
Macrophage Expression of Inflammatory Genes in Response to EMCV Infection

{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Encephalomyocarditis Virus (EMCV)
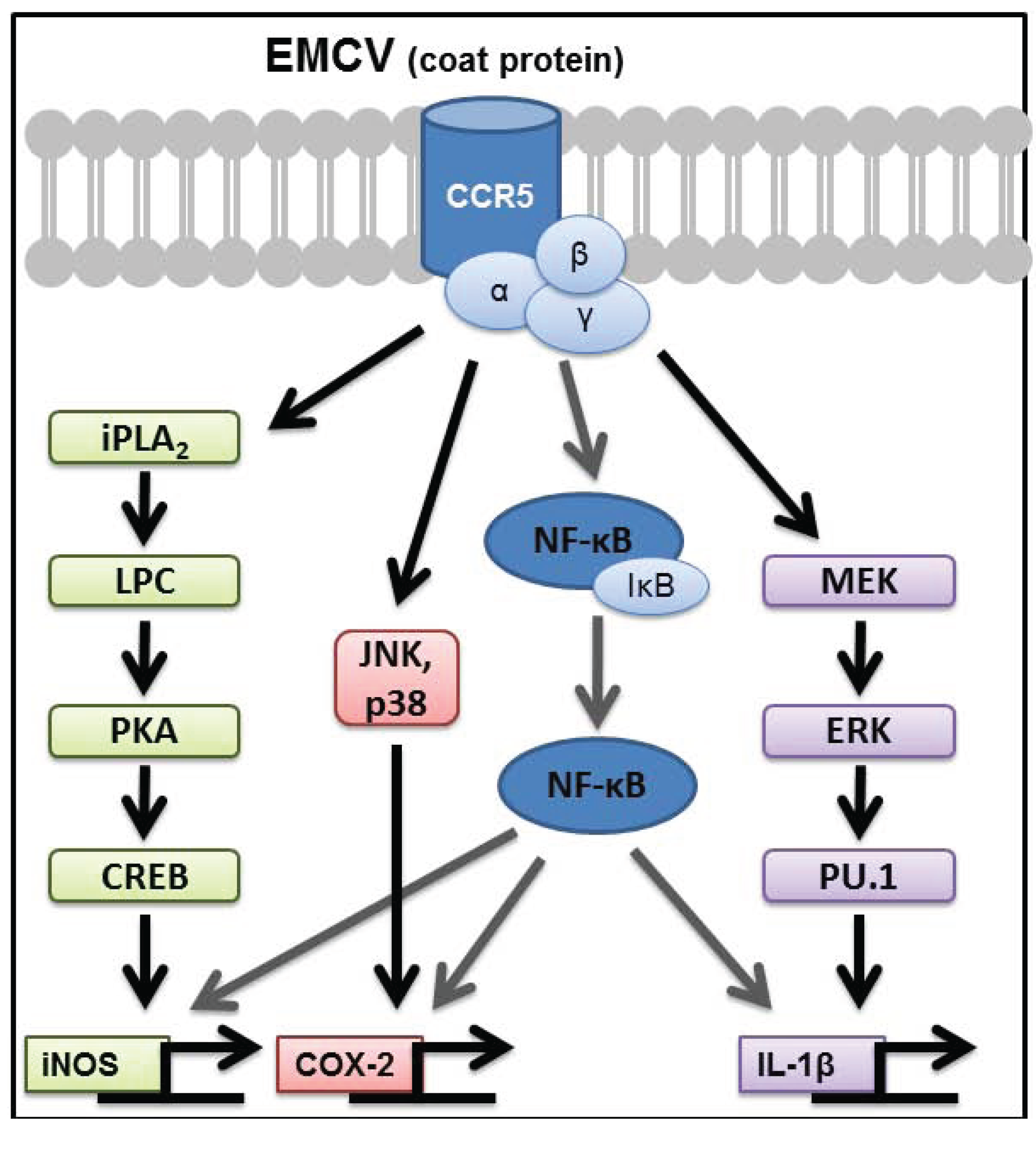
3. Signaling Pathways Regulating Inflammatory Gene Expression in EMCV-Infected Macrophages

4. Additional Signaling Pathways That Contribute to the Regulation of Inflammatory Genes in EMCV-Infected Macrophages
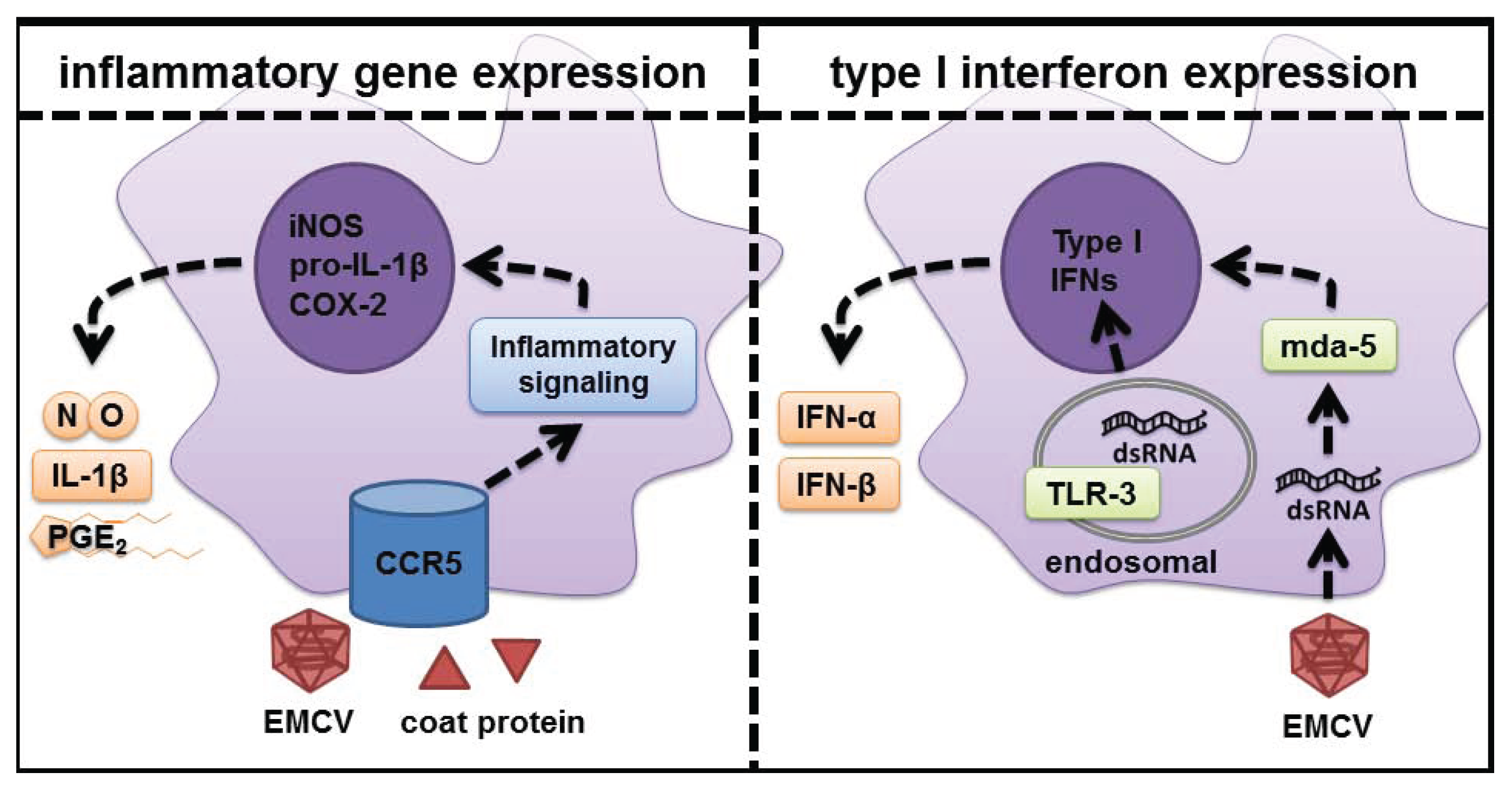
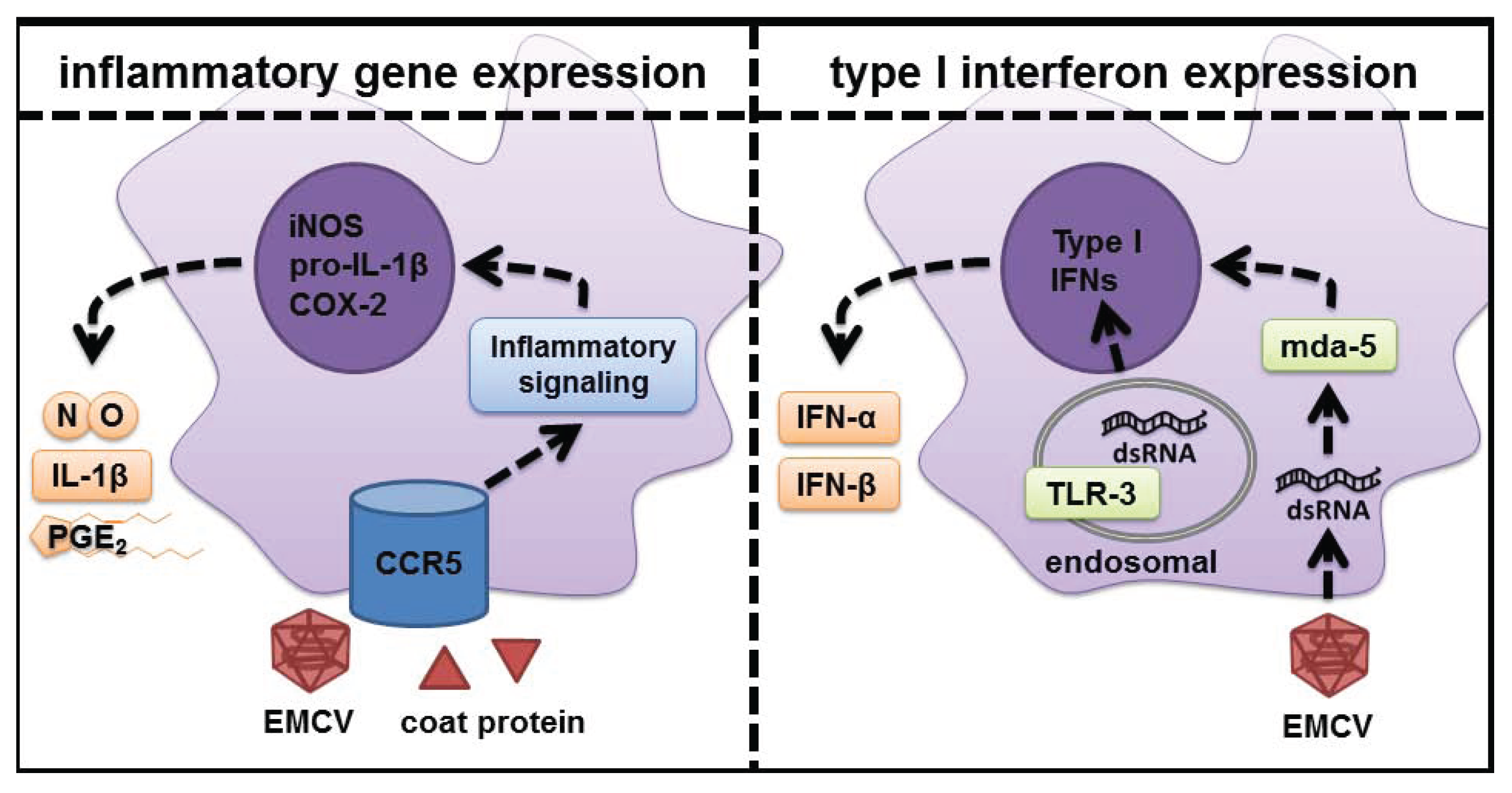
5. Role of dsRNA Sensors in Response to Virus Infection
6. CCR5 as the Signaling Receptor Responsible for Inflammatory Gene Expression in Response to EMCV Infection
7. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Aderem, A.; Underhill, D.M. Mechanisms of phagocytosis in macrophages. Ann. Rev. Immunol. 1999, 17, 593–623. [Google Scholar] [CrossRef] [PubMed]
- Janeway, C.A., Jr.; Medzhitov, R. Innate immune recognition. Ann. Rev. Immunol. 2002, 20, 197–216. [Google Scholar] [CrossRef] [PubMed]
- Takeuchi, O.; Akira, S. Pattern recognition receptors and inflammation. Cell 2010, 140, 805–820. [Google Scholar] [CrossRef] [PubMed]
- Dinarello, C.A. Interleukin-1 in the pathogenesis and treatment of inflammatory diseases. Blood 2011, 117, 3720–3732. [Google Scholar] [CrossRef] [PubMed]
- Dinarello, C.A. Immunological and inflammatory functions of the interleukin-1 family. Ann. Rev. Immunol. 2009, 27, 519–550. [Google Scholar] [CrossRef] [PubMed]
- Ben-Sasson, S.Z.; Hu-Li, J.; Quiel, J.; Cauchetaux, S.; Ratner, M.; Shapira, I.; Dinarello, C.A.; Paul, W.E. IL-1 acts directly on CD4 T cells to enhance their antigen-driven expansion and differentiation. Proc. Natl. Acad. Sci. USA 2009, 106, 7119–7124. [Google Scholar] [CrossRef] [PubMed]
- Marrack, P.; McKee, A.S.; Munks, M.W. Towards an understanding of the adjuvant action of aluminium. Nat. Rev. Immunol. 2009, 9, 287–293. [Google Scholar] [CrossRef] [PubMed]
- Steer, S.A.; Corbett, J.A. The role and regulation of COX-2 during viral infection. Viral Immunol. 2003, 16, 447–460. [Google Scholar] [CrossRef] [PubMed]
- Kalinski, P. Regulation of immune responses by prostaglandin E2. J. Immunol. 2012, 188, 21–28. [Google Scholar] [CrossRef] [PubMed]
- Harris, S.G.; Padilla, J.; Koumas, L.; Ray, D.; Phipps, R.P. Prostaglandins as modulators of immunity. Trends Immunol. 2002, 23, 144–150. [Google Scholar] [CrossRef]
- Karupiah, G.; Xie, Q.W.; Buller, R.M.; Nathan, C.; Duarte, C.; MacMicking, J.D. Inhibition of viral replication by interferon-gamma-induced nitric oxide synthase. Science 1993, 261, 1445–1448. [Google Scholar] [CrossRef] [PubMed]
- Sanders, S.P.; Siekierski, E.S.; Porter, J.D.; Richards, S.M.; Proud, D. Nitric oxide inhibits rhinovirus-induced cytokine production and viral replication in a human respiratory epithelial cell line. J. Virol. 1998, 72, 934–942. [Google Scholar] [PubMed]
- Croen, K.D. Evidence for antiviral effect of nitric oxide. Inhibition of herpes simplex virus type 1 replication. J. Clin. Investig. 1993, 91, 2446–2452. [Google Scholar] [CrossRef] [PubMed]
- Mannick, J.B. The antiviral role of nitric oxide. Res. Immunol. 1995, 146, 693–697. [Google Scholar] [CrossRef]
- Flodstrom, M.; Horwitz, M.S.; Maday, A.; Balakrishna, D.; Rodriguez, E.; Sarvetnick, N. A critical role for inducible nitric oxide synthase in host survival following coxsackievirus B4 infection. Virology 2001, 281, 205–215. [Google Scholar] [CrossRef] [PubMed]
- Saura, M.; Zaragoza, C.; McMillan, A.; Quick, R.A.; Hohenadl, C.; Lowenstein, J.M.; Lowenstein, C.J. An antiviral mechanism of nitric oxide: inhibition of a viral protease. Immunity 1999, 10, 21–28. [Google Scholar] [CrossRef]
- Carocci, M.; Bakkali-Kassimi, L. The encephalomyocarditis virus. Virulence 2012, 3, 351–367. [Google Scholar] [CrossRef] [PubMed]
- Craighead, J.E.; McLane, M.F. Diabetes mellitus: induction in mice by encephalomyocarditis virus. Science 1968, 162, 913–914. [Google Scholar] [CrossRef] [PubMed]
- Yoon, J.W.; Lesniak, M.A.; Fussganger, R.; Notkins, A.L. Genetic differences in susceptibility of pancreatic β cells to virus-induced diabetes mellitus. Nature 1976, 264, 178–180. [Google Scholar] [CrossRef] [PubMed]
- Boucher, D.W.; Hayashi, K.; Rosenthal, J.; Notkins, A.L. Virus-induced diabetes mellitus. III. Influence of the sex and strain of the host. J. Infect. Dis. 1975, 131, 462–466. [Google Scholar] [CrossRef] [PubMed]
- Jun, H.S.; Yoon, J.W. The role of viruses in type I diabetes: Two distinct cellular and molecular pathogenic mechanisms of virus-induced diabetes in animals. Diabetologia 2001, 44, 271–285. [Google Scholar] [CrossRef] [PubMed]
- Baek, H.S.; Yoon, J.W. Role of macrophages in the pathogenesis of encephalomyocarditis virus-induced diabetes in mice. J. Virol. 1990, 64, 5708–5715. [Google Scholar] [PubMed]
- Hirasawa, K.; Takeda, M.; Itagaki, S.; Doi, K. Involvement of macrophages in the development of encephalomyocarditis (EMC) virus-induced diabetes in mice. Exp. Anim. Jpn. Assoc. Lab. Anim. Sci. 1996, 45, 77–80. [Google Scholar] [CrossRef]
- Baek, H.S.; Yoon, J.W. Direct involvement of macrophages in destruction of β-cells leading to development of diabetes in virus-infected mice. Diabetes 1991, 40, 1586–1597. [Google Scholar] [CrossRef] [PubMed]
- Hirasawa, K.; Tsutsui, S.; Takeda, M.; Mizutani, M.; Itagaki, S.; Doi, K. Depletion of Mac1-positive macrophages protects DBA/2 mice from encephalomyocarditis virus-induced myocarditis and diabetes. J. Gen. Virol. 1996, 77, 737–741. [Google Scholar] [CrossRef] [PubMed]
- Yoon, J.W.; McClintock, P.R.; Bachurski, C.J.; Longstreth, J.D.; Notkins, A.L. Virus-induced diabetes mellitus. No evidence for immune mechanisms in the destruction of β-cells by the D-variant of encephalomyocarditis virus. Diabetes 1985, 34, 922–925. [Google Scholar] [CrossRef] [PubMed]
- Kounoue, E.; Izumi, K.; Ogawa, S.; Kondo, S.; Katsuta, H.; Akashi, T.; Niho, Y.; Harada, M.; Tamiya, S.; Kurisaki, H.; et al. The significance of T cells, B cells, antibodies and macrophages against encephalomyocarditis (EMC)-D virus-induced diabetes in mice. Arch. Virol. 2008, 153, 1223–1231. [Google Scholar] [CrossRef] [PubMed]
- Hirasawa, K.; Jun, H.S.; Maeda, K.; Kawaguchi, Y.; Itagaki, S.; Mikami, T.; Baek, H.S.; Doi, K.; Yoon, J.W. Possible role of macrophage-derived soluble mediators in the pathogenesis of encephalomyocarditis virus-induced diabetes in mice. J. Virol. 1997, 71, 4024–4031. [Google Scholar] [PubMed]
- Corbett, J.A.; Tilton, R.G.; Chang, K.; Hasan, K.S.; Ido, Y.; Wang, J.L.; Sweetland, M.A.; Lancaster, J.R., Jr.; Williamson, J.R.; McDaniel, M.L. Aminoguanidine, a novel inhibitor of nitric oxide formation, prevents diabetic vascular dysfunction. Diabetes 1992, 41, 552–556. [Google Scholar] [CrossRef] [PubMed]
- Corbett, J.A.; McDaniel, M.L. The use of aminoguanidine, a selective iNOS inhibitor, to evaluate the role of nitric oxide in the development of autoimmune diabetes. Methods 1996, 10, 21–30. [Google Scholar] [CrossRef] [PubMed]
- Corbett, J.A.; Lancaster, J.R., Jr.; Sweetland, M.A.; McDaniel, M.L. Interleukin-1 β-induced formation of EPR-detectable iron-nitrosyl complexes in islets of Langerhans. Role of nitric oxide in interleukin-1 β-induced inhibition of insulin secretion. J. Biol. Chem. 1991, 266, 21351–21354. [Google Scholar] [PubMed]
- Southern, C.; Schulster, D.; Green, I.C. Inhibition of insulin secretion by interleukin-1β and tumour necrosis factor-α via an L-arginine-dependent nitric oxide generating mechanism. FEBS Lett. 1990, 276, 42–44. [Google Scholar] [CrossRef]
- Welsh, N.; Eizirik, D.L.; Bendtzen, K.; Sandler, S. Interleukin-1β-induced nitric oxide production in isolated rat pancreatic islets requires gene transcription and may lead to inhibition of the Krebs cycle enzyme aconitase. Endocrinology 1991, 129, 3167–3173. [Google Scholar] [CrossRef] [PubMed]
- Mandrup-Poulsen, T. The role of interleukin-1 in the pathogenesis of IDDM. Diabetologia 1996, 39, 1005–1029. [Google Scholar] [CrossRef] [PubMed]
- Corbett, J.A.; McDaniel, M.L. Intraislet release of interleukin 1 inhibits β cell function by inducing β cell expression of inducible nitric oxide synthase. J. Exp. Med. 1995, 181, 559–568. [Google Scholar] [CrossRef] [PubMed]
- Arnush, M.; Heitmeier, M.R.; Scarim, A.L.; Marino, M.H.; Manning, P.T.; Corbett, J.A. IL-1 produced and released endogenously within human islets inhibits β cell function. J. Clin. Investig. 1998, 102, 516–526. [Google Scholar] [CrossRef] [PubMed]
- Arnush, M.; Scarim, A.L.; Heitmeier, M.R.; Kelly, C.B.; Corbett, J.A. Potential role of resident islet macrophage activation in the initiation of autoimmune diabetes. J. Immunol. 1998, 160, 2684–2691. [Google Scholar] [PubMed]
- Lee, Y.S.; Li, N.; Shin, S.; Jun, H.S. Role of nitric oxide in the pathogenesis of encephalomyocarditis virus-induced diabetes in mice. J. Virol. 2009, 83, 8004–8011. [Google Scholar] [CrossRef] [PubMed]
- Lacy, P.E. The intraislet macrophage and type I diabetes. Mt. Sinai J. Med. N. Y. 1994, 61, 170–174. [Google Scholar]
- Chung, Y.H.; Jun, H.S.; Kang, Y.; Hirasawa, K.; Lee, B.R.; van Rooijen, N.; Yoon, J.W. Role of macrophages and macrophage-derived cytokines in the pathogenesis of Kilham rat virus-induced autoimmune diabetes in diabetes-resistant BioBreeding rats. J. Immunol. 1997, 159, 466–471. [Google Scholar] [PubMed]
- Ellerman, K.E.; Richards, C.A.; Guberski, D.L.; Shek, W.R.; Like, A.A. Kilham rat triggers T-cell-dependent autoimmune diabetes in multiple strains of rat. Diabetes 1996, 45, 557–562. [Google Scholar] [CrossRef] [PubMed]
- Guberski, D.L.; Thomas, V.A.; Shek, W.R.; Like, A.A.; Handler, E.S.; Rossini, A.A.; Wallace, J.E.; Welsh, R.M. Induction of type I diabetes by Kilham’s rat virus in diabetes-resistant BB/Wor rats. Science 1991, 254, 1010–1013. [Google Scholar] [CrossRef] [PubMed]
- Mendez, I.I.; Chung, Y.H., II; Jun, H.S.; Yoon, J.W. Immunoregulatory role of nitric oxide in Kilham rat virus-induced autoimmune diabetes in DR-BB rats. J. Immunol. 2004, 173, 1327–1335. [Google Scholar] [CrossRef] [PubMed]
- Jun, H.S.; Yoon, C.S.; Zbytnuik, L.; van Rooijen, N.; Yoon, J.W. The role of macrophages in T cell-mediated autoimmune diabetes in nonobese diabetic mice. J. Exp. Med. 1999, 189, 347–358. [Google Scholar] [CrossRef] [PubMed]
- Jun, H.S.; Santamaria, P.; Lim, H.W.; Zhang, M.L.; Yoon, J.W. Absolute requirement of macrophages for the development and activation of β-cell cytotoxic CD8+ T-cells in T-cell receptor transgenic NOD mice. Diabetes 1999, 48, 34–42. [Google Scholar] [CrossRef] [PubMed]
- Lefkowith, J.; Schreiner, G.; Cormier, J.; Handler, E.S.; Driscoll, H.K.; Greiner, D.; Mordes, J.P.; Rossini, A.A. Prevention of diabetes in the BB rat by essential fatty acid deficiency. Relationship between physiological and biochemical changes. J. Exp. Med. 1990, 171, 729–743. [Google Scholar] [CrossRef] [PubMed]
- Oschilewski, U.; Kiesel, U.; Kolb, H. Administration of silica prevents diabetes in BB-rats. Diabetes 1985, 34, 197–199. [Google Scholar] [CrossRef] [PubMed]
- Wright, J.R., Jr.; Lefkowith, J.B.; Schreiner, G.; Lacy, P.E. Essential fatty acid deficiency prevents multiple low-dose streptozotocin-induced diabetes in CD-1 mice. Proc. Natl. Acad. Sci. USA 1988, 85, 6137–6141. [Google Scholar] [CrossRef] [PubMed]
- Baldwin, A.S., Jr. The NF-κB and I κB proteins: New discoveries and insights. Ann. Rev. Immunol. 1996, 14, 649–683. [Google Scholar] [CrossRef] [PubMed]
- Heitmeier, M.R.; Scarim, A.L.; Corbett, J.A. Double-stranded RNA-induced inducible nitric-oxide synthase expression and interleukin-1 release by murine macrophages requires NF-κB activation. J. Biol. Chem. 1998, 273, 15301–15307. [Google Scholar] [CrossRef] [PubMed]
- Steer, S.A.; Moran, J.M.; Maggi, L.B., Jr.; Buller, R.M.; Perlman, H.; Corbett, J.A. Regulation of cyclooxygenase-2 expression by macrophages in response to double-stranded RNA and viral infection. J. Immunol. 2003, 170, 1070–1076. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Lee, H.S.; Chang, K.T.; Ko, T.H.; Baek, K.J.; Kwon, N.S. Chloromethyl ketones block induction of nitric oxide synthase in murine macrophages by preventing activation of nuclear factor-κB. J. Immunol. 1995, 154, 4741–4748. [Google Scholar] [PubMed]
- Mulsch, A.; Schray-Utz, B.; Mordvintcev, P.I.; Hauschildt, S.; Busse, R. Diethyldithiocarbamate inhibits induction of macrophage NO synthase. FEBS Lett. 1993, 321, 215–218. [Google Scholar] [CrossRef]
- Chen, C.C.; Sun, Y.T.; Chen, J.J.; Chiu, K.T. TNF-α-induced cyclooxygenase-2 expression in human lung epithelial cells: Involvement of the phospholipase C-gamma 2, protein kinase C-α, tyrosine kinase, NF-κB-inducing kinase, and I-kappa B kinase 1/2 pathway. J. Immunol. 2000, 165, 2719–2728. [Google Scholar] [CrossRef] [PubMed]
- Hwang, D.; Jang, B.C.; Yu, G.; Boudreau, M. Expression of mitogen-inducible cyclooxygenase induced by lipopolysaccharide: Mediation through both mitogen-activated protein kinase and NF-κB signaling pathways in macrophages. Biochem. Pharmacol. 1997, 54, 87–96. [Google Scholar] [CrossRef]
- Hiscott, J.; Marois, J.; Garoufalis, J.; D’Addario, M.; Roulston, A.; Kwan, I.; Pepin, N.; Lacoste, J.; Nguyen, H.; Bensi, G.; et al. Characterization of a functional NF-κB site in the human interleukin 1 β promoter: Evidence for a positive autoregulatory loop. Mol. Cell. Biol. 1993, 13, 6231–6240. [Google Scholar] [PubMed]
- Cogswell, J.P.; Godlevski, M.M.; Wisely, G.B.; Clay, W.C.; Leesnitzer, L.M.; Ways, J.P.; Gray, J.G. NF-κB regulates IL-1 β transcription through a consensus NF-κB binding site and a nonconsensus CRE-like site. J. Immunol. 1994, 153, 712–723. [Google Scholar] [PubMed]
- Steer, S.A.; Moran, J.M.; Christmann, B.S.; Maggi, L.B., Jr.; Corbett, J.A. Role of MAPK in the regulation of double-stranded RNA- and encephalomyocarditis virus-induced cyclooxygenase-2 expression by macrophages. J. Immunol. 2006, 177, 3413–3420. [Google Scholar] [CrossRef] [PubMed]
- Maggi, L.B., Jr.; Moran, J.M.; Buller, R.M.; Corbett, J.A. ERK activation is required for double-stranded RNA- and virus-induced interleukin-1 expression by macrophages. J. Biol. Chem. 2003, 278, 16683–16689. [Google Scholar] [CrossRef] [PubMed]
- Martinson, B.D.; Albert, C.J.; Corbett, J.A.; Wysolmerski, R.B.; Ford, D.A. Calcium-independent phospholipase A2 mediates CREB phosphorylation in double-stranded RNA-stimulated endothelial cells. J. Lipid Res. 2003, 44, 1686–1691. [Google Scholar] [CrossRef] [PubMed]
- Maggi, L.B., Jr.; Moran, J.M.; Scarim, A.L.; Ford, D.A.; Yoon, J.W.; McHowat, J.; Buller, R.M.; Corbett, J.A. Novel role for calcium-independent phospholipase A(2) in the macrophage antiviral response of inducible nitric-oxide synthase expression. J. Biol. Chem. 2002, 277, 38449–38455. [Google Scholar] [CrossRef] [PubMed]
- Moran, J.M.; Buller, R.M.; McHowat, J.; Turk, J.; Wohltmann, M.; Gross, R.W.; Corbett, J.A. Genetic and pharmacologic evidence that calcium-independent phospholipase A2β regulates virus-induced inducible nitric-oxide synthase expression by macrophages. J. Biol. Chem. 2005, 280, 28162–28168. [Google Scholar] [CrossRef] [PubMed]
- Kominato, Y.; Galson, D.; Waterman, W.R.; Webb, A.C.; Auron, P.E. Monocyte expression of the human prointerleukin 1 β gene (IL1B) is dependent on promoter sequences which bind the hematopoietic transcription factor Spi-1/PU.1. Mol. Cell. Biol. 1995, 15, 58–68. [Google Scholar] [PubMed]
- Lodie, T.A.; Savedra, R., Jr.; Golenbock, D.T.; van Beveren, C.P.; Maki, R.A.; Fenton, M.J. Stimulation of macrophages by lipopolysaccharide alters the phosphorylation state, conformation, and function of PU.1 via activation of casein kinase II. J. Immunol. 1997, 158, 1848–1856. [Google Scholar] [PubMed]
- Wasylyk, B.; Hagman, J.; Gutierrez-Hartmann, A. Ets transcription factors: nuclear effectors of the Ras-MAP-kinase signaling pathway. Trends Biochem. Sci. 1998, 23, 213–216. [Google Scholar] [CrossRef]
- Williams, S.D.; Ford, D.A. Calcium-independent phospholipase A2 mediates CREB phosphorylation and c-fos expression during ischemia. Am. J. Physiol. Heart circ. Physiol. 2001, 281, H168–H176. [Google Scholar] [PubMed]
- Blair, L.A.; Maggi, L.B., Jr.; Scarim, A.L.; Corbett, J.A. Role of interferon regulatory factor-1 in double-stranded RNA-induced iNOS expression by mouse islets. J. Biol. Chem. 2002, 277, 359–365. [Google Scholar] [CrossRef] [PubMed]
- Freudenburg, W.; Buller, R.M.; Corbett, J.A. Src family kinases participate in the regulation of encephalomyocarditis virus-induced cyclooxygenase-2 expression by macrophages. J. Gen. Virol. 2010, 91, 2278–2285. [Google Scholar] [CrossRef] [PubMed]
- Leu, T.H.; Charoenfuprasert, S.; Yen, C.K.; Fan, C.W.; Maa, M.C. Lipopolysaccharide-induced c-Src expression plays a role in nitric oxide and TNFα secretion in macrophages. Mol. Immunol. 2006, 43, 308–316. [Google Scholar] [CrossRef] [PubMed]
- Stovall, S.H.; Yi, A.K.; Meals, E.A.; Talati, A.J.; Godambe, S.A.; English, B.K. Role of vav1- and src-related tyrosine kinases in macrophage activation by CpG DNA. J. Biol. Chem. 2004, 279, 13809–13816. [Google Scholar] [CrossRef] [PubMed]
- Choi, K.S.; Jun, H.S.; Kim, H.N.; Park, H.J.; Eom, Y.W.; Noh, H.L.; Kwon, H.; Kim, H.M.; Yoon, J.W. Role of Hck in the pathogenesis of encephalomyocarditis virus-induced diabetes in mice. J. Virol. 2001, 75, 1949–1957. [Google Scholar] [CrossRef] [PubMed]
- Vanhaesebroeck, B.; Leevers, S.J.; Ahmadi, K.; Timms, J.; Katso, R.; Driscoll, P.C.; Woscholski, R.; Parker, P.J.; Waterfield, M.D. Synthesis and function of 3-phosphorylated inositol lipids. Ann. Rev. Biochem. 2001, 70, 535–602. [Google Scholar] [CrossRef] [PubMed]
- Vanhaesebroeck, B.; Guillermet-Guibert, J.; Graupera, M.; Bilanges, B. The emerging mechanisms of isoform-specific PI3K signalling. Nat. Rev. Mol. Cell Biol. 2010, 11, 329–341. [Google Scholar] [CrossRef] [PubMed]
- Vanhaesebroeck, B.; Stephens, L.; Hawkins, P. PI3K signalling: The path to discovery and understanding. Nat. Rev. Mol. Cell Biol. 2012, 13, 195–203. [Google Scholar] [CrossRef] [PubMed]
- Freudenburg, W.; Moran, J.M.; Lents, N.H.; Baldassare, J.J.; Buller, R.M.; Corbett, J.A. Phosphatidylinositol 3-kinase regulates macrophage responses to double-stranded RNA and encephalomyocarditis virus. J. Innate Immun. 2010, 2, 77–86. [Google Scholar] [CrossRef] [PubMed]
- Tyner, J.W.; Uchida, O.; Kajiwara, N.; Kim, E.Y.; Patel, A.C.; O’Sullivan, M.P.; Walter, M.J.; Schwendener, R.A.; Cook, D.N.; Danoff, T.M.; et al. CCL5-CCR5 interaction provides antiapoptotic signals for macrophage survival during viral infection. Nat. Med. 2005, 11, 1180–1187. [Google Scholar] [CrossRef] [PubMed]
- Cooray, S. The pivotal role of phosphatidylinositol 3-kinase-Akt signal transduction in virus survival. J. Gen. Virol. 2004, 85, 1065–1076. [Google Scholar] [CrossRef] [PubMed]
- Ehrhardt, C.; Ludwig, S. A new player in a deadly game: Influenza viruses and the PI3K/Akt signalling pathway. Cell. Microbiol. 2009, 11, 863–871. [Google Scholar] [CrossRef] [PubMed]
- Stetson, D.B.; Medzhitov, R. Type I interferons in host defense. Immunity 2006, 25, 373–381. [Google Scholar] [CrossRef] [PubMed]
- Samuel, C.E. The eIF-2α protein kinases, regulators of translation in eukaryotes from yeasts to humans. J. Biol. Chem. 1993, 268, 7603–7606. [Google Scholar] [PubMed]
- Donnelly, N.; Gorman, A.M.; Gupta, S.; Samali, A. The eIF2α kinases: Their structures and functions. Cell. Mol. Life Sci. 2013, 70, 3493–3511. [Google Scholar] [CrossRef] [PubMed]
- Meurs, E.; Chong, K.; Galabru, J.; Thomas, N.S.; Kerr, I.M.; Williams, B.R.; Hovanessian, A.G. Molecular cloning and characterization of the human double-stranded RNA-activated protein kinase induced by interferon. Cell 1990, 62, 379–390. [Google Scholar] [CrossRef]
- Alexopoulou, L.; Holt, A.C.; Medzhitov, R.; Flavell, R.A. Recognition of double-stranded RNA and activation of NF-κB by Toll-like receptor 3. Nature 2001, 413, 732–738. [Google Scholar] [CrossRef] [PubMed]
- Kato, H.; Takeuchi, O.; Sato, S.; Yoneyama, M.; Yamamoto, M.; Matsui, K.; Uematsu, S.; Jung, A.; Kawai, T.; Ishii, K.J.; et al. Differential roles of MDA5 and RIG-I helicases in the recognition of RNA viruses. Nature 2006, 441, 101–105. [Google Scholar] [CrossRef] [PubMed]
- Kato, H.; Takeuchi, O.; Mikamo-Satoh, E.; Hirai, R.; Kawai, T.; Matsushita, K.; Hiiragi, A.; Dermody, T.S.; Fujita, T.; Akira, S. Length-dependent recognition of double-stranded ribonucleic acids by retinoic acid-inducible gene-I and melanoma differentiation-associated gene 5. J. Exp. Med. 2008, 205, 1601–1610. [Google Scholar] [CrossRef] [PubMed]
- Pichlmair, A.; Schulz, O.; Tan, C.P.; Rehwinkel, J.; Kato, H.; Takeuchi, O.; Akira, S.; Way, M.; Schiavo, G.; Reis e Sousa, C. Activation of MDA5 requires higher-order RNA structures generated during virus infection. J. Virol. 2009, 83, 10761–10769. [Google Scholar] [CrossRef] [PubMed]
- Christmann, B.S.; Moran, J.M.; McGraw, J.A.; Buller, R.M.; Corbett, J.A. Ccr5 regulates inflammatory gene expression in response to encephalomyocarditis virus infection. Am. J. Pathol. 2011, 179, 2941–2951. [Google Scholar] [CrossRef] [PubMed]
- Moran, J.M.; Moxley, M.A.; Buller, R.M.; Corbett, J.A. Encephalomyocarditis virus induces PKR-independent mitogen-activated protein kinase activation in macrophages. J. Virol. 2005, 79, 10226–10236. [Google Scholar] [CrossRef] [PubMed]
- Iordanov, M.S.; Wong, J.; Bell, J.C.; Magun, B.E. Activation of NF-κB by double-stranded RNA (dsRNA) in the absence of protein kinase R and RNase L demonstrates the existence of two separate dsRNA-triggered antiviral programs. Mol. Cell. Biol. 2001, 21, 61–72. [Google Scholar] [CrossRef] [PubMed]
- Gitlin, L.; Barchet, W.; Gilfillan, S.; Cella, M.; Beutler, B.; Flavell, R.A.; Diamond, M.S.; Colonna, M. Essential role of mda-5 in type I IFN responses to polyriboinosinic:polyribocytidylic acid and encephalomyocarditis picornavirus. Proc. Natl. Acad. Sci. USA 2006, 103, 8459–8464. [Google Scholar] [CrossRef] [PubMed]
- McCartney, S.A.; Vermi, W.; Lonardi, S.; Rossini, C.; Otero, K.; Calderon, B.; Gilfillan, S.; Diamond, M.S.; Unanue, E.R.; Colonna, M. RNA sensor-induced type I IFN prevents diabetes caused by a β cell-tropic virus in mice. J. Clin. Invest. 2011, 121, 1497–1507. [Google Scholar] [CrossRef] [PubMed]
- Oppermann, M. Chemokine receptor CCR5: Insights into structure, function, and regulation. Cell. Signal. 2004, 16, 1201–1210. [Google Scholar] [CrossRef] [PubMed]
- Raport, C.J.; Gosling, J.; Schweickart, V.L.; Gray, P.W.; Charo, I.F. Molecular cloning and functional characterization of a novel human CC chemokine receptor (CCR5) for RANTES, MIP-1β, and MIP-1α. J. Biol. Chem. 1996, 271, 17161–17166. [Google Scholar] [CrossRef] [PubMed]
- Samson, M.; Labbe, O.; Mollereau, C.; Vassart, G.; Parmentier, M. Molecular cloning and functional expression of a new human CC-chemokine receptor gene. Biochemistry 1996, 35, 3362–3367. [Google Scholar] [CrossRef] [PubMed]
- Alkhatib, G.; Combadiere, C.; Broder, C.C.; Feng, Y.; Kennedy, P.E.; Murphy, P.M.; Berger, E.A. CC CKR5: A RANTES, MIP-1α, MIP-1β receptor as a fusion cofactor for macrophage-tropic HIV-1. Science 1996, 272, 1955–1958. [Google Scholar] [CrossRef] [PubMed]
- Deng, H.; Liu, R.; Ellmeier, W.; Choe, S.; Unutmaz, D.; Burkhart, M.; di Marzio, P.; Marmon, S.; Sutton, R.E.; Hill, C.M.; et al. Identification of a major co-receptor for primary isolates of HIV-1. Nature 1996, 381, 661–666. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.; Gerard, N.P.; Wyatt, R.; Choe, H.; Parolin, C.; Ruffing, N.; Borsetti, A.; Cardoso, A.A.; Desjardin, E.; Newman, W.; et al. CD4-induced interaction of primary HIV-1 gp120 glycoproteins with the chemokine receptor CCR-5. Nature 1996, 384, 179–183. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.; Paxton, W.A.; Choe, S.; Ceradini, D.; Martin, S.R.; Horuk, R.; MacDonald, M.E.; Stuhlmann, H.; Koup, R.A.; Landau, N.R. Homozygous defect in HIV-1 coreceptor accounts for resistance of some multiply-exposed individuals to HIV-1 infection. Cell 1996, 86, 367–377. [Google Scholar] [CrossRef]
- Samson, M.; Libert, F.; Doranz, B.J.; Rucker, J.; Liesnard, C.; Farber, C.M.; Saragosti, S.; Lapoumeroulie, C.; Cognaux, J.; Forceille, C.; et al. Resistance to HIV-1 infection in caucasian individuals bearing mutant alleles of the CCR-5 chemokine receptor gene. Nature 1996, 382, 722–725. [Google Scholar] [CrossRef] [PubMed]
- Racaniello, V.R. Cell receptors for picornaviruses. Curr. Top. Microbiol. Immunol. 1990, 161, 1–22. [Google Scholar] [PubMed]
- Bergelson, J.M.; Shepley, M.P.; Chan, B.M.; Hemler, M.E.; Finberg, R.W. Identification of the integrin VLA-2 as a receptor for echovirus 1. Science 1992, 255, 1718–1720. [Google Scholar] [CrossRef] [PubMed]
- Huber, S.A. VCAM-1 is a receptor for encephalomyocarditis virus on murine vascular endothelial cells. J. Virol. 1994, 68, 3453–3458. [Google Scholar] [PubMed]
- Tomassini, J.E.; Graham, D.; DeWitt, C.M.; Lineberger, D.W.; Rodkey, J.A.; Colonno, R.J. cDNA cloning reveals that the major group rhinovirus receptor on HeLa cells is intercellular adhesion molecule 1. Proc. Natl. Acad. Sci. USA 1989, 86, 4907–4911. [Google Scholar] [CrossRef] [PubMed]
- Floto, R.A.; MacAry, P.A.; Boname, J.M.; Mien, T.S.; Kampmann, B.; Hair, J.R.; Huey, O.S.; Houben, E.N.; Pieters, J.; Day, C.; et al. Dendritic cell stimulation by mycobacterial Hsp70 is mediated through CCR5. Science 2006, 314, 454–458. [Google Scholar] [CrossRef] [PubMed]
- Bukrinsky, M.I.; Nottet, H.S.; Schmidtmayerova, H.; Dubrovsky, L.; Flanagan, C.R.; Mullins, M.E.; Lipton, S.A.; Gendelman, H.E. Regulation of nitric oxide synthase activity in human immunodeficiency virus type 1 (HIV-1)-infected monocytes: Implications for HIV-associated neurological disease. J. Exp. Med. 1995, 181, 735–745. [Google Scholar] [CrossRef] [PubMed]
- Lisi, L.; Tramutola, A.; de Luca, A.; Navarra, P.; Russo, C.D. Modulatory effects of the CCR5 antagonist maraviroc on microglial pro-inflammatory activation elicited by gp120. J. Neurochem. 2012, 120, 106–114. [Google Scholar] [CrossRef] [PubMed]
- Tomkowicz, B.; Lee, C.; Ravyn, V.; Cheung, R.; Ptasznik, A.; Collman, R.G. The Src kinase Lyn is required for CCR5 signaling in response to MIP-1β and R5 HIV-1 gp120 in human macrophages. Blood 2006, 108, 1145–1150. [Google Scholar] [CrossRef] [PubMed]
- Cheung, R.; Ravyn, V.; Wang, L.; Ptasznik, A.; Collman, R.G. Signaling mechanism of HIV-1 gp120 and virion-induced IL-1β release in primary human macrophages. J. Immunol. 2008, 180, 6675–6684. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, T.; Zhu, S.J.; Sumino, H.; Saegusa, S.; Nakahashi, T.; Iwai, K.; Morimoto, S.; Kanda, T. Inhibition of cyclooxygenase-2 enhances myocardial damage in a mouse model of viral myocarditis. Life Sci. 2005, 78, 195–204. [Google Scholar] [CrossRef] [PubMed]
- Platanias, L.C. Mechanisms of type-I- and type-II-interferon-mediated signalling. Nat. Rev. Immunol. 2005, 5, 375–386. [Google Scholar] [CrossRef] [PubMed]
- Sadler, A.J.; Williams, B.R. Interferon-inducible antiviral effectors. Nat. Rev. Immunol. 2008, 8, 559–568. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shaheen, Z.R.; Corbett, J.A. Macrophage Expression of Inflammatory Genes in Response to EMCV Infection. Biomolecules 2015, 5, 1938-1954. https://doi.org/10.3390/biom5031938
Shaheen ZR, Corbett JA. Macrophage Expression of Inflammatory Genes in Response to EMCV Infection. Biomolecules. 2015; 5(3):1938-1954. https://doi.org/10.3390/biom5031938
Chicago/Turabian StyleShaheen, Zachary R., and John A. Corbett. 2015. "Macrophage Expression of Inflammatory Genes in Response to EMCV Infection" Biomolecules 5, no. 3: 1938-1954. https://doi.org/10.3390/biom5031938