Data Pre-Processing for Label-Free Multiple Reaction Monitoring (MRM) Experiments

Abstract
:1. Introduction
2. Data Sets
2.1. Data Set 1: Rat Brian Post Synaptic Density
2.2. Data Set 2: Cysteine String Protein
2.3. Data Set 3: S. Pyogenes
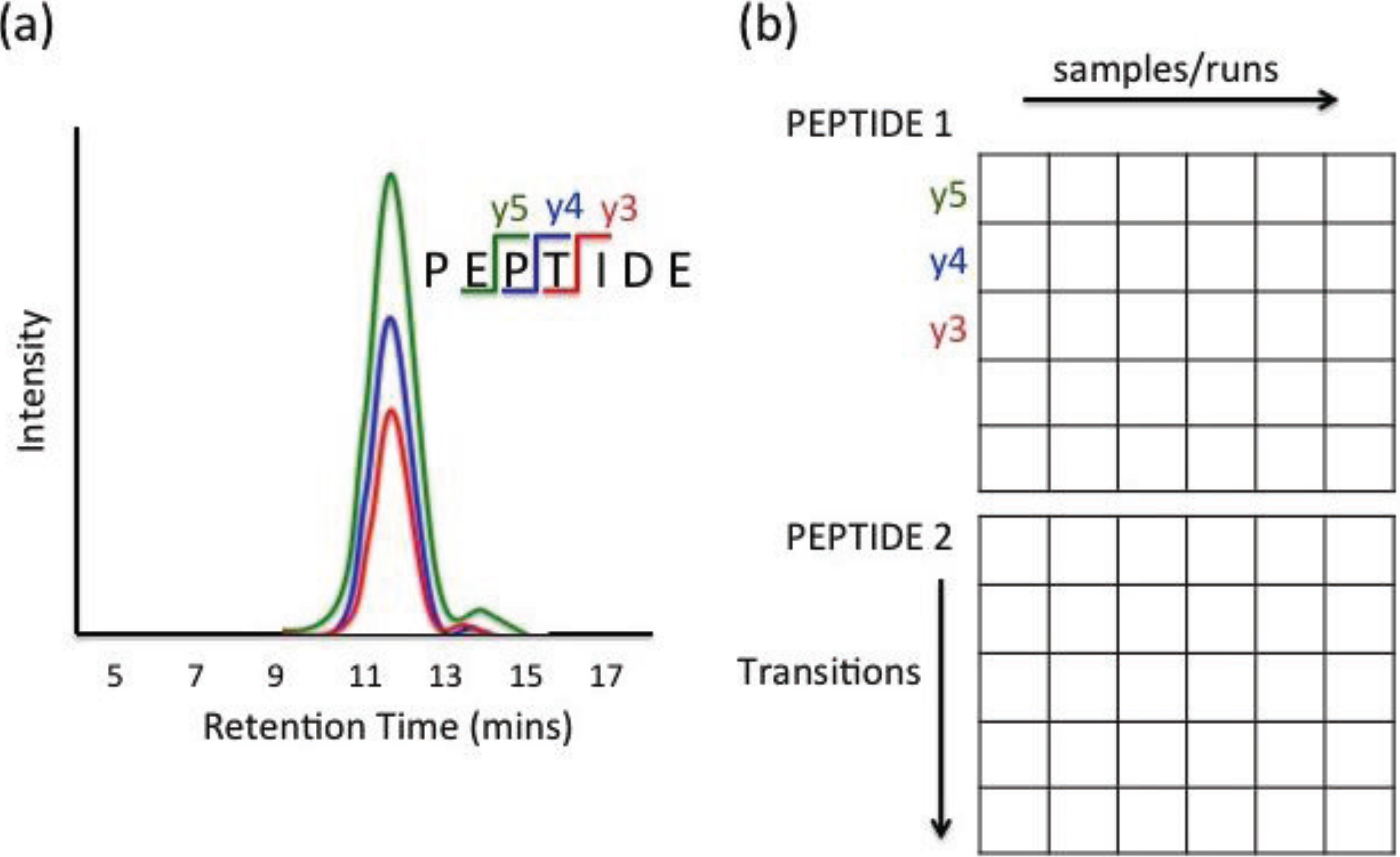
2.4. Data Structure
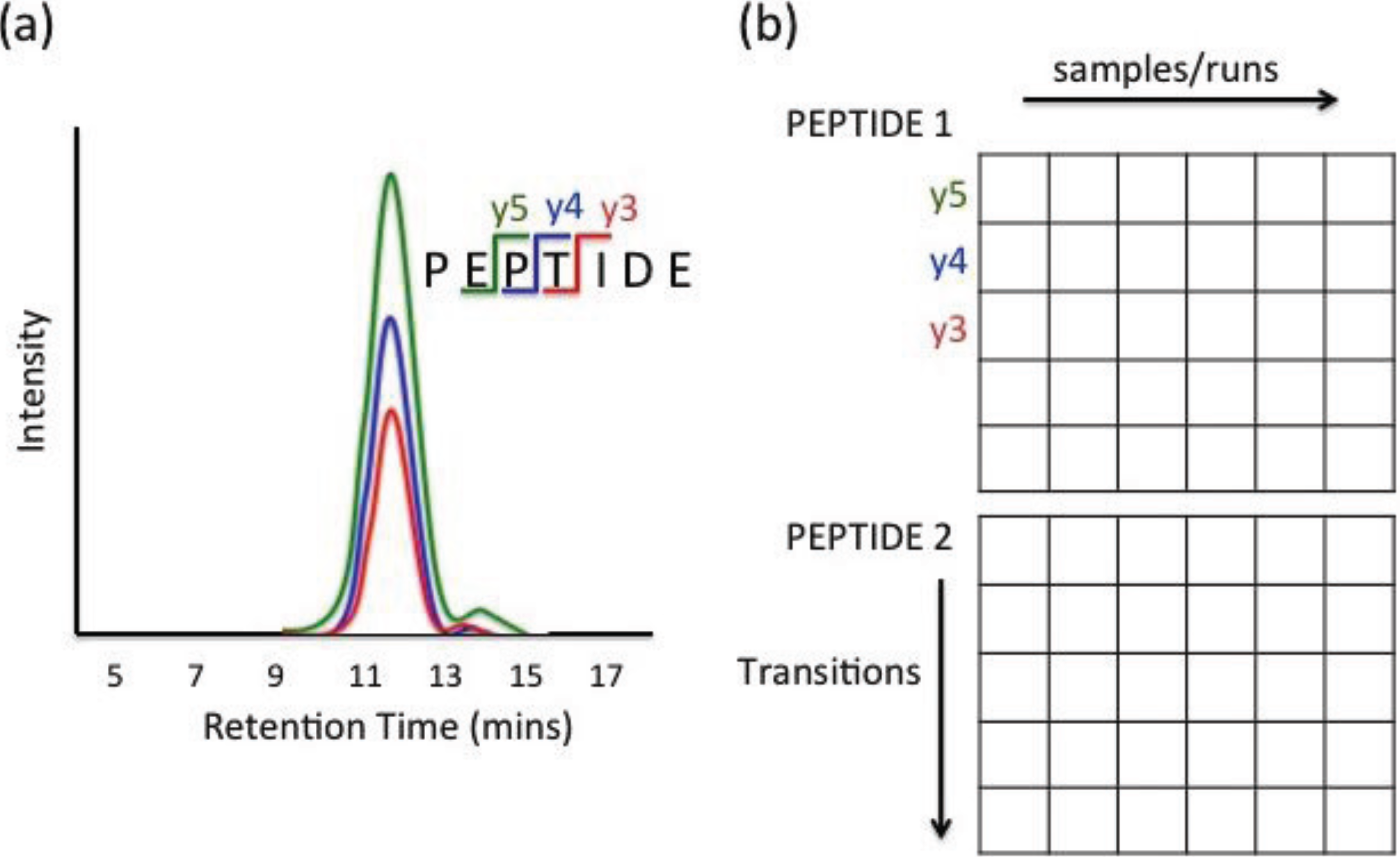
3. Data Pre-Processing
3.1. Quality Assessment
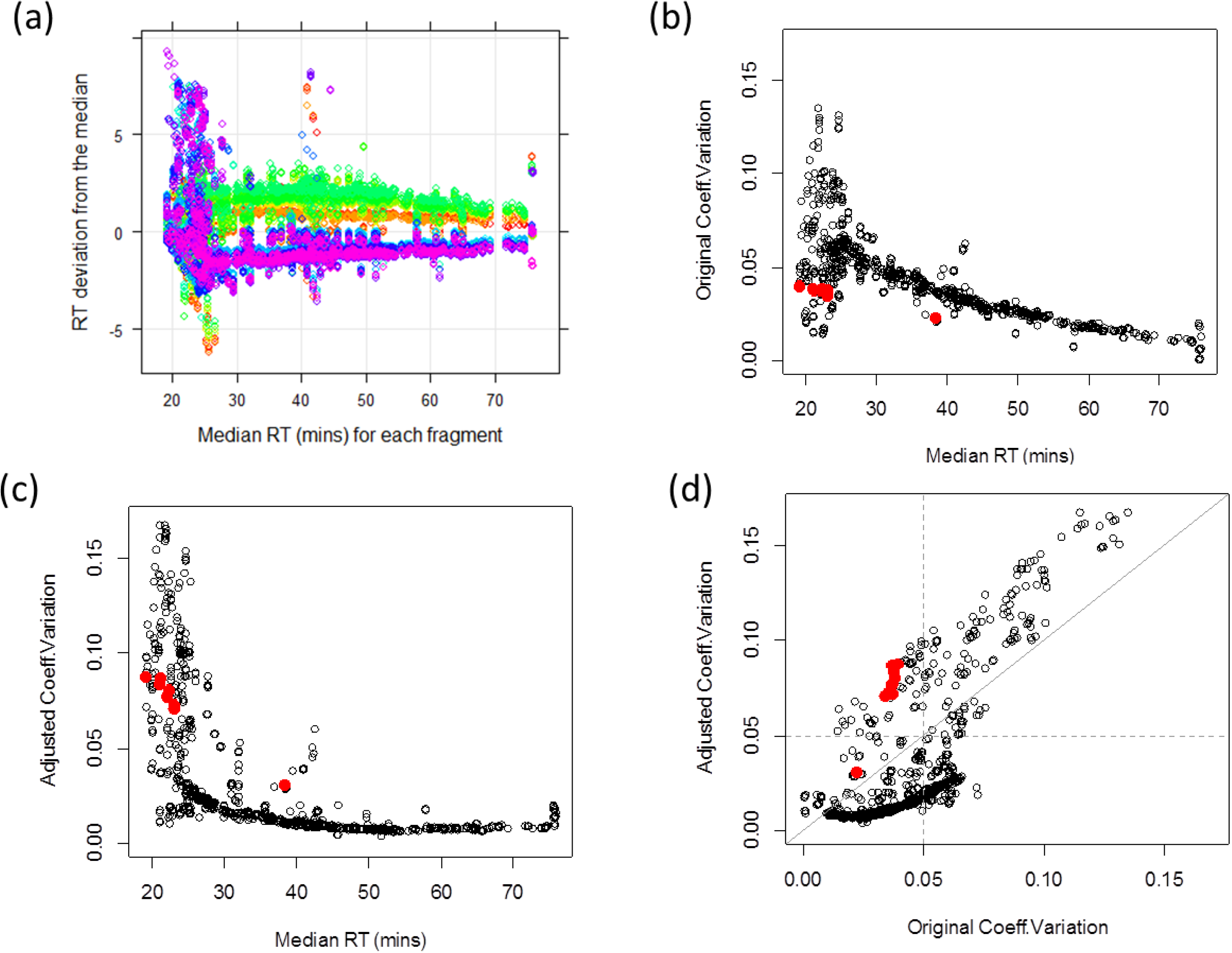
3.1.1. Adjusted Retention Time Deviation and Outlier Detection
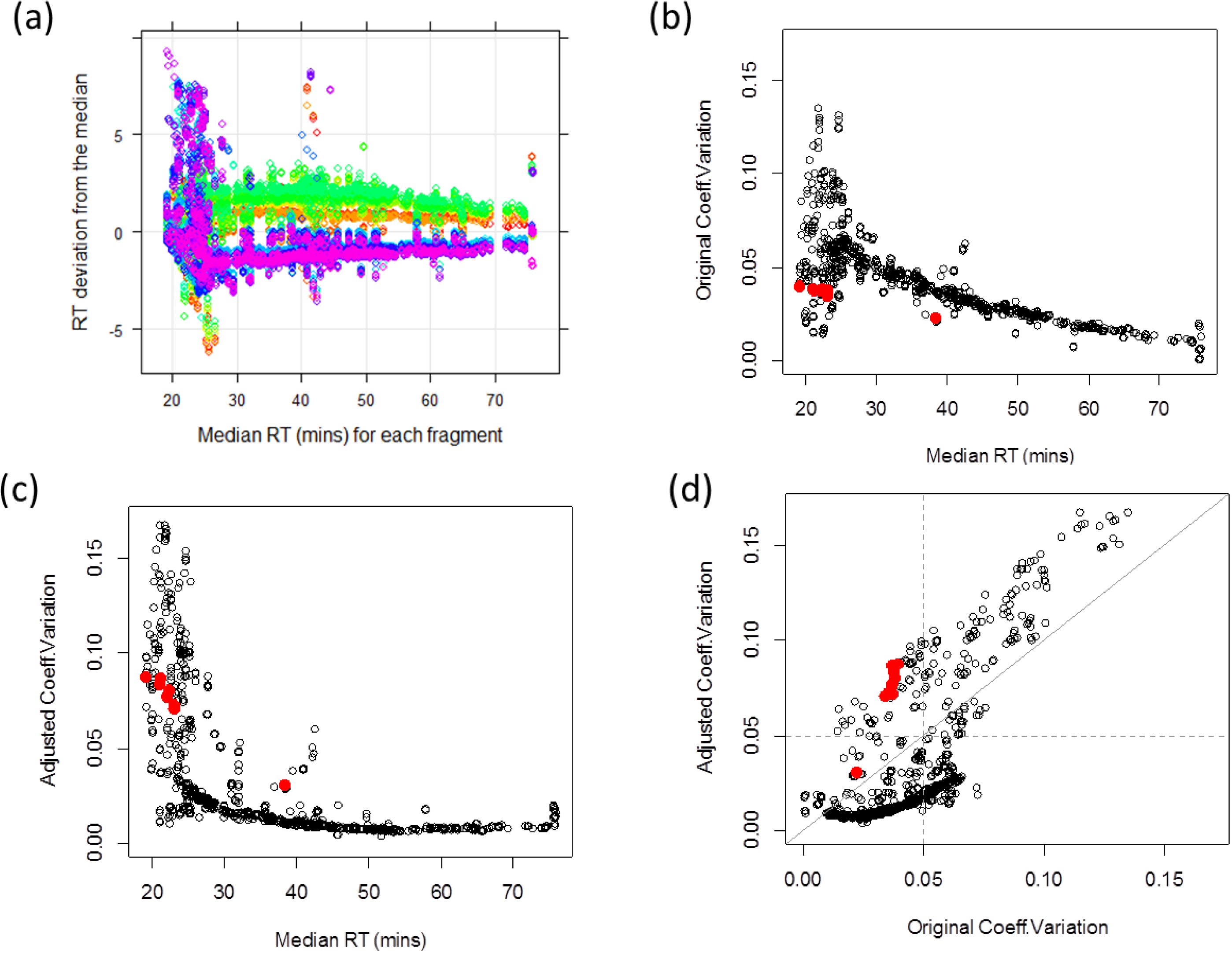
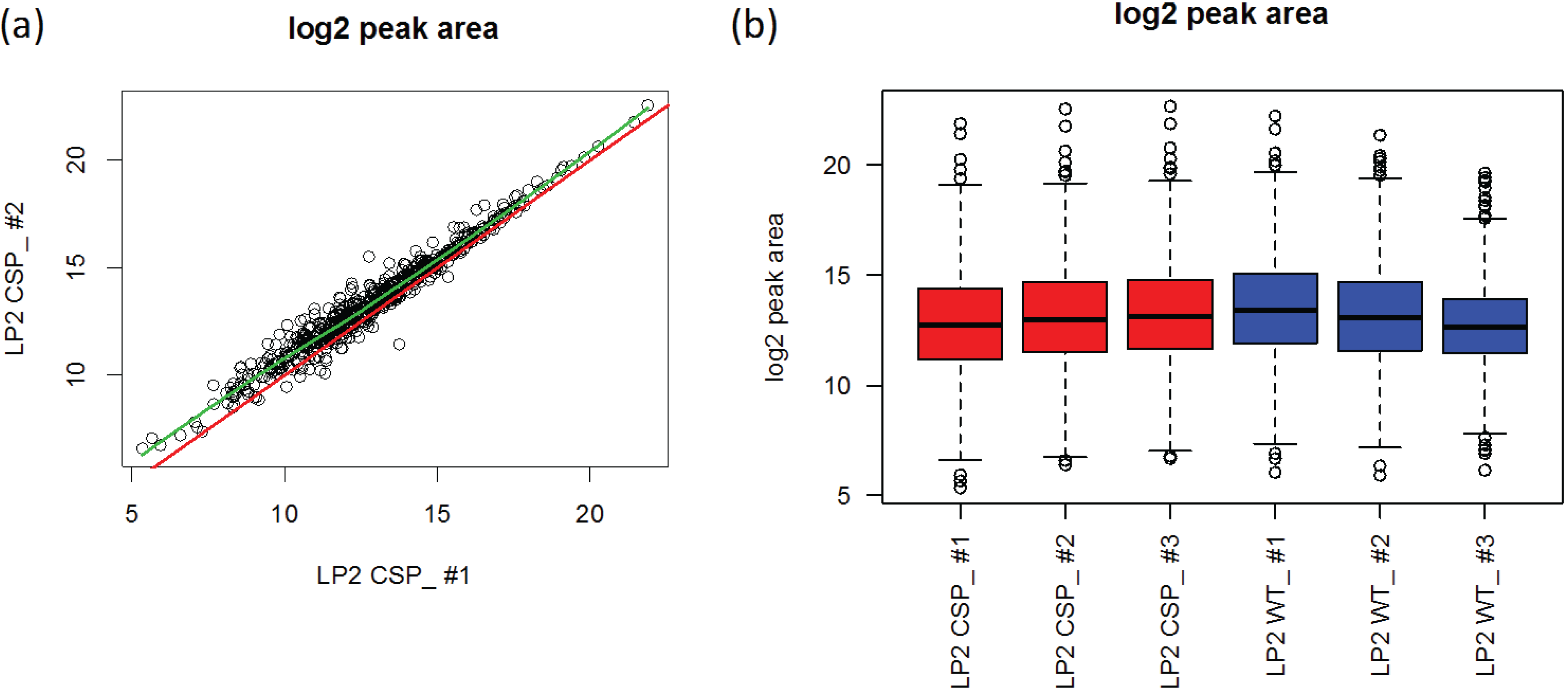
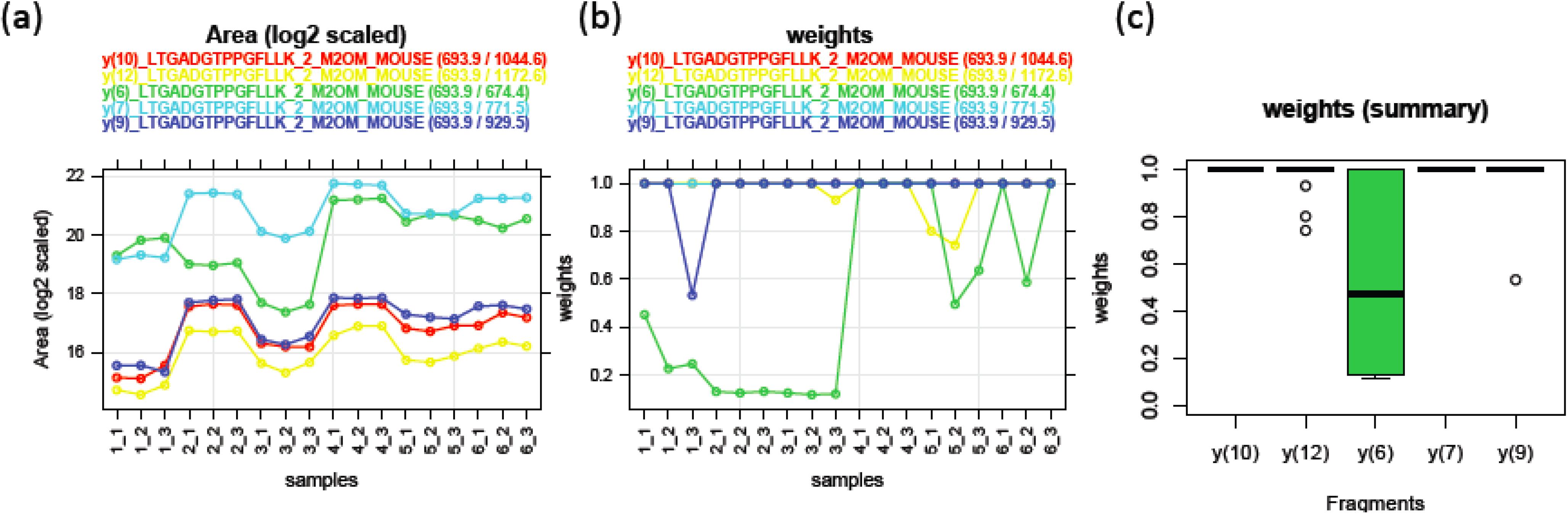
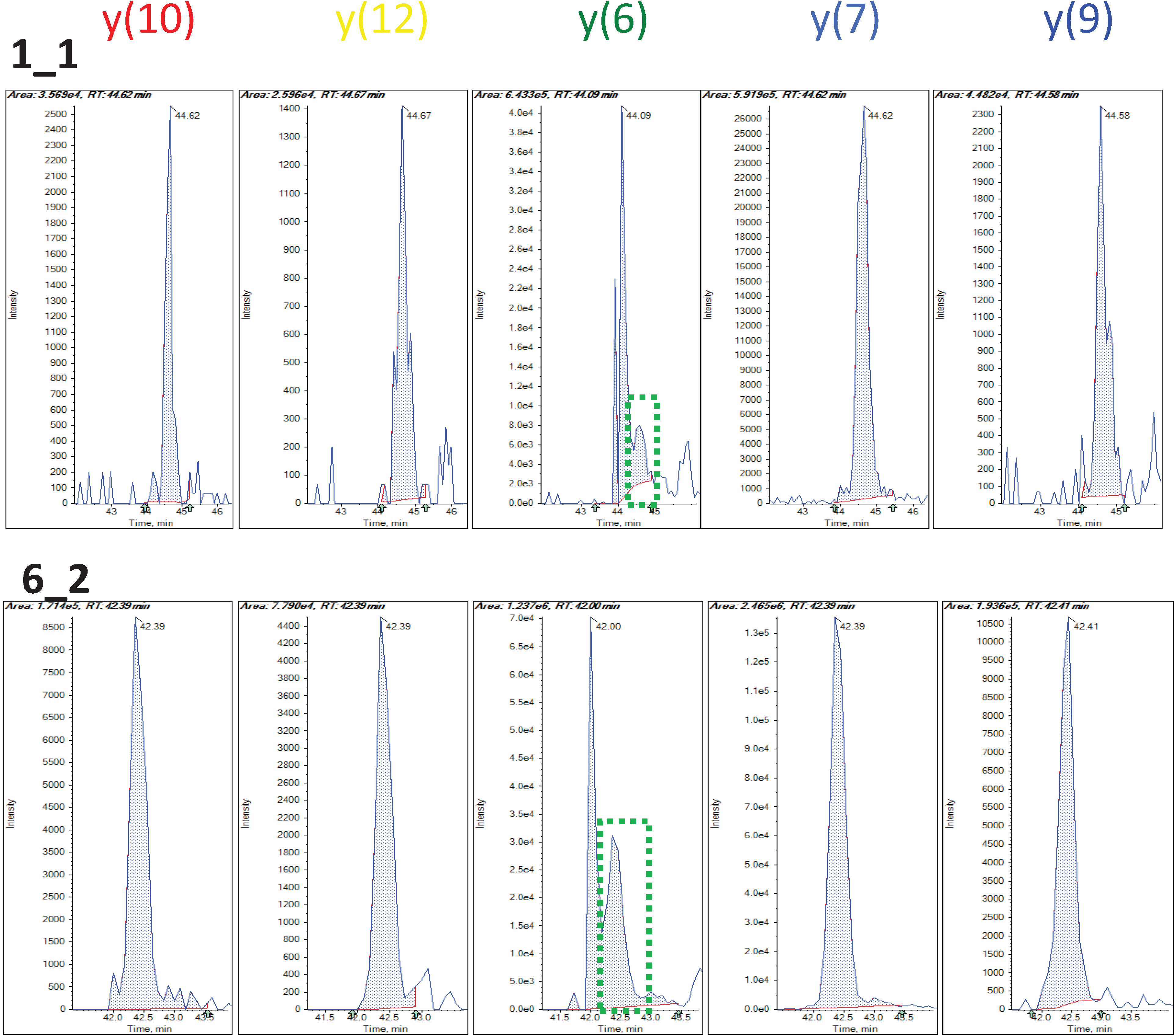
3.1.2. Peptide-Level Intensity Estimation via Robust Linear Regression and Measure of Outlying Peak Area
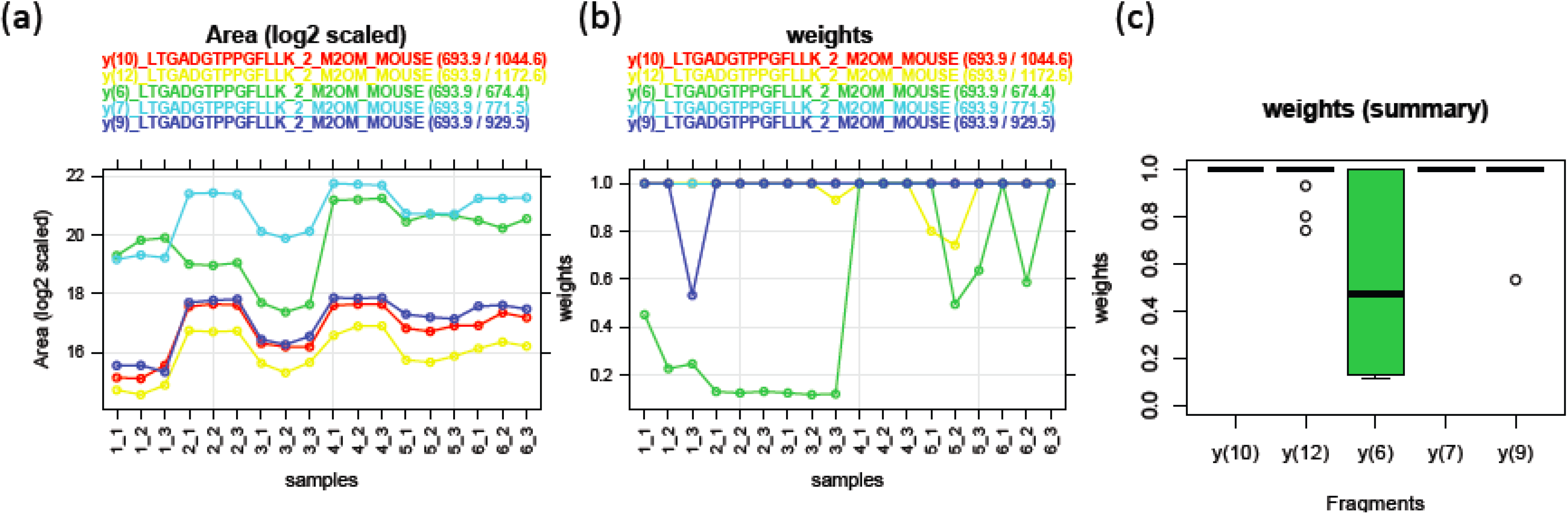
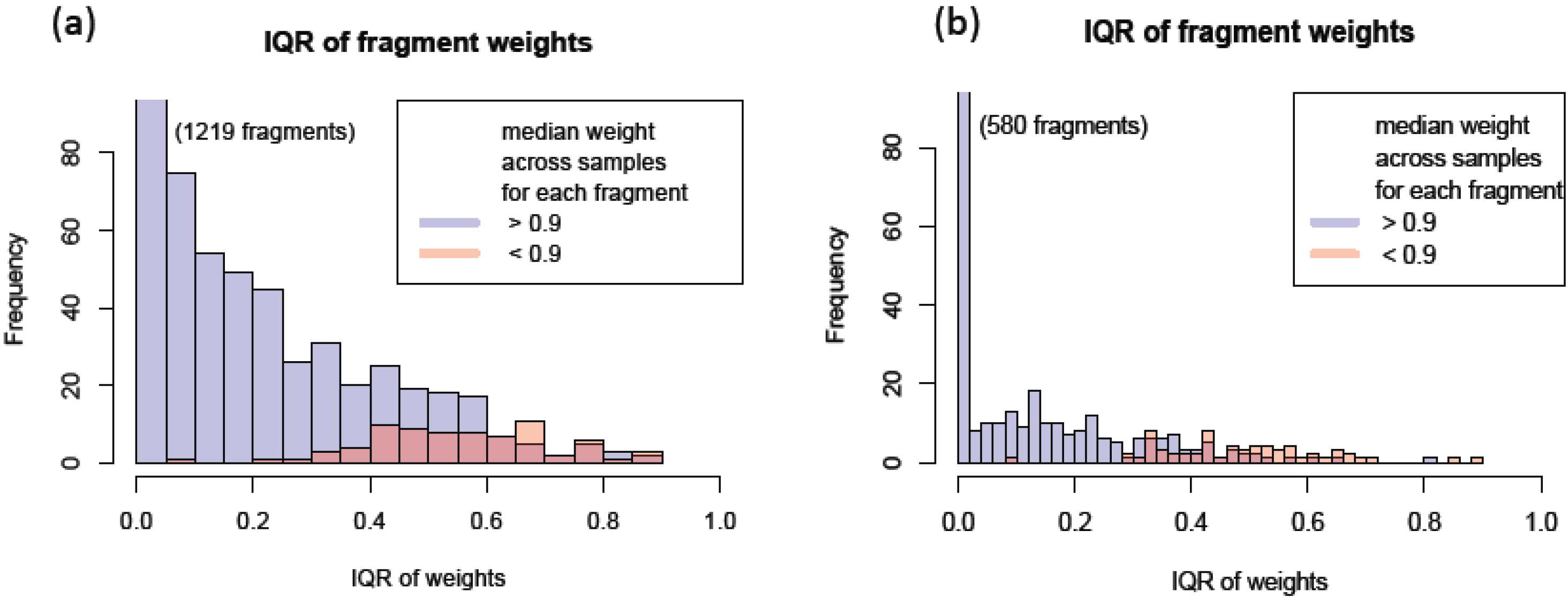
3.1.3. Use of Robust Model to Measure Inconsistent MRM Transitions
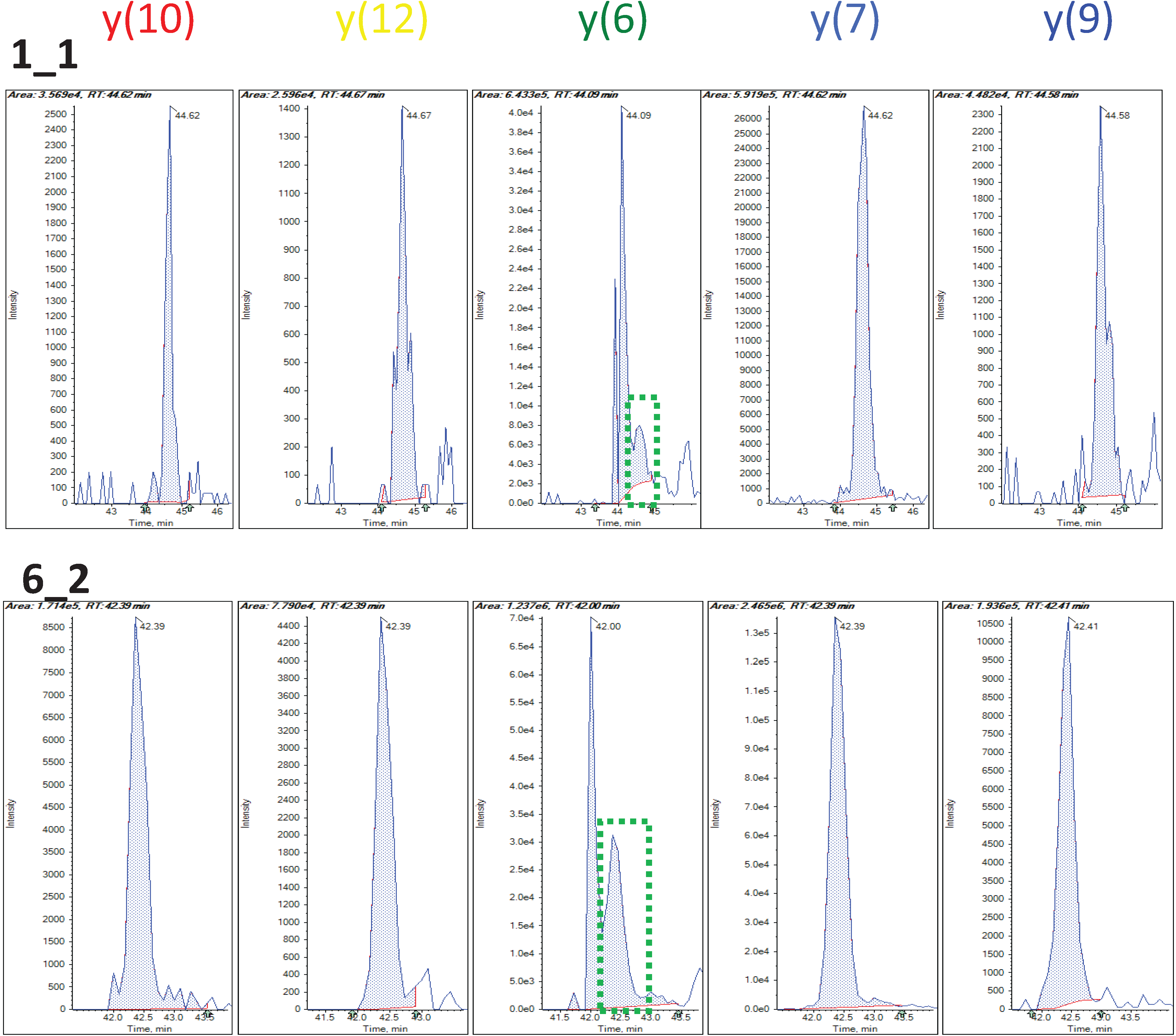
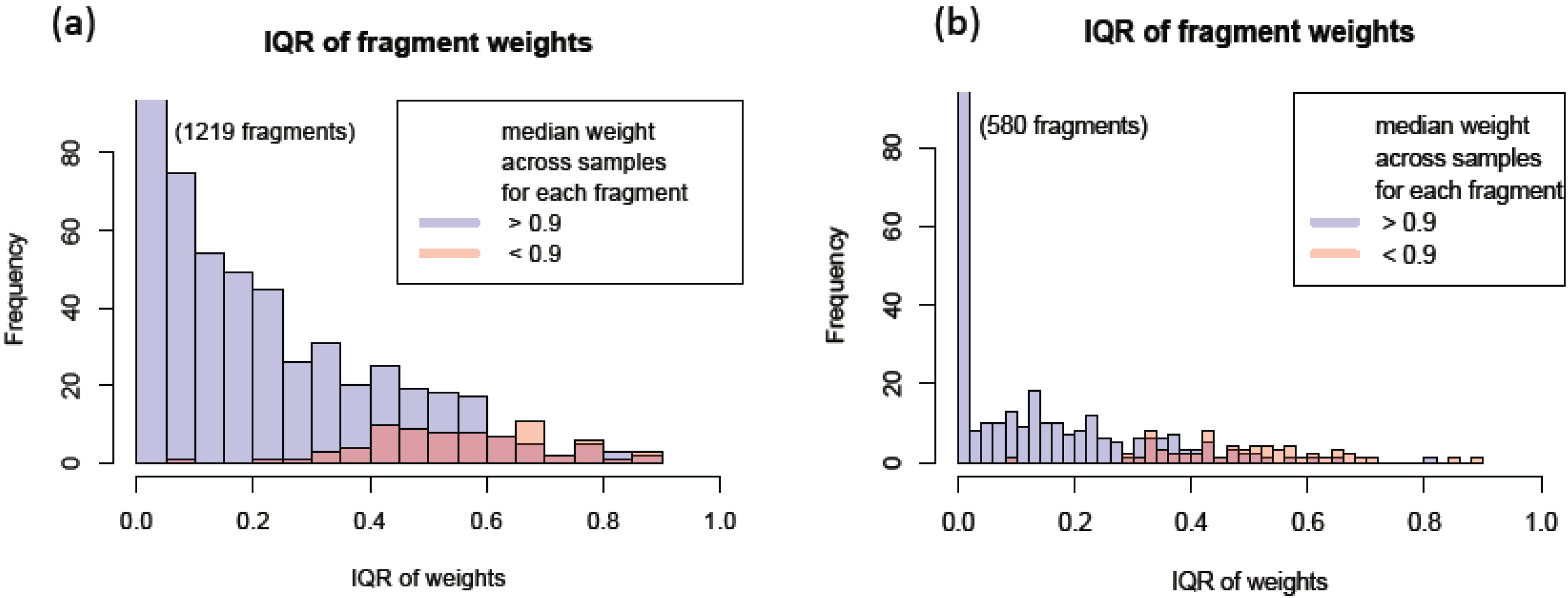
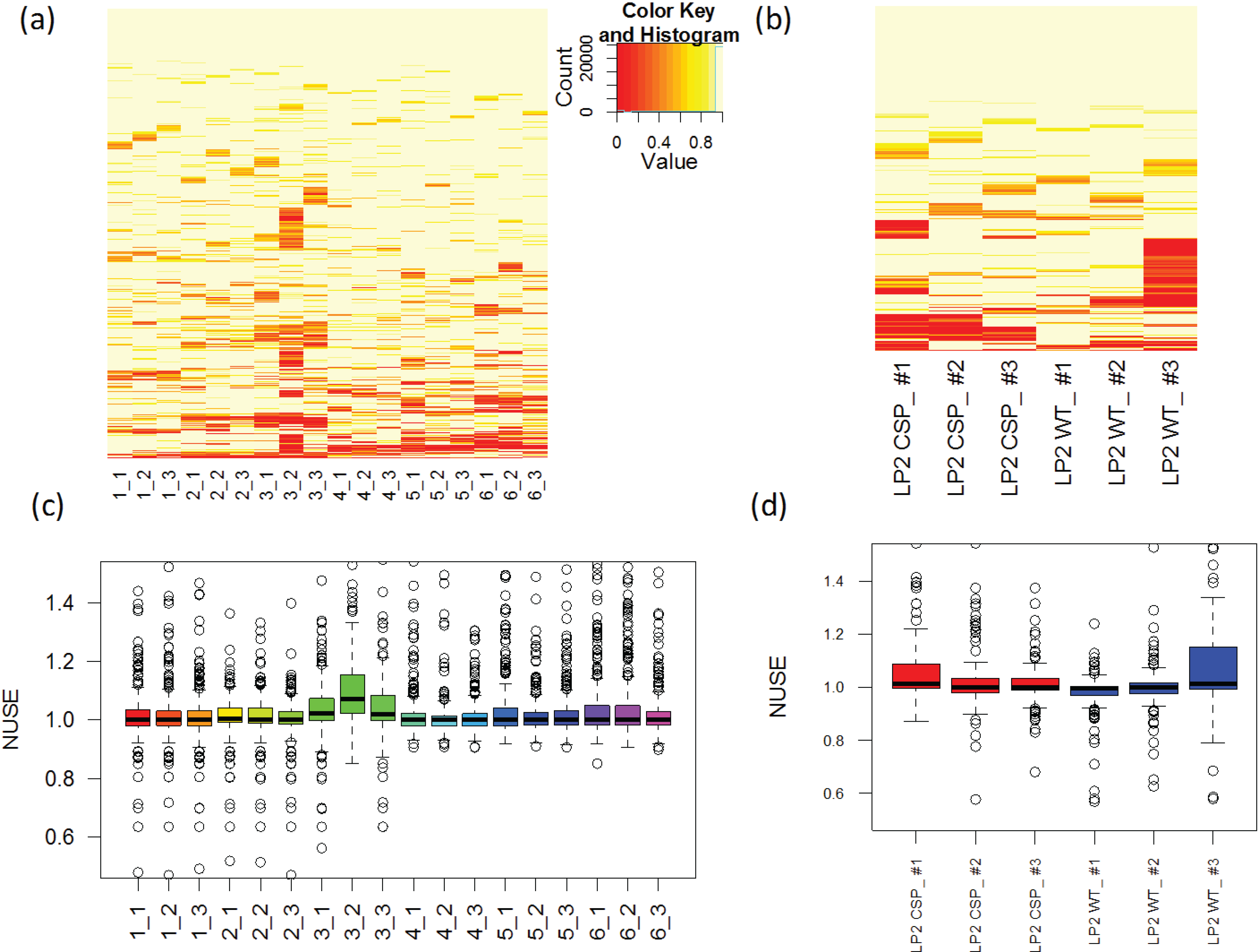
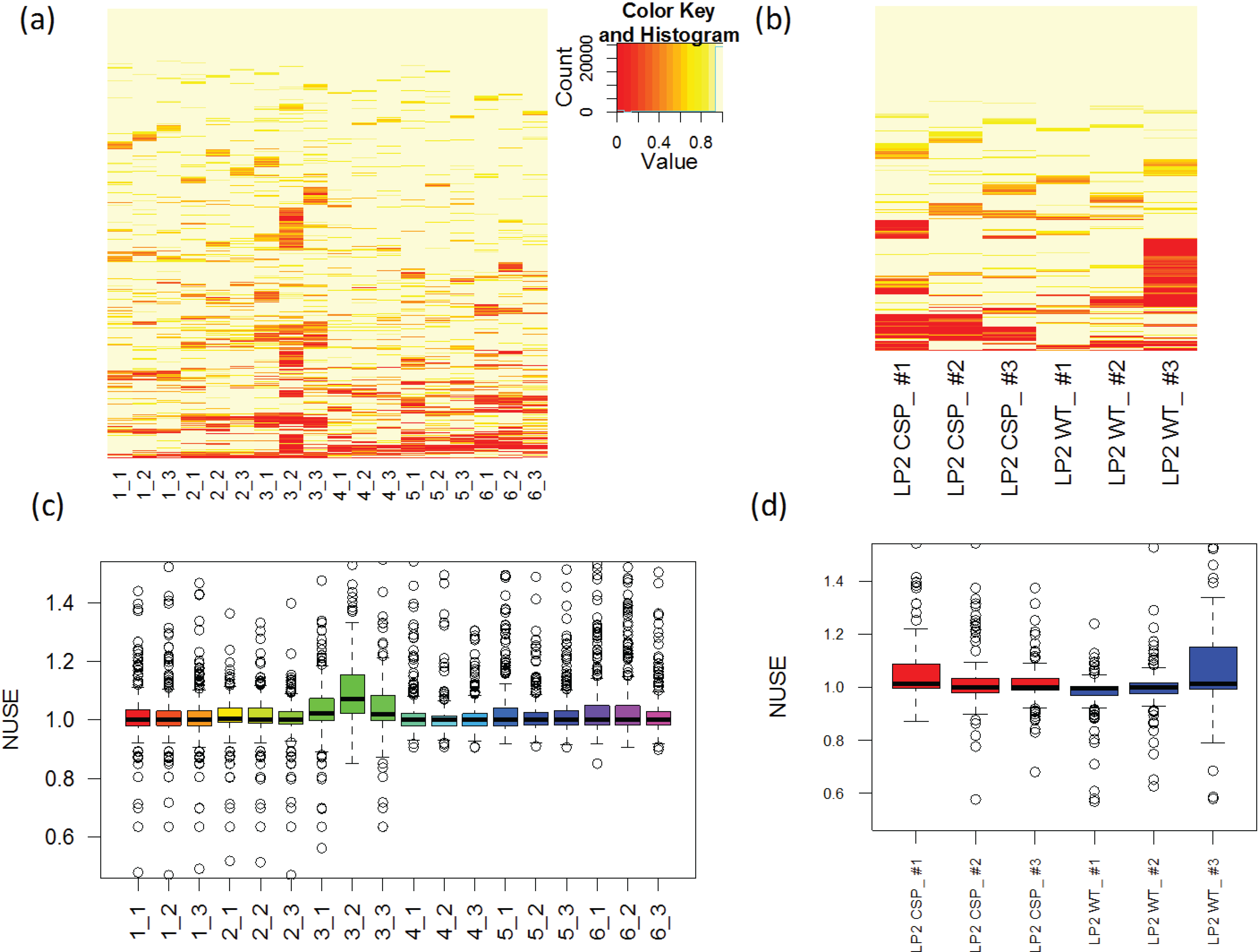
3.1.4. Use of Robust Model to Assess the Sample Quality
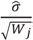 , where
, where 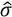 is the estimated standard deviation of error term (Equation (1)) and Wj = Σiwij is a total transition weight [38]. However, this estimate depends on the standard deviation estimate and does not consider heterogeneity among peptides. Furthermore, it does not take into account the variation in the number of transitions per peptide or the presence of poor quality fragments. Normalized unscaled standard error (NUSE) was proposed as a measure of overall sample quality using the weights [38]. Briefly, it first replaces by 1. Then, the authors suggested to normalize the value through dividing it by its median across all samples, i.e.,
is the estimated standard deviation of error term (Equation (1)) and Wj = Σiwij is a total transition weight [38]. However, this estimate depends on the standard deviation estimate and does not consider heterogeneity among peptides. Furthermore, it does not take into account the variation in the number of transitions per peptide or the presence of poor quality fragments. Normalized unscaled standard error (NUSE) was proposed as a measure of overall sample quality using the weights [38]. Briefly, it first replaces by 1. Then, the authors suggested to normalize the value through dividing it by its median across all samples, i.e.,
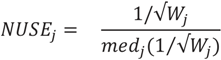
3.2. Data Normalization
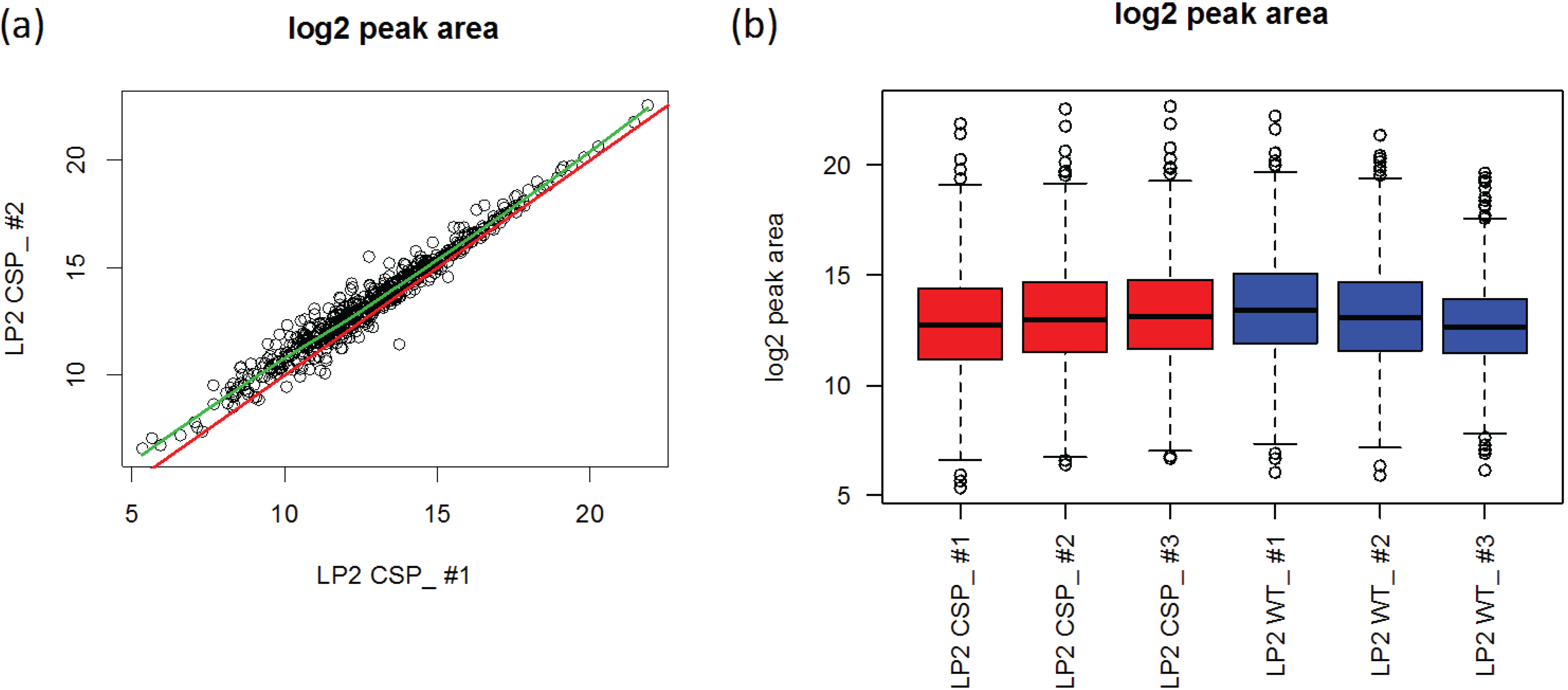
3.2.1. Source of Experimental Artifacts
3.2.2. Using All the Transitions in the Data
3.2.3. Using a Subset of the Data
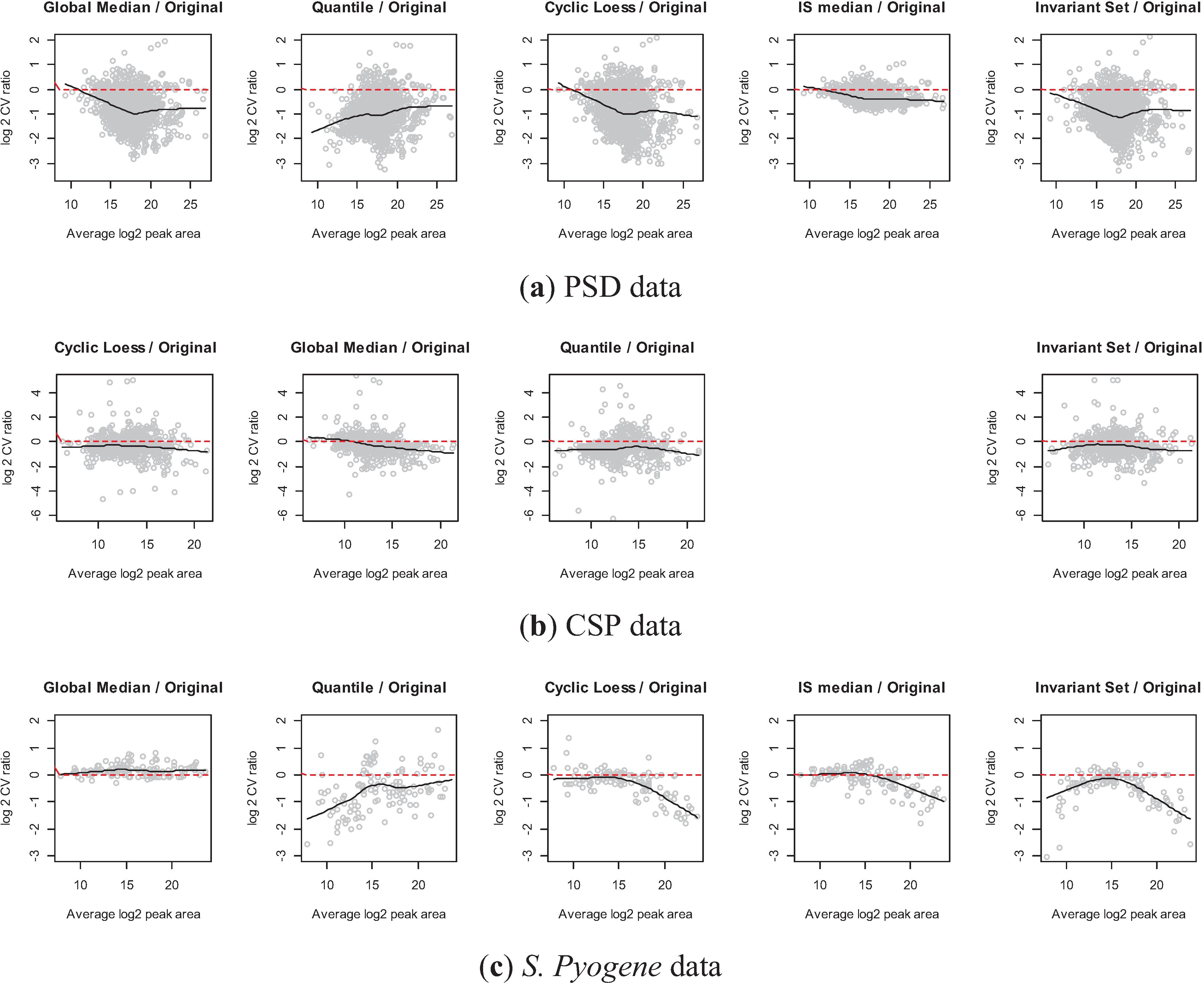
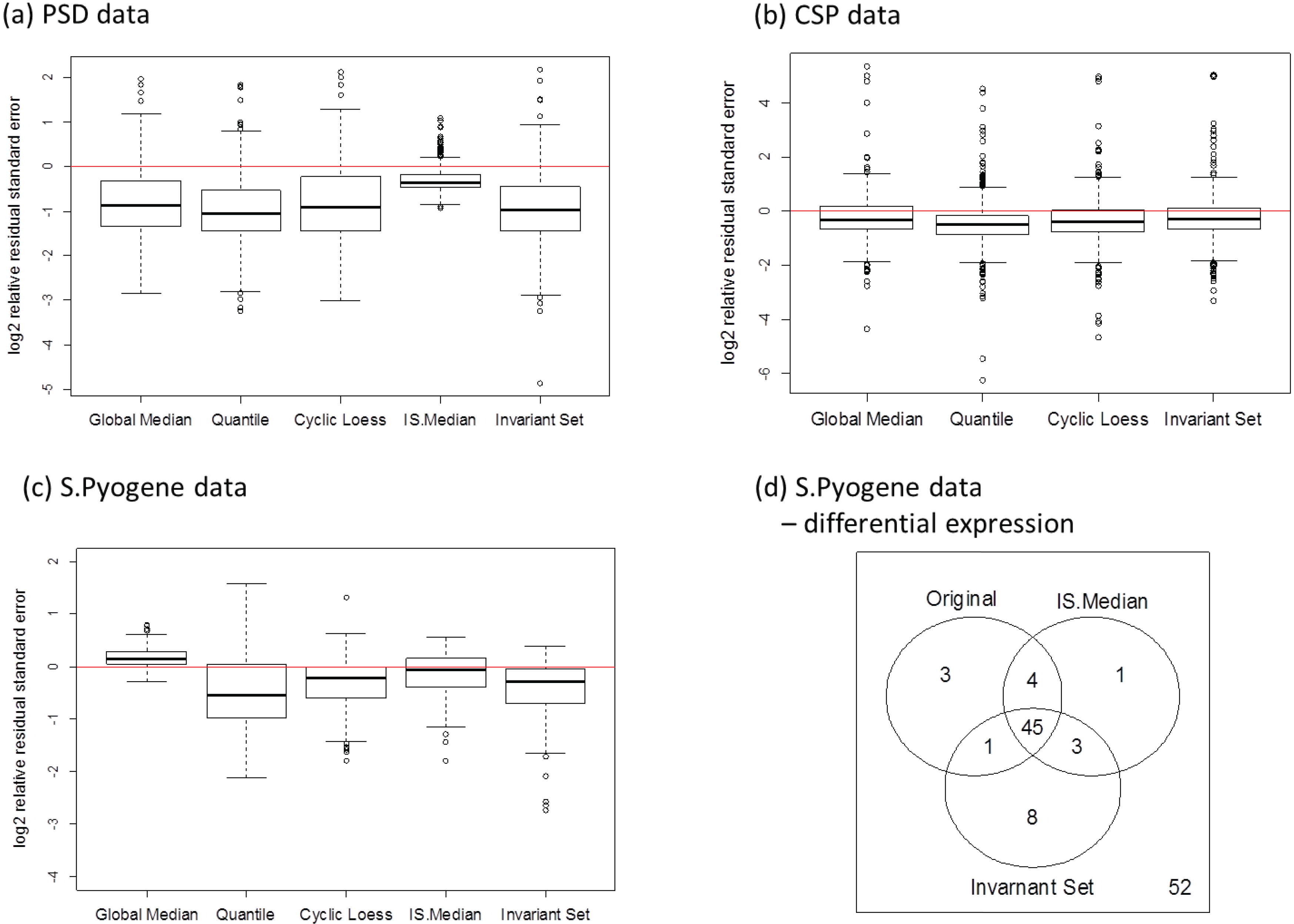
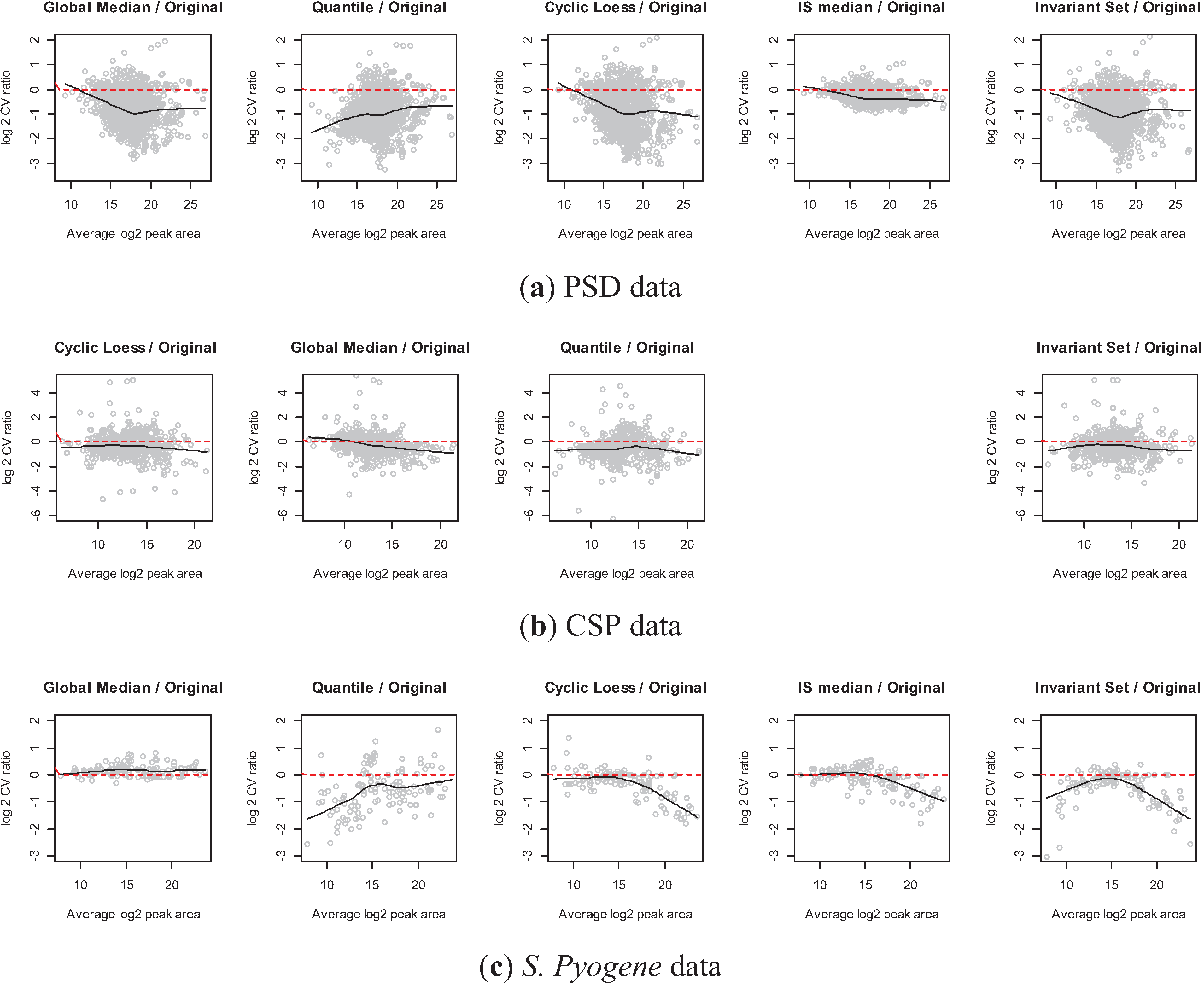
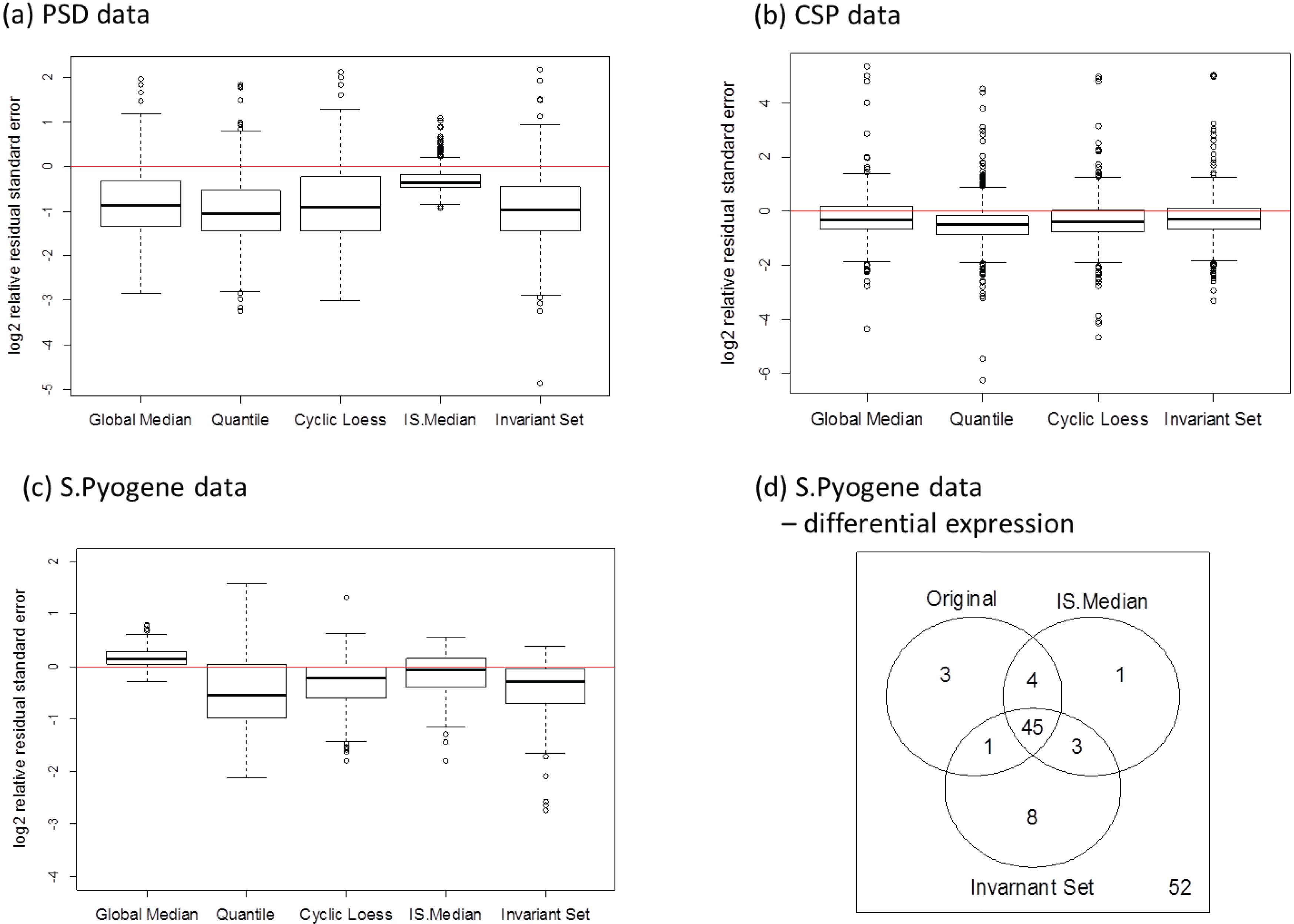
3.2.4. Comparison of the Normalization Performance

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Data Set | Original | Global Median | Quantile | Cyclic Loess | IS.Median | Invariant Set |
|---|---|---|---|---|---|---|
| PSD | 4.18% (1.96%) | 2.15% (1.41%) | 1.97% (1.12%) | 2.12% (1.39%) | 3.09% (1.51%) | 1.99% (1.29%) |
| CSP | 4.04% (2.82%) | 3.47% (2.75%) | 2.85% (2.03%) | 3.26% (2.16%) | NA | 3.29% (2.43%) |
| S. Pyogene | 3.05% (2.78%) | 3.47% (2.84%) | 2.34% (2.02%) | 2.85% (3.15%) | 3.12% (3.32%) | 2.69% (2.74%) |
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References and Notes
- Ong, S.; Blagoev, B.; Kratchmarova, I.; Kristensen, D.; Steen, H.; Pandey, A.; Mann, M. Stable isotope labeling by amino acids in cell culture, silac, as a simple and accurate approach to expression proteomics. Mol. Cell. Proteomics 2002, 1, 376–378. [Google Scholar] [CrossRef]
- Ross, P.; Huang, Y.; Marchese, J.; Williamson, B.; Parker, K.; Hattan, S.; Khainovski, N.; Pillai, S.; Dey, S.; Daniels, S.; et al. Multiplexed protein quantitation in saccharomyces cerevisiae using amine-reactive isobaric tagging reagents. Mol. Cell. Proteomics 2004, 3, 1154–1169. [Google Scholar] [CrossRef]
- Asara, J.; Christofk, H.; Freimark, L.; Cantley, L. A label-free quantification method by ms/ms tic compared to silac and spectral counting in a proteomics screen. Proteomics 2008, 8, 994–999. [Google Scholar] [CrossRef]
- Luo, R.; Colangelo, C.M.; Sessa, W.C.; Zhao, H. Bayesian analysis of itraq data with nonrandom missingness: Identification of differentially expressed proteins. Stat. Biosci. 2009, 1, 228–245. [Google Scholar] [CrossRef]
- Gerber, S.A.; Rush, J.; Stemman, O.; Kirschner, M.W.; Gygi, S.P. Absolute quantification of proteins and phosphoproteins from cell lysates by tandem ms. Proc. Natl. Acad. Sci. USA 2003, 100, 6940–6945. [Google Scholar] [CrossRef]
- Dupuis, A.; Hennekinne, J.A.; Garin, J.; Brun, V. Protein standard absolute quantification (psaq) for improved investigation of staphylococcal food poisoning outbreaks. Proteomics 2008, 8, 4633–4636. [Google Scholar] [CrossRef]
- Wolf-Yadlin, A.; Hautaniemi, S.; Lauffenburger, D.A.; White, F.M. Multiple reaction monitoring for robust quantitative proteomic analysis of cellular signaling networks. Proc. Natl. Acad. Sci. USA 2007, 104, 5860–5865. [Google Scholar] [CrossRef]
- Domon, B.; Aebersold, R. Mass spectrometry and protein analysis. Science 2006, 312, 212–217. [Google Scholar] [CrossRef]
- Picotti, P.; Bodenmiller, B.; Mueller, L.N.; Domon, B.; Aebersold, R. Full dynamic range proteome analysis of s. Cerevisiae by targeted proteomics. Cell 2009, 138, 795–806. [Google Scholar] [CrossRef]
- Addona, T.A.; Abbatiello, S.E.; Schilling, B.; Skates, S.J.; Mani, D.R.; Bunk, D.M.; Spiegelman, C.H.; Zimmerman, L.J.; Ham, A.J.; Keshishian, H.; et al. Multi-site assessment of the precision and reproducibility of multiple reaction monitoring-based measurements of proteins in plasma. Nat. Biotechnol. 2009, 27, 633–641. [Google Scholar] [CrossRef]
- Zhang, H.; Liu, Q.; Zimmerman, L.J.; Ham, A.J.; Slebos, R.J.; Rahman, J.; Kikuchi, T.; Massion, P.P.; Carbone, D.P.; Billheimer, D.; et al. Methods for peptide and protein quantitation by liquid chromatography-multiple reaction monitoring mass spectrometry. Mol. Cell. Proteomics 2011, 10, M110 006593. [Google Scholar] [CrossRef]
- MacLean, B.; Tomazela, D.M.; Shulman, N.; Chambers, M.; Finney, G.L.; Frewen, B.; Kern, R.; Tabb, D.L.; Liebler, D.C.; MacCoss, M.J. Skyline: An open source document editor for creating and analyzing targeted proteomics experiments. Bioinformatics 2010, 26, 966–968. [Google Scholar] [CrossRef]
- Abbatiello, S.E.; Mani, D.R.; Keshishian, H.; Carr, S.A. Automated detection of inaccurate and imprecise transitions in peptide quantification by multiple reaction monitoring mass spectrometry. Clin. Chem. 2010, 56, 291–305. [Google Scholar] [CrossRef]
- Reiter, L.; Rinner, O.; Picotti, P.; Huttenhain, R.; Beck, M.; Brusniak, M.Y.; Hengartner, M.O.; Aebersold, R. Mprophet: Automated data processing and statistical validation for large-scale srm experiments. Nat. Methods 2011, 8, 430–435. [Google Scholar] [CrossRef]
- Bolstad, B.M.; Irizarry, R.A.; Astrand, M.; Speed, T.P. A comparison of normalization methods for high density oligonucleotide array data based on variance and bias. Bioinformatics 2003, 19, 185–193. [Google Scholar] [CrossRef]
- Tseng, G.C.; Oh, M.K.; Rohlin, L.; Liao, J.C.; Wong, W.H. Issues in cdna microarray analysis: Quality filtering, channel normalization, models of variations and assessment of gene effects. Nucleic Acids Res. 2001, 29, 2549–2557. [Google Scholar] [CrossRef]
- Ballman, K.V.; Grill, D.E.; Oberg, A.L.; Therneau, T.M. Faster cyclic loess: Normalizing rna arrays via linear models. Bioinformatics 2004, 20, 2778–2786. [Google Scholar] [CrossRef]
- Smyth, G.K.; Speed, T. Normalization of cdna microarray data. Methods 2003, 31, 265–273. [Google Scholar] [CrossRef]
- Gagnon-Bartsch, J.A.; Speed, T.P. Using control genes to correct for unwanted variation in microarray data. Biostatistics 2012, 13, 539–552. [Google Scholar] [CrossRef]
- Chang, C.Y.; Picotti, P.; Huttenhain, R.; Heinzelmann-Schwarz, V.; Jovanovic, M.; Aebersold, R.; Vitek, O. Protein significance analysis in selected reaction monitoring (srm) measurements. Mol. Cell. Proteomics 2012, 11, M111 014662. [Google Scholar] [CrossRef]
- Teleman, J.; Karlsson, C.; Waldemarson, S.; Hansson, K.; James, P.; Malmstrom, J.; Levander, F. Automated selected reaction monitoring software for accurate label-free protein quantification. J. Proteome Res. 2012, 11, 3766–3773. [Google Scholar] [CrossRef]
- Colangelo, C.; Abbott, T.; Shifman, M.; Ivosev, G.; Chung, L.; Sakaue, F.; Cox, D.; Tate, S.A.; Nairn, A.; Rinehart, J.; Williams, K. Development of targeted proteomics assays. In Proceedings of the 60th ASMS Conference on Mass Spectrometry and Allied Topics, Vancouver, Canada, 20–24 May 2012.
- Colangelo, C.M.; Ivosev, G.; Chung, L.; Abbott, T.; Shifman, M.; Sakaue, F.; Cox, D.; Kitchen, R.; Burton, L.; Tate, S.A.; Gulcicek, E.; Bonner, R.; Rinehart, J.; Nairn, A.C.; Williams, K. Development of a highly automated and multiplexed targeted proteome pipeline and assay for 112 rat brain synaptic proteins. Mol. Cell. Proteomics 2014. submitted for publication. [Google Scholar]
- Multiquant™ 2.1 software, 2.1 (research version); AB SCIEX: Concord, Canada, 2011.
- Shifman, M.; Li, Y.; Colangelo, C.; Stone, K.; Wu, T.; Cheung, K.; Miller, P.; Williams, K. Yped: A web-accessible database system for protein expression analysis. J. Proteome Res. 2007, 6, 4019–4024. [Google Scholar] [CrossRef]
- Shifman, M.A.; Sun, K.; Colangelo, C.M.; Cheung, K.H.; Miller, P.; Williams, K. Yped: A proteomics database for protein expression analysis. AMIA Annu. Symp. Proc. 2005, 2005, 1111. [Google Scholar]
- Shifman, M.; Li, Y.; Colangelo, C.; Stone, K.; Wu, T.; Cheung, K.; Miller, P.; Williams, K. Yale protein expression database. Available online: http://yped.med.yale.edu/repository/ViewSeriesMenu.do?series_id=4522&series_name=Data+Pre-processing+for+label-free+multiple+reaction+monitoring+(MRM)+experiments/ (accessed on 6 January 2014).
- Chandra, S.; Gallardo, G.; Fernandez-Chacon, R.; Schluter, O.M.; Sudhof, T.C. Alpha-synuclein cooperates with cspalpha in preventing neurodegeneration. Cell 2005, 123, 383–396. [Google Scholar]
- Fernandez-Chacon, R.; Wolfel, M.; Nishimune, H.; Tabares, L.; Schmitz, F.; Castellano-Munoz, M.; Rosenmund, C.; Montesinos, M.L.; Sanes, J.R.; Schneggenburger, R.; et al. The synaptic vesicle protein csp alpha prevents presynaptic degeneration. Neuron 2004, 42, 237–251. [Google Scholar] [CrossRef]
- Zhang, Y.Q.; Henderson, M.X.; Colangelo, C.M.; Ginsberg, S.D.; Bruce, C.; Wu, T.; Chandra, S.S. Identification of cspalpha clients reveals a role in dynamin 1 regulation. Neuron 2012, 74, 136–150. [Google Scholar] [CrossRef]
- Cox, D.M.; Tate, S.; Duchoslav, E. Automated ion selection and method building for mrm based protein validation and quantification. In Proceedings of ASMS, Indianapolis, IN, USA, 3–7 June 2007; p. MP501.
- Unwin, R.D.; Griffiths, J.R.; Whetton, A.D. A sensitive mass spectrometric method for hypothesis-driven detection of peptide post-translational modifications: Multiple reaction monitoring-initiated detection and sequencing (midas). Nat. Protoc. 2009, 4, 870–877. [Google Scholar]
- Burton, L.; Ivosev, G.; Lau, A.; Bonner, R. A novel algorithm for quantitative lc peak integration. In Proceedings of ASMS, Philadelphia, PA, USA, 31 May–4 June 2009; p. Poster I-224.
- Cunningham, M.W. Pathogenesis of group a streptococcal infections. Clin. Microbiol. Rev. 2000, 13, 470–511. [Google Scholar] [CrossRef]
- The swedish storage initiative (swestore). Available online: http://webdav.swegrid.se/snic/bils/lu_proteomics/pub/anubis_data.zip (accessed on 30 April 2013).
- Colangelo, C.M.; Chung, L.; Bruce, C.; Cheung, K.H. Review of software tools for design and analysis of large scale mrm proteomic datasets. Methods 2013, 61, 287–298. [Google Scholar] [CrossRef]
- Irizarry, R.A.; Hobbs, B.; Collin, F.; Beazer-Barclay, Y.D.; Antonellis, K.J.; Scherf, U.; Speed, T.P. Exploration, normalization, and summaries of high density oligonucleotide array probe level data. Biostatistics 2003, 4, 249–264. [Google Scholar] [CrossRef]
- Brettschneider, J.; Collin, F.; Bolstad, B.M.; Speed, T.P. Quality assessment for short oligonucleotide microarray data. Technometrics 2008, 50, 241–264. [Google Scholar] [CrossRef]
- Yang, Y.H.; Dudoit, S.; Luu, P.; Lin, D.M.; Peng, V.; Ngai, J.; Speed, T.P. Normalization for cdna microarray data: A robust composite method addressing single and multiple slide systematic variation. Nucleic Acids Res 2002, 30, e15. [Google Scholar] [CrossRef]
- Ejigu, B.A.; Valkenborg, D.; Baggerman, G.; Vanaerschot, M.; Witters, E.; Dujardin, J.C.; Burzykowski, T.; Berg, M. Evaluation of normalization methods to pave the way towards large-scale lc-ms-based metabolomics profiling experiments. OMICS 2013, 17, 473–485. [Google Scholar] [CrossRef]
- Mar, J.C.; Kimura, Y.; Schroder, K.; Irvine, K.M.; Hayashizaki, Y.; Suzuki, H.; Hume, D.; Quackenbush, J. Data-driven normalization strategies for high-throughput quantitative rt-pcr. BMC Bioinformatics 2009, 10, 110. [Google Scholar] [CrossRef]
- Qureshi, R.; Sacan, A. A novel method for the normalization of microrna rt-pcr data. BMC Med. Genomics 2013, 6, S14. [Google Scholar] [CrossRef]
- Martins-de-Souza, D.; Alsaif, M.; Ernst, A.; Harris, L.W.; Aerts, N.; Lenaerts, I.; Peeters, P.J.; Amess, B.; Rahmoune, H.; Bahn, S.; et al. The application of selective reaction monitoring confirms dysregulation of glycolysis in a preclinical model of schizophrenia. BMC Res. Notes 2012, 5, 146. [Google Scholar] [CrossRef]
- Schadt, E.E.; Li, C.; Ellis, B.; Wong, W.H. Feature extraction and normalization algorithms for high-density oligonucleotide gene expression array data. J. Cell Biochem. Suppl. 2001, 84, 120–125. [Google Scholar] [CrossRef]
- Ferretti, J.J.; McShan, W.M.; Ajdic, D.; Savic, D.J.; Savic, G.; Lyon, K.; Primeaux, C.; Sezate, S.; Suvorov, A.N.; Kenton, S.; et al. Complete genome sequence of an m1 strain of streptococcus pyogenes. Proc. Natl. Acad. Sci. USA 2001, 98, 4658–4663. [Google Scholar] [CrossRef]
- Hongyu Zhao’s lab statistical genomics and proteomics. Available online: http://zhaocenter.org/ (accessed on 5 May 2014).
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Chung, L.M.; Colangelo, C.M.; Zhao, H. Data Pre-Processing for Label-Free Multiple Reaction Monitoring (MRM) Experiments. Biology 2014, 3, 383-402. https://doi.org/10.3390/biology3020383
Chung LM, Colangelo CM, Zhao H. Data Pre-Processing for Label-Free Multiple Reaction Monitoring (MRM) Experiments. Biology. 2014; 3(2):383-402. https://doi.org/10.3390/biology3020383
Chicago/Turabian StyleChung, Lisa M., Christopher M. Colangelo, and Hongyu Zhao. 2014. "Data Pre-Processing for Label-Free Multiple Reaction Monitoring (MRM) Experiments" Biology 3, no. 2: 383-402. https://doi.org/10.3390/biology3020383
APA StyleChung, L. M., Colangelo, C. M., & Zhao, H. (2014). Data Pre-Processing for Label-Free Multiple Reaction Monitoring (MRM) Experiments. Biology, 3(2), 383-402. https://doi.org/10.3390/biology3020383