R-Based Software for the Integration of Pathway Data into Bioinformatic Algorithms

Abstract
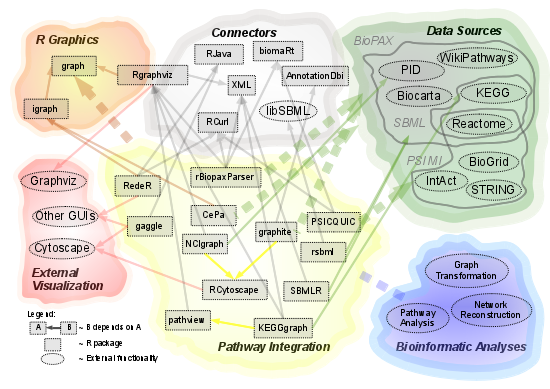
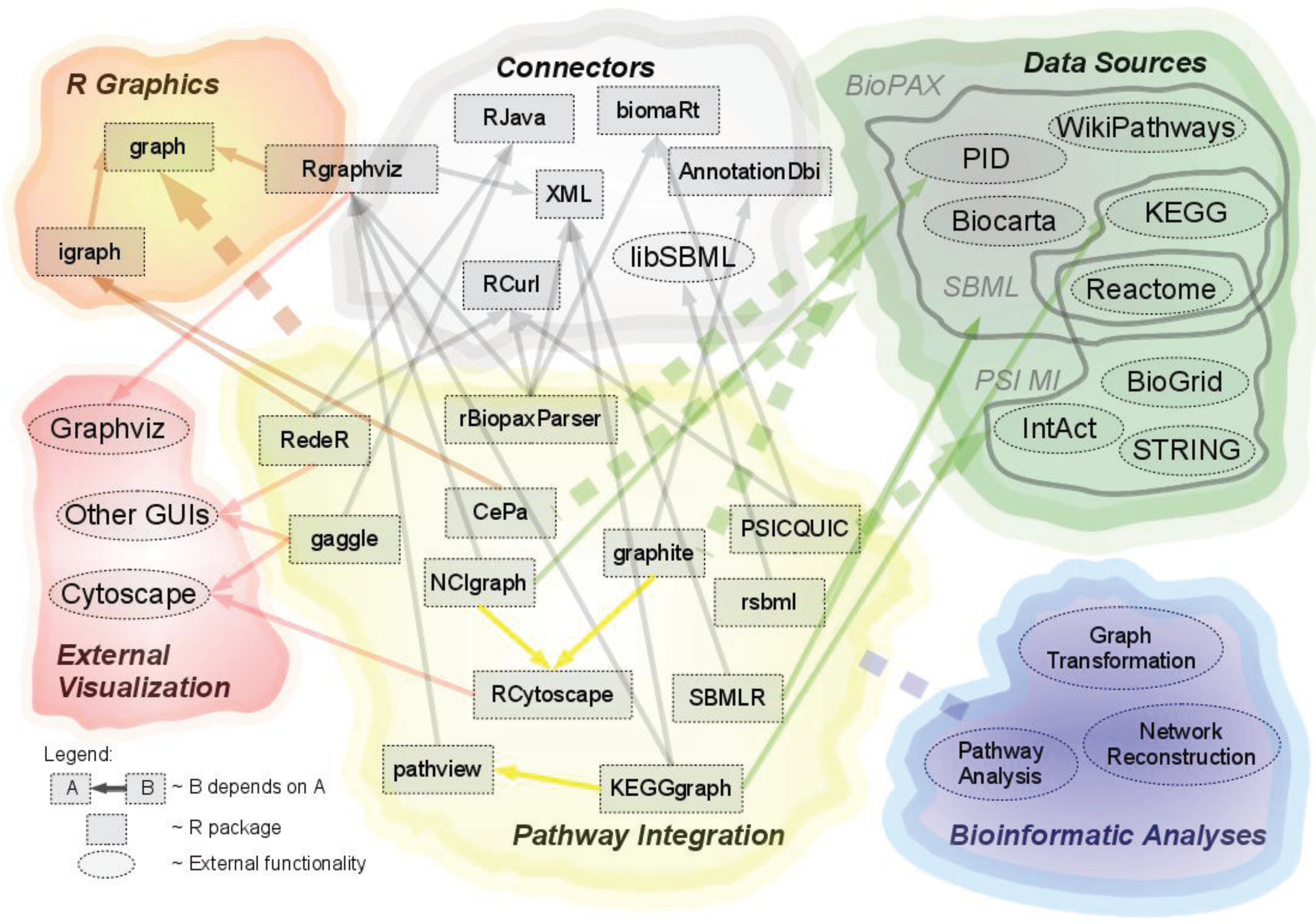
:
1. Introduction
1.1. Modeling Pathway Knowledge
1.2. Pathway Databases
1.3. Tools for Pathway Curation and Analysis
1.4. R Framework for Statistical Computing
2. Methods Section
2.1. Overview of Available Packages

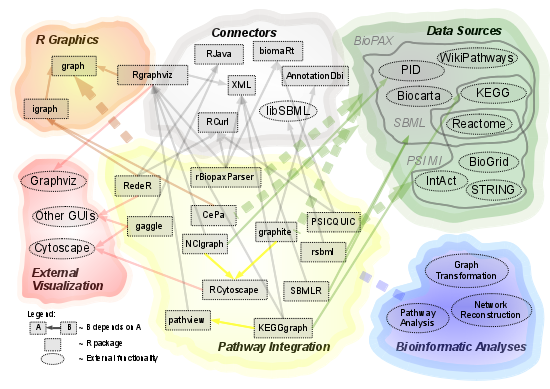{kind=link}
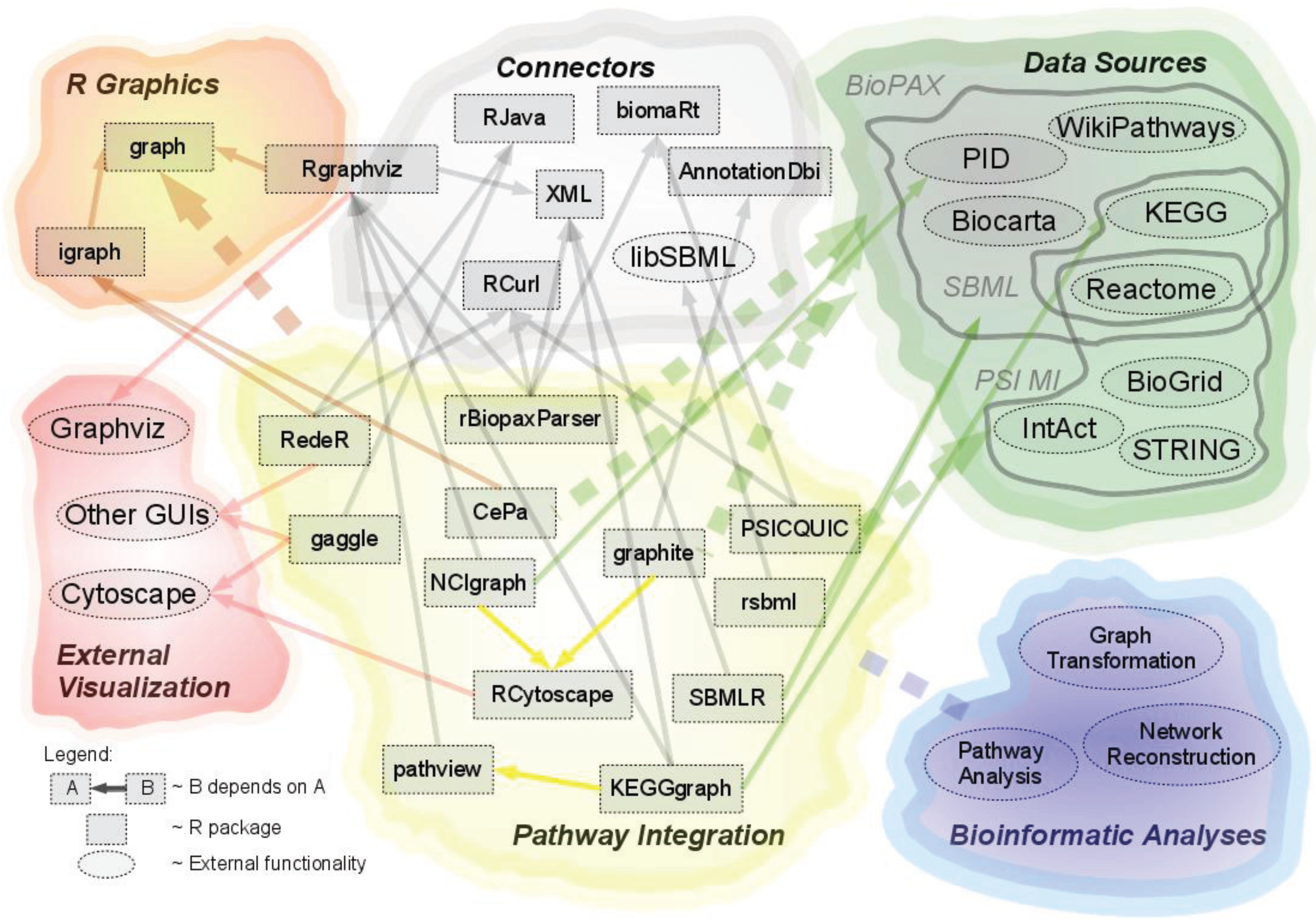{kind=link}
| Package Name | Data Source | Data Import | Dependencies | Further Analyses | Visualization |
|---|---|---|---|---|---|
| rBiopaxParser | generic BioPAX parser; all BioPAX databases | gene sets, directed graphs, full annotation | XML, biomaRt | Rgraphviz | |
| graphite | includes KEGG, BioCarta, PID, Reactome, SPIKE | gene sets, directed graphs, mapping and converting IDs | AnnotationDbi | Pathway analyses: clipper, SPIA | Cytoscape |
| NCIgraph | load PID data via Cytoscape | graph objects with directed edges | Java, Cytoscape | Rgraphviz | |
| pathview | load data via KEGGgraph | gene sets with graph layout annotation | KEGGgraph | Pathway analyses: gage | Rgraphviz + native KEGG |
| KEGGgraph | generic KGML parser, KEGG | graph objects with directed edges | XML, biomaRt | Rgraphviz | |
| RedeR | igraph objects | Java | Java GUI | ||
| SBMLR | generic SBML parser, limited functionality | list of SBML class instances | XML | deSolve | - |
| rsbml | generic SBML parser | graph objects | libSBML | SBML ODE Solver Library (SOSLib) | Rgraphviz |
| RCytoscape | load data via Cytoscape, R | graphNEL objects | Java, Cytoscape | Cytoscape | |
| Gaggle | load data via Gaggle server | graph objects with directed edges | Gaggle | - | |
| CePa | includes KEGG, BioCarta, PID, Reactome | igraph objects | igraph | Pathway analyses.GSEA, ORA | igraph |
| PSICQUIC | PSI MI-QL compliant databases | list of interactions | RCurl |
2.2. Source of Integrated Pathway Data
2.3. Internal Data Model
2.4. Dependency on External Tools
2.5. Integration with Further Analysis Steps
2.6. Visualization of Pathway Data
3. Summary
4. Conclusions
Author Contributions
Conflicts of Interest
References and Notes
- Beißbarth, T.; Speed, T.P. GOstat: Find statistically overrepresented Gene Ontologies within a group of genes. Bioinformatics 2004, 20, 1464–1465. [Google Scholar] [CrossRef]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; Mesirov, J.P. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef]
- Tarca, A.L.; Draghici, S.; Khatri, P.; Hassan, S.S.; Mittal, P.; Kim, J.; Kim, C.J.; Kusanovic, J.P.; Romero, R. A novel signaling pathway impact analysis. Bioinformatics 2009, 25, 75–82. [Google Scholar] [CrossRef]
- Martini, P.; Sales, G.; Massa, M.S.; Chiogna, M.; Romualdi, C. Along signal paths: an empirical gene set approach exploiting pathway topology. Nucleic Acids Res. 2012, 41, 19. [Google Scholar]
- Dutta, B.; Wallqvist, A.; Reifman, J. PathNet: A tool for pathway analysis using topological information. Source Code Biol. Med. 2012, 7, 10. [Google Scholar] [CrossRef]
- Beissbarth, T. Interpreting Experimental Results Using Gene Ontologies. In Methods in Enzymology; Kimmel, A., Oliver, B., Eds.; Academic Press: Salt Lake City, UT, USA, 2006; Volume 411, pp. 340–352. [Google Scholar]
- Fröhlich, H.; Beißbarth, T.; Tresch, A.; Kostka, D.; Jacob, J.; Spang, R.; Markowetz, F. Analyzing gene perturbation screens with nested effects models in R and bioconductor. Bioinformatics 2008, 24, 2549–2550. [Google Scholar] [CrossRef]
- Gade, S.; Porzelius, C.; Fälth, M.; Brase, J.C.; Wuttig, D.; Kuner, R.; Binder, H.; Sültmann, H.; Beißbarth, T. Graph based fusion of miRNA and mRNA expression data improves clinical outcome prediction in prostate cancer. BMC Bioinformatics 2011, 12, 488. [Google Scholar] [CrossRef]
- Johannes, M.; Brase, J.C.; Fröhlich, H.; Gade, S.; Gehrmann, M.; Fälth, M.; Sültmann, H.; Beißbarth, T. Integration of pathway knowledge into a reweighted recursive feature elimination approach for risk stratification of cancer patients. Bioinformatics 2010, 26, 2136–2144. [Google Scholar] [CrossRef]
- Kanehisa, M.; Goto, S.; Kawashima, S.; Okuno, Y.; Hattori, M. The KEGG resource for deciphering the genome. Nucleic Acids Res. 2004, 32, D277–D280. [Google Scholar] [CrossRef]
- Hucka, M.; Finney, A.; Sauro, H.M.; Bolouri, H.; Doyle, J.C.; Kitano, H.; Arkin, A.P.; Bornstein, B.J.; Bray, D.; Cornish-Bowden, A.; Cuellar, A.A.; Dronov, S.; Gilles, E.D.; Ginkel, M.; Gor, V.; Goryanin, I.I.; Hedley, W.J.; Hodgman, T.C.; Hofmeyr, J.-H.; Hunter, P.J.; Juty, N.S.; Kasberger, J.L.; Kremling, A.; Kummer, U.; Novère, N.L.; Loew, L.M.; Lucio, D.; Mendes, P.; Minch, E.; Mjolsness, E.D.; Nakayama, Y.; Nelson, M.R.; Nielsen, P.F.; Sakurada, T.; Schaff, J.C.; Shapiro, B.E.; Shimizu, T.S.; Spence, H.D.; Stelling, J.; Takahashi, K.; Tomita, M.; Wagner, J.; Wang, J. The systems biology markup language (SBML): A medium for representation and exchange of biochemical network models. Bioinformatics 2003, 19, 524–531. [Google Scholar] [CrossRef]
- Demir, E.; Cary, M.P.; Paley, S.; Fukuda, K.; Lemer, C.; Vastrik, I.; Wu, G.; D’Eustachio, P.; Schaefer, C.; Luciano, J.; Schacherer, F.; Martinez-Flores, I.; Hu, Z.; Jimenez-Jacinto, V.; Joshi-Tope, G.; Kandasamy, K.; Lopez-Fuentes, A.C.; Mi, H.; Pichler, E.; Rodchenkov, I.; Splendiani, A.; Tkachev, S.; Zucker, J.; Gopinath, G.; Rajasimha, H.; Ramakrishnan, R.; Shah, I.; Syed, M.; Anwar, N.; Babur, Ö.; Blinov, M.; Brauner, E.; Corwin, D.; Donaldson, S.; Gibbons, F.; Goldberg, R.; Hornbeck, P.; Luna, A.; Murray-Rust, P.; Neumann, E.; Ruebenacker, O.; Samwald, M.; Van Iersel, M.; Wimalaratne, S.; Allen, K.; Braun, B.; Whirl-Carrillo, M.; Cheung, K.-H.; Dahlquist, K.; Finney, A.; Gillespie, M.; Glass, E.; Gong, L.; Haw, R.; Honig, M.; Hubaut, O.; Kane, D.; Krupa, S.; Kutmon, M.; Leonard, J.; Marks, D.; Merberg, D.; Petri, V.; Pico, A.; Ravenscroft, D.; Ren, L.; Shah, N.; Sunshine, M.; Tang, R.; Whaley, R.; Letovksy, S.; Buetow, K.H.; Rzhetsky, A.; Schachter, V.; Sobral, B.S.; Dogrusoz, U.; McWeeney, S.; Aladjem, M.; Birney, E.; Collado-Vides, J.; Goto, S.; Hucka, M.; Novère, N.L.; Maltsev, N.; Pandey, A.; Thomas, P.; Wingender, E.; Karp, P.D.; Sander, C.; Bader, G.D. The BioPAX community standard for pathway data sharing. Nat. Biotechnol. 2010, 28, 935–942. [Google Scholar] [CrossRef]
- Cary, M.P.; Bader, G.D.; Sander, C. Pathway information for systems biology. FEBS Lett. 2005, 579, 1815–1820. [Google Scholar] [CrossRef]
- Hermjakob, H.; Montecchi-Palazzi, L.; Bader, G.; Wojcik, J.; Salwinski, L.; Ceol, A.; Moore, S.; Orchard, S.; Sarkans, U.; von Mering, C.; Roechert, B.; Poux, S.; Jung, E.; Mersch, H.; Kersey, P.; Lappe, M.; Li, Y.; Zeng, R.; Rana, D.; Nikolski, M.; Husi, H.; Brun, C.; Shanker, K.; Grant, S.G. N.; Sander, C.; Bork, P.; Zhu, W.; Pandey, A.; Brazma, A.; Jacq, B.; Vidal, M.; Sherman, D.; Legrain, P.; Cesareni, G.; Xenarios, I.; Eisenberg, D.; Steipe, B.; Hogue, C.; Apweiler, R. The HUPO PSI’s Molecular Interaction format?a community standard for the representation of protein interaction data. Nat. Biotechnol. 2004, 22, 177–183. [Google Scholar] [CrossRef]
- Kerrien, S.; Orchard, S.; Montecchi-Palazzi, L.; Aranda, B.; Quinn, A.F.; Vinod, N.; Bader, G.D.; Xenarios, I.; Wojcik, J.; Sherman, D.; Tyers, M.; Salama, J.J.; Moore, S.; Ceol, A.; Chatr-aryamontri, A.; Oesterheld, M.; Stümpflen, V.; Salwinski, L.; Nerothin, J.; Cerami, E.; Cusick, M.E.; Vidal, M.; Gilson, M.; Armstrong, J.; Woollard, P.; Hogue, C.; Eisenberg, D.; Cesareni, G.; Apweiler, R.; Hermjakob, H. Broadening the horizon—Level 2.5 of the HUPO-PSI format for molecular interactions. BMC Biol. 2007, 5, 44. [Google Scholar] [CrossRef]
- Aranda, B.; Blankenburg, H.; Kerrien, S.; Brinkman, F.S. L.; Ceol, A.; Chautard, E.; Dana, J.M.; De Las Rivas, J.; Dumousseau, M.; Galeota, E.; Gaulton, A.; Goll, J.; Hancock, R.E. W.; Isserlin, R.; Jimenez, R.C.; Kerssemakers, J.; Khadake, J.; Lynn, D.J.; Michaut, M.; O’Kelly, G.; Ono, K.; Orchard, S.; Prieto, C.; Razick, S.; Rigina, O.; Salwinski, L.; Simonovic, M.; Velankar, S.; Winter, A.; Wu, G.; Bader, G.D.; Cesareni, G.; Donaldson, I.M.; Eisenberg, D.; Kleywegt, G.J.; Overington, J.; Ricard-Blum, S.; Tyers, M.; Albrecht, M.; Hermjakob, H. PSICQUIC and PSISCORE: Accessing and scoring molecular interactions. Nat. Methods 2011, 8, 528–529. [Google Scholar] [CrossRef] [Green Version]
- Bader, G.D.; Cary, M.P.; Sander, C. Pathguide: a Pathway Resource List. Nucleic Acids Res. 2006, 34, D504–D506. [Google Scholar] [CrossRef]
- Croft, D.; O?Kelly, G.; Wu, G.; Haw, R.; Gillespie, M.; Matthews, L.; Caudy, M.; Garapati, P.; Gopinath, G.; Jassal, B.; Jupe, S.; Kalatskaya, I.; Mahajan, S.; May, B.; Ndegwa, N.; Schmidt, E.; Shamovsky, V.; Yung, C.; Birney, E.; Hermjakob, H.; D'Eustachio, P.; Stein, L. Reactome: A database of reactions, pathways and biological processes. Nucleic Acids Res. 2011, 39, D691–D697. [Google Scholar] [CrossRef]
- Schaefer, C.F.; Anthony, K.; Krupa, S.; Buchoff, J.; Day, M.; Hannay, T.; Buetow, K.H. PID: The Pathway Interaction Database. Nucleic Acids Res. 2009, 37, D674–D679. [Google Scholar] [CrossRef]
- Kelder, T.; van Iersel, M.P.; Hanspers, K.; Kutmon, M.; Conklin, B.R.; Evelo, C.T.; Pico, A.R. WikiPathways: building research communities on biological pathways. Nucleic Acids Res. 2011, 40, D1301–D1307. [Google Scholar]
- Bauer-Mehren, A.; Furlong, L.I.; Sanz, F. Pathway databases and tools for their exploitation: Benefits, current limitations and challenges. Mol. Syst. Biol. 2009, 5, 290. [Google Scholar]
- Funahashi, A.; Morohashi, M.; Kitano, H.; Tanimura, N. CellDesigner: A process diagram editor for gene-regulatory and biochemical networks. BIOSILICO 2003, 1, 159–162. [Google Scholar] [CrossRef]
- Iersel, M.P. van; Kelder, T.; Pico, A.R.; Hanspers, K.; Coort, S.; Conklin, B.R.; Evelo, C. Presenting and exploring biological pathways with PathVisio. BMC Bioinformatics 2008, 9, 399. [Google Scholar] [CrossRef]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A Software Environment for Integrated Models of Biomolecular Interaction Networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]
- Shannon, P.T.; Grimes, M.; Kutlu, B.; Bot, J.J.; Galas, D.J. RCytoscape: Tools for exploratory network analysis. BMC Bioinformatics 2013, 14, 217. [Google Scholar] [CrossRef]
- Lotia, S.; Montojo, J.; Dong, Y.; Bader, G.D.; Pico, A.R. Cytoscape app store. Bioinforma. Oxf. Engl. 2013, 29, 1350–1351. [Google Scholar] [CrossRef]
- Suderman, M.; Hallett, M. Tools for visually exploring biological networks. Bioinformatics 2007, 23, 2651–2659. [Google Scholar] [CrossRef]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2013. [Google Scholar]
- Fröhlich, H.; Fellmann, M.; Sültmann, H.; Poustka, A.; Beissbarth, T. Estimating large-scale signaling networks through nested effect models with intervention effects from microarray data. Bioinformatics 2008, 24, 2650–2656. [Google Scholar] [CrossRef]
- Bender, C.; Henjes, F.; Fröhlich, H.; Wiemann, S.; Korf, U.; Beißbarth, T. Dynamic deterministic effects propagation networks: learning signalling pathways from longitudinal protein array data. Bioinformatics 2010, 26, i596–i602. [Google Scholar] [CrossRef]
- Carey, V.J.; Gentry, J.; Whalen, E.; Gentleman, R. Network structures and algorithms in Bioconductor. Bioinformatics 2005, 21, 135–136. [Google Scholar] [CrossRef]
- Hornik, K. The Comprehensive R Archive Network. Wiley Interdiscip. Rev. Comput. Stat. 2012, 4, 394–398. [Google Scholar] [CrossRef]
- Gentleman, R.C.; Carey, V.J.; Bates, D.M.; Bolstad, B.; Dettling, M.; Dudoit, S.; Ellis, B.; Gautier, L.; Ge, Y.; Gentry, J.; Hornik, K.; Hothorn, T.; Huber, W.; Iacus, S.; Irizarry, R.; Leisch, F.; Li, C.; Maechler, M.; Rossini, A.J.; Sawitzki, G.; Smith, C.; Smyth, G.; Tierney, L.; Yang, J.Y.; Zhang, J. Bioconductor: open software development for computational biology and bioinformatics. Genome Biol. 2004, 5, R80. [Google Scholar] [CrossRef] [Green Version]
- Lang, D.T. The Omegahat Environment: New Possibilities for Statistical Computing. J. Comput. Graph. Stat. 2000, 9, 423–451. [Google Scholar]
- Ellson, J.; Gansner, E.; Koutsofios, L.; North, S.C.; Woodhull, G. Graphviz—Open Source Graph Drawing Tools. In Graph Drawing; Mutzel, P., Jünger, M., Leipert, S., Eds.; Springer: Berlin /Heidelberg, Germany, 2002; pp. 483–484. [Google Scholar]
- Shannon, P.T.; Reiss, D.J.; Bonneau, R.; Baliga, N.S. The Gaggle: An open-source software system for integrating bioinformatics software and data sources. BMC Bioinformatics 2006, 7, 176. [Google Scholar] [CrossRef]
- Sales, G.; Calura, E.; Cavalieri, D.; Romualdi, C. graphite - a Bioconductor package to convert pathway topology to gene network. BMC Bioinformatics 2012, 7, 176. [Google Scholar]
- Nishimura, D. BioCarta. Biotech Softw. Internet Rep. 2001, 2, 117–120. [Google Scholar] [CrossRef]
- Paz, A.; Brownstein, Z.; Ber, Y.; Bialik, S.; David, E.; Sagir, D.; Ulitsky, I.; Elkon, R.; Kimchi, A.; Avraham, K.B.; Shiloh, Y.; Shamir, R. SPIKE: A database of highly curated human signaling pathways. Nucleic Acids Res. 2011, 39, D793–D799. [Google Scholar] [CrossRef]
- Gu, Z.; Wang, J. CePa: An R package for finding significant pathways weighted by multiple network centralities. Bioinformatics 2013, 29, 658–660. [Google Scholar]
- Zhang, J.D.; Wiemann, S. KEGGgraph: A graph approach to KEGG PATHWAY in R and bioconductor. Bioinformatics 2009, 25, 1470–1471. [Google Scholar] [CrossRef]
- Luo, W.; Brouwer, C. Pathview: An R/Bioconductor package for pathway-based data integration and visualization. Bioinformatics 2013, 29, 1830–1831. [Google Scholar] [CrossRef]
- Radivoyevitch, T. A two-way interface between limited Systems Biology Markup Language and R. BMC Bioinformatics 2004, 5, 190. [Google Scholar] [CrossRef] [Green Version]
- Bornstein, B.J.; Keating, S.M.; Jouraku, A.; Hucka, M. LibSBML: An API Library for SBML. Bioinformatics 2008, 24, 880–881. [Google Scholar] [CrossRef]
- del-Toro, N.; Dumousseau, M.; Orchard, S.; Jimenez, R.C.; Galeota, E.; Launay, G.; Goll, J.; Breuer, K.; Ono, K.; Salwinski, L.; Hermjakob, H. A new reference implementation of the PSICQUIC web service. Nucleic Acids Res. 2013, 41, W601–W606. [Google Scholar] [CrossRef]
- Kramer, F.; Bayerlová, M.; Klemm, F.; Bleckmann, A.; Beißbarth, T. rBiopaxParser—An R package to parse, modify and visualize BioPAX data. Bioinformatics 2013, 29, 520–522. [Google Scholar] [CrossRef]
- Wang, X.; Terfve, C.; Rose, J.C.; Markowetz, F. HTSanalyzeR: An R/Bioconductor package for integrated network analysis of high-throughput screens. Bioinformatics 2011, 27, 879–880. [Google Scholar] [CrossRef]
- Smyth, G.K. Limma: Linear models for microarray data. In Bioinformatics and Computational Biology Solutions Using R and Bioconductor; Springer: Amsterdam, Netherlands, 2005; pp. 397–420. [Google Scholar]
- Wu, D.; Smyth, G.K. Camera: A competitive gene set test accounting for inter-gene correlation. Nucleic Acids Res. 2012, 40, e133–e133. [Google Scholar] [CrossRef]
- Geistlinger, L.; Csaba, G.; Küffner, R.; Mulder, N.; Zimmer, R. From sets to graphs: towards a realistic enrichment analysis of transcriptomic systems. Bioinformatics 2011, 27, i366–i373. [Google Scholar] [CrossRef]
- Chiromatzo, A.O.; Oliveira, T.Y.K.; Pereira, G.; Costa, A.Y.; Montesco, C.A.E.; Gras, D.E.; Yosetake, F.; Vilar, J.B.; Cervato, M.; Prado, P.R.R.; Cardenas, R.G.C.C.L.; Cerri, R.; Borges, R.L.; Lemos, R.N.; Alvarenga, S.M.; Perallis, V.R.C.; Pinheiro, D.G.; Silva, I.T.; Brandão, R.M.; Cunha, M.A.V.; Giuliatti, S.; Silva, W.A. miRNApath: A database of miRNAs, target genes and metabolic pathways. Genet. Mol. Res. GMR 2007, 6, 859–865. [Google Scholar]
- Tian, L.; Greenberg, S.A.; Kong, S.W.; Altschuler, J.; Kohane, I.S.; Park, P.J. Discovering statistically significant pathways in expression profiling studies. Proc. Natl. Acad. Sci. USA 2005, 102, 13544–13549. [Google Scholar] [CrossRef]
- ReactomePA. Available online: http://www.bioconductor.org/packages/2.13/bioc/html/ReactomePA.html (accessed on 28 November 2013).
- Baumbach, J.; Tauch, A.; Rahmann, S. Towards the integrated analysis, visualization and reconstruction of microbial gene regulatory networks. Brief. Bioinform. 2009, 10, 75–83. [Google Scholar]
- Zacher, B.; Abnaof, K.; Gade, S.; Younesi, E.; Tresch, A.; Fröhlich, H. Joint Bayesian inference of condition-specific miRNA and transcription factor activities from combined gene and microRNA expression data. Bioinformatics 2012, 28, 1714–1720. [Google Scholar] [CrossRef]
- Müssel, C.; Hopfensitz, M.; Kestler, H.A. BoolNet—An R package for generation, reconstruction and analysis of Boolean networks. Bioinformatics 2010, 26, 1378–1380. [Google Scholar] [CrossRef]
- Machné, R.; Finney, A.; Müller, S.; Lu, J.; Widder, S.; Flamm, C. The SBML ODE Solver Library: A native API for symbolic and fast numerical analysis of reaction networks. Bioinformatics 2006, 22, 1406–1407. [Google Scholar] [CrossRef]
- Soetaert, K.; Petzoldt, T.; Setzer, R.W. Solving differential equations in R: Package deSolve. J. Stat. Softw. 2010, 33, 1–25. [Google Scholar]
- Jacob, L.; Neuvial, P.; Dudoit, S. More power via graph-structured tests for differential expression of gene networks. Ann. Appl. Stat. 2012, 6, 561–600. [Google Scholar] [CrossRef]
- Castro, M.A.; Wang, X.; Fletcher, M.N.; Meyer, K.B.; Markowetz, F. RedeR: R/Bioconductor package for representing modular structures, nested networks and multiple levels of hierarchical associations. Genome Biol. 2012, 13, R29. [Google Scholar] [CrossRef]
- Huang, D.W.; Sherman, B.T.; Tan, Q.; Kir, J.; Liu, D.; Bryant, D.; Guo, Y.; Stephens, R.; Baseler, M.W.; Lane, H.C.; Lempicki, R.A. DAVID Bioinformatics Resources: Expanded annotation database and novel algorithms to better extract biology from large gene lists. Nucleic Acids Res. 2007, 35, W169–W175. [Google Scholar] [CrossRef]
- Sales, G.; Calura, E.; Martini, P.; Romualdi, C. Graphite Web: Web tool for gene set analysis exploiting pathway topology. Nucleic Acids Res. 2013, 41, W89–W97. [Google Scholar] [CrossRef]
- Wrzodek, C.; Büchel, F.; Ruff, M.; Dräger, A.; Zell, A. Precise generation of systems biology models from KEGG pathways. BMC Syst. Biol. 2013, 7, 15. [Google Scholar] [CrossRef]
- Strömbäck, L.; Lambrix, P. Representations of molecular pathways: an evaluation of SBML, PSI MI and BioPAX. Bioinformatics 2005, 21, 4401–4407. [Google Scholar] [CrossRef]
- Büchel, F.; Wrzodek, C.; Mittag, F.; Dräger, A.; Eichner, J.; Rodriguez, N.; Novère, N.L.; Zell, A. Qualitative translation of relations from BioPAX to SBML qual. Bioinformatics 2012, 28, 2648–2653. [Google Scholar] [CrossRef]
- Keating, S.M.; Le Novère, N. Supporting SBML as a model exchange format in software applications. Methods Mol. Biol. 2013, 1021, 201–225. [Google Scholar] [CrossRef]
- Ruebenacker, O.; Moraru, I.I.; Schaff, J.C.; Blinov, M.L. Integrating BioPAX pathway knowledge with SBML models. IET Syst. Biol. 2009, 3, 317–328. [Google Scholar] [CrossRef]
- Vlaic, S.; Hoffmann, B.; Kupfer, P.; Weber, M.; Dräger, A. GRN2SBML: Automated encoding and annotation of inferred gene regulatory networks complying with SBML. Bioinforma. Oxf. Engl. 2013, 29, 2216–2217. [Google Scholar] [CrossRef]
- Webb, R.L.; Ma’ayan, A. Sig2BioPAX: Java tool for converting flat files to BioPAX Level 3 format. Source Code Biol. Med. 2011, 6, 5. [Google Scholar] [CrossRef]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Kramer, F.; Bayerlová, M.; Beißbarth, T. R-Based Software for the Integration of Pathway Data into Bioinformatic Algorithms. Biology 2014, 3, 85-100. https://doi.org/10.3390/biology3010085
Kramer F, Bayerlová M, Beißbarth T. R-Based Software for the Integration of Pathway Data into Bioinformatic Algorithms. Biology. 2014; 3(1):85-100. https://doi.org/10.3390/biology3010085
Chicago/Turabian StyleKramer, Frank, Michaela Bayerlová, and Tim Beißbarth. 2014. "R-Based Software for the Integration of Pathway Data into Bioinformatic Algorithms" Biology 3, no. 1: 85-100. https://doi.org/10.3390/biology3010085