase Methylates DNA Processively with High Preference for Hemimethylated Target Sites. J. Biol. Chem. 2004, 279, 48350–48359. [Google Scholar] [CrossRef] [Green Version]
- Lanciano, S.; Cristofari, G. Measuring and Interpreting Transposable Element Expression. Nat. Rev. Genet. 2020, 21, 721–736. [Google Scholar] [CrossRef]
- Gualandi, N.; Iperi, C.; Esposito, M.; Ansaloni, F.; Gustincich, S.; Sanges, R. Meta-Analysis Suggests That Intron Retention Can Affect Quantification of Transposable Elements from RNA-Seq Data. Biology 2022, 11, 826. [Google Scholar] [CrossRef]
- Deininger, P.; Morales, M.E.; White, T.B.; Baddoo, M.; Hedges, D.J.; Servant, G.; Srivastav, S.; Smither, M.E.; Concha, M.; DeHaro, D.L.; et al. A Comprehensive Approach to Expression of L1 Loci. Nucleic Acids Res. 2017, 45, e31. [Google Scholar] [CrossRef] [Green Version]
- Tiwari, B.; Jones, A.E.; Caillet, C.J.; Das, S.; Royer, S.K.; Abrams, J.M. P53 Directly Represses Human LINE1 Transposons. Genes Dev. 2020, 34, 1439–1451. [Google Scholar] [CrossRef]
- Belgnaoui, S.M.; Gosden, R.G.; Semmes, O.J.; Haoudi, A. Human LINE-1 Retrotransposon Induces DNA Damage and Apoptosis in Cancer Cells. Cancer Cell Int. 2006, 6, 13. [Google Scholar] [CrossRef]
- De Cecco, M.; Ito, T.; Petrashen, A.P.; Elias, A.E.; Skvir, N.J.; Criscione, S.W.; Caligiana, A.; Brocculi, G.; Adney, E.M.; Boeke, J.D.; et al. L1 Drives IFN in Senescent Cells and Promotes Age-Associated Inflammation. Nature 2019, 566, 73–78. [Google Scholar] [CrossRef] [PubMed]
- Roush, S.; Slack, F.J. The Let-7 Family of MicroRNAs. Trends Cell Biol. 2008, 18, 505–516. [Google Scholar] [CrossRef] [PubMed]
- Ha, M.; Kim, V.N. Regulation of MicroRNA Biogenesis. Nat. Rev. Mol. Cell Biol. 2014, 15, 509–524. [Google Scholar] [CrossRef] [PubMed]
- Wee, L.M.; Flores-Jasso, C.F.; Salomon, W.E.; Zamore, P.D. Argonaute Divides Its RNA Guide into Domains with Distinct Functions and RNA-Binding Properties. Cell 2012, 151, 1055–1067. [Google Scholar] [CrossRef] [Green Version]
- Sadic, D.; Schmidt, K.; Groh, S.; Kondofersky, I.; Ellwart, J.; Fuchs, C.; Theis, F.J.; Schotta, G. Atrx Promotes Heterochromatin Formation at Retrotransposons. EMBO Rep. 2015, 16, 836–850. [Google Scholar] [CrossRef] [Green Version]
- Loinger, A.; Shemla, Y.; Simon, I.; Margalit, H.; Biham, O. Competition between Small RNAs: A Quantitative View. Biophys. J. 2012, 102, 1712–1721. [Google Scholar] [CrossRef] [Green Version]
- Wylie, A.; Jones, A.E.; D’Brot, A.; Lu, W.-J.; Kurtz, P.; Moran, J.V.; Rakheja, D.; Chen, K.S.; Hammer, R.E.; Comerford, S.A.; et al. P53 Genes Function to Restrain Mobile Elements. Genes Dev. 2016, 30, 64–77. [Google Scholar] [CrossRef] [Green Version]
- Adlakha, Y.K.; Saini, N. MicroRNA-128 Downregulates Bax and Induces Apoptosis in Human Embryonic Kidney Cells. Cell Mol. Life Sci. 2011, 68, 1415–1428. [Google Scholar] [CrossRef]
- Vickers, T.A.; Lima, W.F.; Nichols, J.G.; Crooke, S.T. Reduced Levels of Ago2 Expression Result in Increased SiRNA Competition in Mammalian Cells. Nucleic Acids Res. 2007, 35, 6598–6610. [Google Scholar] [CrossRef] [Green Version]
- Jachowicz, J.W.; Bing, X.; Pontabry, J.; Bošković, A.; Rando, O.J.; Torres-Padilla, M.-E. LINE-1 Activation after Fertilization Regulates Global Chromatin Accessibility in the Early Mouse Embryo. Nat. Genet. 2017, 49, 1502–1510. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Esposito, M.; Gualandi, N.; Spirito, G.; Ansaloni, F.; Gustincich, S.; Sanges, R. Transposons Acting as Competitive Endogenous RNAs: In-Silico Evidence from Datasets Characterised by L1 Overexpression. Biomedicines 2022, 10, 3279. https://doi.org/10.3390/biomedicines10123279
Esposito M, Gualandi N, Spirito G, Ansaloni F, Gustincich S, Sanges R. Transposons Acting as Competitive Endogenous RNAs: In-Silico Evidence from Datasets Characterised by L1 Overexpression. Biomedicines. 2022; 10(12):3279. https://doi.org/10.3390/biomedicines10123279
Chicago/Turabian StyleEsposito, Mauro, Nicolò Gualandi, Giovanni Spirito, Federico Ansaloni, Stefano Gustincich, and Remo Sanges. 2022. "Transposons Acting as Competitive Endogenous RNAs: In-Silico Evidence from Datasets Characterised by L1 Overexpression" Biomedicines 10, no. 12: 3279. https://doi.org/10.3390/biomedicines10123279