Sequence-Based Analysis of Structural Organization and Composition of the Cultivated Sunflower (Helianthus annuus L.) Genome

Abstract
:
1. Introduction
2. Experimental
2.1. Sequencing and Assembly of the BAC Clones
2.2. Identification and Annotation of the Repetitive Fraction of the Genome
2.2.1. Mathematically Derived Repeats
2.2.2. Class I LTR-Retrotransposons
2.2.3. Coverage of LTR-RTs in the Genome
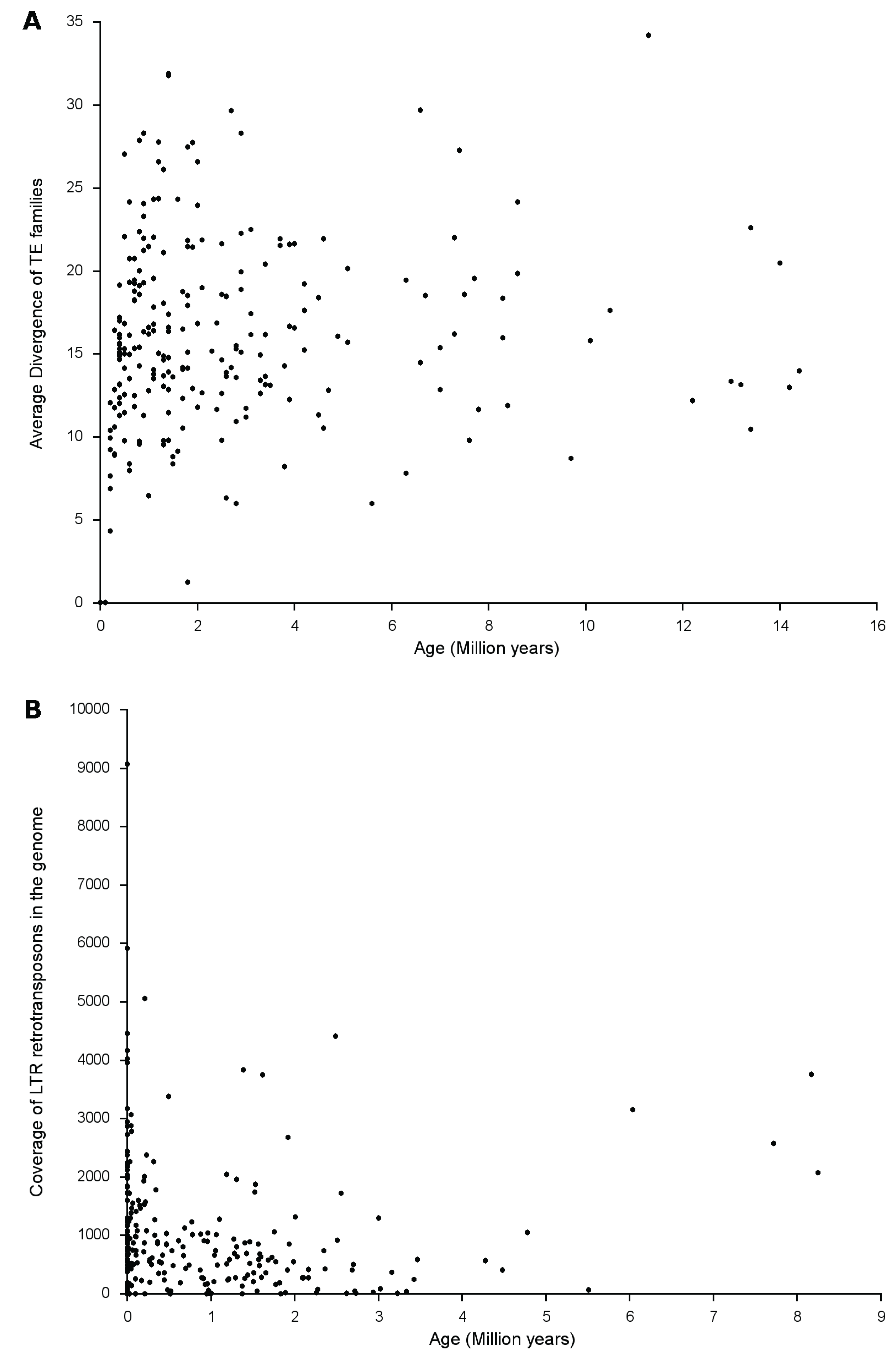
2.2.4. Estimation of Insertion Age of LTR-RTs and Other TE Families
2.3. Identification and Annotation of the Genic Content of the Genome
Gene Ontology (GO) Annotation
2.4. Transcriptome Analysis
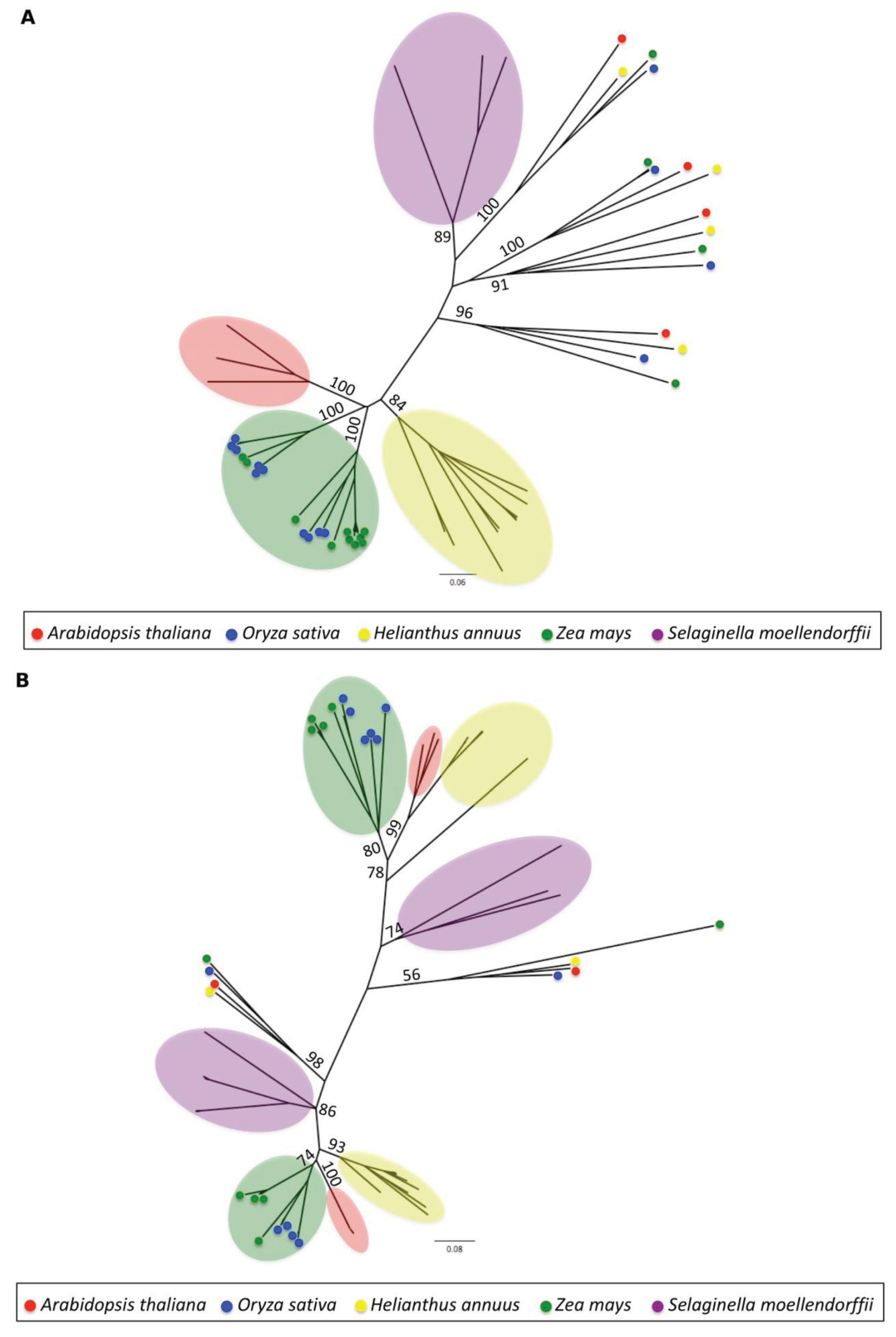
2.5. Phylogenetic Analysis
3. Results
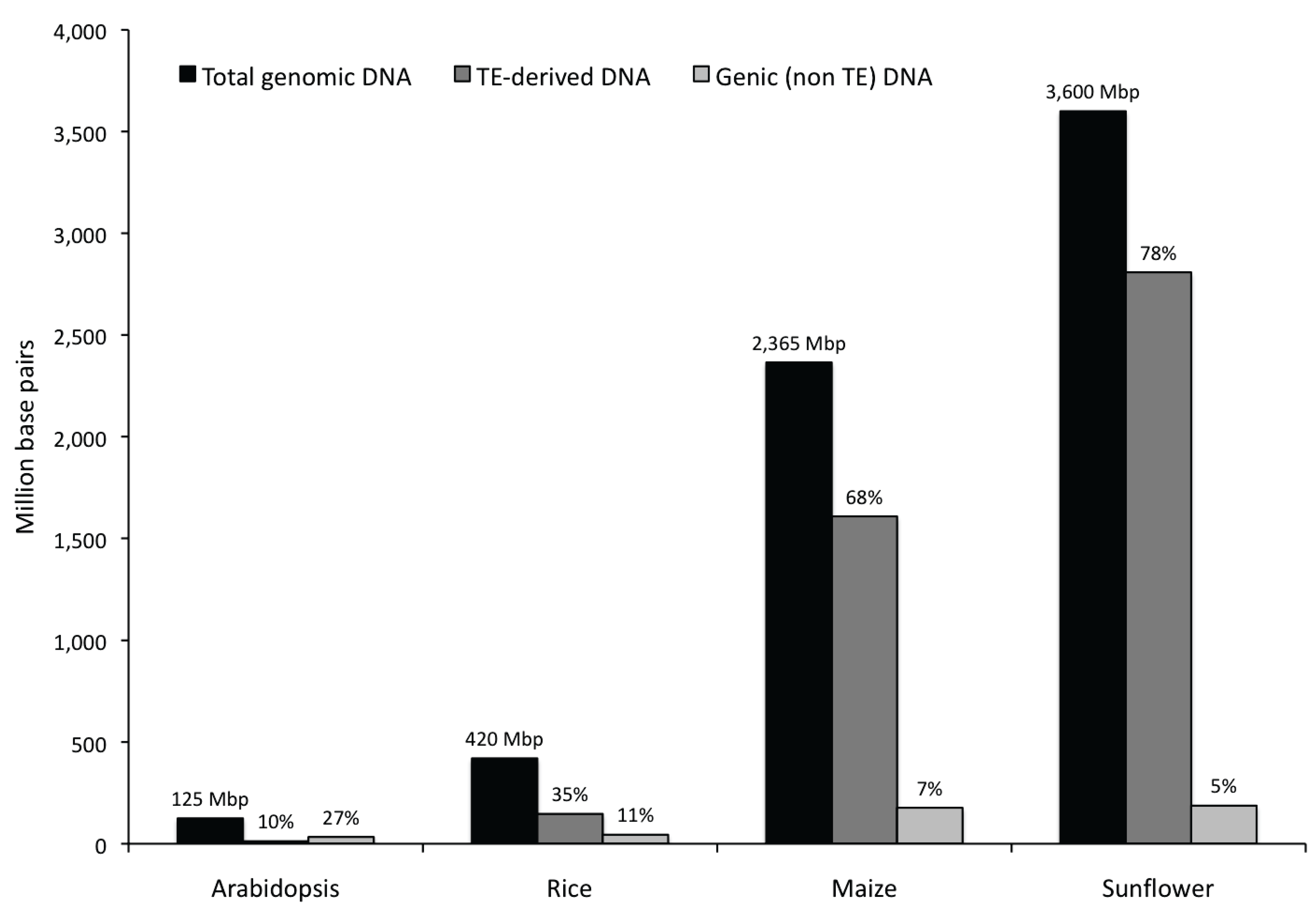
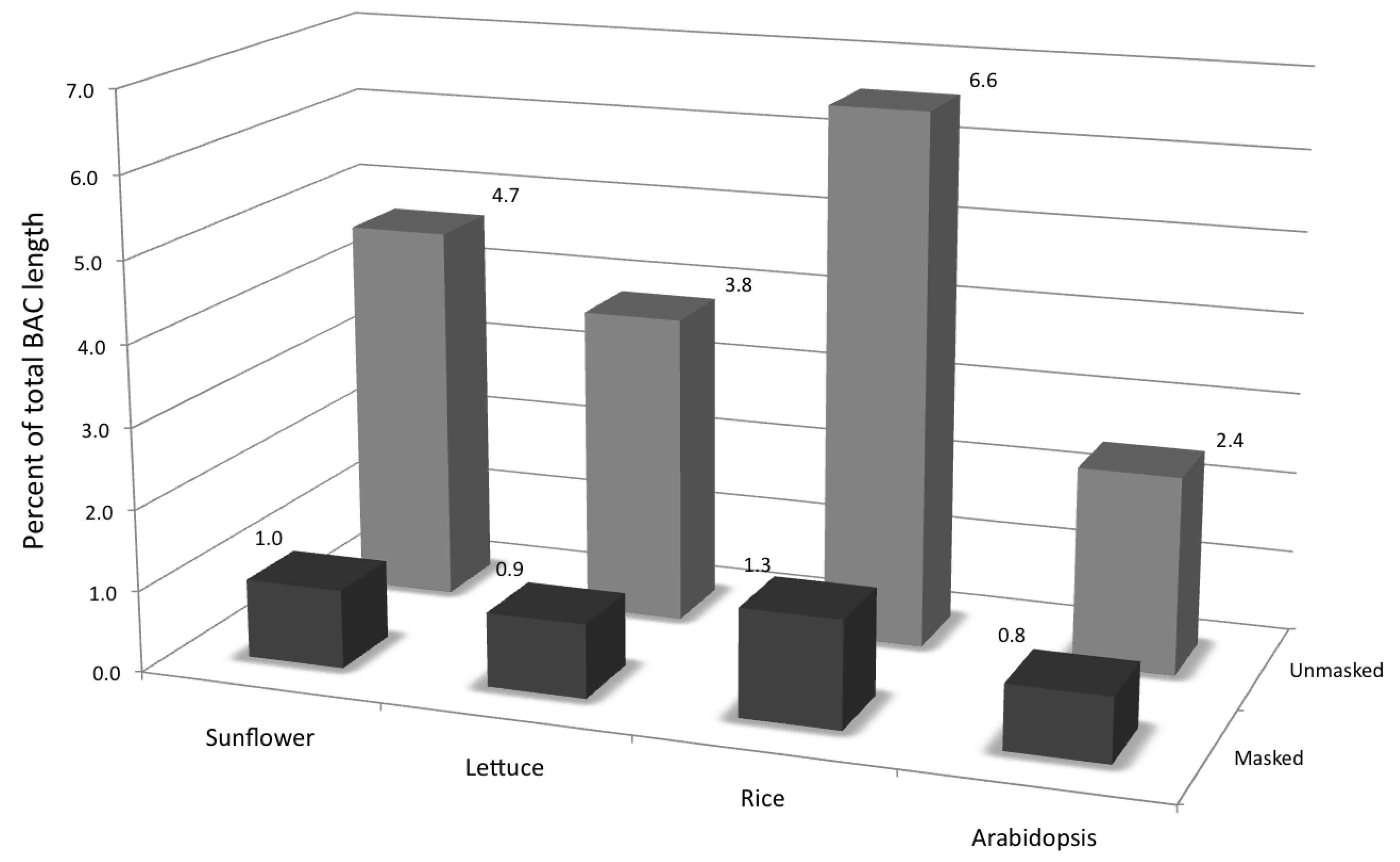
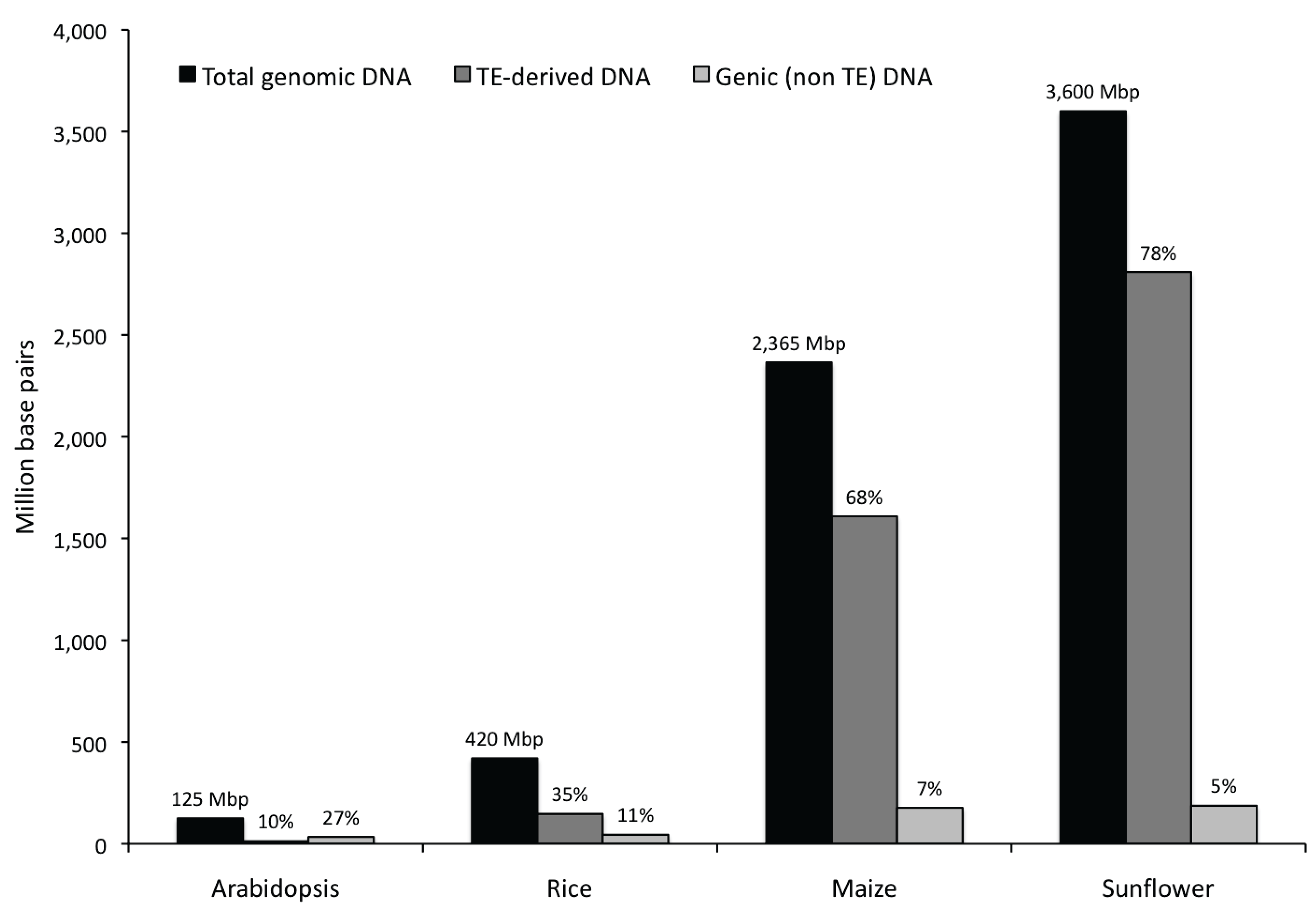
3.1. Abundance, Distribution, Amplification and Divergence of Repetitive Elements in the Sunflower Genome
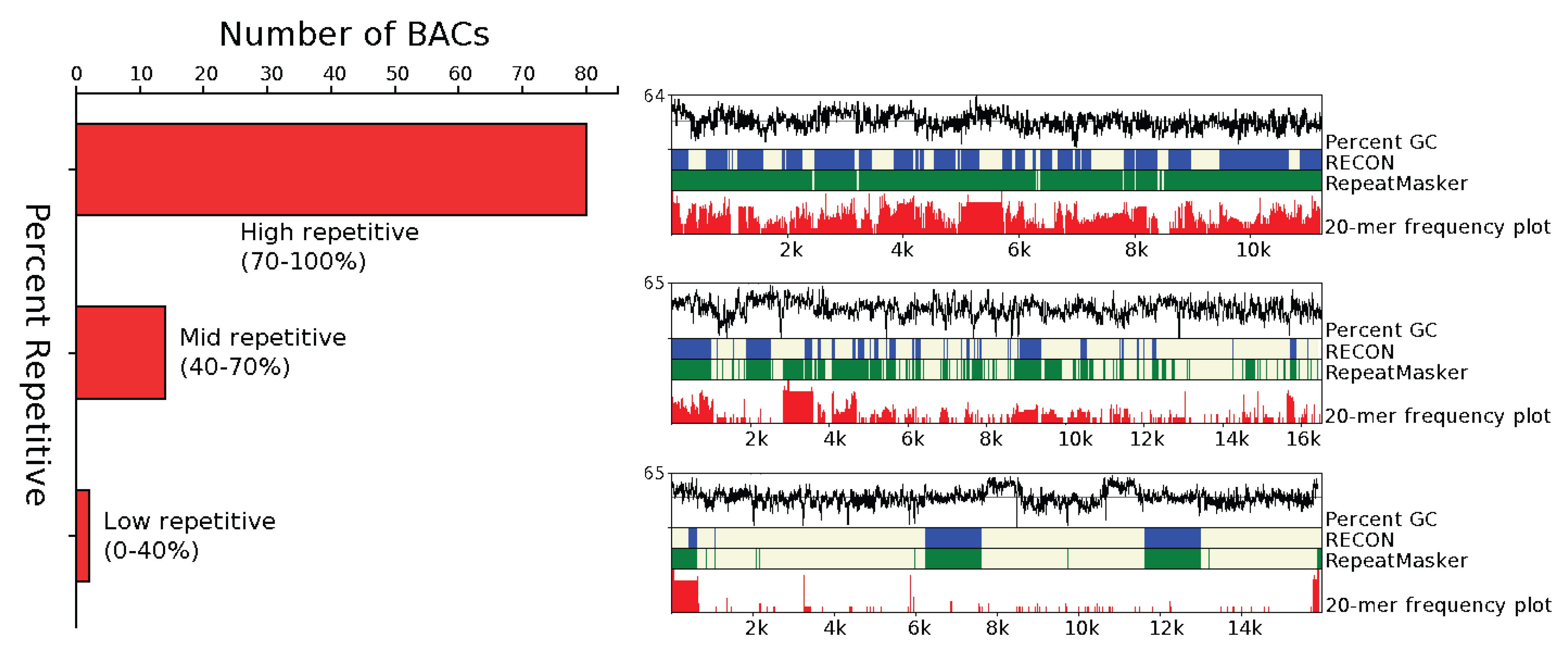
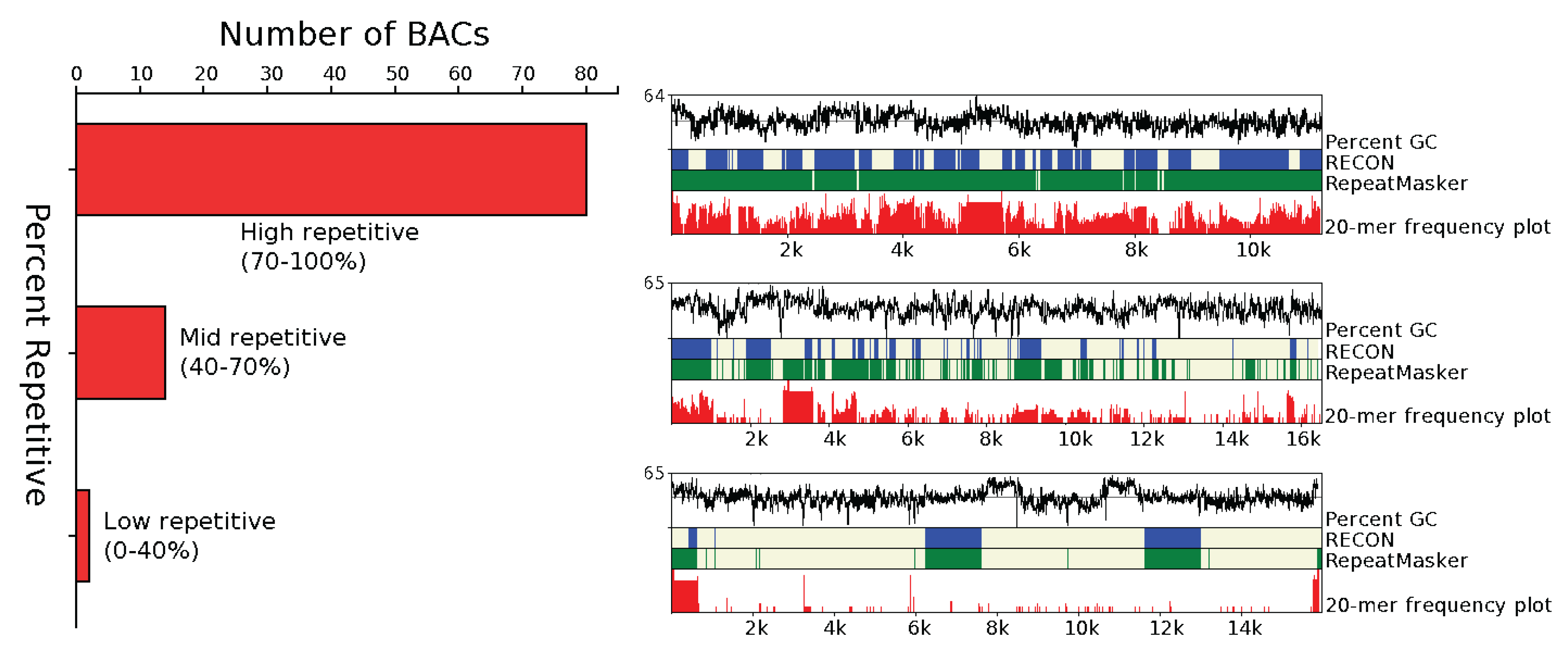
3.1.1. Abundance

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| REPEAT CLASS | REPEAT TYPE | TOTAL NUCLEOTIDES IN THE BAC DATASET (bp) | TOTAL NUCLEOTIDES IN THE GENOME (bp) | $ PERCENT OF THE GENOME | PERCENT OF TOTAL REPETITIVE |
|---|---|---|---|---|---|
| Classs I Retrotransposons | |||||
| Ty1-Copia | 2,014,560 | 515,864,483,658 | 14.33 | 16.28 | |
| Ty3-Gypsy | 2,633,637 | 674,390,333,943 | 18.73 | 21.28 | |
| LINEs, SINEs | 14,316 | 3,665,870,437 | 0.10 | 0.12 | |
| * Unclassified | 2,307,645 | 590,914,192,871 | 16.41 | 18.65 | |
| ** Novel | 2,463,529 | 630,831,107,319 | 17.52 | 19.91 | |
| Subtotal | 9,433,687 | 2,415,665,988,229 | 67.10 | 76.24 | |
| Class II DNA Transposons | |||||
| # Non-MITEs | 19,451 | 4,980,779,958 | 0.14 | 0.16 | |
| MITEs | 3,442 | 881,386,284 | 0.02 | 0.03 | |
| Unclassified | 32,398 | 8,296,093,212 | 0.23 | 0.26 | |
| Subtotal | 55,291 | 14,158,259,454 | 0.39 | 0.45 | |
| Ribosomal DNA | 5S, 45S | 34,127 | 8,738,834,899 | 0.24 | 0.28 |
| Centromeric Repeats | 32,353 | 8,284,570,149 | 0.23 | 0.26 | |
| Telomeric Repeats | 1,281 | 328,023,193 | 0.01 | 0.01 | |
| SSRs and Low Complexity | 160,912 | 41,204,424,685 | 1.14 | 1.30 | |
| Unclassified Repeats | 1,194,287 | 305,818,762,705 | 8.49 | 9.65 | |
| ** Other Novel Repeats | 1,462,325 | 374,454,735,061 | 10.40 | 11.82 | |
| Total Repetitive | 12,374,263 | 3,168,653,598,375 | $$ 88.02 | ||
| Total number of sequences | 6286 |
|---|---|
| Number of sequences not clustered | 97 |
| Number of clusters | 1335 |
| Clusters with >5 sequences | 346 |
| Clusters with >20 sequences | 27 |
| Clusters with >50 sequences | 8 |
| Clusters with >100 sequences | 4 |
| Number of sequences in the largest cluster | 139 |
3.1.2. Distribution
3.1.3. Amplification and Divergence
3.1.4. Phylogenetic Analysis


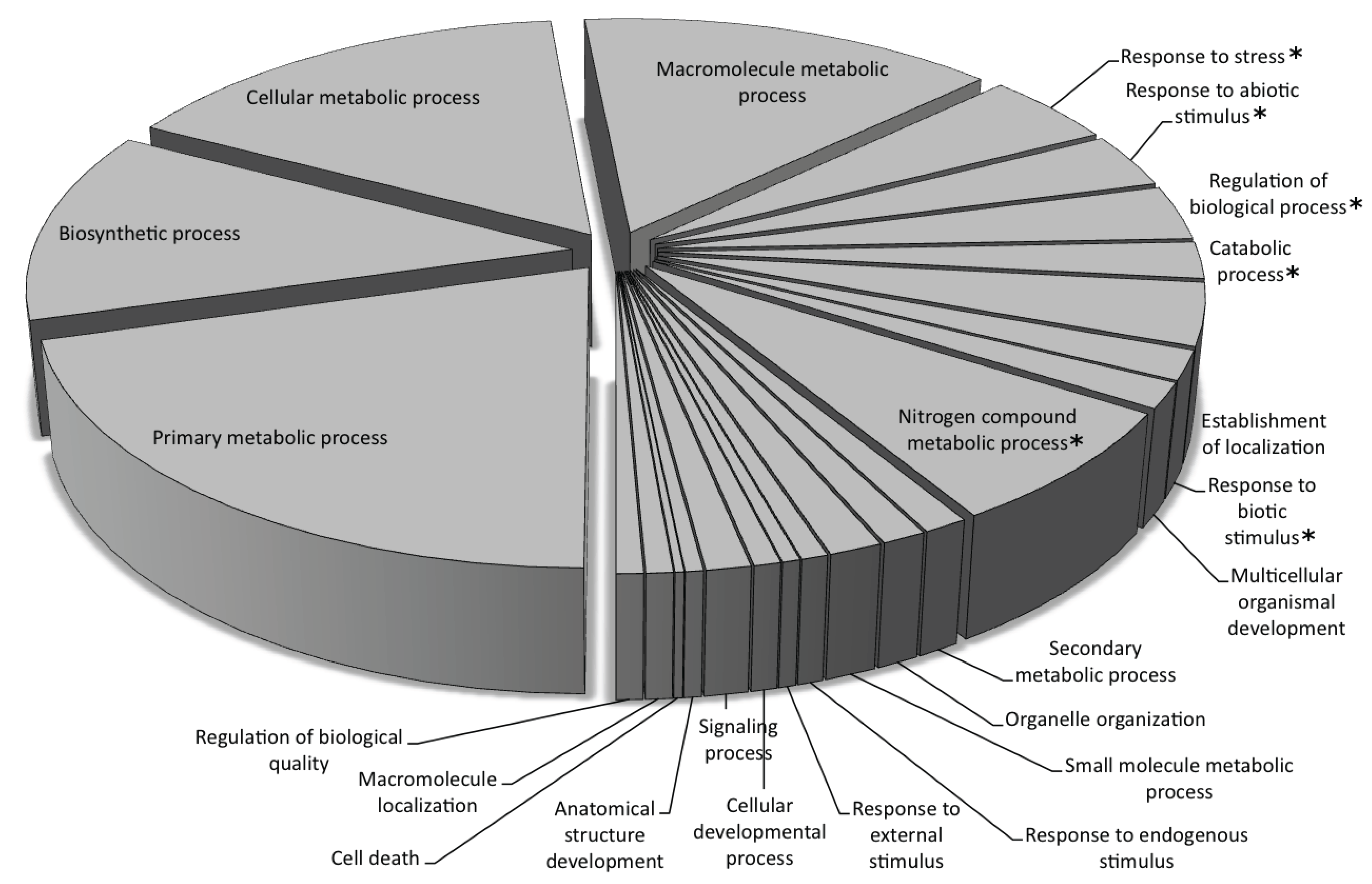
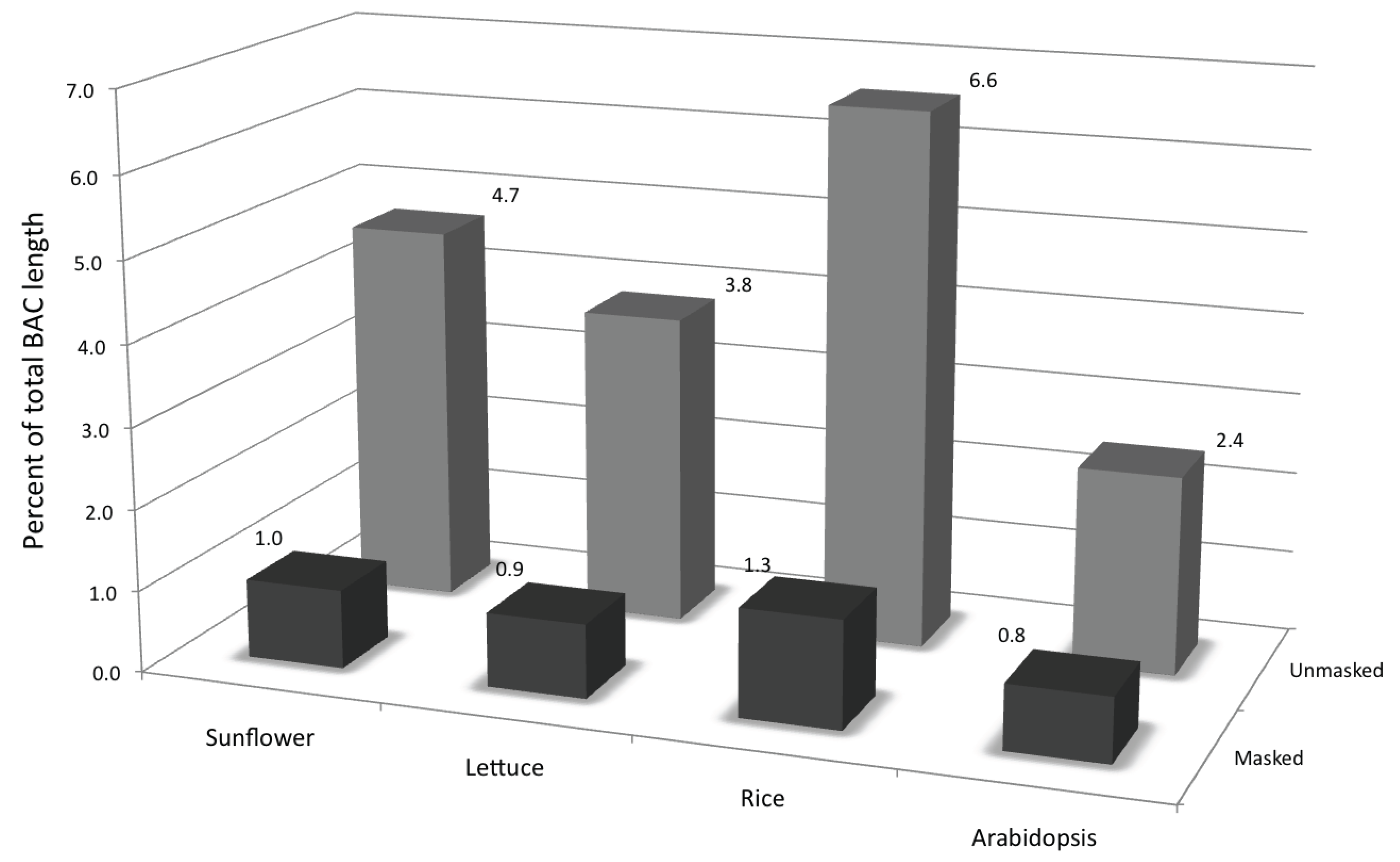
3.2. Genic Content of the Sunflower Genome
| BAC Statistics | Unmasked | Repeat-masked |
|---|---|---|
| Length (Mb) | 14.1 | 3.4 |
| GC content (%) | 39 | 36.8 |
| Number of predicted genes | 2,321 | 643 |
| BlastN against the Helianthus EST db [proportion of total] | 816 [0.4] | 209 [0.3] |
| BlastX against NCBI nr db [proportion of total] | 979 [0.4] | 304 [0.5] |
| BlastN and BlastX [proportion of total] | 551 [0.2] | 160 [0.2] |
| Average gene size (kb) | 3.6 | 3.0 |
| Gene GC content (%) | 41.9 | 40.0 |
| Minimum gene density (bp/gene) | 1,011 | 4,644 |
| Maximum gene density (bp/gene) | 8,042 | 215,568 |
| Average exon size (bp) | 507.6 | 360.1 |
| Exon size/gene (bp) | 640 | 483.8 |
| Number of exons/gene | 4.7 | 4.1 |
| Exon GC content (%) | 44.3 | 42.7 |
| Average intron size (bp) | 339.7 | 487.1 |
| Intron size/gene (bp) | 390.9 | 618.9 |
| Number of introns/gene | 3.7 | 3.1 |
| Intron GC content (%) | 38.5 | 35.2 |
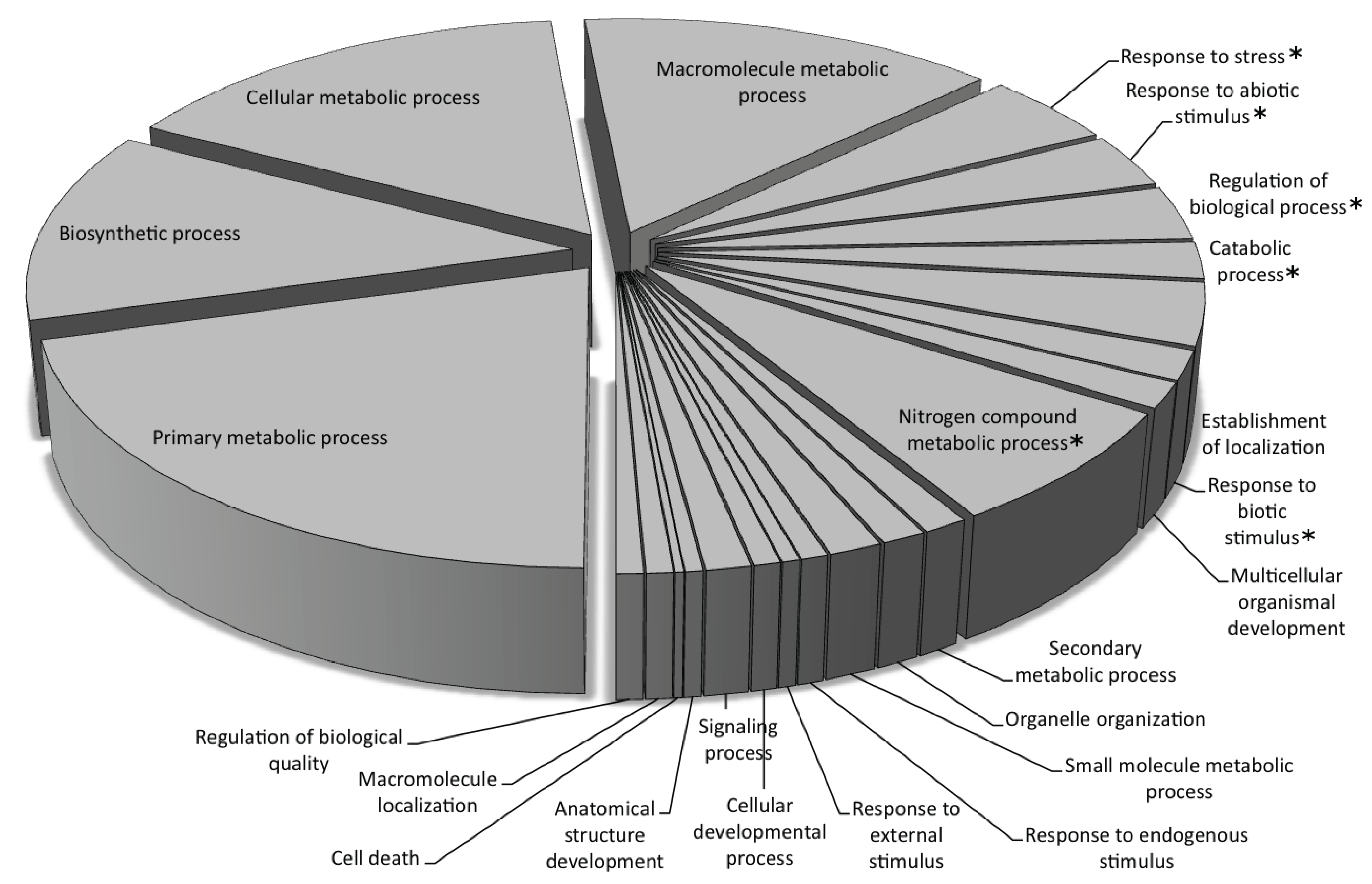
Gene Ontology Annotation of the Predicted Genes
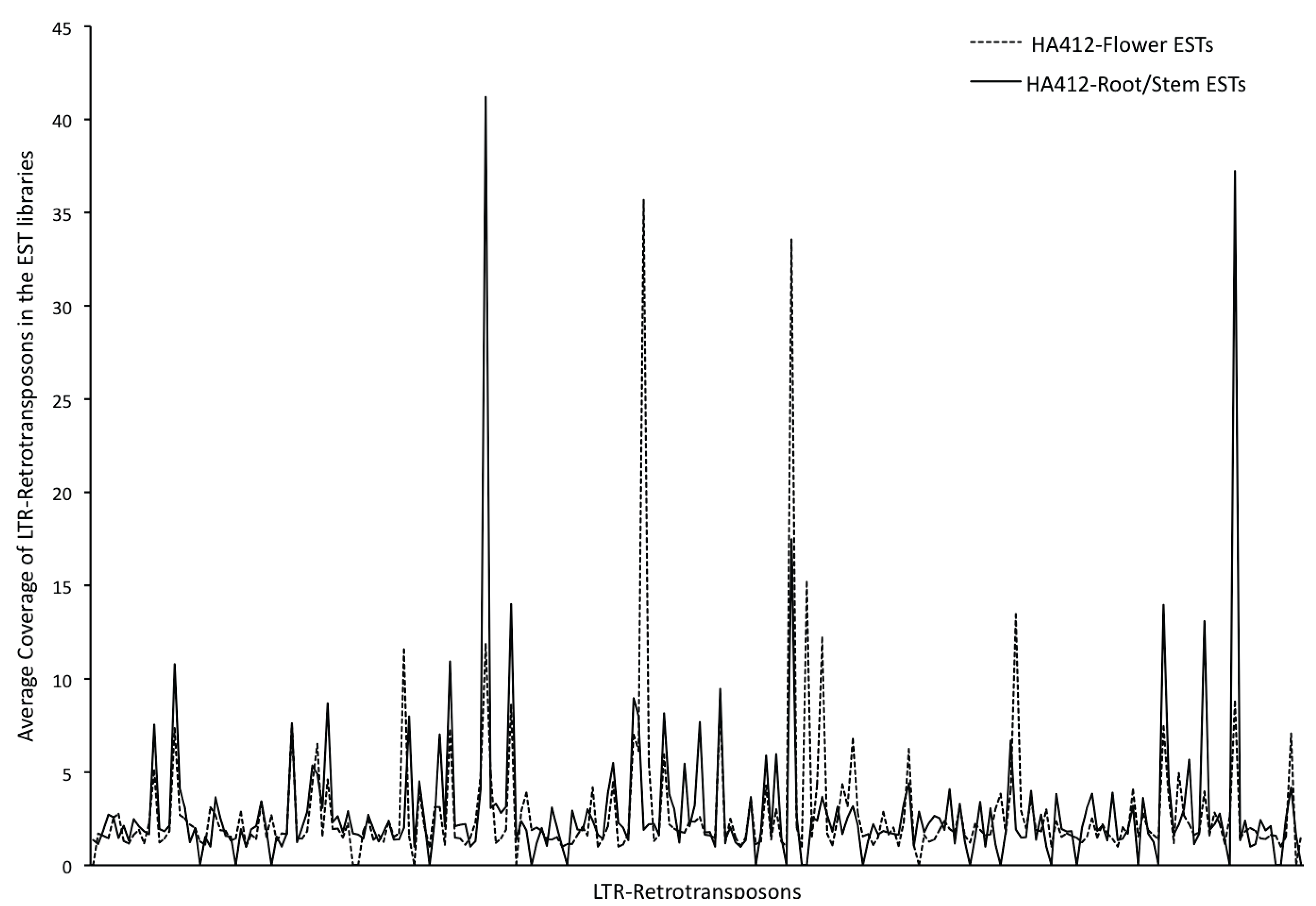
3.3. Transcriptional Activity of the Repetitive Elements in the Sunflower Genome
4. Discussion

5. Conclusions
Supplementary Files
Acknowledgements
Author Contributions
Conflicts of Interest
References
- FAO. Available online: http://www.fao.org/ (accessed on 13 August 2013).
- Harter, A.; Gardener, K.; Falush, D.; Lentz, D.; Bye, R.; Rieseberg, L. Origin of extant domesticated sunflowers in eastern north america. Nature 2004, 430, 201–205. [Google Scholar] [CrossRef]
- Blackman, B.; Scascitelli, M.; Kane, N.; Luton, H.; Rasmussen, D.; Bye, R.; Lentz, D.; Rieseberg, L. Sunflower domestication alleles support single domestication center in eastern north america. Proc. Natl. Acad. Sci. USA 2011, 108, 14360–14365. [Google Scholar]
- Smith, B.D. Origins of agriculture in eastern north-america. Science 1989, 246, 1566–1571. [Google Scholar]
- Stevens, P. Angiosperm Phylogeny. Available online: http://www.Mobot.Org/mobot/research/apweb/ (accessed on 15 August 2013).
- Baack, E.; Whitney, K.; Rieseberg, L. Hybridization and genome size evolution: Timing and magnitude of nuclear DNA content increases in helianthus homoploid hybrid species. New Phytol. 2005, 167, 623–630. [Google Scholar] [CrossRef]
- Cavallini, A.; Natali, L.; Zuccolo, A.; Giordani, T.; Jurman, I.; Ferrillo, V.; Vitacolonna, N.; Sarri, V.; Cattonaro, F.; Ceccarelli, M.; et al. Analysis of transposons and repeat composition of the sunflower (helianthus annuus l.) genome. Theor. Appl. Genet. 2009, 120, 491–508. [Google Scholar]
- Kane, N.C.; Gill, N.; King, M.G.; Bowers, J.E.; Berges, H.; Gouzy, J.; Bachlava, E.; Langlade, N.B.; Lai, Z.; Stewart, M. Progress towards a reference genome for sunflower. Botany 2011, 89, 429–437. [Google Scholar] [CrossRef]
- Staton, S.E.; Bakken, B.H.; Blackman, B.K.; Chapman, M.A.; Kane, N.C.; Tang, S.; Ungerer, M.C.; Knapp, S.J.; Rieseberg, L.H.; Burke, J.M. The sunflower (helianthus annuus l.) genome reflects a recent history of biased accumulation of transposable elements. Plant J. 2012, 72, 142–153. [Google Scholar] [CrossRef]
- Natali, L.; Cossu, R.; Barghini, E.; Giordani, T.; Buti, M.; Mascagni, F.; Morgante, M.; Gill, N.; Kane, N.; Rieseberg, L.; et al. The repetitive component of the sunflower genome: Comparative analyses using different approaches for assembling ngs reads. BMC Genomics 2013, 14, 686–699. [Google Scholar] [CrossRef]
- Xiao, H.; Jiang, N.; Schaffner, E.; Stockinger, E.; van der Knaap, E. A retrotransposon-mediated gene duplication underlies morphological variation of tomato fruit. Science 2008, 319, 1527–1530. [Google Scholar] [CrossRef]
- Studer, A.; Zhao, Q.; Ross-Ibarra, J.; Doebley, J. Identification of a functional transposon insertion in the maize domestication gene tb1. Nat. Genet. 2011, 43, 1160–1163. [Google Scholar] [CrossRef]
- Devos, K.; Brown, J.; Bennetzen, J. Genome size reduction through illegitimate recombination counteracts genome. Genome Res. 2002, 12, 1075–1079. [Google Scholar] [CrossRef]
- Piegu, B.; Guyot, R.; Picault, N.; Roulin, A.; Sanyal, A.; Kim, H.; Collura, K.; Brar, D.S.; Jackson, S.; Wing, R.A.; et al. Doubling genome size without polyploidization: Dynamics of retrotransposition-driven genomic expansions in oryza australiensis, a wild relative of rice. Genome Res. 2006, 16, 1262–1269. [Google Scholar] [CrossRef]
- Shapiro, J. Evolution: A View from the 21st Century; FT Press Science: Upper Saddle River, NJ, USA, 2011. [Google Scholar]
- Rieseberg, L.H. Chromosomal rearrangements and speciation. Trends Ecol. Evol. 2001, 16, 351–358. [Google Scholar] [CrossRef]
- Faria, R.; Navarro, A. Chromosomal speciation revisited: Rearranging theory with pieces of evidence. Trends Ecol. Evol. 2011, 25, 660–669. [Google Scholar] [CrossRef]
- Lynch, M. The frailty of adaptive hypotheses for the origins of organismal complexity. Proc. Natl. Acad. Sci. USA 2007, 104, 8597–8604. [Google Scholar] [CrossRef]
- Lynch, M.; Conery, J.S. The origins of genome complexity. Science 2003, 302, 1401–1404. [Google Scholar] [CrossRef]
- Lai, J.; Dey, N.; Kim, C.-S.; Bharti, A.K.; Rudd, S.; Mayer, K.F.X.; Larkins, B.A.; Becraft, P.; Messing, J. Characterization of the maize endosperm transcriptome and its comparison to the rice genome. Genome Res. 2004, 14, 1932–1937. [Google Scholar] [CrossRef]
- Messing, J.; Bharti, A.K.; Karlowski, W.M.; Gundlach, H.; Kim, H.R.; Yu, Y.; Wei, F.; Fuks, G.; Soderlund, C.A.; Mayer, K.F.X.; et al. Sequence composition and genome organization of maize. Proc. Natl. Acad. Sci. USA 2004, 101, 14349–14354. [Google Scholar] [CrossRef]
- Barker, M.S.; Kane, N.C.; Matvienko, M.; Kozik, A.; Michelmore, R.W.; Knapp, S.J.; Rieseberg, L.H. Multiple paleopolyploidizations during the evolution of the compositae reveal parallel patterns of duplicate gene retention after millions of years. Mol. Biol. Evol. 2008, 25, 2445–2455. [Google Scholar] [CrossRef]
- Osborn, T.C.; Chris Pires, J.; Birchler, J.A.; Auger, D.L.; Jeffery Chen, Z.; Lee, H.-S.; Comai, L.; Madlung, A.; Doerge, R.W.; Colot, V.; et al. Understanding mechanisms of novel gene expression in polyploids. Trends Genet. 2003, 19, 141–147. [Google Scholar]
- Adams, K.L.; Wendel, J.F. Polyploidy and genome evolution in plants. Curr. Opin. Plant Biol. 2005, 8, 135–141. [Google Scholar] [CrossRef]
- Scascitelli, M.; Cognet, M.; Adams, K.L. An interspecific plant hybrid shows novel changes in parental splice forms of genes for splicing factors. Genetics 2010, 184, 975–983. [Google Scholar] [CrossRef]
- The Arabidopsis Genome Initiative. Analysis of the genome sequence of the flowering plant arabidopsis thaliana. Nature 2000, 408, 796–815. [Google Scholar] [CrossRef]
- International Rice Genome Sequencing Project. The map-based sequence of the rice genome. Nature 2005, 436, 793–800. [Google Scholar] [CrossRef]
- Schnable, P.S.; Ware, D.; Fulton, R.S.; Stein, J.C.; Wei, F.; Pasternak, S.; Liang, C.; Zhang, J.; Fulton, L.; Graves, T.A.; et al. The b73 maize genome: Complexity, diversity, and dynamics. Science 2009, 326, 1112–1115. [Google Scholar] [CrossRef]
- Schmutz, J.; Cannon, S.B.; Schlueter, J.; Ma, J.; Mitros, T.; Nelson, W.; Hyten, D.L.; Song, Q.; Thelen, J.J.; Cheng, J.; et al. Genome sequence of the palaeopolyploid soybean. Nature 2010, 463, 178–183. [Google Scholar] [CrossRef]
- Banks, J.A.; Nishiyama, T.; Hasebe, M.; Bowman, J.L.; Gribskov, M.; dePamphilis, C.; Albert, V.A.; Aono, N.; Aoyama, T.; Ambrose, B.A.; et al. The selaginella genome identifies genetic changes associated with the evolution of vascular plants. Science 2011, 332, 960–963. [Google Scholar] [CrossRef]
- Wicker, T.; Sabot, F.; Hua-Van, A.; Bennetzen, J.L.; Capy, P.; Chalhoub, B.; Flavell, A.; Leroy, P.; Morgante, M.; Panaud, O.; et al. A unified classification system for eukaryotic transposable elements. Nat. Rev. Genet. 2007, 8, 973–982. [Google Scholar] [CrossRef]
- Santini, S.; Cavallini, A.; Natali, L.; Minelli, S.; Maggini, F.; Cionini, P. Ty1/copia- and ty3/gypsy-like DNA sequences in helianthus species. Chromosoma 2002, 111, 192–200. [Google Scholar] [CrossRef]
- Natali, L.; Santini, S.; Giordani, T.; Minelli, S.; Maestrini, P.; Cionini, P.G.; Cavallini, A. Distribution of ty3-gypsy- and ty1-copia-like DNA sequences in the genus helianthus and other asteraceae. Genome 2006, 49, 64–72. [Google Scholar] [CrossRef]
- Vukich, M.; Giordani, T.; Natali, L.; Cavallini, A. Copia and gypsy retrotransposons activity in sunflower (helianthus annuus l.). BMC Plant Biol. 2009, 9, 150. [Google Scholar] [CrossRef]
- Staton, S.E.; Ungerer, M.C.; Moore, R.C. The genomic organization of ty3/gypsy-like retrotransposons in helianthus (asteraceae) homoploid hybrid species. Am. J. Bot. 2009, 96, 1646–1655. [Google Scholar] [CrossRef]
- Ungerer, M.C.; Strakosh, S.C.; Zhen, Y. Genome expansion in three hybrid sunflower species is associated with retrotransposon proliferation. Curr. Biol. 2006, 16, R872–R873. [Google Scholar] [CrossRef]
- Ungerer, M.; Strakosh, S.; Stimpson, K. Proliferation of ty3/gypsy-like retrotransposons in hybrid sunflower taxa inferred from phylogenetic data. BMC Biol. 2009, 7, 40. [Google Scholar] [CrossRef]
- Kawakami, T.; Strakosh, S.C.; Zhen, Y.; Ungerer, M.C. Different scales of ty1/copia-like retrotransposon proliferation in the genomes of three diploid hybrid sunflower species. Heredity 2010, 104, 341–350. [Google Scholar] [CrossRef]
- CNRGV: The French Plant Genome Resource Center. Available online: http://cnrgv.toulouse.inra.fr/en/library/sunflower/ (accessed on 14 October 2012).
- Huang, X.; Madan, A. Cap3: A DNA sequence assembly program. Genome Res. 1999, 9, 868–877. [Google Scholar] [CrossRef]
- CLC Genomics Workbench. Available online: http://www.clcbio.com/products/clc-genomics-workbench/ (accessed on 13 April 2011).
- Boetzer, M.; Henkel, C.V.; Jansen, H.J.; Butler, D.; Pirovano, W. Scaffolding pre-assembled contigs using sspace. Bioinformatics 2011, 27, 578–579. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Fast and accurate short read alignment with burrows-wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R.; 1000 Genome Project Data Processing Subgroup. The sequence alignment/map format and samtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar]
- Bao, Z.; Eddy, S.R. Automated de novo identification of repeat sequence families in sequenced genomes. Genome Res. 2002, 12, 1269–1276. [Google Scholar] [CrossRef]
- Price, A.L.; Jones, N.C.; Pevzner, P.A. De novo identification of repeat families in large genomes. Bioinformatics 2005, 21, i351–i358. [Google Scholar] [CrossRef]
- Smit, A.F.A.; Hubley, R.; Green, P. Repeatmasker open-3.0. Available online: http://www.repeatmasker.org/ (accessed on 29 August 2013).
- Altschul, S.F.; Madden, T.L.; Schaffer, A.A.; Zhang, J.; Zhang, Z.; Miller, W.; Lipman, D.J. Gapped blast and psi-blast: A new generation of protein database search programs. Nucleic Acids Res. 1997, 25, 3389–3402. [Google Scholar] [CrossRef]
- TransposonPsi. Available online: http://transposonpsi.sourceforge.net/ (accessed on 12 December 2012).
- Ouyang, S.; Buell, C.R. The tigr plant repeat databases: A collective resource for the identification of repetitive sequences in plants. Nucleic Acids Res. 2004, 32, D360–D363. [Google Scholar] [CrossRef]
- Blastclust. Available online: ftp://ftp.ncbi.nih.gov/blast/documents/blastclust.html/ (accessed on 15 August 2013).
- Kurtz, S.; Narechania, A.; Stein, J.; Ware, D. A new method to compute k-mer frequencies and its application to annotate large repetitive plant genomes. BMC Genomics 2008, 9, 517. [Google Scholar] [CrossRef]
- Abouelhoda, M.I.; Kurtz, S.; Ohlebusch, E. Replacing suffix trees with enhanced suffix arrays. J. Discret. Algorithms 2004, 2, 53–86. [Google Scholar] [CrossRef]
- Xu, Z.; Wang, H. Ltr_finder: An efficient tool for the prediction of full-length ltr retrotransposons. Nucleic Acids Res. 2007, 35, W265–W268. [Google Scholar] [CrossRef]
- Sonnhammer, E.L.L.; Durbin, R. A dot-matrix program with dynamic threshold control suited for genomic DNA and protein sequence analysis. Gene 1995, 167, GC1–GC10. [Google Scholar] [CrossRef]
- Thompson, J.D.; Higgins, D.G.; Gibson, T.J. Clustal w: Improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994, 22, 4673–4680. [Google Scholar] [CrossRef]
- TE nest. Available online: http://www.plantgdb.org/tool/TE_nest/ (accessed on 20 August 2013).
- Huang, X.; Miller, W. Lalign-find the best local alignments between two sequences. Adv. Appl. Math. 1991, 12, 373. [Google Scholar]
- Librado, P.; Rozas, J. Dnasp v5: A software for comprehensive analysis of DNA polymorphism data. Bioinformatics 2009, 25, 1451–1452. [Google Scholar] [CrossRef]
- Kimura, M. A simple method for estimating evolutionary rates of base substitutions through comparative studies of nucleotide sequences. J. Mol. Evol. 1980, 16, 111–120. [Google Scholar] [CrossRef]
- Kapitonov, V.; Jurka, J. The age of alu subfamilies. J. Mol. Evol. 1996, 42, 59–65. [Google Scholar] [CrossRef]
- Smit, A.F.A.; Toth, G.; Riggs, A.D.; Jurka, J. Ancestral, mammalian-wide subfamilies of line-1 repetitive sequences. J. Mol. Biol. 1995, 246, 401–417. [Google Scholar] [CrossRef]
- Feschotte, C.; Pritham, E.J. Computational analysis and paleogenomics of interspersed repeats in eukaryotes. In Computational genomics: Current Methods; Stojanovic, N., Ed.; Horizon Bioscience: Wymondham, Norfolk, UK, 2007; pp. 31–53. [Google Scholar]
- Stanke, M.; Diekhans, M.; Baertsch, R.; Haussler, D. Using native and syntenically mapped cdna alignments to improve de novo gene finding. Bioinformatics 2008, 24, 637–644. [Google Scholar] [CrossRef]
- Conesa, A.; Gotz, S.; Garcia-Gomez, J.M.; Terol, J.; Talon, M.; Robles, M. Blast2go: A universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 2005, 21, 3674–3676. [Google Scholar] [CrossRef]
- The Compositae Est Assembly. Available online: http://cgpdb.ucdavis.edu/asteraceae_assembly/ (accessed on September 29 2012).
- Msu Rice Genome Annotation Project. Available online: ftp://ftp.plantbiology.msu.edu/pub/data/ Eukaryotic_Projects/o_sativa/annotation_dbs/pseudomolecules/version_7.0/all.dir/ (accessed on 16 September 2012).
- The Arabidopsis Information Resource (Tair). Available online: ftp://ftp.Arabidopsis.org/home/ tair/Sequences/whole_chromosomes/ (accessed on 16 September 2012).
- The Maize Genome Sequencing Project. Available online: http://ftp.maizesequence.org/ (accessed on 16 September 2012).
- Selaginella Moellendorffii v1.0. Available online: http://genome.jgi-psf.org/Selmo1/Selmo1.download.ftp.html/ (accessed on 16 September 2012).
- Edgar, R.C. Muscle: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef]
- Waterhouse, A.M.; Procter, J.B.; Martin, D.M.A.; Clamp, M.L.; Barton, G.J. Jalview version 2-a multiple sequence alignment editor and analysis workbench. Bioinformatics 2009, 25, 1189–1191. [Google Scholar] [CrossRef]
- Zuckerkandl, E.; Pauling, L. Evolutionary Divergence and Convergence in Proteins; Academic Press: New York, NY, USA, 1965. [Google Scholar]
- Saitou, N.; Nei, M. The neighbor-joining method: A new method for reconstructing phylogenetic trees. Mol. Biol. Evol. 1987, 4, 406–425. [Google Scholar]
- Tamura, K.; Peterson, D.; Peterson, N.; Stecher, G.; Nei, M.; Kumar, S. Mega5: Molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol. Biol. Evol. 2011, 28, 2731–2739. [Google Scholar] [CrossRef]
- Daniel, W.W. Applied Nonparametric Statistics; PWS Kent: Boston, MA, USA, 1990. [Google Scholar]
- SanMiguel, P.; Gaut, B.S.; Tikhonov, A.; Nakajima, Y.; Bennetzen, J.L. The paleontology of intergene retrotransposons of maize. Nat. Genet. 1998, 20, 43–45. [Google Scholar] [CrossRef]
- SanMiguel, P.; Tikhonov, A.; Jin, Y.-K.; Motchoulskaia, N.; Zakharov, D.; Melake-Berhan, A.; Springer, P.S.; Edwards, K.J.; Lee, M.; Avramova, Z.; et al. Nested retrotransposons in the intergenic regions of the maize genome. Science 1996, 274, 765–768. [Google Scholar] [CrossRef]
- Bennetzen, J.L.; SanMiguel, P.; Chen, M.; Tikhonov, A.; Francki, M.; Avramova, Z. Grass genomes. Proc. Natl. Acad. Sci. USA 1998, 95, 1975–1978. [Google Scholar]
- Jurka, J.; Milosavljevic, A. Reconstruction and analysis of human alu genes. J. Mol. Evol. 1991, 32, 105–121. [Google Scholar] [CrossRef]
- Meyers, B.C.; Tingey, S.V.; Morgante, M. Abundance, distribution, and transcriptional activity of repetitive elements in the maize genome. Genome Res. 2001, 11, 1660–1676. [Google Scholar] [CrossRef]
- Omnimapfree. Available online: http://www.omnimapfree.org/ (accessed on 14 October 2012).
- Gill, N.; SanMiguel, P.; Dhillon, B.; Abernathy, B.; Kim, H.; Stein, L.; Ware, D.; Wing, R.; Jackson, S. Dynamic oryza genomes: Repetitive DNA sequences as genome modeling agents. Rice 2010, 3, 251–269. [Google Scholar] [CrossRef]
- Ammiraju, J.S.S.; Zuccolo, A.; Yu, Y.; Song, X.; Piegu, B.; Chevalier, F.; Walling, J.G.; Ma, J.; Talag, J.; Brar, D.S.; et al. Evolutionary dynamics of an ancient retrotransposon family provides insights into evolution of genome size in the genus oryza. Plant J. 2007, 52, 342–351. [Google Scholar] [CrossRef]
- SanMiguel, P.; Bennetzen, J.L. Evidence that a recent increase in maize genome size was caused by the massive amplification of intergene retrotransposons. Ann. Bot. 1998, 82, 37–44. [Google Scholar] [CrossRef]
- Hawkins, J.S.; Kim, H.; Nason, J.D.; Wing, R.A.; Wendel, J.F. Differential lineage-specific amplification of transposable elements is responsible for genome size variation in gossypium. Genome Res. 2006, 16, 1252–1261. [Google Scholar] [CrossRef]
- Ma, J.; Bennetzen, J.L. Rapid recent growth and divergence of rice nuclear genomes. Proc. Natl. Acad. Sci. USA 2004, 101, 12404–12410. [Google Scholar] [CrossRef]
- Sprent, P.; Smeeton, N.C. Applied Nonparametric Statistical Methods, 4th ed.; Taylor & Francis: Boca Raton, FL, USA, 2007. [Google Scholar]
- Thon, M.; Pan, H.; Diener, S.; Papalas, J.; Taro, A.; Mitchell, T.; Dean, R. The role of transposable element clusters in genome evolution and loss of synteny in the rice blast fungus magnaporthe oryzae. Genome Biol. 2006, 7, R16. [Google Scholar] [CrossRef]
- Wei, L.; Xiao, M.; An, Z.; Ma, B.; Mason, A.S.; Qian, W.; Li, J.; Fu, D. New insights into nested long terminal repeat retrotransposons in brassica species. Mol. Plant 2013, 6, 470–482. [Google Scholar] [CrossRef]
- Gao, C.; Xiao, M.; Ren, X.; Hayward, A.; Yin, J.; Wu, L.; Fu, D.; Li, J. Characterization and functional annotation of nested transposable elements in eukaryotic genomes. Genomics 2012, 100, 222–230. [Google Scholar] [CrossRef]
- McClintock, B. The Discovery and Character of Transposable Elements: The Collected Papers (1938–1984) of Barbara Mcclintock; Garland Publishing, Incorporated: New York, NY, USA, 1987. [Google Scholar]
- McClintock, B. The significance of responses of the genome to challenge. Science 1984, 226, 792–801. [Google Scholar]
- Shapiro, J. Mobile DNA and evolution in the 21st century. Mobile DNA 2010, 1, 4. [Google Scholar] [CrossRef]
- Ma, J.; SanMiguel, P.; Lai, J.; Messing, J.; Bennetzen, J.L. DNA rearrangement in orthologous orp regions of the maize, rice and sorghum genomes. Genetics 2005, 170, 1209–1220. [Google Scholar] [CrossRef]
- Vitte, C.; Panaud, O. Formation of solo-ltrs through unequal homologous recombination counterbalances amplifications of ltr retrotransposons in rice oryza sativa l. Mol. Biol. Evol. 2003, 20, 528–540. [Google Scholar] [CrossRef]
- Kulathinal, R.J.; Stevison, L.S.; Noor, M.A.F. The genomics of speciation in drosophila: Diversity, divergence, and introgression estimated using low-coverage genome sequencing. PLoS Genet. 2009, 5, e1000550. [Google Scholar] [CrossRef]
- Bennetzen, J.L.; Kellogg, E.A. Do plants have a one-way ticket to genomic obesity? Plant Cell Online 1997, 9, 1509–1514. [Google Scholar]
- Vitte, C.; Panaud, O. Ltr retrotransposons and flowering plant genome size: Emergence of the increase/decrease model. Cytogenet. Genome Res. 2005, 110, 91–107. [Google Scholar] [CrossRef]
Supplementary Materials
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Gill, N.; Buti, M.; Kane, N.; Bellec, A.; Helmstetter, N.; Berges, H.; Rieseberg, L.H. Sequence-Based Analysis of Structural Organization and Composition of the Cultivated Sunflower (Helianthus annuus L.) Genome. Biology 2014, 3, 295-319. https://doi.org/10.3390/biology3020295
Gill N, Buti M, Kane N, Bellec A, Helmstetter N, Berges H, Rieseberg LH. Sequence-Based Analysis of Structural Organization and Composition of the Cultivated Sunflower (Helianthus annuus L.) Genome. Biology. 2014; 3(2):295-319. https://doi.org/10.3390/biology3020295
Chicago/Turabian StyleGill, Navdeep, Matteo Buti, Nolan Kane, Arnaud Bellec, Nicolas Helmstetter, Hélène Berges, and Loren H. Rieseberg. 2014. "Sequence-Based Analysis of Structural Organization and Composition of the Cultivated Sunflower (Helianthus annuus L.) Genome" Biology 3, no. 2: 295-319. https://doi.org/10.3390/biology3020295
APA StyleGill, N., Buti, M., Kane, N., Bellec, A., Helmstetter, N., Berges, H., & Rieseberg, L. H. (2014). Sequence-Based Analysis of Structural Organization and Composition of the Cultivated Sunflower (Helianthus annuus L.) Genome. Biology, 3(2), 295-319. https://doi.org/10.3390/biology3020295