Carbonic Anhydrase IX (CAIX), Cancer, and Radiation Responsiveness
by
 ,
,
Carol Ward
1,2,* ,
,
James Meehan
1,3,
Mark Gray
1,2,
Ian H. Kunkler
1,
Simon P. Langdon
1 and 
David J. Argyle
2 1
Cancer Research UK Edinburgh Centre and Division of Pathology Laboratory, Institute of Genetics and Molecular Medicine, University of Edinburgh, Edinburgh EH4 2XU, UK
2
Royal (Dick) School of Veterinary Studies and Roslin Institute, The University of Edinburgh, Easter Bush, Midlothian EH25 9RG, UK
3
Institute of Sensors, Signals and Systems, School of Engineering and Physical Sciences, Heriot-Watt University, Edinburgh EH14 4AS, UK
*
Author to whom correspondence should be addressed.
Metabolites 2018, 8(1), 13; https://doi.org/10.3390/metabo8010013
Submission received: 12 January 2018
/
Revised: 6 February 2018
/
Accepted: 7 February 2018
/
Published: 10 February 2018
(This article belongs to the Special Issue Carbonic Anhydrases and Metabolism)
{kind=link}
{kind=link}
{kind=link}
Abstract
:Carbonic anhydrase IX has been under intensive investigation as a therapeutic target in cancer. Studies demonstrate that this enzyme has a key role in pH regulation in cancer cells, allowing these cells to adapt to the adverse conditions of the tumour microenviroment. Novel CAIX inhibitors have shown efficacy in both in vitro and in vivo pre-clinical cancer models, adversely affecting cell viability, tumour formation, migration, invasion, and metastatic growth when used alone. In co-treatments, CAIX inhibitors may enhance the effects of anti-angiogenic drugs or chemotherapy agents. Research suggests that these inhibitors may also increase the response of tumours to radiotherapy. Although many of the anti-tumour effects of CAIX inhibition may be dependent on its role in pH regulation, recent work has shown that CAIX interacts with several of the signalling pathways involved in the cellular response to radiation, suggesting that pH-independent mechanisms may also be an important basis of its role in tumour progression. Here, we discuss these pH-independent interactions in the context of the ability of CAIX to modulate the responsiveness of cancer to radiation.
1. Introduction
During growth, many solid tumours develop areas of low oxygen tension, or hypoxia, caused by malformation of the tumour vasculature and the increasing distance of tumour cells from the capillary bed. In tissues, O2 concentrations of 2–9% are typical, O2 concentrations ≤2% O2 are defined as hypoxic, and ≤0.02% are defined as severely hypoxic [1]. The diffusion distance of O2 from capillaries is approximately 100–200 μm; tumour cells situated further than this become hypoxic, as oxygen gradients develop in the tumour [2,3]. Circulation in the tumour is often cyclic, causing periods of acute or chronic hypoxia [4]. Tumour pH also falls with increased distance from blood vessels, with decreases from 7.4 to 6.0 measured around 300 μm from the vasculature [5]. However, the intracellular pH (pHi) of tumour cells is maintained between 7.0 and 7.4, by the actions of pH regulating proteins [6]. Poor perfusion also inhibits the removal of waste metabolites from the tumour, allowing acidosis to develop within the tumour microenvironment (TME). Hypoxia and acidosis cause major problems in cancer treatment, contributing to increased levels of resistance to both radiotherapy and chemotherapy [7].
2. Survival Strategies
To survive in these adverse conditions, cancer cells must adapt or die [8]. As one of the mechanisms of adaptation, cells in hypoxic conditions activate the transcription factor hypoxia inducible factor-1 (HIF-1), consisting of a heterodimer constructed from α and β subunits. In normal cellular O2 concentrations, the α subunit is rapidly degraded. The oxygen-dependent activation of prolyl hydroxylases causes hydroxylation of two proline residues (402 and 564) on HIF-1α, allowing interaction with an E3 ubiquitin ligase, VHL (the Von Hippel-Lindau factor), which targets this subunit for destruction in the proteasome [9]. This does not occur in hypoxic conditions and instead HIF-1α is stabilised and interacts with the HIF-1β subunit forming HIF-1, which activates gene transcription after nuclear translocation. Hypoxia-independent mechanisms for HIF-1α stabilisation additionally occur [10,11,12]. Other proteins also regulate HIF-1 activation, for example, factor inhibiting hypoxia inducible factor-1 (FIH-1), which prevents full activation of HIF-1 in moderate hypoxia. This protein maintains its activity in low O2 concentrations [13,14] by impairing the interaction between the C-terminal transactivation domain of HIF-1α and its co-activator proteins, causing partial activation of HIF-1 [15]. FIH-1 itself is inhibited in severe hypoxia, or by membrane type-1 matrix metalloproteinase/(MMP14), allowing full HIF-1 activation [13,16].
HIF-1 regulates the expression of genes involved in glycolysis, angiogenesis, pH regulation, migration, epithelial-mesenchymal transition (EMT), and invasion [17,18,19], many of which aid cancer progression. For example, EMT involves E-cadherin loss, which allows cancer cells to disperse and develop a migratory and invasive phenotype, and is also linked to increased resistance to chemotherapy and radiotherapy [20]. Hypoxia via HIF-1 causes E-cadherin loss by stimulating the lysyl oxidase (LOX)-Snail pathway [21]. LOX inhibition decreases the motility and invasiveness of cancer cells in hypoxia and also reduces metastasis in vivo [22]. Hypoxia also interferes with the homologous recombination, non-homologous end-joining, and mismatch repair DNA pathways, and inhibits the G1/S cell cycle checkpoint. This increases DNA errors and causes chromosomal instability [6,21].
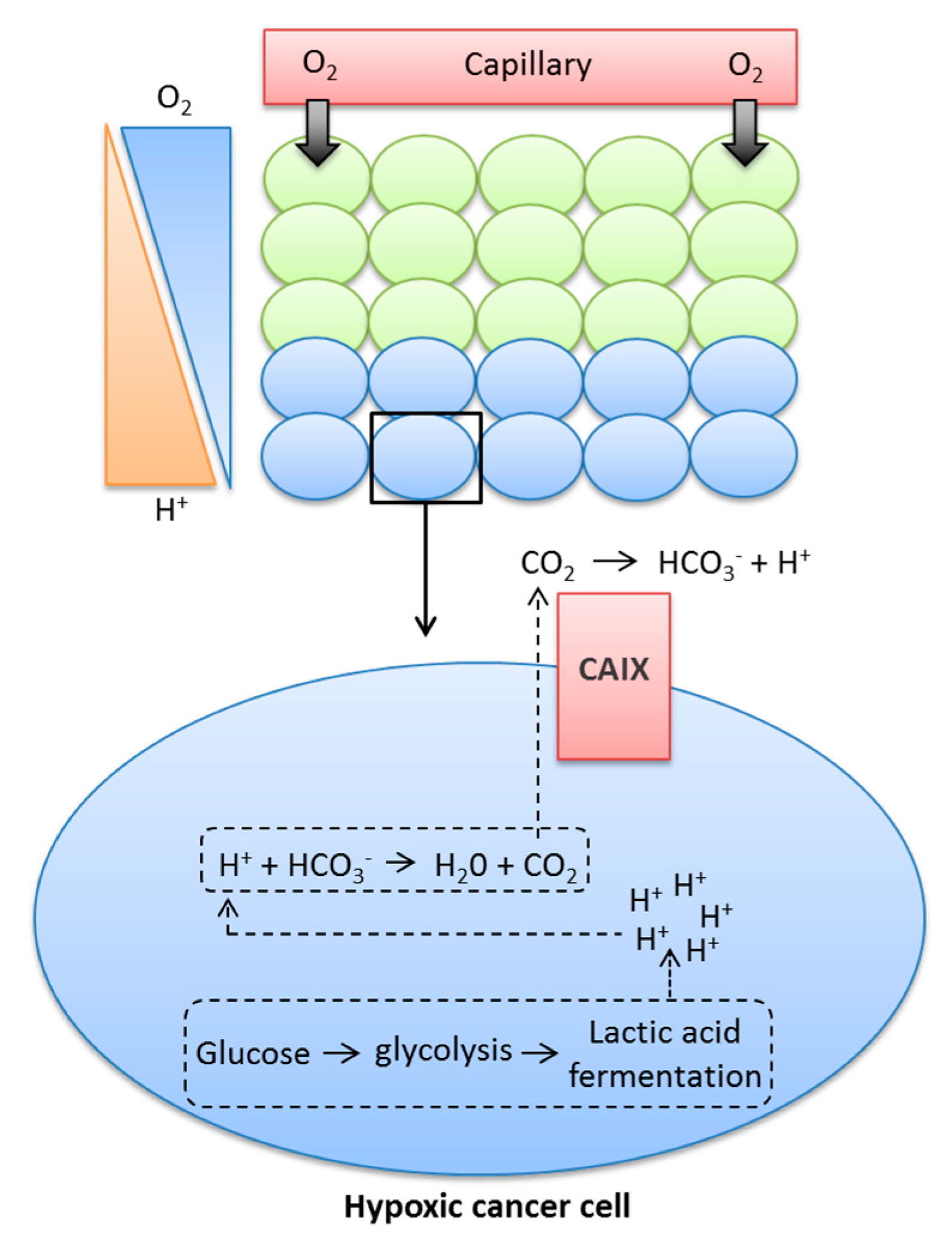
Cancer cells use aerobic glycolysis for energy and to provide components for cell growth and proliferation, even in normoxic conditions, causing higher rates of glycolysis and increased production of CO2, H+, and lactate [8,23]. These metabolic by-products must be removed from the cell to prevent the pHi becoming acidic, and thus maintain a slightly alkaline pHi consistent with survival. Early studies using D2O in yeast demonstrated that active transport mechanisms are likely to be dependent on protons, since deuterons could not substitute for protons in these processes [24], and further illustrated the role of alkaline pHi in transformation, tumorigenicity, and proliferation [25,26]. Tumour cells can maintain their pHi through increased expression and activation of pH regulatory proteins, some of which are HIF1-dependent, such as monocarboxylate transporter 4 (MCT4), which exports lactate and H+ from tumour cells, or carbonic anhydrase IX (CAIX), an enzyme that accelerates the conversion of CO2 and H2O to HCO3− and H+ [18,27,28,29]. HCO3− is transported back into tumour cells via HCO3− transporters and used to buffer pHi [28,30]. The role of CAIX is shown in Figure 1.
As a result of low O2 levels, hypoxic cancer cells are required to undergo lactic acid fermentation for the production of energy, a process that leads to the production of H+ ions. If these H+ ions are allowed to build up in the cytoplasm, they can lead to changes in pH, which can be detrimental to the cell. The metabolic acids generated within the cell can react with HCO3−, leading to the production of H2O and CO2. Membrane-permeant CO2 is a form in which much acid is removed from cancer cells. CAIX facilitates CO2 diffusion out of the cell by catalysing the extracellular hydration of CO2, leading to the production of H+ and HCO3−. CAIX therefore maintains a steeper efflux gradient for CO2, leading to a more alkaline intracellular pH, while also causing the acidification of the extracellular milieu.
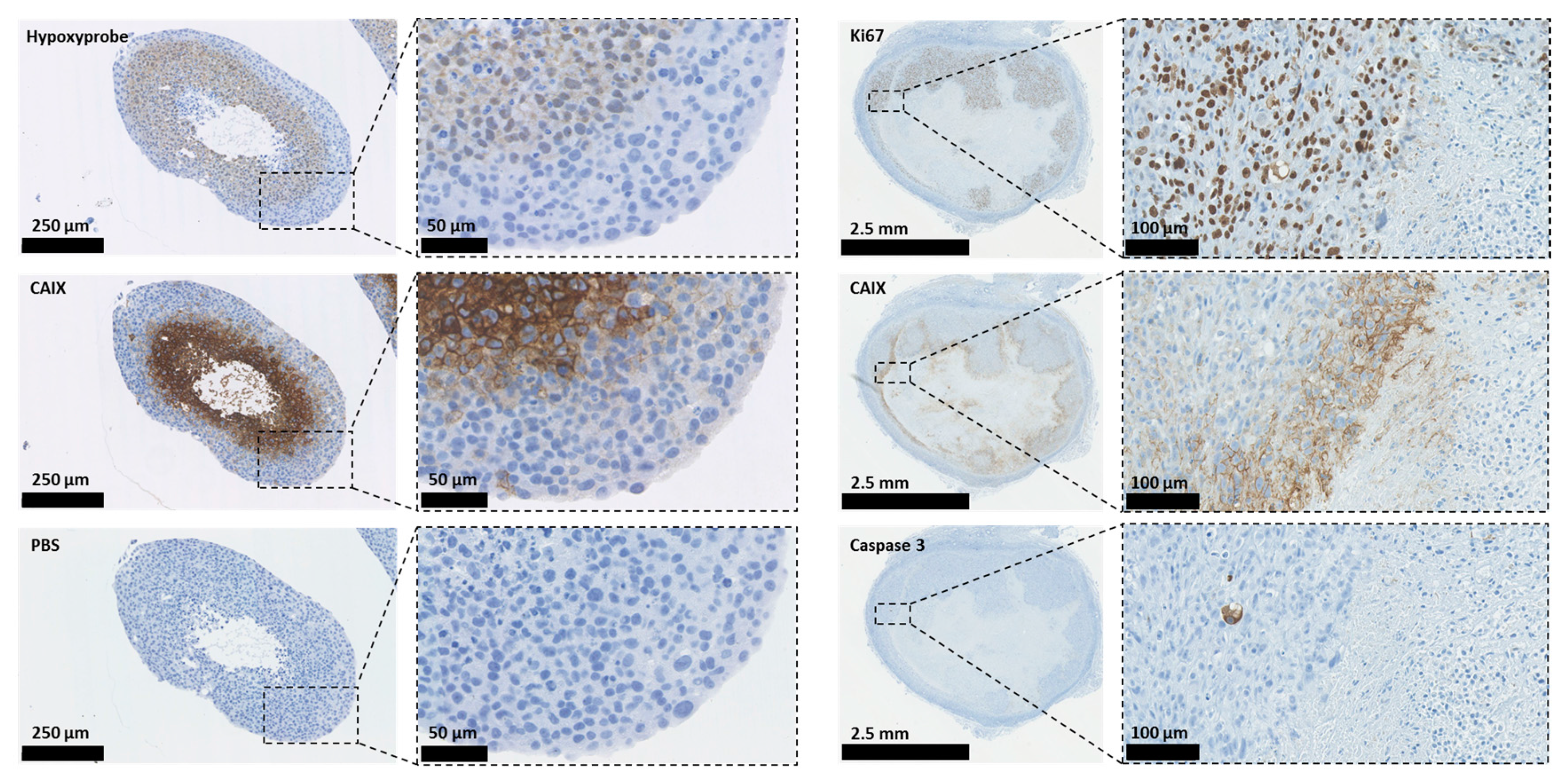
Carbonic anhydrases (CAs) are ubiquitous metalloenzymes that catalyse the reversible formation of HCO3− and H+ ions from H2O and CO2 [31]. At least 16 different isoforms of CAs have been isolated from mammals and differ in terms of cellular location, activity, and tissue locations. One CA, CAVI, is secreted, two (CAVA and VB) are found in the mitochondria, five are cytosolic (CAs I, II, III, VII, and XIII), and five are found on membranes (CA IV, IX, XII, XIV, and XV); of these, CAIX and CAXII have been shown to play an important role in cancer progression [32,33]. Some CAs have been shown to operate as part of transport ‘metabolons’ to increase the effectiveness of HCO3−- and H+-transporters [34,35,36,37]. This contributes to the maintenance of an alkaline pHi in tumour cells and an acidic pHe in the TME, which supports tumour growth, invasion, metastasis, and resistance to both chemotherapy and radiotherapy [5,6,38,39,40]. For example, in tumours, the most invasive regions are those exhibiting the lowest pH, which causes activation and increases expression of proteinases and metalloproteases that degrade components of the extracellular matrix (ECM), facilitating invasion and migration [5,41,42,43]. Alkaline pHi causes resistance to apoptotic stimuli because caspase activation occurs in acidic pHi conditions [44]; it also increases both DNA synthesis and cell proliferation, allowing tumour growth and progression [6,8,45,46]. Figure 2 illustrates the expression of CAIX, proliferation, the hypoxic region, and apoptotic staining in human 3D breast cancer models.
In Figure 2, the illustrations on the left demonstrate overlapping staining for CAIX and the hypoxic marker hypoxyprobe-1 in 3D spheroid cultures of HBL-100 human breast cancer cells. The right-hand illustrations demonstrate staining for CAIX, Ki67 (a proliferation marker), and caspase-3 (apoptosis) in a xenograft model of MDA-MB-231 human breast cancer cells.
Multiple studies have demonstrated a role for CAIX in pH regulation in cancer cells [28,47,48,49,50]. Targeting CAIX is proposed as a logical strategy for anti-cancer therapy, since it is an extracellular target, mainly associated with malignant growth, and is largely absent from most healthy tissue, with the exception of the gastro-intestinal tract and stomach [32,38,47,51,52]. CAIX staining in tumours is associated with poor prognosis and progression in several types of cancer, and in a series of lymph node-positive breast tumours it was found to correlate with metastasis [40,53,54,55,56,57,58]. Knockdown of CAIX in murine models leads to few phenotypic abnormalities other than gastric hyperplasia, inferring limited toxicity issues in normal tissue [59,60].
3. CAIX Inhibition as a Cancer Therapy
The effectiveness of CAIX inhibition as an anti-cancer strategy has been demonstrated in many pre-clinical studies using various cancer models. CAIX inhibitors negatively affect cancer cell viability and migration, as well as collagen destruction and invasion, and hinder both tumour formation and metastic growth in murine models, suggesting that increased expression and activity of CAIX in a cancer will adversly affect progression and prognosis. Many groups have now validated a crucial role for CAIX in growth, migration, invasion, and metastasis of tumours [40,52,61,62,63,64,65,66,67,68].
In vitro investigations have demonstrated that inhibition of CAIX using siRNA or CAIX inhibitors decreased the invasiveness of renal and ovarian cancer cells, while also reducing the amount of cells invading from human breast carcinoma spheroids [40,66,69,70]. A novel class of sulfamate CAIX inhibitors reduced the invasion, proliferation, and migration of human breast cancer cells, and also exhibited the capacity to reverse established invasion in a model consisting of breast tumour tissue from naïve biopsies [40,63,66,71]. CAIX knockdown significantly reduced the proliferation and survival of cancer cells under both normoxic and hypoxic conditions [40,66,72].
In vivo studies have shown that knockdown of CAIX can decrease tumour volume in both breast and colon cancer xenografts. Additive results could be obtained in co-treatments alongside antiangiogenic therapy. Knockdown also inhibited lung metastasis in breast cancer models [52,63,64]. CAIX overexpression studies in a colon cancer model demonstrated increased rates of tumour growth and expression of Ki-67, a marker of proliferation [64]. CAIX inhibitors also slowed tumour growth in breast cancer xenografts by decreasing proliferation and increasing cell death [40]. One of these inhibitors also exhibited anti-metastatic effects in a xenograft model of human breast cancer [73]. Interestingly, the use of CAIX inhibitors in in vivo systems did not lead to any reports of non-specific toxicity [40,69,73,74]. The development of novel CAIX inhibitors and clinical trials has been reviewed recently [8,28,29,30,75].
4. Other Possible Functions for CAIX in Cancer Progression
Although the main function of CAIX in cancer is as a regulator of pHi, several studies show that other possible mechanisms may be linked to this enzyme, thereby expanding its role in tumour progression. A recent study found novel roles for CAIX in tumour cell migration and MMP14-mediated invasion [67]. Interactions were identified with β1 integrins, metabolic transporters, integrin-associated protein CD98hc, and MMP14. CAIX appears to associate with MMP14 through phosphorylation sites in the intracellular domain of CAIX and can increase the degradation of collagen by MMP14 through providing the H+ the protease needs for its catalytic activity [67].
Interestingly, another recent report has linked MMP14 to the invasive capacity of breast cancer cell lines [66]. This study showed that although HIF-1α levels increased in hypoxic conditions, the expression of CAIX was variable between cell lines, and was only markedly upregulated in MCF-7 cells, a non-invasive cell line, after exposure to chronic hypoxia. HIF-1α gene expression can be constrained by FIH-1, which in turn can be inhibited by MMP14 [16]. Although MCF-7 cells expressed high FIH-1 levels, they lacked the active form of MMP14, suggesting that in this cell line, FIH-1 is able to prevent full HIF-1 activation in acute hypoxia, but not in chronic hypoxia [66]. This is in agreement with prior studies showing that FIH-1 suppression increases CAIX expression in hepatoma and osteosarcoma cell lines [14]. In the MCF-7 models, FIH-1 knockdown increased CAIX expression in hypoxic cells [66]. Taken together, these results suggest a strong link between hypoxia, HIF-1, FIH-1, CAIX, and MMP14 in the regulation of cancer cell invasive potential [14,16,66,67].
CAIX can decrease binding of E-cadherin to the cytoskeleton and affect cell adhesion [76] while also increasing the metastatic potential of tumour cells by effects on the activity of Rho-GTPase [77]. It similarly interacts with DKK1 protein, and thus the Rho/ROCK pathway, activating paxillin and stimulating migration [77]. CAIX can be phosphorylated at Thr-443 by Protein Kinase A during hypoxia, causing activation of CAIX and facilitating migration via increased transcription of proteins involved in cytoskeletal organisation and EMT [77,78,79]. Overexpression of CAIX also modulates Rho/ROCK signalling (which is pH sensitive), activates paxillin, increases focal adhesion turnover, cell migration, and activation of the FAK/PI3K/mTOR/p70S6K signalling pathway [77,80]. Conversely, CAIX inhibition impedes ROCK1 and decreases invasion [68,81].
5. Radiation
Radiotherapy is used to treat approximately 50% of cancer patients, either alone or in concert with chemotherapy or surgery [82]. It aims to eliminate cancer cells from the primary tumour, regional lymph nodes, or oligometastatic disease whilst limiting normal tissue damage. Radiation responses depend on the production of free radicals and intermediate ions that cause DNA damage in the form of single-strand (SSBs) or double-strand (DSBs) breaks in DNA. DBSs are the most effective in terms of inflicting cell damage and activate the DNA damage response (DDR) pathway that regulates whether the cell repairs DNA or undergoes cell death [83,84].
DSBs in DNA are detected by ataxia telangiectasia mutated (ATM) and ataxia telangiectasia and Rad3-related (ATR) kinases, which activate signalling pathways that stimulate cell cycle checkpoints and DNA repair. H2AX, once phosphorylated by ATM, recruits DNA repair proteins to the damaged area, and cyclins and cyclin-dependent kinases (CDKs) at G1/S and G2/M interphases delay cell division while DNA is repaired. The damage is restored by either homologous recombination or nonhomologous end joining (NHEJ) repair pathways. If DNA is not repaired, the damage results in cell death.
However, some tumours may either acquire or possess intrinsic radioresistance, and a major clinical advantage would be achieved in cancer treatment if new approaches to sensitizing these tumours to radiotherapy were developed [84,85]. Studies in the 1950s acertained the role of hypoxia in radioresistance and, conversely, the role that O2 plays in radiation responses [86,87,88]. Maximal cell kill in response to radiotherapy needs O2 to form free radicals that damage DNA, and stabilise or ‘fix’ radiation-induced DNA damage [89]. This causes changes in the DNA that cannot be repaired, leading to cell death if the cell tries to undergo cell division [1,90]. Hypoxic cells can be 2 to 3 times more resistant to the same dose of radiation, because fewer DSBs are stabilised [88]. Another factor is the decrease in cell proliferation caused by hypoxia, as DNA damage is higher in rapidly dividing cells [5]. The phase of the cell cycle also affects radiation responses, with cells in G2/M and G1 phases the most radiosensitive and those in the S phase more radioresistant [91,92]. Increased acidification decreases the effectiveness of radiation; cells cultured in acidic media are more resistant to radiation, with acidic pHe shown to reduce fixation of radiation-induced DNA damage, inhibit radiation-induced apoptosis, and delay G2/M-phase arrest allowing more time for treated cells to repair DNA damage, thus increasing radioresistance [7,93,94,95,96,97,98,99].
6. CAIX Inhibition and Radiation
Studies have shown that CAIX can influence the response of cancers to radiation [53,100]. The knockdown of hypoxia-induced CAIX, or CAIX and CAXII together, sensitised tumour cells to radiation by decreasing the number of cells in the radioresistant S phase in both in vitro and in vivo models. This knockdown caused decreased intracellular pH values, which were found to correlate with enhanced cell death, suggesting that active CAIX is protecting cells against radiation by preserving an alkaline pHi, since ectopic CAIX expression increased cell survival after radiation treatment [29,62]. Recent studies have also shown the ability of CAIX inhibitors to sensitise renal cell carcinoma to radiation by increasing radiation-induced apoptosis [101], which is one mechanism known to be involved in the therapeutic effect of radiotherapy [102]. CAIX inhibitors similarly enhanced radiation sensitivity when used in combination in a breast cancer model; proteomic studies indicated that co-treatment increased expression of pro-apoptotic proteins and reduced expression of anti-apoptotic proteins [66]. Other pre-clinical data using xenograft models have shown that tumours expressing high levels of CAIX were less responsive to radiation, but that CAIX inhibitors could significantly increase radiosensitivity [103,104]. For example, the co-treatment of CAIX inhibitors with radiation in a colon HT29 mouse xenograft model demonstrated an improved therapeutic effect [103]. Although this was not apparent in in vitro studies, a novel class of sulfamate CAIX inhibitors enhanced the effects of radiation in a colorectal model, both in vitro and in vivo [103,104]. The use of isotopic substitution experiments could give insight into whether CAIX inhibition induces radiation sensitivity by increasing intracellular H+ concentrations [24].
7. CAIX and Radiation Responses, and Other Mechanisms
Although most research suggests that CAIX influences cancer responses via pH regulation, this enzyme can also interact with other mechanisms involved in cellular reactions to radiation, suggesting that additional factors may be involved in its ability to radiosensitize cancer cells. Radiation triggers several signalling cascades known to be involved in cell survival such as the PI3K/AKT and ERK pathways; this occurs through activation of the epidermal growth factor receptor (EGFR) [105,106]. EGFR can influence radiation responses by binding to DNA-dependent protein kinases (DNA-PK), enhancing their activity and thus DNA repair [106,107,108]. Radiation induces nuclear accumulation of EGFR, where it is involved in the relaxation of chromatin, allowing DNA repair proteins access and thus enhancing resistance to radiation [109]. Decreased expression of EGFR or AKT has been shown to increase radiation sensitivity in human cancer cells [110]. Activation of EGFR by epidermal growth factor causes phosphorylation of CAIX on Tyr449, which in turn can activate the PI3K/AKT pathway by interacting with the p85 regulatory subunit [111]. This suggests that radiation itself may activate survival pathways through a mechanism that at least partially involves CAIX. PI3K/Akt is one of the pathways stimulated by radiation that is known to be involved in the inhibition of cell death via apoptosis; further, several studies have also linked overexpression and activation of EGFR with radiation resistance in cancer [112,113,114,115,116,117,118]. EGFR inhibitors can sensitise cancer cells to radiation both in vitro and in vivo, with positive results also observed in a Phase III trial in head and neck cancer [119,120]. Whether this response to radiation is in part through the prevention of CAIX phosphorlyation and activation of PI3K/AKT is currently unclear.
NF-κB activity is stimulated by hypoxia and acidic pH [121,122,123,124,125]. It is also activated by radiation and has a key role in radiation resistance and cell survival [126,127,128,129,130]. CAIX can interact with the NF-κB signalling pathway via a mechanism involving β1-integrin. Expression of β1 integrin is increased in various cancers, where it is involved in cell survival, proliferation, apoptosis, invasion, metastasis, and resistance to both chemotherapy and radiotherapy [131,132,133,134,135]. Studies have shown that cancer cells can be sensitized to radiotherapy by targeting β1 integrins [136,137]. The radioresistance mediated by β1 integrin is regulated by NF-κB, which increases β1-integrin expression, but conversely, inhibition of β1-integrin can inhibit the transcriptional activity of NF-κB [138]. A recent study demonstrated an interaction between CAIX and β1 integrin in tumour cells [67]; therefore this is another possible mode of interaction between CAIX and radiation responses. The downmodulation of β1 integrin can synergistically inhibit Akt-mediated survival in breast cancer cell lines and enhance radiotherapy in breast cancer xenografts [134,136,139]. A further study has shown that CAIX is required for the activation of NF-κB in hypoxia and can, via this interaction, stimulate the production of G-CSF to promote movement of granulocytic myeloid-derived suppressor cells (MDSC) to the metastatic niche of the lung [140]. Interestingly, G-CSF is strongly linked with protection from radiation damage [141,142], and high expression of the receptor is associated with poor response to radiotherapy in rectal cancer [143].
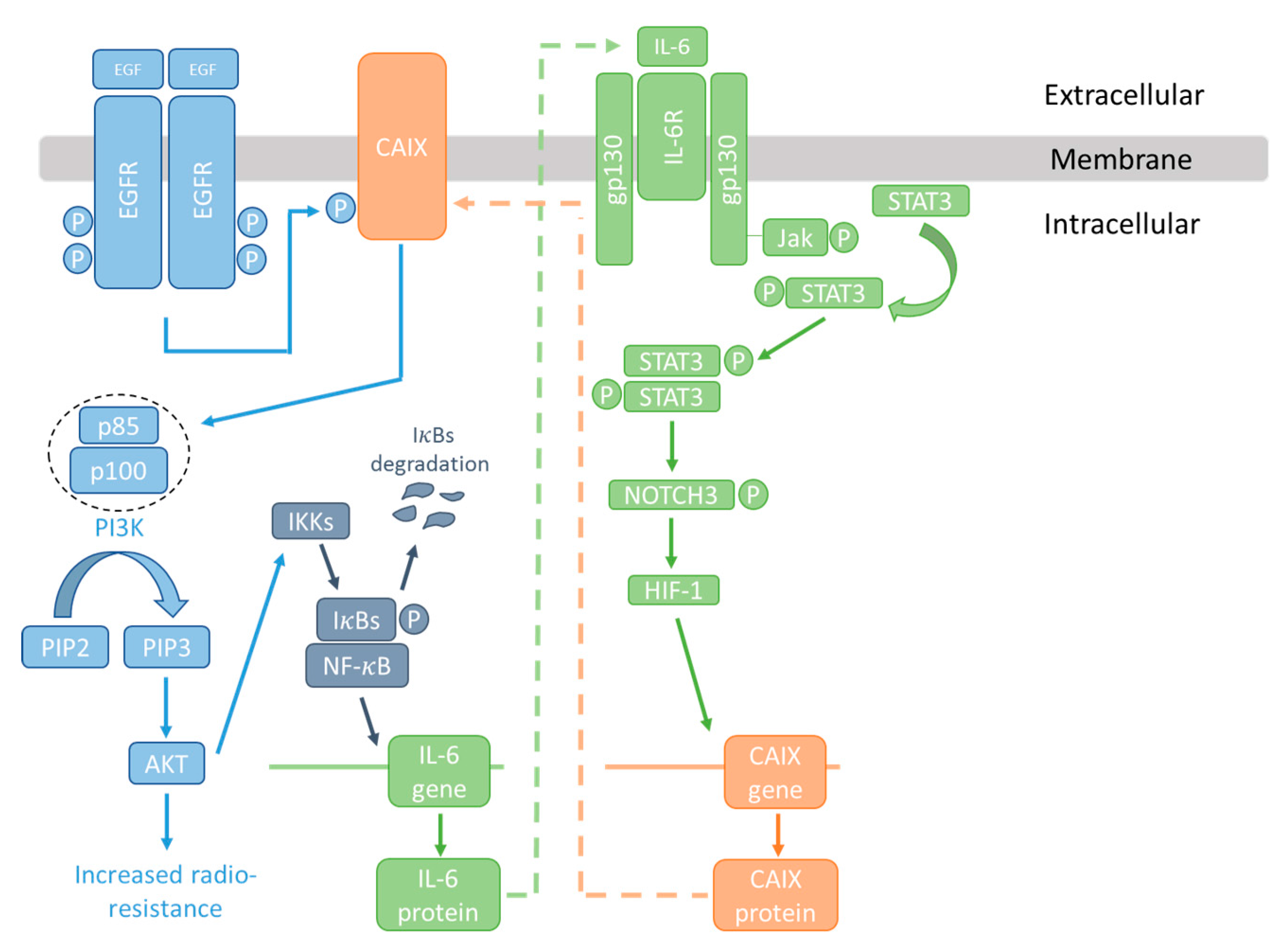
Signal transducer and activator of transcription 3 (Stat3) is overexpressed in many cancers; STAT3 has been shown to be involved in the regulation of CAIX expression in several studies [144,145]. Inhibition of STAT3 has been found to increase radiation sensitivity in cancer cells, and to inhibit radiation-induced progression in glioma [146,147,148]. IL-6 promotes tumour growth and invasion through STAT3 activation [140], and has likewise been linked to radiation resistance [149,150]. IL-6 is also an NF-κB responsive gene [151]. Taken together, it is therefore possible that CAIX is part of an IL-6-STAT3-NF-κB signalling axis involved in radiation resistance as illustrated in Figure 3. These interactions appear to be independent of the pH-regulating roles of CAIX.
EGF induces phosphorylation of CAIX via the EGFR, allowing interaction with the p85 regulatory subunit of PI3K and activation of survival pathways. AKT can in turn activate I-κB kinase (IKK) and the NF-κB transcription factor, causing the production of IL-6. IL-6 can, via STAT3 and HIF, increase CAIX expression. Overexpression of CAIX, EGFR, STAT3, and IL-6, and activation of NF-κB, EGFR, and STAT3, have all been linked with radiation resistance.
The increased presence of lactate in the TME has been linked to both radiation and chemoresistance [36,152,153], which may be due to the antioxidant effects of lactate in the case of radioresistance [154]. The secretion of excess lactic acid from the cell is regulated by MCTs, such as the HIF1-inducible MCT4, which operates almost solely to export lactate, or MCT1, which also transports other monocarboxylates [18,155,156,157]. Both MCT1 and 4 co-transport H+ with lactate [18]. MCT1 expression is increased in various cancers such as ovarian, prostate, breast, and colorectal cancers, where it correlates with progression and poor patient prognosis [158,159,160]. In xenografts of lung, colorectal, or small cell lung cancer, MCT1 inhibitors decreased lactate secretion and increased radiosensitivity [161,162].
The MCT1/4 accessory molecule CD147 is required for plasma membrane expression of these transporters; if targeted, it can decrease expression of these MCTs and inhibit tumour growth in an in vivo model [163]; therefore, CD147 should also be linked to radiation responses. Studies show that CD147 can promote radioresistance in hepatocellular carcinoma cells in vitro and in vivo, and it has been linked to radioresistance in cervical cancer [164,165,166]. Interestingly, CD147 has also been demonstrated to interact with integrin β1 in hepatocellular carcinoma, causing activation of the FAK pathway and increasing malignancy of these cells.
Could CAIX inhibition affect lactate secretion from cancer cells and thus sensitise resistant tissues to radiation? In an interesting study, it was found that the increased lactate efflux from hypoxic breast cancer cells was not due to amplified expression of MCTs, but to a hypoxia-induced upregulation of CAIX, via a mechanism that was independent of its catalytic activity [37]. Other studies have also demonstrated that CAs have effects on cells that are not dependent on the catalytic activity of these enzymes. For example, it has been demonstrated that both the cytosolic CAII and CAIV can enhance the activity of monocarboxylate transporters 1 and 4 in a non-enzymatic manner, thus increasing lactate flux [34,36,167,168]. Jamali et al. showed that knock-down of CAIX decreased lactate flux by approximately 50%. This study proposed that CAIX may function as a ‘proton-collecting/distributing antenna’ that accelerates proton transfer and requires an extracellular location for CAIX, and that could facilitate both MCT1 and MCT4 activity. They further suggested that this collaboration between MCTs and CAIX would not be inhibited by compounds that specifically target CAIX catalytic activity [37].
Recently, lactate has been shown to induce the expression of CAIX in normoxic cancer cells both in vitro and in vivo in a mechanism involving both HIF-1 and specificity protein (SP-1) transcription factors [169]. The major mechanism of CAIX increase appeared to be through redox-dependent stabilisation of HIF-1α. Again, this suggests a possible signalling loop in which hypoxic cells produce lactate that can increase CAIX expression via HIF, and which in turn allows increased lactate export via CAIX/MCT co-operation and thus increased radiation resistance.
Hypoxia induces dedifferentiation of cancer cells to become phenotypically more stem cell-like [170]. It has been proposed that CAIX may be involved in this process, as CAIX expression has been shown to correlate well with that of CD44, a breast cancer stem cell marker [171,172,173]. Radioresistance is considered an inherent characteristic of cancer stem cells [174,175,176]. It has been suggested that such cells may repair DNA more effectively after radiation, since they express high levels of genes associated with DNA damage repair [177,178,179,180]. Inhibition of CAIX depletes the number of breast cancer stem cells in tumour hypoxic subvolumes, and therefore CAIX inhibitors may be useful to treat the radioresistant cancer stem cell population. Furthermore, it has been inferred that CAIX is required to maintain the stemness phenotype within the hypoxic niche of breast tumors [65]. This may be due to the possible mechanisms outlined above, or to the effect of CAIX on the acidic TME, since extracellular acidosis has also been linked to the development of ‘stemness’ [181]. Interestingly, increased lactate concentrations can also cause cancer cells to develop a cancer stem cell phenotype [182]. In pancreatic cancer stem cells, STAT3 is activated, but STAT3 inhibition decreases both radioresistance and stem cell numbers in pancreatic cancer [148]. Therefore, since both lactate and STAT3 activation can increase expression of CAIX [144,145,169], it is certainly possible that the effect of CAIX inhibitors on radiation sensitivity of cancer stem cells is due to the interactions of CAIX with lactate and STAT3.
8. Conclusions
CAIX is an attractive target for the treatment of cancer [8,31,32,33]. Data suggests that CAIX inhibition is a therapeutic strategy that could interfere with cancer cell proliferation, migration, and invasion, while in vivo studies demonstrate that metastatic growth could also be limited. While evidence indicates that the effectiveness of this inhibition is through interference with pH regulation in cancer cells, recent studies show that CAIX can interact with many other signalling pathways and mechanisms known to be active in cancer cells, many of which appear to influence the response of cancer cells to radiation. These pathways are not mutually exclusive, and sensitivity to radiation could be determined by additive or synergistic interactions between pH-dependent and independent mechanisms, which suggests that CAIX may have many important roles in cancer cells that could potentially be exploited therapeutically, particularly by radiation oncologists.
Acknowledgments
This work was supported by funding from the UK Engineering and Physical Sciences Research Council, through the IMPACT programme Grant (EP/K-34510/1). No funds were received for covering the costs to publish in open access.
Author Contributions
C.W., S.P.L., I.H.K., and D.J.A. conceptualized and designed the structure of the article; C.W. wrote the paper; J.M. and M.G. performed the immunohistochemistry and designed the figures; C.W., J.M., M.G., I.H.K., and D.J.A. were involved in the critical reading and editing of the manuscript. All authors have read and approved the final version of the manuscript.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Bertout, J.A.; Patel, S.A.; Simon, M.C. The impact of O2 availability of human cancer. Nat. Rev. Cancer 2008, 8, 967–975. [Google Scholar] [CrossRef] [PubMed]
- Secomb, T.W.; Dewhirst, M.W.; Pries, A.R. Structural adaptation of normal and tumour vascular networks. Basic Clin. Pharmacol. Toxicol. 2012, 110, 63–69. [Google Scholar] [CrossRef] [PubMed]
- Vaupel, P.; Mayer, A. Hypoxia in tumors: Pathogenesis-related classification, characterization of hypoxia subtypes, and associated biological and clinical implications. Adv. Exp. Med. Biol. 2014, 812, 19–24. [Google Scholar] [CrossRef] [PubMed]
- Michiels, C.; Tellier, C.; Feron, O. Cycling hypoxia: A key feature of the tumor microenvironment. Biochim. Biophys. Acta 2016, 1866, 76–86. [Google Scholar] [CrossRef] [PubMed]
- Gatenby, R.A.; Smallbone, K.; Maini, P.K.; Rose, F.; Averill, J.; Nagle, R.B.; Worrall, L.; Gillies, R.J. Cellular adaptations to hypoxia and acidosis during somatic evolution of breast cancer. Br. J. Cancer 2007, 97, 646–653. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Webb, B.A.; Chimenti, M.; Jacobson, M.P.; Barber, D.L. Dysregulated pH: A perfect storm for cancer progression. Nat. Rev. Cancer 2011, 11, 671–677. [Google Scholar] [CrossRef] [PubMed]
- Vaupel, P. Tumor microenvironmental physiology and its implications for radiation oncology. Semin. Radiat. Oncol. 2004, 14, 198–206. [Google Scholar] [CrossRef] [PubMed]
- Ward, C.; Langdon, S.P.; Mullen, P.; Harris, A.L.; Harrison, D.J.; Supuran, C.T.; Kunkler, I.H. New strategies for targeting the hypoxic tumour microenvironment in breast cancer. Cancer Treat. Rev. 2013, 39, 171–179. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.L.; Jiang, B.; Rue, E.A.; Semenza, G.L. Hypoxia-inducible factor 1 is a basic-helix-loop-helix-PAS heterodimer regulated by cellular O2 tension. Proc. Natl. Acad. Sci. USA 1995, 92, 5510–5514. [Google Scholar] [CrossRef] [PubMed]
- Doe, M.R.; Ascano, J.M.; Kaur, M.; Cole, M.D. Myc posttranslationally induces HIF1 protein and target gene expression in normal and cancer cells. Cancer Res. 2012, 72, 949–957. [Google Scholar] [CrossRef] [PubMed]
- Semenza, G.L. Targeting HIF-1 for cancer therapy. Nat. Rev. Cancer 2003, 3, 721–732. [Google Scholar] [CrossRef] [PubMed]
- Lu, H.; Forbes, R.A.; Verma, A. Hypoxia-inducible factor 1 activation by aerobic glycolysis implicates the Warburg effect in carcinogenesis. J. Biol. Chem. 2002, 277, 23111–23115. [Google Scholar] [CrossRef] [PubMed]
- Tian, Y.M.; Yeoh, K.K.; Lee, M.K.; Eriksson, T.; Kessler, B.M.; Kramer, H.B.; Edelmann, M.J.; Willam, C.; Pugh, C.W.; Schofield, C.J.; et al. Differential sensitivity of hypoxia inducible factor hydroxylation sites to hypoxia and hydroxylase inhibitors. J. Biol. Chem. 2011, 286, 13041–13051. [Google Scholar] [CrossRef] [PubMed]
- Stolze, I.P.; Tian, Y.M.; Appelhoff, R.J.; Turley, H.; Wykoff, C.C.; Gleadle, J.M.; Ratcliffe, P.J. Genetic analysis of the role of the asparaginyl hydroxylase factor inhibiting hypoxia-inducible factor (FIH) in regulating hypoxia-inducible factor (HIF) transcriptional target genes. J. Biol. Chem. 2004, 279, 42719–42725. [Google Scholar] [CrossRef] [PubMed]
- Dayan, F.; Mazure, N.M.; Brahimi-Horn., M.C.; Pouyssegur, J. A dialogue between the hypoxia-inducible factor and the tumor microenvironment. Cancer Microenviron. 2008, 1, 53–68. [Google Scholar] [CrossRef] [PubMed]
- Sakamoto, T.; Seiki, M. A membrane protease regulates energy production in macrophages by activating hypoxia-inducible factor-1 via a non-proteolytic mechanism. J. Biol. Chem. 2010, 285, 29951–29964. [Google Scholar] [CrossRef] [PubMed]
- Wykoff, C.C.; Beasley, N.J.; Watson, P.H.; Turner, K.J.; Pastorek, J.; Sibtain, A.; Wilson, G.D.; Turley, H.; Talks, K.L.; Maxwell, P.H.; et al. Hypoxia-inducible expression of tumor-associated carbonic anhydrases. Cancer Res. 2000, 60, 7075–7083. [Google Scholar] [PubMed]
- Ullah, M.S.; Davies, A.J.; Halestrap, A.P. The plasma membrane lactate transporter MCT4, but not MCT1, is up-regulated by hypoxia through a HIF-1α-dependent mechanism. J. Biol. Chem. 2006, 281, 9030–9037. [Google Scholar] [CrossRef] [PubMed]
- Potter, C.; Harris, A.L. Hypoxia inducible carbonic anhydrase IX, marker of tumour hypoxia, survival pathway and therapy target. Cell Cycle 2004, 3, 164–167. [Google Scholar] [CrossRef] [PubMed]
- Marie-Egyptienne, D.T.; Lohse, I.; Hill, R.P. Cancer stem cells, the epithelial to mesenchymal transition (EMT) and radioresistance: Potential role of hypoxia. Cancer Lett. 2013, 341, 63–72. [Google Scholar] [CrossRef] [PubMed]
- Bristow, R.G.; Hill, R.P. Hypoxia and metabolism. Hypoxia, DNA repair and genetic instability. Nat. Rev. Cancer 2008, 8, 180–192. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.H.; Su, W.C.; Lin, P.W.; Guo, H.R.; Lee, W.Y. Hypoxia-inducible factor-1α correlates with MET and metastasis in node-negative breast cancer. Breast Cancer Res. Treat. 2007, 103, 167–175. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Kotyk, A.; Dvorakova, M.; Koryta, J. Deuterons cannot replace protons in active transport processes in yeast. FEBS Lett. 1990, 264, 203–205. [Google Scholar] [CrossRef]
- Perona, R.; Serrano, R. Increased pH and tumorigenicity of fibroblasts expressing a yeast proton pump. Nature 1988, 334, 438–440. [Google Scholar] [CrossRef] [PubMed]
- Perona, R.; Portillo, F.; Giraldez, F.; Serrano, R. Transformation and pH homeostasis of fibroblasts expressing yeast H+-ATPase containing site-directed mutations. Mol. Cell. Biol. 1990, 10, 4110–4115.27. [Google Scholar] [CrossRef] [PubMed]
- Pinheiro, C.; Longatto-Filho, A.; Azevedo-Silva, J.; Casal, M.; Schmitt, F.C.; Baltazar, F. Role of monocarboxylate transporters in human cancers: State of the art. J. Bioenerg. Biomembr. 2012, 44, 127–139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Swietach, P.; Hulikova, A.; Vaughan-Jones, R.D.; Harris, A.L. New insights into the physiological role of carbonic anhydrase IX in tumour pH regulation. Oncogene 2010, 29, 6509–6521. [Google Scholar] [CrossRef] [PubMed]
- Chiche, J.; Brahimi-Horn, M.C.; Pouyssegur, J. Tumour hypoxia induces a metabolic shift causing acidosis: A common feature in cancer. J. Cell. Mol. Med. 2010, 4, 771–794. [Google Scholar] [CrossRef] [PubMed]
- Parks, S.K.; Chiche, J.; Pouyssegur, J. pH control mechanisms of tumor survival and growth. J. Cell Physiol. 2011, 226, 299–308. [Google Scholar] [CrossRef] [PubMed]
- Supuran, C.T. Carbonic anhydrase inhibitors. Bioorg. Med. Chem. Lett. 2010, 20, 3467–3474. [Google Scholar] [CrossRef] [PubMed]
- Neri, D.; Supuran, C.T. Interfering with pH regulation in tumors as a therapeutic strategy. Nat. Rev. Drug Discov. 2011, 10, 767–777. [Google Scholar] [CrossRef] [PubMed]
- Supuran, C.T. Carbonic anhydrases: Novel therapeutic applications for inhibitors and activators. Nat. Rev. Drug Discov. 2008, 7, 168–181. [Google Scholar] [CrossRef] [PubMed]
- Becker, H.M.; Klier, M.; Schüler, C.; McKenna, R.; Deitmer, J.W. Intramolecular proton shuttle supports not only catalytic but also noncatalytic function of carbonic anhydrase II. Proc. Natl. Acad. Sci. USA 2011, 108, 3071–3076. [Google Scholar] [CrossRef] [PubMed]
- Deitmer, J.W.; Becker, H.M. Transport metabolons with carbonic anhydrases. Front. Physiol. 2013, 4, 291. [Google Scholar] [CrossRef] [PubMed]
- Klier, M.; Andes, F.T.; Deitmer, J.W.; Becker, H.M. Intracellular and extracellular carbonic anhydrases cooperate non-enzymaticaly to enhance activity of monocarboxylate transporters. J. Biol. Chem. 2014, 289, 2765–2775. [Google Scholar] [CrossRef] [PubMed]
- Jamali, S.; Klier, M.; Ames, S.; Felipe Barros, L.; McKenna, R.; Deiter, J.W.; Becker, H.M. Hypoxia-induced carbonic anhydrase IX facilitates lactate flux in human breast cancer cells by non-catalytic function. Sci. Rep. 2015, 5, 13605. [Google Scholar] [CrossRef] [PubMed]
- Parks, S.K.; Chiche, J.; Pouyssegur, J. Disrupting proton dynamics and energy metabolism for cancer therapy. Nat. Rev. Cancer 2013, 13, 611–623. [Google Scholar] [CrossRef] [PubMed]
- Gillies, R.J.; Verduzco, D.; Gatenby, R.A. Evolutionary dynamics of carcinogenesis and why targeted therapy does not work. Nat. Rev. Cancer 2012, 12, 487–493. [Google Scholar] [CrossRef] [PubMed]
- Ward, C.; Meehan, J.; Mullen, P.; Supuran, C.; Dixon, J.M.; Thomas, J.S.; Winum, J.Y.; Lambin, P.; Dubois, L.; Pavathaneni, N.K.; et al. Evaluation of carbonic anhydrase IX as a therapeutic target for inhibition of breast cancer invasion and metastasis using a series of in vitro breast cancer models. Oncotarget 2015, 6, 24856–24870. [Google Scholar] [CrossRef] [PubMed]
- Rofstad, E.K.; Mathiesen, B.; Kindem, K.; Galappathi, K. Acidic extracellular pH promotes experimental metastasis of human melanoma cells in athymic nude mice. Cancer Res. 2006, 66, 6699–6707. [Google Scholar] [CrossRef] [PubMed]
- Kato, Y.; Ozawa, S.; Tsukuda, M.; Kubota, E.; Miyazaki, K.; St-Pierre, Y.; Hata, R. Acidic extracellular pH increases calcium influx-triggered phospholipase D activity along with acidic spingomyelinase activation to induce matrix metalloproteinase-9 expression in mouse metastatic melanoma. FEBS J. 2007, 274, 3171–3183. [Google Scholar] [CrossRef] [PubMed]
- Gatenby, R.A.; Gawlinski, E.T.; Gmitro, A.F.; Kaylor, B.; Gillies, R.J. Acid-mediated tumor invasion; a multidisciplinary study. Cancer Res. 2006, 66, 5216–5223. [Google Scholar] [CrossRef] [PubMed]
- Matsuyama, S.; Llopis, J.; Deveraux, Q.L.; Tsien, R.Y.; Reed, J.C. Changes in intramitochondrial and cytosolic pH: Early events that modulate caspase activation during apoptosis. Nat. Cell Biol. 2000, 2, 318–325. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.H.; Cragoe, E.J., Jr.; Edwards, A.M. Control of hepatocyte DNA synthesis by intracellular pH and its role in the action of tumor promoters. J. Cell. Physiol. 2003, 195, 61–69. [Google Scholar] [CrossRef] [PubMed]
- Schreiber, R. Ca2+ signalling, intracellular pH and cell volume in cell proliferation. J. Membr. Biol. 2005, 205, 129–137. [Google Scholar] [CrossRef] [PubMed]
- Svastova, E.; Hulikova, A.; Rafajoba, M.; Zat’ovivova, M.; Gibadulinova, A.; Casini, A.; Cecchi, A.; Scozzafava, A.; Supuran, C.T.; Pastorek, J.; et al. Hypoxia activates the capacity of tumor-associated carbonic anhydrase IX to acidify extracellular pH. FEBS Lett. 2004, 577, 439–445. [Google Scholar] [CrossRef] [PubMed]
- Swietach, P.; Vaughan-Jones, R.D.; Harris, A.L.; Hulikova, A. The chemistry, physiology and pathology of pH in cancer. Philos. Trans. R. Soc. Lond. Biol. Sci. 2014, 369, 20130099. [Google Scholar] [CrossRef] [PubMed]
- Hulikova, A.; Vaughan-Jones, R.D.; Swietach, P. Dual role of CO2/HCO3− buffer in the regulation of intracellular pH of three-dimensional tumor growths. J. Biol. Chem. 2011, 286, 13815–13826. [Google Scholar] [CrossRef] [PubMed]
- Pouysségur, J.; Dayan, F.; Mazure, N.M. Hypoxia signalling in cancer and approaches to enforce tumour regression. Nature 2006, 441, 437–443. [Google Scholar] [CrossRef] [PubMed]
- Pastoreková, S.; Parkkila, S.; Parkkila, A.K.; Opavsky, R.; Zelnik, V.; Saarnio, J.; Pastorek, J. Carbonic anhydrase IX, MN/CA IX: Analysis of stomach complementary DNA sequence and expression in human and rat alimentary tracts. Gastroenterology 1997, 112, 398–408. [Google Scholar] [CrossRef] [PubMed]
- Chiche, J.; Ilc, K.; Laferriere, J.; Trottier, E.; Dayan, F.; Mazure, N.M.; Brahimi-Horn, M.C.; Pouysségur, J. Hypoxia-inducible carbonic anhydrase IX and XII promote tumor cell growth by counteracting acidosis through the regulation of the intracellular pH. Cancer Res. 2009, 69, 358–368. [Google Scholar] [CrossRef] [PubMed]
- Koukourakis, M.I.; Giatromanolaki, A.; Sivridis, E.; Simopoulos, K.; Pastorek, J.; Wykoff, C.C.; Gatter, K.C.; Harris, A.L. Hypoxia-regulated carbonic anhydrase-9 (CA9) relates to poor vascularization and the resistance of squamous head and neck cancer to chemoradiotherapy. Clin. Cancer Res. 2001, 11, 3399–3403. [Google Scholar]
- Korkeila, E.; Talvinen, K.; Jaakkola, P.M.; Minn, H.; Syrjanen, K.; Sundstrom, J.; Pyrhonen, S. Expression of carbonic anhydrase IX suggests poor outcome in rectal cancer. Br. J. Cancer 2009, 100, 874–880. [Google Scholar] [CrossRef] [PubMed]
- Tan, E.Y.; Yan, M.; Campo, L.; Han, C.; Takano, E.; Turley, H.; Candiloro, I.; Pezzella, F.; Gatter, K.C.; Millar, E.K.; et al. The key hypoxia regulated gene CAIX is upregulated in basal-like breast tumours and is associated with resistance to chemotherapy. Br. J. Cancer 2009, 100, 405–411. [Google Scholar] [CrossRef] [PubMed]
- Chia, S.K.; Wykoff, C.C.; Watson, P.H.; Han, C.; Leek, R.D.; Pastorek, J.; Gatter, K.C.; Ratcliffe, P.; Harris, A.L. Prognostic significance of a novel hypoxia-regulated marker, carbonic anhydrase IX, invasive breast carcinoma. J. Clin. Oncol. 2001, 19, 3660–3668. [Google Scholar] [CrossRef] [PubMed]
- Bartosova, M.; Parkkila, S.; Pohlodek, K.; Karttunen, T.J.; Galbavy, S.; Mucha, V.; Harris, A.L.; Pastorek, J.; Pastorekova, S. Expression of carbonic anhydrase IX in breast is associated with malignant tissues and is related to overexpression of c-erbB2. J. Pathol. 2002, 197, 314–321. [Google Scholar] [CrossRef] [PubMed]
- Generali, D.; Fox, S.B.; Berruti, A.; Brizzi, M.P.; Campo, L.; Bonardi, S.; Wigfield, S.M.; Bruzzi, P.; Bersiga, A.; Allevi, G.; et al. Role of carbonic anhydrase IX expression in prediction of the efficacy and outcome of primary epirubicin/tamoxifen therapy for breast cancer. Endocr. Relat. Cancer 2006, 13, 921–930. [Google Scholar] [CrossRef] [PubMed]
- Gut, M.O.; Parkkila, S.; Vernerová, Z.; Rohde, E.; Závada, J.; Höcker, M.; Pastorek, J.; Karttunen, T.; Gibadulinová, A.; Závadová, Z.; et al. Gastric hyperplasia in mice with targeted disruption of the carbonic anhydrase gene Car9. Gastroenterology 2002, 123, 1889–1903. [Google Scholar] [CrossRef] [PubMed]
- Leppilampi, M.; Karttunen, J.; Kivela, J.; Gut, M.O.; Pastorekova, S.; Pastorek, J.; Parkkila, S. Gastric pit cell hyperplasia and glandular atrophy in carbonic anhydrase IX knockout mice: Studies on two strains C57/BL6 and BALB/C. Transgenic Res. 2005, 14, 655–663. [Google Scholar] [CrossRef] [PubMed]
- Morris, J.C.; Chiche, J.; Grellier, C.; Lopez, M.; Bornaghi, L.F.; Maresca, A.; Supuran, C.T.; Pouysségur, J.; Poulsen, S.A. Targeting hypoxic tumor cell viability with carbohydrate-based carbonic anhydrase IX and XII inhibitors. J. Med. Chem. 2011, 54, 6905–6918. [Google Scholar] [CrossRef] [PubMed]
- Doyen, J.; Parks, S.K.; Marcie, S.; Pouyssegur, J.; Chiche, J. Knock-down of hypoxia-induced carbonic anhydrases IX and XII radiosensitizes tumor cells by increasing intracellular acidosis. Front. Oncol. 2013, 2, 199.2013. [Google Scholar] [CrossRef] [PubMed]
- Lou, Y.; McDonald, P.C.; Oloumi, A.; Chia, S.; Ostlund, C.; Ahamdi, A.; Kyle, A.; Auf dem Keller, U.; Leung, S.; Huntsman, D.; et al. Targeting tumor hypoxia: Suppression of breast tumor growth and metastasis by novel carbonic anhydrase IX inhibitors. Cancer Res. 2011, 71, 3364–3376. [Google Scholar] [CrossRef] [PubMed]
- McIntyre, A.; Patiar, S.; Wigfield, S.; Li, J.; Ledaki, I.; Turley, H.; Leek, R.; Snell, C.; Gatter, K.; Sly, W.S.; et al. Carbonic anhydrase IX promotes tumor growth and necrosis in vivo and inhibition enhances anti-VEGF therapy. Clin. Cancer Res. 2012, 18, 3100–3111. [Google Scholar] [CrossRef] [PubMed]
- Lock, F.E.; McDonald, P.C.; Lou, Y.; Serrano, I.; Chafe, S.C.; Ostlund, C.; Aparicio, S.; Winum, J.Y.; Supuran, C.T.; Dedhar, S. Targeting carbonic anhydrase IX depletes breast cancer stem cells within the hypoxic niche. Oncogene 2013, 32, 5210–5219. [Google Scholar] [CrossRef] [PubMed]
- Meehan, J.; Ward, C.; Turnbull, A.; Bukowski-Wills, J.; Finch, A.J.; Jarman, E.J.; Xintaropoulou, C.; Martinez-Perez, C.; Gray, M.; Pearson, M.; et al. Inhibition of pH regulation as a therapeutic strategy in hypoxic human breast cancer cells. Oncotarget 2017, 8, 42857–42875. [Google Scholar] [CrossRef] [PubMed]
- Swayampakula, M.; McDonald, P.; Vallejo, M.; Coyaud, E.; Chafe, S.C.; Westerbeck, A.; Venkateswaran, G.; Shankar, J.; Gao, G.; Laurent, E.M.N.; et al. The interactome of metabolic enzyme carbonic anhydrase IX reveals novel roles in tumor cell migration and invadopodia/MMP14-mediated invasion. Oncogene 2017, 36, 6244–6261. [Google Scholar] [CrossRef] [PubMed]
- Radvak, P.; Repic, M.; Svastova, E.; Takacova, M.; Csaderova, L.; Strnad, H.; Pastorek, J.; Pastorekova, S.; Kopacek, J. Suppression of carbonic anhydrase IX leads to aberrant focal adhesion and decreased invasion of tumor cells. Oncol. Rep. 2013, 29, 1147–1153. [Google Scholar] [CrossRef] [PubMed]
- Sansone, P.; Storci, G.; Tavolari, S.; Guarnieri, T.; Giovannini, C.; Taffurelli, M.; Ceccarelli, C.; Santini, D.; Paterini, P.; Marcu, K.B.; et al. IL-6 triggers malignant features in mammospheres from human ductal breast carcinoma and normal mammary gland. J. Clin. Investig. 2007, 117, 3988–4002. [Google Scholar] [CrossRef] [PubMed]
- Parkkila, S.; Rajaniemi, H.; Parkkila, A.K.; Kivela, J.; Waheed, A.; Pastorekova, S.; Pastorek, J.; Sly, W.S. Carbonic anhydrase inhibitor suppresses invasion of renal cancer cells in vitro. Proc. Natl. Acad. Sci. USA 2000, 97, 2220–2224. [Google Scholar] [CrossRef] [PubMed]
- Winum, J.Y.; Carta, F.; Ward, C.; Mullen, P.; Harrison, D.; Langdon, S.P.; Cecchi, A.; Scozzafava, A.; Kunkler, I.; Supuran, C.T. Ureido-substituted sulfamates show potent carbonic anhydrase IX inhibitory and antiproliferative activities against breast cancer cell lines. Bioorg. Med. Chem. Lett. 2012, 22, 4681–4685. [Google Scholar] [CrossRef] [PubMed]
- Robertson, N.; Potter, C.; Harris, A.L. Role of carbonic anhydrase IX in human tumor cell growth, survival and invasion. Cancer Res. 2004, 64, 6160–6165. [Google Scholar] [CrossRef] [PubMed]
- Gieling, R.G.; Babur, M.; Mamnani, L.; Burrows, N.; Telfer, B.A.; Carta, F.; Winum, J.Y.; Scozzafava, A.; Supuran, C.T.; Williams, K.J. Antimetastatic effect of sulfamate carbonic anhydrase IX inhibitors in breast carcinoma xenografts. J. Med. Chem. 2012, 55, 5591–6000. [Google Scholar] [CrossRef] [PubMed]
- Pacchiano, F.; Carta, F.; McDonald, P.C.; Lou, Y.; Vullo, D.; Scozzafava, A.; Dedhar, S.; Supuran, C.T. Ureido-substituted benzenesulfonamides potently inhibit carbonic anhydrase IX and show antimetastatic activity in a model of breast cancer metastasis. J. Med. Chem. 2011, 54, 1896–1902. [Google Scholar] [CrossRef] [PubMed]
- Supuran, C.T. Carbonic Anhydrase Inhibition and the Management of Hypoxic Tumors. Metabolites 2017, 7, 48. [Google Scholar] [CrossRef]
- Svastová, E.; Zilka, N.; Zat’ovicová, M.; Gibadulinová, A.; Ciampor, F.; Pastorek, J.; Pastoreková, S. Carbonic anhydrase IX reduces E-cadherin-mediated adhesion of MDCK cells via interaction with catenin. Exp. Cell Res. 2003, 290, 332–345. [Google Scholar] [CrossRef]
- Shin, H.J.; Rho, S.B.; Jung, D.; Han, I.O.; Oh, E.S.; Kim, J.Y. Carbonic anhydrase IX (CA9) modulates tumor-associated cell migration and invasion. J. Cell Sci. 2011, 124, 1077–1087. [Google Scholar] [CrossRef] [PubMed]
- Hulikova, A.; Zatovicova, M.; Svastova, E.; Ditte, P.; Brasseur, R.; Kettmann, R.; Supuran, C.T.; Kopacek, J.; Pastorek, J.; Pastorekova, S. Intact intracellular tail is critical for proper functioning of the tumour-associated, hypoxia-regulated carbonic anhydrase IX. FEBS Lett. 2009, 583, 3563–3568. [Google Scholar] [CrossRef] [PubMed]
- Ditte, P.; Dequiedt, F.; Svastova, E.; Hulikova, A.; Ohradanova-Repic, A.; Zatovicova, M.; Csaderova, L.; Kopacek, J.; Supuran, C.T.; Pastorekova, S.; et al. Phosphorylation of carbonic anhydrase IX controls its ability to mediate extracellular acidification in hypoxic tumors. Cancer Res. 2011, 71, 7558–7567. [Google Scholar] [CrossRef] [PubMed]
- Worthylake, R.A.; Burridge, K. RhoA and ROCK promote migration by limiting membrane protrusions. J. Biol. Chem. 2003, 278, 13578–13584. [Google Scholar] [CrossRef] [PubMed]
- Csaderova, L.; Debreova, M.; Radvak, P.; Stano, M.; Vrestiakova, M.; Kopacek, J.; Pastorekova, S.; Svastova, E. The effect of carbonic anhydrase IX on focal contacts during cell spreading and migration. Front. Physiol. 2013, 4, 271. [Google Scholar] [CrossRef] [PubMed]
- Thariat, J.; Hannoun-Levi, J.M.; Sun Myint, A.; Vuong, T.; Gérard, J.P. Past, present, and future of radiotherapy for the benefit of patients. Nat. Rev. Clin. Oncol. 2013, 10, 52–60. [Google Scholar] [CrossRef] [PubMed]
- Kavanagh, J.N.; Redmond, K.M.; Schettino, G.; Prise, K.M. DNA double strand break repair: A radiation perspective. Antioxid. Redox Signal. 2013, 18, 2458–2472. [Google Scholar] [CrossRef] [PubMed]
- Raleigh, D.R.; Haas-Kogan, D.A. Molecular targets and mechanisms of radiosensitization using DNA damage response pathways. Future Oncol. 2013, 9, 219–233. [Google Scholar] [CrossRef] [PubMed]
- Curtin, N.J. DNA repair dysregulation from cancer driver to therapeutic target. Nat. Rev. Cancer 2012, 12, 801–817. [Google Scholar] [CrossRef] [PubMed]
- Deschner, E.E.; Gray, L.H. Influence of oxygen tension on X-ray-induced chromosomal damage in Ehrlich ascites tumor cells irradiated in vitro and in vivo. Radiat. Res. 1959, 11, 115–146. [Google Scholar] [CrossRef] [PubMed]
- Dewey, D.L. Effect of oxygen and nitric oxide on the radio-sensitivity of human cells in tissue culture. Nature 1960, 186, 780–782. [Google Scholar] [CrossRef] [PubMed]
- Gray, L.H. The initiation and development of cellular damage by ionizing radiations; the thirty-second Silvanus Thompson Memorial Lecture. Br. J. Radiol. 1953, 26, 609–618. [Google Scholar] [CrossRef] [PubMed]
- Brown, J.M. Tumor hypoxia in cancer therapy. Methods Enzymol. 2007, 435, 297–321. [Google Scholar] [CrossRef] [PubMed]
- Eriksson, D.; Stigbrand, T. Radiation-induced cell death mechanisms. Tumour Biol. 2010, 31, 363–372. [Google Scholar] [CrossRef] [PubMed]
- Hwang, H.S.; Davis, T.W.; Houghton, J.A.; Kinsella, T.J. Radiosensitivity of thymidylate synthase-deficient human tumor cells is affected by progression through the G1 restriction point into S-phase: Implications for fluoropyrimidine radiosensitization. Cancer Res. 2000, 60, 92–100. [Google Scholar] [PubMed]
- Pawlik, T.M.; Keyomarsi, K. Role of cell cycle in mediating sensitivity to radiotherapy. Int. J. Radiat. Oncol. Biol. Phys. 2004, 59, 928–942. [Google Scholar] [CrossRef] [PubMed]
- Holahan, E.V.; Stuart, P.K.; Dewey, W.C. Enhancement of survival of CHO cells by acidic pH after x irradiation. Radiat. Res. 1982, 89, 433–435. [Google Scholar] [CrossRef] [PubMed]
- Rottinger, E.M.; Mendonca, M.; Gerweck, L.E. Modification of pH induced cellular inactivation by irradiation-glial cells. Int. J. Radiat. Oncol. Biol. Phys. 1980, 6, 1659–1662. [Google Scholar] [CrossRef]
- Freeman, M.L.; Holahan, E.V.; Highfield, D.P.; Raaphorst, G.P.; Spiro, I.J.; Dewey, W.C. The effect of pH on hyperthermic and X-ray induced cell killing. Int. J. Radiat. Oncol. Biol. Phys. 1981, 7, 211–216. [Google Scholar] [CrossRef]
- Freeman, M.L.; Sierra, E. An acidic extracellular environment reduces the fixation of radiation damage. Radiat. Res. 1984, 97, 154–161. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.S.; Park, H.J.; Lyons, J.C.; Griffin, R.J.; Auger, E.A.; Song, C.W. Radiation-induced apoptosis in different pH environments in vitro. Int. J. Radiat. Oncol. Biol. Phys. 1997, 38, 1079–1087. [Google Scholar] [CrossRef]
- Ojeda, F.; Skardova, I.; Guarda, M.I.; Maldonado, C.; Folch, H. Radiation-induced apoptosis in thymocytes: pH sensitization. J. Biosci. 1996, 51, 432–434. [Google Scholar]
- Park, H.J.; Lee, S.H.; Chung, H.; Rhee, Y.H.; Lim, B.U.; Ha, S.W.; Griffin, R.J.; Lee, H.S.; Song, C.W.; Choi, E.K. Influence of environmental pH on G2-phase arrest caused by ionizing radiation. Radiat. Res. 2003, 159, 86–93. [Google Scholar] [CrossRef]
- Koukourakis, M.I.; Bentzen, S.M.; Giatromanolaki, A.; Wilson, G.D.; Daley, F.M.; Saunders, M.I.; Dische, S.; Sivridis, E.; Harris, A.L. Endogenous markers of two separate hypoxia response pathways (hypoxia inducible factor 2 alpha and carbonic anhydrase 9) are associated with radiotherapy failure in head and neck cancer patients recruited in the CHART randomized trial. J. Clin. Oncol. 2006, 24, 727–735. [Google Scholar] [CrossRef] [PubMed]
- Duivenvoorden, W.C.; Hopmans, S.N.; Gallino, D.; Farrell, T.; Gerdes, C.; Glennie, D.; Lukka, H.; Pinthus, J.H. Inhibition of carbonic anhydrase IX (CA9) sensitizes renal cell carcinoma to ionizing radiation. Oncol. Rep. 2015, 34, 1968–1976. [Google Scholar] [CrossRef] [PubMed]
- Balcer-Kubiczek, E.K. Apoptosis in radiation therapy: A double-edged sword. Exp. Oncol. 2012, 34, 277–285. [Google Scholar] [PubMed]
- Dubois, L.; Peeters, S.; Lieuwes, N.G.; Geusens, N.; Thiry, A.; Wigfield, S.; Carta, F.; McIntyre, A.; Scozzafava, A.; Dogne, J.M.; et al. Specific inhibition of carbonic anhydrase IX activity enhances the in vivo therapeutic effect of tumor irradiation. Radiother. Oncol. 2011, 99, 424–431. [Google Scholar] [CrossRef] [PubMed]
- Dubois, L.; Peeters, S.G.; van Kuijk, S.J.; Yaromina, A.; Lieuwes, N.G.; Sarava, R.; Biemans, R.; Rami, M.; Parvathaneni, N.K.; Vullo, D.; et al. Targeting carbonic anhydrase IX by nitroimidazole based sulfamides enhances the therapeutic effect of tumor irradiation: A new concept of dual targeting drugs. Radiother. Oncol. 2013, 108, 523–528. [Google Scholar] [CrossRef] [PubMed]
- Dent, P.; Yacoub, A.; Contessa, J.; Caron, R.; Amorino, G.; Valerie, K.; Hagan, M.P.; Grant, S.; Schmidt-Ullrich, R. Stress and radiation-induced activation of multiple intracellular signaling pathways. Radiat. Res. 2003, 159, 283–300. [Google Scholar] [CrossRef]
- Brand, T.M.; Iida, M.; Luthar, N.; Starr, M.M.; Huppert, E.J.; Wheeler, D.L. Nuclear EGFR as a molecular target in cancer. Radiother. Oncol. 2013, 108, 370–377. [Google Scholar] [CrossRef] [PubMed]
- Dittmann, K.; Mayer, C.; Kehlbach, R.; Rodemann, H.P. Radiation-induced caveolin-1 associated EGFR internalization is linked with nuclear EGFR transport and activation of DNA-PK. Mol. Cancer 2008, 7, 69. [Google Scholar] [CrossRef] [PubMed]
- Dittmann, K.; Mayer, C.; Fehrenbacher, B.; Schaller, M.; Raju, U.; Milas, L.; Chen, D.J.; Kehlbach, R.; Rodemann, H.P. Radiation-induced epidermal growth factor receptor nuclear import is linked to activation of DNA-dependent protein kinase. J. Biol. Chem. 2005, 280, 31182–31189. [Google Scholar] [CrossRef] [PubMed]
- Dittmann, K.; Mayer, C.; Fehrenbacher, B.; Schaller, M.; Kehlbach, R.; Rodemann, H.P. Nuclear epidermal growth factor receptor modulates cellular radio-sensitivity by regulation of chromatin access. Radiother. Oncol. 2011, 99, 317–322. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.M.; Choi, E.J.; Kim, I.A. microrna-7 increases radiosensitivity of human cancer cells with activated EGFR-associated signaling. Radiother. Oncol. 2011, 101, 171–176. [Google Scholar] [CrossRef] [PubMed]
- Dorai, T.; Sawczuk, I.S.; Pastorek, J.; Wiernik, P.H.; Dutcher, J.P. The role of carbonic anhydrase IX overexpression in kidney cancer. Eur. J. Cancer 2005, 41, 2935–2947. [Google Scholar] [CrossRef] [PubMed]
- Ogawa, K.; Yoshioka, Y.; Isohashi, F.; Seo, Y.; Yoshida, K.; Yamazaki, H. Radiotherapy targeting cancer stem cells: Current views and future perspectives. Anticancer Res. 2013, 33, 747–754. [Google Scholar] [PubMed]
- Pang, L.Y.; Sanders, L.; Argyle, D.J. Epidermal growth factor receptor activity is elevated in glioma cancer stem cells and is required to maintain chemotherapy and radiation resistance. Oncotarget 2017, 8, 72494–72512. [Google Scholar] [CrossRef] [PubMed]
- Higgins, G.S.; Krause, M.; McKenna, W.G.; Baumann, M. Personalized radiation oncology: Epidermal growth factor and other receptor tyrosine kinase inhibitors. Recent Results Cancer Res. 2016, 198, 107–122. [Google Scholar] [CrossRef] [PubMed]
- Baumann, M.; Krause, M.; Dikomey, E.; Dittmann, K.; Dorr, W.; Kasten-Pisula, U.; Rodemann, H.P. EGFR-targeted anti-cancer drugs in radiotherapy: Preclinical evaluation of mechanisms. Radiother. Oncol. 2007, 83, 238–248. [Google Scholar] [CrossRef] [PubMed]
- Skvortsova, I.; Skvortsov, S.; Stasyk, T.; Raju, U.; Popper, B.A.; Schiestl, B.; von Guggenberg, E.; Neher, A.; Bonn, G.K.; Huber, L.A.; et al. Intracellular signaling pathways regulating radioresistance of human prostate carcinoma cells. Proteomics 2008, 8, 4521–4533. [Google Scholar] [CrossRef] [PubMed]
- Miyaguchi, M.; Takeuchi, T.; Morimoto, K.; Kubo, T. Correlation of epidermal growth factor receptor and radiosensitivity in human maxillary carcinoma cell lines. Acta Otolaryngol. 1998, 118, 428–431. [Google Scholar] [PubMed]
- Sheridan, M.T.; O’Dwyer, T.; Seymour, C.B.; Mothersill, C.E. Potential indicators of radiosensitivity in squamous cell carcinoma of the head and neck. Radiat. Oncol. Investig. 1997, 5, 180–186. [Google Scholar] [CrossRef]
- Nyati, M.K.; Morgan, M.A.; Feng, F.Y.; Lawrence, T.S. Integration of EGFR inhibitors with radiochemotherapy. Nat. Rev. Cancer 2006, 6, 876–885. [Google Scholar] [CrossRef] [PubMed]
- Thariat, J.; Milas, L.; Ang, K.K. Integrating radiotherapy with epidermal growth factor receptor antagonists and other molecular therapeutics for the treatment of head and neck cancer. Int. J. Radiat. Oncol. Biol. Phys. 2007, 69, 974–984. [Google Scholar] [CrossRef] [PubMed]
- Perkins, N.D. The diverse and complex roles of NF-B subunits in cancer. Nat. Rev. Cancer 2012, 12, 121–132. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Fidler, I.J. Acidic pH-induced elevation in interleukin 8 expression by human ovarian carcinoma cells. Cancer Res. 2000, 60, 4610–4616. [Google Scholar] [PubMed]
- Fukumura, D.; Xu, L.; Chen, Y.; Gohongi, T.; Seed, B.; Jain, R.K. Hypoxia and acidosis independently up-regulate vascular endothelial growth factor transcription in brain tumors in vivo. Cancer Res. 2001, 61, 6020–6024. [Google Scholar] [PubMed]
- Dunn, S.M.; Coles, L.S.; Lang, R.K.; Gerondakis, S.; Vadas, M.A.; Shannon, M.F. Requirement for nuclear factor (NF)-κB p65 and NF-interleukin-6 binding elements in the tumor necrosis factor response region of the granulocyte colony-stimulating factor promoter. Blood 1994, 83, 2469–2479. [Google Scholar] [PubMed]
- Bellocq, A.; Suberville, S.; Philippe, C.; Bertrand, F.; Perez, J.; Fouqueray, B.; Cherqui, G.; Baud, L. Low environmental pH is responsible for the induction of nitric-oxide synthase in macrophages. Evidence for involvement of nuclear factor-κB activation. J. Biol. Chem. 1998, 273, 5086–5092. [Google Scholar] [CrossRef] [PubMed]
- Kim, B.Y.; Kim, K.A.; Kwon, O.; Kim, S.O.; Kim, M.S.; Kim, B.S.; Oh, W.K.; Kim, G.D.; Jung, M.; Ahn, J.S. NF-κB inhibition radiosensitizes Ki-Ras-transformed cells to ionizing radiation. Carcinogenesis 2005, 26, 1395–1403. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, K.M.; Dong, S.; Fan, M.; Li, J.J. Nuclear Factor-κB p65 inhibits mitogen-activated protein kinase signaling pathway in radioresistant breast cancer cells. Mol. Cancer Res. 2006, 4, 945–955. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, K.M.; Li, J.J. NF-κB-mediated adaptive resistance to ionizing radiation. Free Radic. Biol. Med. 2008, 44, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Cataldi, A.; Rapino, M.; Centurione, L.; Sabatini, N.; Grifone, G.; Garaci, F.; Rana, R. NF-κB activation plays an antiapoptotic role in human leukemic K562 cells exposed to ionizing radiation. J. Cell. Biochem. 2003, 89, 956–963. [Google Scholar] [CrossRef] [PubMed]
- Guo, G.; Yan-Sanders, Y.; Lyn-Cook, B.D.; Wang, T.; Tamae, D.; Ogi, J.; Khaletskiy, A.; Li, Z.; Weydert, C.; Longmate, J.A.; et al. Manganese superoxide dismutase-mediated gene expression in radiation-induced adaptive responses. Mol. Cell. Biol. 2003, 23, 2362–2378. [Google Scholar] [CrossRef] [PubMed]
- Shaw, L.M. Integrin function in breast carcinoma progression. J. Mammary Gland Biol. Neoplasia 1999, 4, 367–376. [Google Scholar] [CrossRef] [PubMed]
- Sethi, T.; Rintoul, R.C.; Moore, S.M.; MacKinnon, A.C.; Salter, D.; Choo, C.; Chilvers, E.R.; Dransfield, I.; Donnelly, S.C.; Strieter, R.; et al. Extracellular matrix proteins protect small cell lung cancer cells against apoptosis: A mechanism for small cell lung cancer growth and drug resistance in vivo. Nat. Med. 1999, 5, 662–668. [Google Scholar] [CrossRef] [PubMed]
- Aoudjit, F.; Vuori, K. Integrin signaling inhibits paclitaxel-induced apoptosis in breast cancer cells. Oncogene 2001, 20, 4995–5004. [Google Scholar] [CrossRef] [PubMed]
- Nam, J.M.; Onodera, Y.; Bissell, M.J.; Park, C.C. Breast cancer cells in three dimensional culture display an enhanced radioresponse after coordinate targeting of integrin 51 and fibronectin. Cancer Res. 2010, 70, 5238–5248. [Google Scholar] [CrossRef] [PubMed]
- Eke, I.; Deuse, Y.; Hehlgans, S.; Gurtner, K.; Krause, M.; Baumann, M.; Shevchenko, A.; Sandfort, V.; Cordes, N. (1) Integrin/FAK/cortactin signaling is essential for human head and neck cancer resistance to radiotherapy. J. Clin. Investig. 2011, 122, 1529–1540. [Google Scholar] [CrossRef] [PubMed]
- Park, C.C.; Zhang, H.J.; Yao, E.S.; Park, C.J.; Bissell, M.J. β1 integrin inhibition dramatically enhances radiotherapy efficacy in human breast cancer xenografts. Cancer Res. 2008, 68, 4398–4405. [Google Scholar] [CrossRef] [PubMed]
- Cordes, N.; Seidler, J.; Durzok, R.; Geinitz, H.; Brakebusch, C. 1-integrin-mediated signaling essentially contributes to cell survival afterradiation-induced genotoxic injury. Oncogene 2006, 25, 1378–1390. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, K.M.; Zhang, H.; Park, C.C. NF-κB Regulates radioresistance mediated by 1-integrin in three-dimensional culture of breast cancer cells. Cancer Res. 2013, 73, 3737–3748. [Google Scholar] [CrossRef] [PubMed]
- Nam, J.M.; Chung, Y.; Hsu, H.C.; Park, C.C. 1 integrin targeting to enhance radiation therapy. Int. J. Radiat. Biol. 2009, 85, 923–928. [Google Scholar] [CrossRef] [PubMed]
- Chafe, S.C.; Lou, Y.; Sceneay, J.; Vallejo, M.; Hamilton, M.J.; McDonald, P.C.; Bennewith, K.L.; Moller, A.; Dedhar, S. Carbonic anhydrase IX promotes myeloid-derived suppressor cell mobilization and establishment of a metastatic niche by stimulating G-CSF production. Cancer Res. 2015, 75, 996–1008. [Google Scholar] [CrossRef] [PubMed]
- Singh, V.K.; Fatanmi, O.O.; Singh, P.K.; Whitnall, M.H. Role of radiation-induced granulocyte colony-stimulating factor in recovery from whole body gamma-irradiation. Cytokine 2012, 58, 406–414. [Google Scholar] [CrossRef] [PubMed]
- Waddick, K.G.; Song, C.W.; Souza, L.; Uckun, F.M. Comparative analysis of the in vivo radioprotective effects of recombinant granulocyte colony-stimulating factor (G-CSF), recombinant granulocyte-macrophage CSF, and their combination. Blood 1991, 77, 2364–2371. [Google Scholar] [PubMed]
- Yang, X.; Huang, P.; Wang, F.; Xu, Z. Expression of granulocyte colony-stimulating factor receptor in rectal cancer. World J. Gastroenterol. 2014, 20, 1074–1078. [Google Scholar] [CrossRef] [PubMed]
- Studebaker, A.W.; Storci, G.; Werbeck, J.L.; Sansone, P.; Sasser, A.K.; Tavolari, S.; Huang, T.; Chan, M.W.; Marini, F.C.; Rosol, T.J.; et al. Fibroblasts isolated from common sites of breast cancer metastasis enhance cancer cell growth rates and invasiveness in an interleukin-6-dependent manner. Cancer Res. 2008, 68, 9087–9095. [Google Scholar] [CrossRef] [PubMed]
- Schoppmann, S.F.; Jesch, B.; Friedrich, J.; Jomrich, G.; Maroske, F.; Birner, P. Phosphorylation of signal transducer and activator of transcription 3 (STAT3) correlates with Her-2 status, carbonic anhydrase 9 expression and prognosis in esophageal cancer. Clin. Exp. Metastasis 2012, 29, 615–624. [Google Scholar] [CrossRef] [PubMed]
- Bonner, J.A.; Trummell, H.Q.; Willey, C.D.; Plants, B.A.; Raisch, K.P. Inhibition of STAT-3 results in radiosensitization of human squamous cell carcinoma. Radiother. Oncol. 2009, 92, 339–344. [Google Scholar] [CrossRef]
- Lau, J.; Ilkhanizadeh, S.; Wang, S.; Miroshnikova, Y.A.; Salvatierra, N.A.; Wong, R.A.; Schmidt, C.; Weaver, V.M.; Weiss, W.A.; Persson, A.I. See comment in PubMed commons below STAT3 blockade inhibits radiation-induced malignant progression in glioma. Cancer Res. 2015, 75, 4302–4311. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Tang, W.; Marquez, R.T.; Li, K.; Highfill, C.A.; He, F.; Lian, J.; Lin, J.; Fuchs, J.R.; Ji, M.; et al. Overcoming chemo/radio-resistance of pancreatic cancer by inhibiting STAT3 signaling. Oncotarget 2016, 7, 11708–11723. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.T.; Chen, M.F.; Chen, W.C.; Hsieh, C.C. The role of IL-6 in the radiation response of prostate cancer. Radiat. Oncol. 2013, 8, 159. [Google Scholar] [CrossRef] [PubMed]
- Matsuoka, Y.; Nakayama, H.; Yoshida, R.; Hirosue, A.; Nagata, M.; Tanaka, T.; Kawahara, K.; Sakata, J.; Arita, H.; Nakashima, H.; et al. IL-6 controls resistance to radiation by suppressing oxidative stress via the Nrf2-antioxidant pathway in oral squamous cell carcinoma. Br. J. Cancer 2016, 115, 1234–1244. [Google Scholar] [CrossRef] [PubMed]
- Karin, M.; Greten, F.R. NF-κB: Linking inflammation and immunity to cancer development and progression. Nat. Rev. Immunol. 2005, 5, 749–759. [Google Scholar] [CrossRef] [PubMed]
- Sattler, U.G.; Meyer, S.S.; Quennet, V.; Hoerner, C.; Knoerzer, H.; Fabian, C.; Yaromina, A.; Zips, D.; Walenta, S.; Baumann, M.; et al. Glycolytic metabolism and tumour response to fractionated irradiation. Radiother. Oncol. 2010, 94, 102–109. [Google Scholar] [CrossRef] [PubMed]
- Hirschhaeuser, F.; Sattler, U.G.; Mueller-Klieser, W. Lactate: A metabolic key player in cancer. Cancer Res. 2011, 71, 6921–6925. [Google Scholar] [CrossRef] [PubMed]
- Groussard, C.; Morel, I.; Chevanne, M.; Monnier, M.; Cillard, J.; Delamarche, A. Free radical scavenging and antioxidant effects of lactate ion: An in vitro study. J. Appl. Physiol. 2000, 89, 169–175. [Google Scholar] [CrossRef] [PubMed]
- Halestrap, A.P. The monocarboxylate transporter family—Structure and functional characterization. IUBMB Life 2012, 64, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Garcia, C.K.; Goldstein, J.L.; Pathak, R.K.; Anderson, R.G.; Brown, M.S. Molecular characterization of a membrane transporter for lactate, pyruvate, and other monocarboxylates: Implications for the Cori cycle. Cell 1994, 76, 865–873. [Google Scholar] [CrossRef]
- Dimmer, K.S.; Friedrich, B.; Lang, F.; Deitmer, J.W.; Bröer, S. The low-affinity monocarboxylate transporter MCT4 is adapted to the export of lactate in highly glycolytic cells. Biochem. J. 2000, 350, 219–227. [Google Scholar] [CrossRef] [PubMed]
- Pertega-Gomes, N.; Vizcain, J.R.; Miranda-Goncalves, V.; Pinheiro, C.; Silva, J.; Pereira, H.; Monteiro, P.; Henrique, R.M.; Reis, R.M.; Lopes, C.; et al. Monocarboxylate transporter 4 (MCT4) and CD147 overexpression is associated with poor prognosis in prostate cancer. BMC Cancer 2011, 11, 312. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doyen, J.; Trastour, C.; Ettore, F.; Peyrottes, I.; Toussant, N.; Gal, J.; Ilc, K.; Roux, D.; Parks, S.K.; Ferrero, J.M.; et al. Expression of hypoxia-inducible monocarboxylate transporter MCT4 is increased in triple negative breast cancer and correlates independently with clinical outcome. Biochem. Biophys. Res. Commun. 2014, 451, 54–61. [Google Scholar] [CrossRef] [PubMed]
- Pinheiro, C.; Longatto-Filho, A.; Scapulatempo, C.; Ferreira, L.; Martins, S.; Pellerin, L.; Rodrigues, M.; Alves, V.A.; Schmitt, F.; Baltazar, F. Increased expression of monocarboxylate transporters 1, 2, and 4 in colorectal carcinomas. Virchows Arch. 2008, 452, 139–146. [Google Scholar] [CrossRef] [PubMed]
- Sonveaux, P.; Vegran, F.; Schroeder, T.; Wergin, M.C.; Verrax, J.; Rabbani, Z.N.; De Saedeleer, C.J.; Kennedy, K.M.; Diepart, C.; Jordan, B.F.; et al. Targeting lactate-fueled respiration selectively kills hypoxic tumor cells in mice. J. Clin. Investig. 2008, 118, 3930–3942. [Google Scholar] [CrossRef] [PubMed]
- Bola, B.M.; Chadwick, A.L.; Michopoulos, F.; Blount, K.G.; Telfer, B.A.; Williams, K.J.; Smith, P.D.; Critchlow, S.E.; Stratford, I.J. Inhibition of monocarboxylate transporter-1 (MCT1) by AZD3965 enhances radiosensitivity by reducing lactate transport. Mol. Cancer Ther. 2014, 13, 2805–2816. [Google Scholar] [CrossRef] [PubMed]
- Le Floch, R.; Chiche, J.; Marchiq, I.; Naiken, T.; Ilc, K.; Murray, C.M.; Critchlow, S.E.; Roux, D.; Simon, M.P.; Pouysségur, J. CD147 subunit of lactate/H+ symporters MCT1 and hypoxia-inducible MCT4 is critical for energetics and growth of glycolytic tumors. Proc. Natl. Acad. Sci. USA 2011, 108, 16663–16668. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Li, Y.; Dang, Y.Z.; Gao, H.X.; Jiang, J.L.; Chen, Z.N. HAB18G/CD147 promotes radioresistance in hepatocellular carcinoma cells: A potential role for integrin 1 signaling. Mol. Cancer Ther. 2015, 14, 553–563. [Google Scholar] [CrossRef] [PubMed]
- Ju, X.Z.; Yang, J.M.; Zhou, X.Y.; Li, Z.T.; Wu, X.H. EMMPRIN expression as a prognostic factor in radiotherapy of cervical cancer. Clin. Cancer Res. 2008, 14, 494–501. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.Q.; Chen, X.; Xie, X.X.; Zhou, Q.; Li, K.; Li, S.; Shen, L.F.; Su, J. Co-expression of CD147 and GLUT-1 indicates radiation resistance and poor prognosis in cervical squamous cell carcinoma. Int. J. Clin. Exp. Pathol. 2014, 7, 1651–1666. [Google Scholar] [PubMed]
- Becker, H.M.; Deitmer, J.W. Nonenzymatic proton handling by carbonic anhydrase II during H+-lactate cotransport via monocarboxylate transporter 1. J. Biol. Chem. 2008, 283, 21655–21667. [Google Scholar] [CrossRef] [PubMed]
- Stridh, M.; Alt, M.D.; Wittmann, S.; Heidtmann, H.; Aggarwal, M.; Riederer, B.; Seidler, U.; Wennemuth, G.; McKenna, R.; Deitmer, J.W.; et al. Lactate flux in astrocytes is enhanced by a non-catalytic action of carbonic anhydrase II. J. Physiol. 2012, 590, 2333–2351. [Google Scholar] [CrossRef] [PubMed]
- Panisova, E.; Kery, M.; Sedlakova, O.; Brisson, L.; Debreova, M.; Sboarina, M.; Sonveaux, P.; Pastorekova, S.; Svastova, E. Lactate stimulates CA IX expression in normoxic cancer cells. Oncotarget 2017, 8, 77819–77838. [Google Scholar] [CrossRef] [PubMed]
- Axelson, H.; Fredlund, E.; Ovenberger, M.; Landberg, G.; Pahlman, S. Hypoxia-induced dedifferentiation of tumor cells—A mechanism behind heterogeneity and aggressiveness of solid tumors. Semin. Cell Dev. Biol. 2005, 16, 554–563. [Google Scholar] [CrossRef] [PubMed]
- Sansone, P.; Storci, G.; Giovannini, C.; Pandolfi, S.; Pianetti, S.; Taffurelli, M.; Santini, D.; Ceccarelli, C.; Chieco, P.; Bonafé, M. p66Shc/Notch-3 interplay controls self-renewal and hypoxia survival in human stem/progenitor cells of the mammary gland expanded in vitro as mammospheres. Stem Cells 2007, 25, 807–815. [Google Scholar] [CrossRef] [PubMed]
- Storci, G.; Sansone, P.; Trere, D.; Tavolari, S.; Taffurelli, M.; Ceccarelli, C.; Guarnieri, T.; Paterini, P.; Pariali, M.; Montanaro, L.; et al. The basal-like breast carcinoma phenotype is regulated by SLUG gene expression. J. Pathol. 2008, 214, 25–37. [Google Scholar] [CrossRef] [PubMed]
- Currie, M.J.; Beardsley, B.E.; Harris, G.C.; Gunningham, S.P.; Dachs, G.U.; Dijkstra, B.; Morrin, H.R.; Wells, J.E.; Robinson, B.A. Immunohistochemical analysis of cancer stem cell markers in invasive breast carcinoma and associated ductal carcinoma in situ: Relationships with markers of tumor hypoxia and microvascularity. Hum. Pathol. 2013, 44, 402–411. [Google Scholar] [CrossRef] [PubMed]
- Phillips, T.M.; McBride, W.H.; Pajonk, F. The response of CD24−/low/CD44+ breast cancer-initiating cells to radiation. J. Natl. Cancer Inst. 2006, 8, 1777–1785. [Google Scholar] [CrossRef] [PubMed]
- Chang, L.; Graham, P.; Hao, J.; Ni, J.; Deng, J.; Bucci, J.; Malouf, D.; Gillatt, D.; Li, Y. Cancer stem cells and signaling pathways in radioresistance. Oncotarget 2016, 7, 11002–11017. [Google Scholar] [CrossRef] [PubMed]
- Ogawa, K.; Murayama, S.; Mori, M. Predicting the tumour response to radiotherapy using microarray analysis. Oncol. Rep. 2007, 18, 1243–1248. [Google Scholar] [PubMed]
- Woodward, W.A.; Chen, M.S.; Behbod, F.; Alfaro, M.P.; Buchholz, T.A.; Rosen, J.M. WNT/catenin mediates radiation resistance of mouse mammary progenitor cells. Proc. Natl. Acad. Sci. USA 2007, 104, 618–623. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Behbod, F.; Atkinson, R.L.; Landis, M.D.; Kittrell, F.; Edwards, D.; Medina, D.; Tsimelzon, A.; Hilsenbeck, S.; Green, J.E.; et al. Identification of tumor-initiating cells in a p53-null mouse model of breast cancer. Cancer Res. 2008, 68, 4674–4682. [Google Scholar] [CrossRef] [PubMed]
- Desai, A.; Webb, B.; Gerson, S.L. CD133+ cells contribute to radioresistance via altered regulation of DNA repair genes in human lung cancer cells. Radiother. Oncol. 2014, 110, 538–545. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Zhang, F.; Tsai, Y.; Yang, X.; Yang, L.; Duan, S.; Wang, X.; Keng, P.; Lee, S.O. IL-6 signaling promotes DNA repair and prevents apoptosis in CD133+ stem-like cells of lung cancer after radiation. Radiat. Oncol. 2015, 10, 227. [Google Scholar] [CrossRef] [PubMed]
- Hjelmeland, A.B.; Wu, Q.; Heddleston, J.M.; Choudhary, G.S.; MacSwords, J.; Lathia, J.D.; McLendon, R.; Lindner, D.; Sloan, A.; Rich, J.N. Acidic stress promotes a glioma stem cell phenotype. Cell Death Differ. 2011, 18, 829–840. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Outschoorn, U.E.; Prisco, M.; Ertel, A.; Tsirigos, A.; Lin, Z.; Pavlides, S.; Wang, C.; Flomenberg, N.; Knudsen, E.S.; Howell, A.; et al. Ketones and lactate increase cancer cell “stemness,” driving recurrence, metastasis and poor clinical outcome in breast cancer: Achieving personalized medicine via Metabolo-Genomics. Cell Cycle 2011, 10, 1271–1286. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Contribution of CAIX to the movement of glycolytic protons from inside the cytoplasm to the extracellular milieu.
Figure 1.
Contribution of CAIX to the movement of glycolytic protons from inside the cytoplasm to the extracellular milieu.
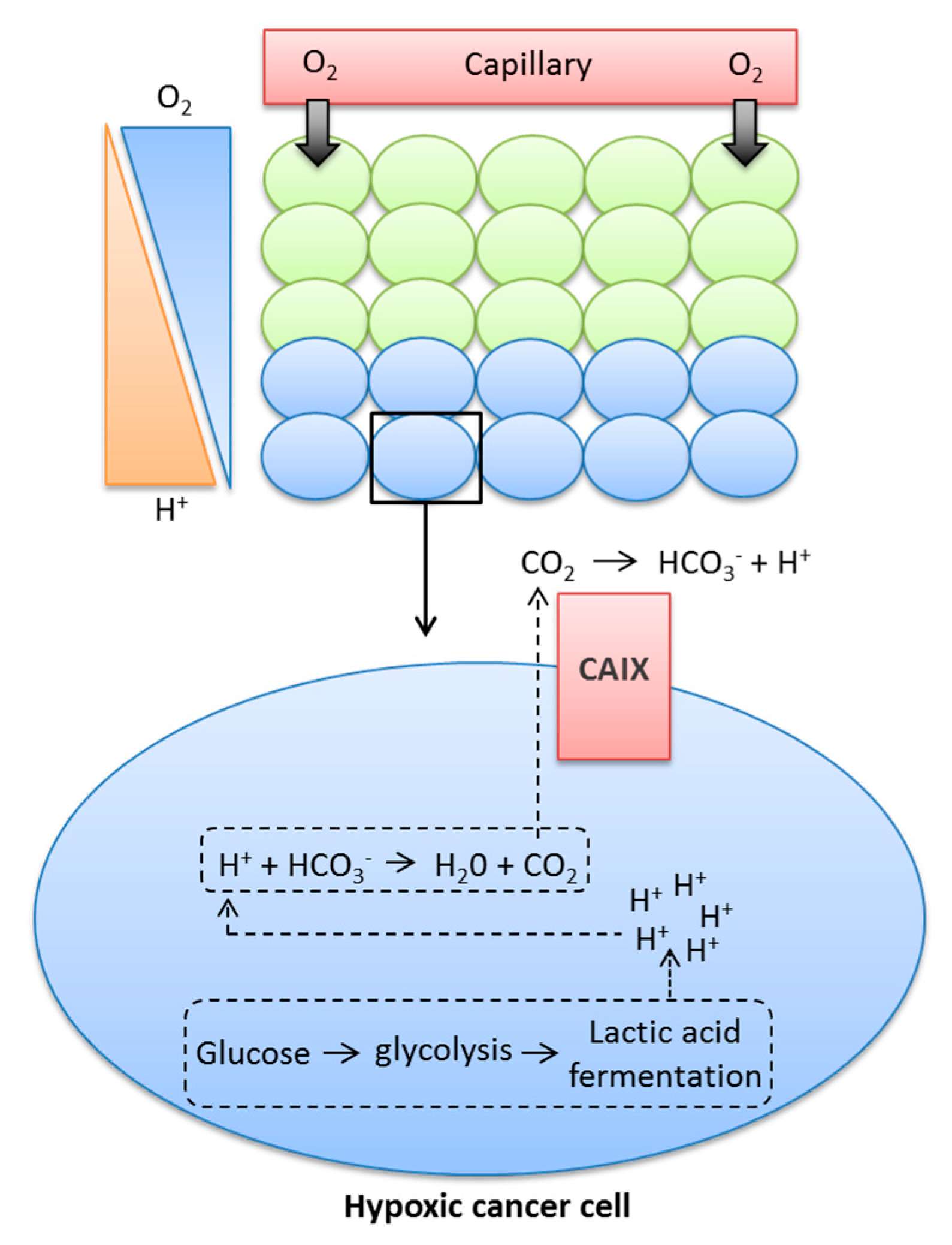
Figure 2.
The identification of hypoxic areas and the expression of CAIX, proliferation, and apoptotic markers in 3D human breast cancer models.
Figure 2.
The identification of hypoxic areas and the expression of CAIX, proliferation, and apoptotic markers in 3D human breast cancer models.
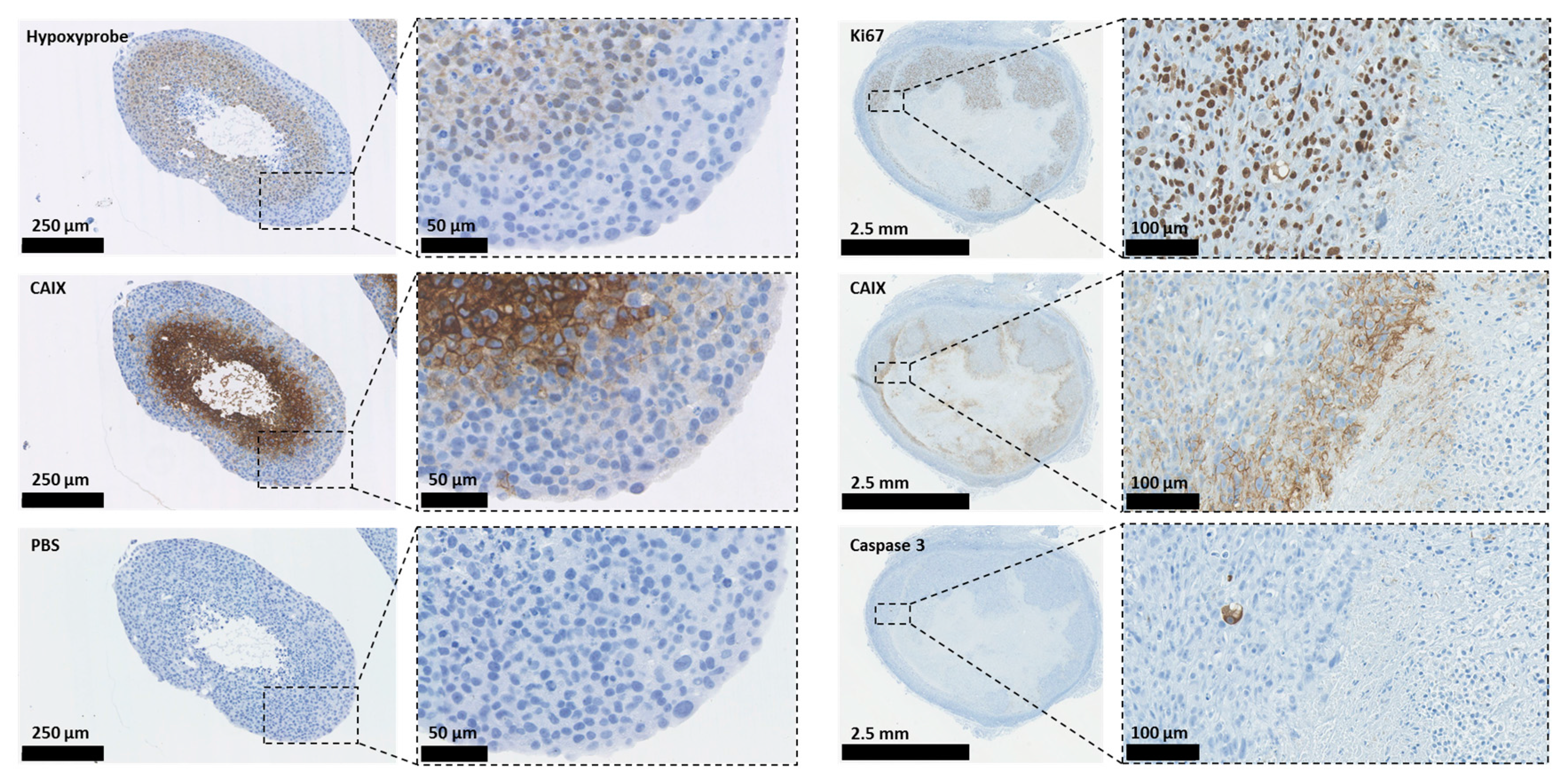
Figure 3.
The CAIX/NF-κB/IL-6 signalling node and radioresistance.

© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Ward, C.; Meehan, J.; Gray, M.; Kunkler, I.H.; Langdon, S.P.; Argyle, D.J. Carbonic Anhydrase IX (CAIX), Cancer, and Radiation Responsiveness. Metabolites 2018, 8, 13. https://doi.org/10.3390/metabo8010013
AMA Style
Ward C, Meehan J, Gray M, Kunkler IH, Langdon SP, Argyle DJ. Carbonic Anhydrase IX (CAIX), Cancer, and Radiation Responsiveness. Metabolites. 2018; 8(1):13. https://doi.org/10.3390/metabo8010013
Chicago/Turabian StyleWard, Carol, James Meehan, Mark Gray, Ian H. Kunkler, Simon P. Langdon, and David J. Argyle. 2018. "Carbonic Anhydrase IX (CAIX), Cancer, and Radiation Responsiveness" Metabolites 8, no. 1: 13. https://doi.org/10.3390/metabo8010013
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.