Insights into Chromatin Structure and Dynamics in Plants

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
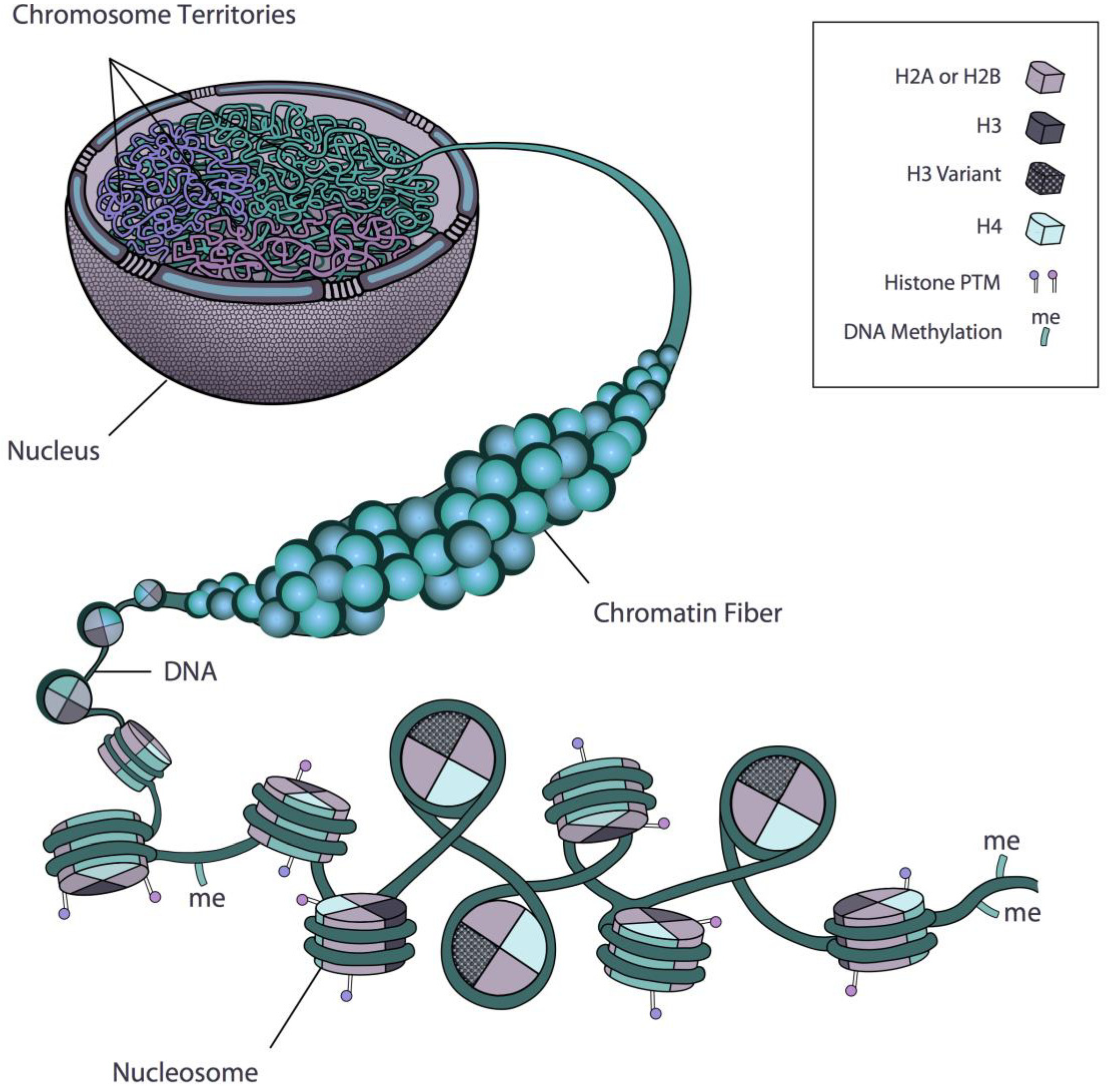
:1. Introduction: Chromatin and Nuclear Architecture
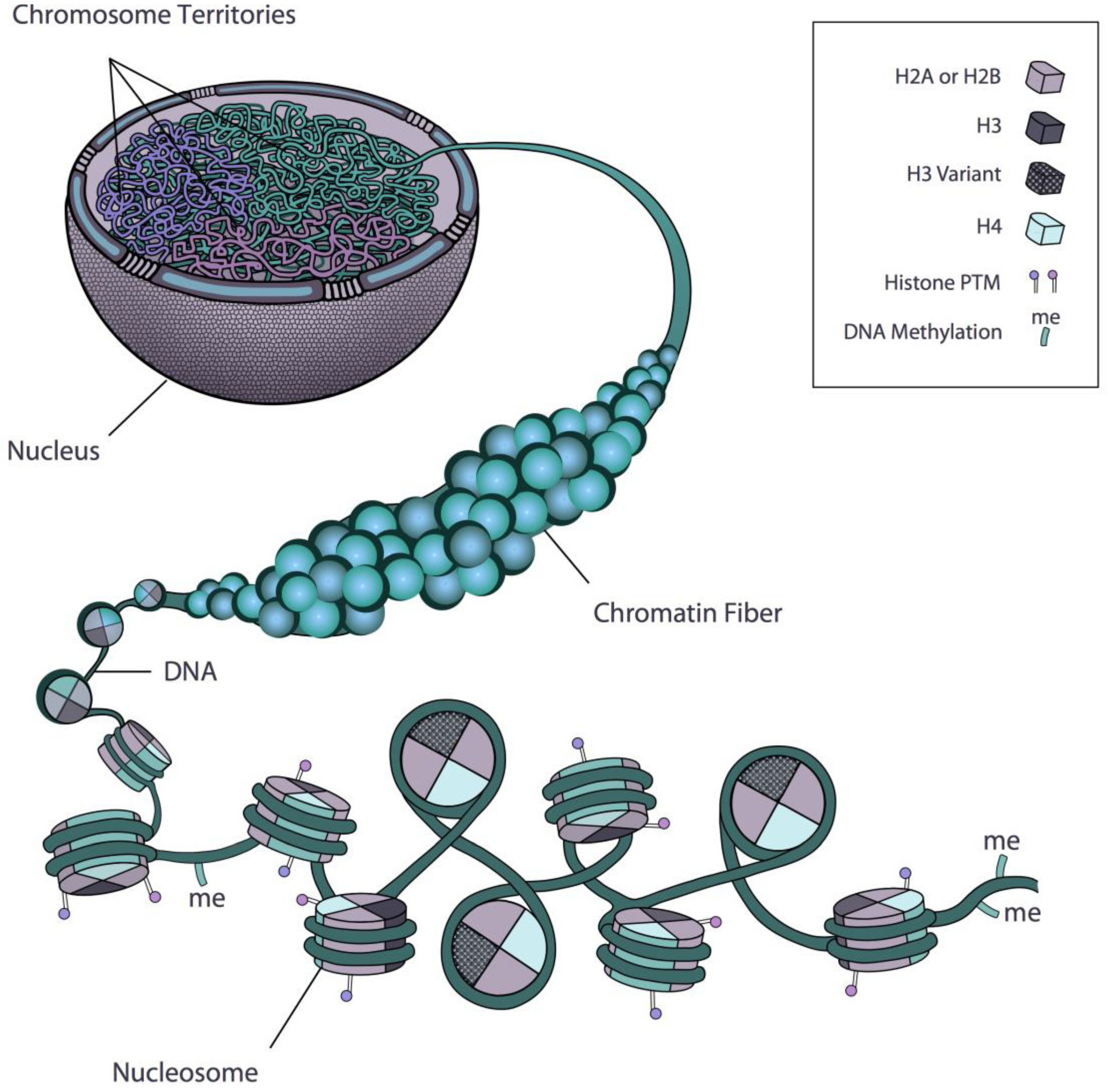
2. Regulation of Gene Expression at the Level of the Nucleosome
2.1. DNA Methylation
2.2. Histone Modifications
2.2.1. Histone Acetylation
2.2.2. Histone Methylation
2.3. Histone Variants
2.3.1. CenH3
2.3.2. H3.3
2.3.3. H2A.Z
2.4. ATP-Dependent Remodelling Complexes
3. Regulation of Gene Expression by Higher-Order Chromatin Organization
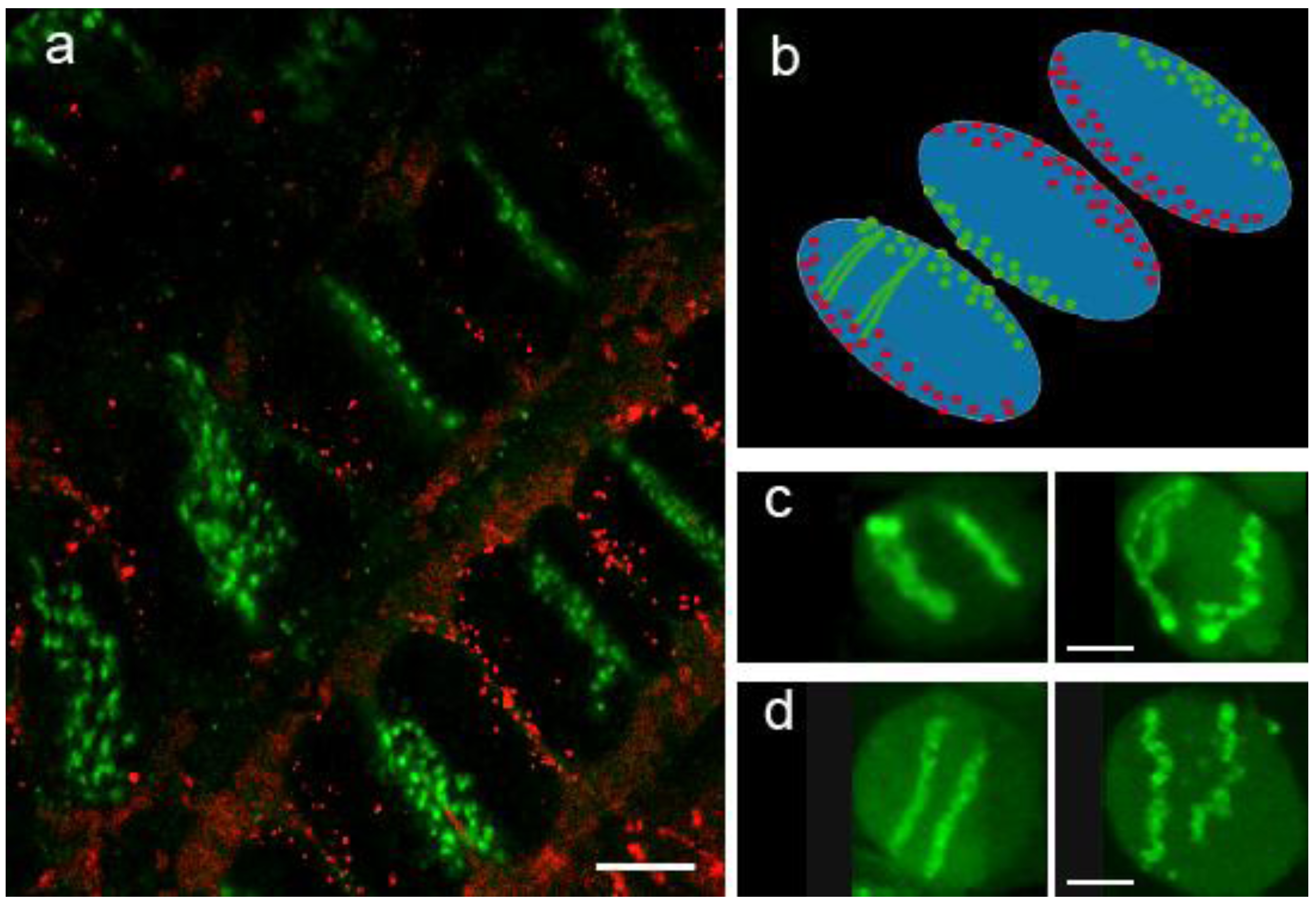
3.1. Chromosome Territories
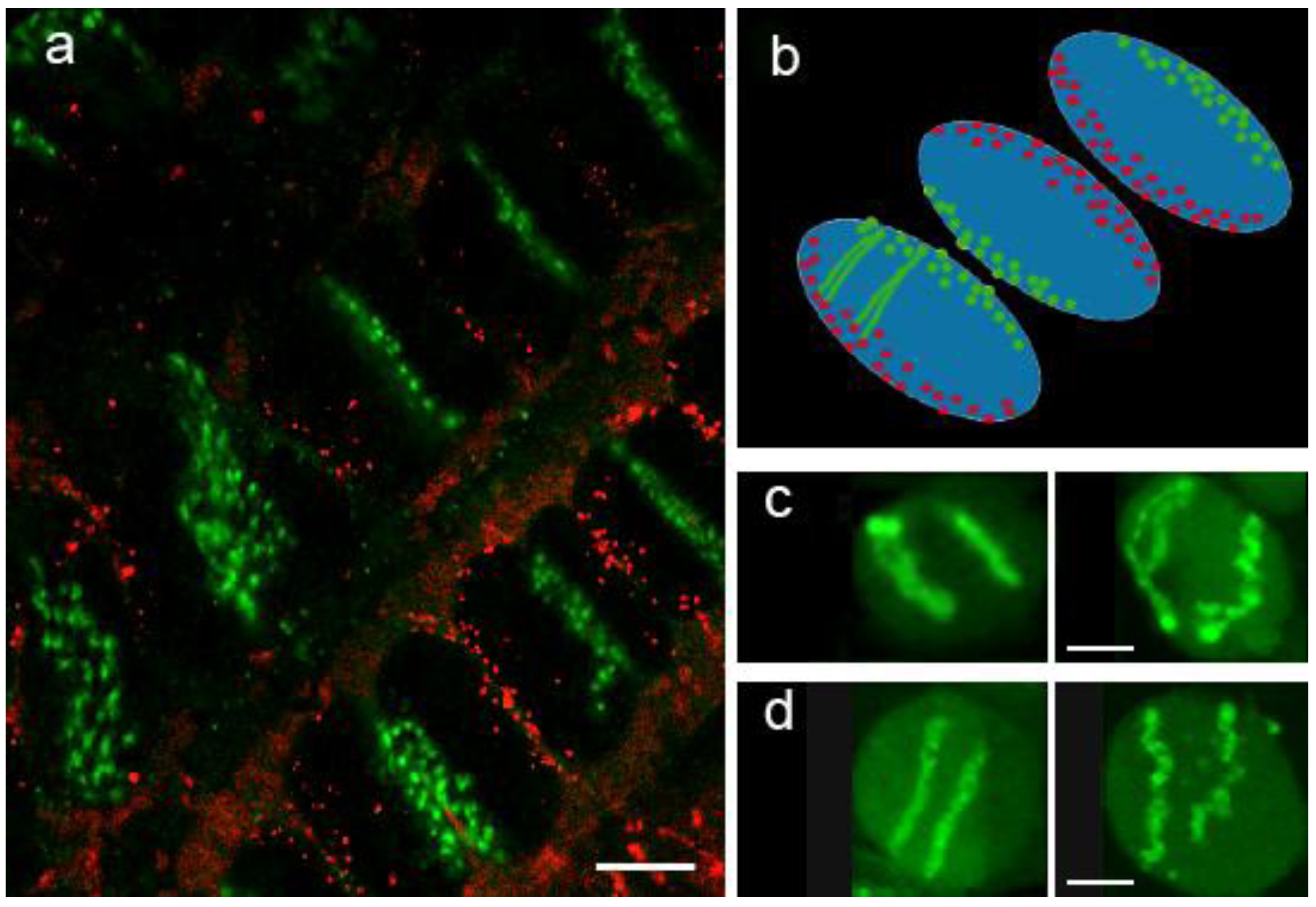
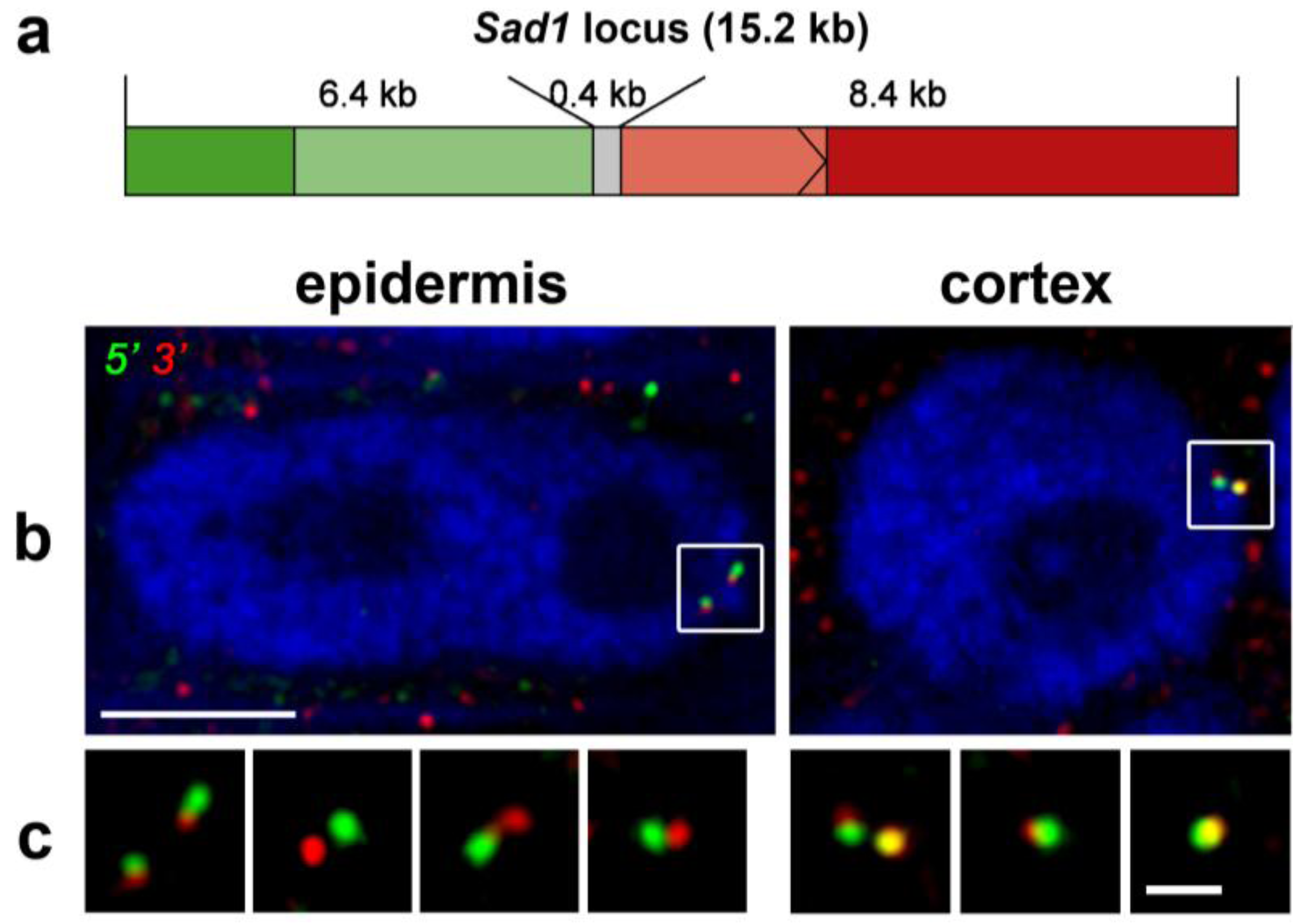
3.2. Chromatin Conformation Capture (3C)
4. Live-Cell Imaging Approaches to Studying Chromatin Dynamics
4.1. Measuring Histone Mobility—FRAP
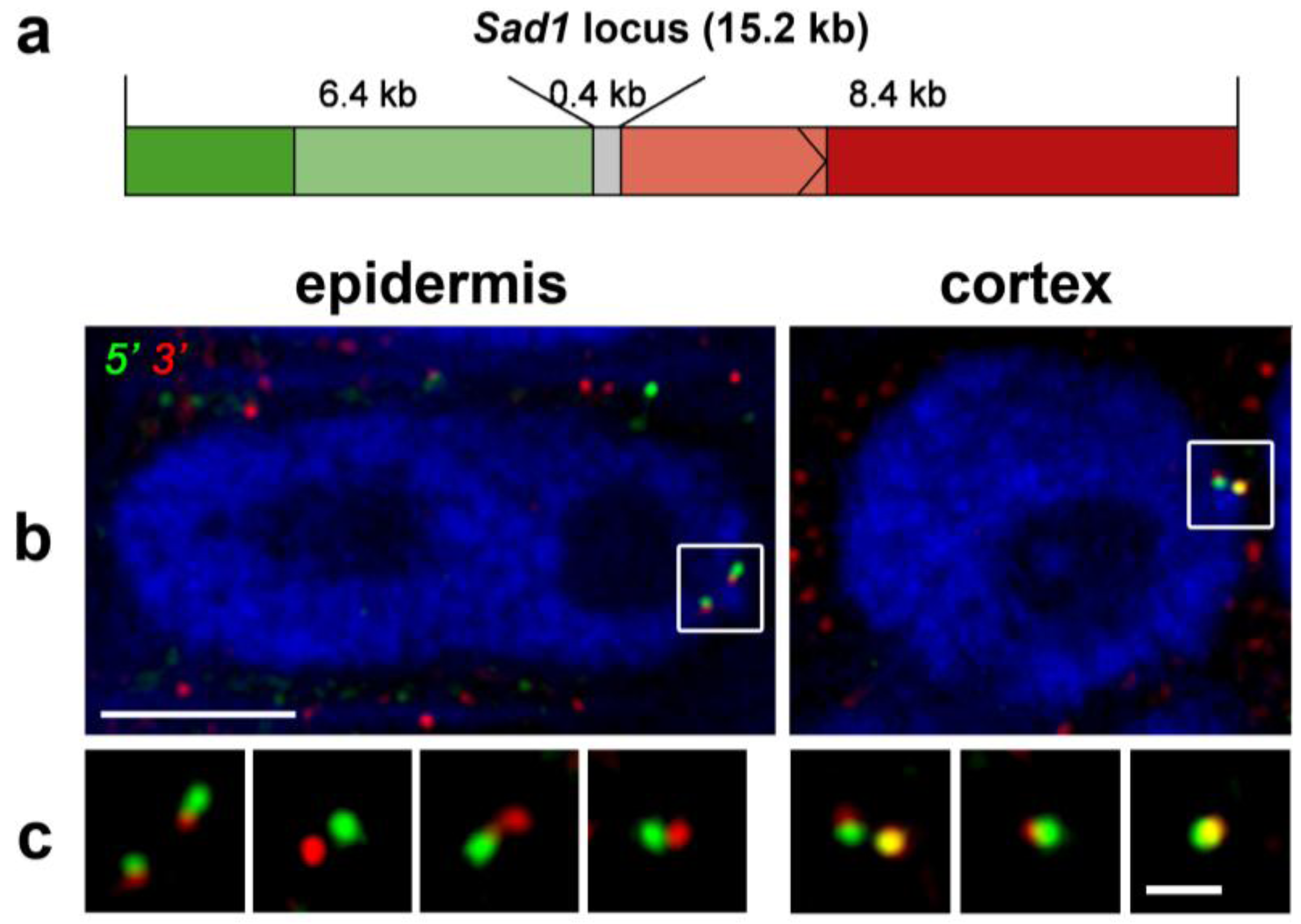
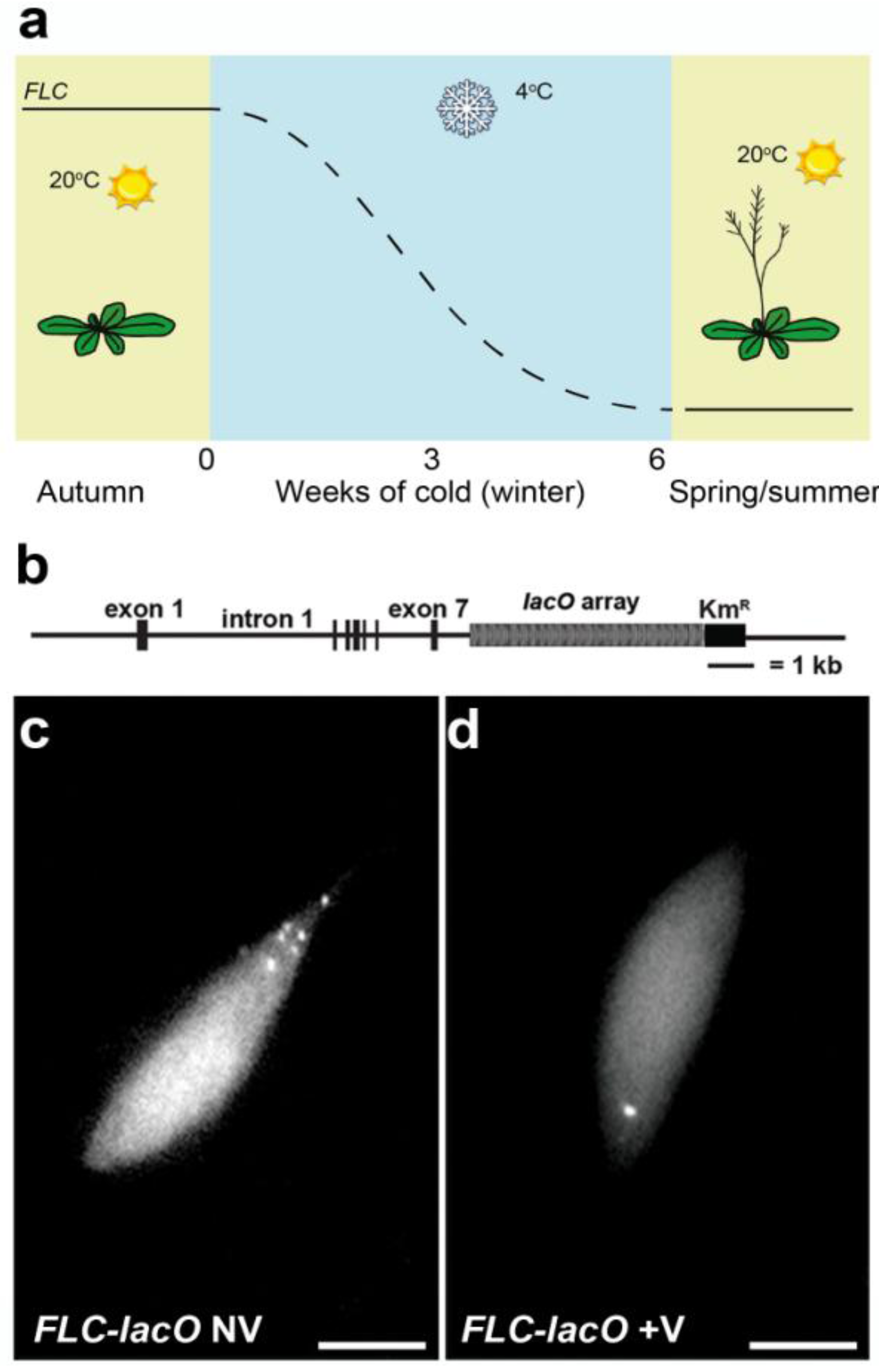
4.2. Single Particle Tracking to Study Gene Positioning—Lac Operon System
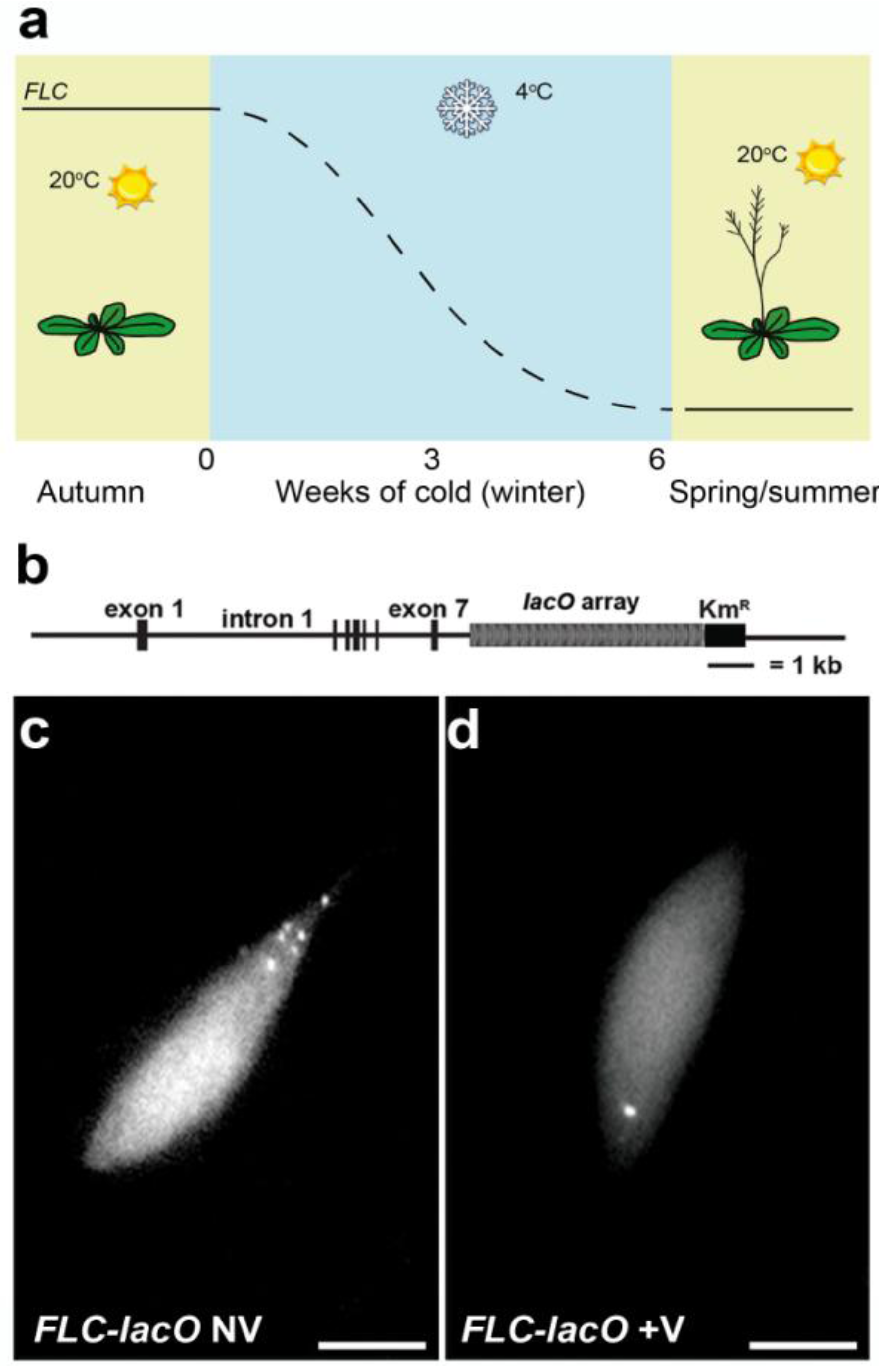
5. Conclusions
Acknowledgments
Conflicts of Interest
References
- Kornberg, R.D. Chromatin structure: A repeating unit of histones and DNA. Science 1974, 184, 868–871. [Google Scholar]
- Locklear, L., Jr.; Ridsdale, J.A.; Bazett-Jones, D.P.; Davie, J.R. Ultrastructure of transcriptionally competent chromatin. Nucleic Acids Res. 1990, 18, 7015–7024. [Google Scholar] [CrossRef]
- Fussner, E.; Ching, R.W.; Bazett-Jones, D.P. Living without 30 nm chromatin fibers. Trends Biochem. Sci. 2011, 36, 1–6. [Google Scholar]
- Woodcock, C.L.; Safer, J.P.; Stanchfield, J.E. Structural repeating units in chromatin. I. Evidence for their general occurrence. Exp. Cell Res. 1976, 97, 101–110. [Google Scholar] [CrossRef]
- Finch, J.T.; Klug, A. Solenoidal model for superstructure in chromatin. Proc. Natl. Acad. Sci. USA 1976, 73, 1897–1901. [Google Scholar] [CrossRef]
- Robinson, P.J.; Fairall, L.; Huynh, V.A.; Rhodes, D. EM measurements define the dimensions of the “30-nm” chromatin fiber: Evidence for a compact, interdigitated structure. Proc. Natl. Acad. Sci. USA 2006, 103, 6506–6511. [Google Scholar] [CrossRef]
- Widom, J.; Klug, A. Structure of the 300A chromatin filament: X-ray diffraction from oriented samples. Cell 1985, 43, 207–213. [Google Scholar] [CrossRef]
- Woodcock, C.L.; Grigoryev, S.A.; Horowitz, R.A.; Whitaker, N. A chromatin folding model that incorporates linker variability generates fibers resembling the native structures. Proc. Natl. Acad. Sci. USA 1993, 90, 9021–9025. [Google Scholar] [CrossRef]
- Schalch, T.; Duda, S.; Sargent, D.F.; Richmond, T.J. X-ray structure of a tetranucleosome and its implications for the chromatin fibre. Nature 2005, 436, 138–141. [Google Scholar] [CrossRef]
- Williams, S.P.; Langmore, J.P. Small angle x-ray scattering of chromatin. Radius and mass per unit length depend on linker length. Biophys. J. 1991, 59, 606–618. [Google Scholar] [CrossRef]
- Zentgraf, H.; Franke, W.W. Differences of supranucleosomal organization in different kinds of chromatin: Cell type-specific globular subunits containing different numbers of nucleosomes. J. Cell Biol. 1984, 99, 272–286. [Google Scholar] [CrossRef]
- Van Holde, K.; Zlatanova, J. Chromatin higher order structure: Chasing a mirage? J. Biol. Chem. 1995, 270, 8373–8376. [Google Scholar] [CrossRef]
- Scheffer, M.P.; Eltsov, M.; Frangakis, A.S. Evidence for short-range helical order in the 30-nm chromatin fibers of erythrocyte nuclei. Proc. Natl. Acad. Sci. USA 2011, 108, 16992–16997. [Google Scholar] [CrossRef]
- Grigoryev, S.A.; Woodcock, C.L. Chromatin organization—The 30 nm fiber. Exp. Cell Res. 2012, 318, 1448–1455. [Google Scholar] [CrossRef]
- Heitz, E. Das heteromchromatin der moose. I. Jahrb. Wiss. Botan. 1928, 69, 762–818. [Google Scholar]
- Sun, F.L.; Cuaycong, M.H.; Elgin, S.C. Long-range nucleosome ordering is associated with gene silencing in Drosophila melanogaster pericentric heterochromatin. Mol. Cell. Biol. 2001, 21, 2867–2879. [Google Scholar] [CrossRef]
- Fransz, P.; Soppe, W.; Schubert, I. Heterochromatin in interphase nuclei of Arabidopsis thaliana. Chromosom. Res. 2003, 11, 227–240. [Google Scholar] [CrossRef]
- DeRisi, J.L.; Iyer, V.R.; Brown, P.O. Exploring the metabolic and genetic control of gene expression on a genomic scale. Science 1997, 278, 680–686. [Google Scholar] [CrossRef]
- De Nooijer, S.; Wellink, J.; Mulder, B.; Bisseling, T. Non-specific interactions are sufficient to explain the position of heterochromatic chromocenters and nucleoli in interphase nuclei. Nucleic Acids Res. 2009, 37, 3558–3568. [Google Scholar] [CrossRef]
- Solovei, I.; Grandi, N.; Knoth, R.; Volk, B.; Cremer, T. Positional changes of pericentromeric heterochromatin and nucleoli in postmitotic Purkinje cells during murine cerebellum development. Cytogenet. Genome Res. 2004, 105, 302–310. [Google Scholar] [CrossRef]
- Kireeva, M.L.; Walter, W.; Tchernajenko, V.; Bondarenko, V.; Kashlev, M.; Studitsky, V.M. Nucleosome remodeling induced by RNA polymerase II: Loss of the H2A/H2B dimer during transcription. Mol. Cell 2002, 9, 541–552. [Google Scholar] [CrossRef]
- Lippman, Z.; Gendrel, A.V.; Black, M.; Vaughn, M.W.; Dedhia, N.; McCombie, W.R.; Lavine, K.; Mittal, V.; May, B.; Kasschau, K.D.; et al. Role of transposable elements in heterochromatin and epigenetic control. Nature 2004, 430, 471–476. [Google Scholar] [CrossRef]
- Arabidopsis Genome, I. Analysis of the genome sequence of the flowering plant Arabidopsis thaliana. Nature 2000, 408, 796–815. [Google Scholar] [CrossRef]
- Lister, R.; Pelizzola, M.; Dowen, R.H.; Hawkins, R.D.; Hon, G.; Tonti-Filippini, J.; Nery, J.R.; Lee, L.; Ye, Z.; Ngo, Q.M.; et al. Human DNA methylomes at base resolution show widespread epigenomic differences. Nature 2009, 462, 315–322. [Google Scholar] [CrossRef]
- Ramsahoye, B.H.; Biniszkiewicz, D.; Lyko, F.; Clark, V.; Bird, A.P.; Jaenisch, R. Non-CpG methylation is prevalent in embryonic stem cells and may be mediated by DNA methyltransferase 3a. Proc. Natl. Acad. Sci. USA 2000, 97, 5237–5242. [Google Scholar]
- Varley, K.E.; Gertz, J.; Bowling, K.M.; Parker, S.L.; Reddy, T.E.; Pauli-Behn, F.; Cross, M.K.; Williams, B.A.; Stamatoyannopoulos, J.A.; Crawford, G.E.; et al. Dynamic DNA methylation across diverse human cell lines and tissues. Genome Res. 2013, 23, 555–567. [Google Scholar] [CrossRef]
- Saze, H.; Kakutani, T. Differentiation of epigenetic modifications between transposons and genes. Curr. Opin. Plant Biol. 2011, 14, 81–87. [Google Scholar] [CrossRef]
- Zemach, A.; McDaniel, I.E.; Silva, P.; Zilberman, D. Genome-wide evolutionary analysis of eukaryotic DNA methylation. Science 2010, 328, 916–919. [Google Scholar] [CrossRef]
- Feng, S.; Cokus, S.J.; Zhang, X.; Chen, P.Y.; Bostick, M.; Goll, M.G.; Hetzel, J.; Jain, J.; Strauss, S.H.; Halpern, M.E.; et al. Conservation and divergence of methylation patterning in plants and animals. Proc. Natl. Acad. Sci. USA 2010, 107, 8689–8694. [Google Scholar] [CrossRef]
- Kankel, M.W.; Ramsey, D.E.; Stokes, T.L.; Flowers, S.K.; Haag, J.R.; Jeddeloh, J.A.; Riddle, N.C.; Verbsky, M.L.; Richards, E.J. Arabidopsis MET1 cytosine methyltransferase mutants. Genetics 2003, 163, 1109–1122. [Google Scholar]
- Cao, X.; Jacobsen, S.E. Role of the arabidopsis DRM methyltransferases in de novo DNA methylation and gene silencing. Curr. Biol. CB 2002, 12, 1138–1144. [Google Scholar]
- Chinnusamy, V.; Zhu, J.K. RNA-directed DNA methylation and demethylation in plants. Sci. China Ser. C Life Sci. Chin. Acad. Sci. 2009, 52, 331–343. [Google Scholar] [CrossRef]
- Law, J.A.; Jacobsen, S.E. Establishing, maintaining and modifying DNA methylation patterns in plants and animals. Nat. Rev. Genet. 2010, 11, 204–220. [Google Scholar] [CrossRef]
- Matzke, M.; Kanno, T.; Huettel, B.; Daxinger, L.; Matzke, A.J. Targets of RNA-directed DNA methylation. Curr. Opin. Plant Biol. 2007, 10, 512–519. [Google Scholar] [CrossRef]
- Wassenegger, M.; Heimes, S.; Riedel, L.; Sanger, H.L. RNA-directed de novo methylation of genomic sequences in plants. Cell 1994, 76, 567–576. [Google Scholar] [CrossRef]
- Castel, S.E.; Martienssen, R.A. RNA interference in the nucleus: Roles for small RNAs in transcription, epigenetics and beyond. Nat. Rev. Genet. 2013, 14, 100–112. [Google Scholar] [CrossRef]
- Lindroth, A.M.; Cao, X.; Jackson, J.P.; Zilberman, D.; McCallum, C.M.; Henikoff, S.; Jacobsen, S.E. Requirement of CHROMOMETHYLASE3 for maintenance of CpXpG methylation. Science 2001, 292, 2077–2080. [Google Scholar] [CrossRef]
- Zemach, A.; Kim, M.Y.; Hsieh, P.H.; Coleman-Derr, D.; Eshed-Williams, L.; Thao, K.; Harmer, S.L.; Zilberman, D. The Arabidopsis nucleosome remodeler DDM1 allows DNA methyltransferases to access H1-containing heterochromatin. Cell 2013, 153, 193–205. [Google Scholar] [CrossRef]
- Jeddeloh, J.A.; Stokes, T.L.; Richards, E.J. Maintenance of genomic methylation requires a SWI2/SNF2-like protein. Nat. Genet. 1999, 22, 94–97. [Google Scholar] [CrossRef]
- Kanno, T.; Aufsatz, W.; Jaligot, E.; Mette, M.F.; Matzke, M.; Matzke, A.J. A SNF2-like protein facilitates dynamic control of DNA methylation. EMBO Rep. 2005, 6, 649–655. [Google Scholar] [CrossRef]
- Woo, H.R.; Dittmer, T.A.; Richards, E.J. Three SRA-domain methylcytosine-binding proteins cooperate to maintain global CpG methylation and epigenetic silencing in Arabidopsis. PLoS Genet. 2008, 4, e1000156. [Google Scholar] [CrossRef]
- Pelizzola, M.; Ecker, J.R. The DNA methylome. FEBS Lett. 2011, 585, 1994–2000. [Google Scholar] [CrossRef]
- Lister, R.; Ecker, J.R. Finding the fifth base: Genome-wide sequencing of cytosine methylation. Genome Res. 2009, 19, 959–966. [Google Scholar] [CrossRef]
- Suzuki, M.M.; Bird, A. DNA methylation landscapes: Provocative insights from epigenomics. Nat. Rev. Genet. 2008, 9, 465–476. [Google Scholar] [CrossRef]
- Cosgrove, M.S.; Wolberger, C. How does the histone code work? Biochem. Cell Biol. 2005, 83, 468–476. [Google Scholar] [CrossRef]
- Jenuwein, T.; Allis, C.D. Translating the histone code. Science 2001, 293, 1074–1080. [Google Scholar] [CrossRef]
- Berger, S.L. Histone modifications in transcriptional regulation. Curr. Opin. Genet. Dev. 2002, 12, 142–148. [Google Scholar] [CrossRef]
- Turner, B.M. Cellular memory and the histone code. Cell 2002, 111, 285–291. [Google Scholar] [CrossRef]
- Strahl, B.D.; Allis, C.D. The language of covalent histone modifications. Nature 2000, 403, 41–45. [Google Scholar] [CrossRef]
- Lee, J.S.; Smith, E.; Shilatifard, A. The language of histone crosstalk. Cell 2010, 142, 682–685. [Google Scholar] [CrossRef]
- Kouzarides, T. Chromatin modifications and their function. Cell 2007, 128, 693–705. [Google Scholar] [CrossRef]
- Bernatavichute, Y.V.; Zhang, X.; Cokus, S.; Pellegrini, M.; Jacobsen, S.E. Genome-wide association of histone H3 lysine nine methylation with CHG DNA methylation in Arabidopsis thaliana. PLoS One 2008, 3, e3156. [Google Scholar] [CrossRef]
- Gendrel, A.V.; Lippman, Z.; Martienssen, R.; Colot, V. Profiling histone modification patterns in plants using genomic tiling microarrays. Nat. Methods 2005, 2, 213–218. [Google Scholar] [CrossRef]
- Gregory, B.D.; Yazaki, J.; Ecker, J.R. Utilizing tiling microarrays for whole-genome analysis in plants. Plant J. Cell Mol. Biol. 2008, 53, 636–644. [Google Scholar] [CrossRef]
- Martienssen, R.A.; Doerge, R.W.; Colot, V. Epigenomic mapping in Arabidopsis using tiling microarrays. Chromosom. Res. Int. J. Mol. Supramol. Evol. Asp. Chromosom. Biol. 2005, 13, 299–308. [Google Scholar] [CrossRef]
- Tran, R.K.; Henikoff, J.G.; Zilberman, D.; Ditt, R.F.; Jacobsen, S.E.; Henikoff, S. DNA methylation profiling identifies CG methylation clusters in Arabidopsis genes. Curr. Biol. CB 2005, 15, 154–159. [Google Scholar] [CrossRef]
- Zhang, X.; Bernatavichute, Y.V.; Cokus, S.; Pellegrini, M.; Jacobsen, S.E. Genome-wide analysis of mono-, di- and trimethylation of histone H3 lysine 4 in Arabidopsis thaliana. Genome Biol. 2009, 10, R62. [Google Scholar] [CrossRef]
- He, G.; Elling, A.A.; Deng, X.W. The epigenome and plant development. Annu. Rev. Plant Biol. 2011, 62, 411–435. [Google Scholar] [CrossRef]
- Mockler, T.C.; Chan, S.; Sundaresan, A.; Chen, H.; Jacobsen, S.E.; Ecker, J.R. Applications of DNA tiling arrays for whole-genome analysis. Genomics 2005, 85, 1–15. [Google Scholar] [CrossRef]
- Allfrey, V.G.; Faulkner, R.; Mirsky, A.E. Acetylation and Methylation of Histones and Their Possible Role in the Regulation of Rna Synthesis. Proc. Natl. Acad. Sci. USA 1964, 51, 786–794. [Google Scholar] [CrossRef]
- Roth, S.Y.; Denu, J.M.; Allis, C.D. Histone acetyltransferases. Annu. Rev. Biochem. 2001, 70, 81–120. [Google Scholar] [CrossRef]
- Santos, A.P.; Abranches, R.; Stoger, E.; Beven, A.; Viegas, W.; Shaw, P.J. The architecture of interphase chromosomes and gene positioning are altered by changes in DNA methylation and histone acetylation. J. Cell Sci. 2002, 115, 4597–4605. [Google Scholar] [CrossRef]
- Probst, A.V.; Fagard, M.; Proux, F.; Mourrain, P.; Boutet, S.; Earley, K.; Lawrence, R.J.; Pikaard, C.S.; Murfett, J.; Furner, I.; et al. Arabidopsis histone deacetylase HDA6 is required for maintenance of transcriptional gene silencing and determines nuclear organization of rDNA repeats. Plant Cell 2004, 16, 1021–1034. [Google Scholar] [CrossRef]
- Hebbes, T.R.; Clayton, A.L.; Thorne, A.W.; Crane-Robinson, C. Core histone hyperacetylation co-maps with generalized DNase I sensitivity in the chicken beta-globin chromosomal domain. EMBO J. 1994, 13, 1823–1830. [Google Scholar]
- Jeppesen, P.; Turner, B.M. The inactive X chromosome in female mammals is distinguished by a lack of histone H4 acetylation, a cytogenetic marker for gene expression. Cell 1993, 74, 281–289. [Google Scholar] [CrossRef]
- Boggs, B.A.; Connors, B.; Sobel, R.E.; Chinault, A.C.; Allis, C.D. Reduced levels of histone H3 acetylation on the inactive X chromosome in human females. Chromosoma 1996, 105, 303–309. [Google Scholar] [CrossRef]
- Shogren-Knaak, M.; Ishii, H.; Sun, J.M.; Pazin, M.J.; Davie, J.R.; Peterson, C.L. Histone H4-K16 acetylation controls chromatin structure and protein interactions. Science 2006, 311, 844–847. [Google Scholar] [CrossRef]
- Sterner, D.E.; Berger, S.L. Acetylation of histones and transcription-related factors. Microbiol. Mol. Biol. Rev. 2000, 64, 435–459. [Google Scholar] [CrossRef]
- Khochbin, S.; Verdel, A.; Lemercier, C.; Seigneurin-Berny, D. Functional significance of histone deacetylase diversity. Curr. Opin. Genet. Dev. 2001, 11, 162–166. [Google Scholar] [CrossRef]
- Abranches, R.; Beven, A.F.; Aragon-Alcaide, L.; Shaw, P.J. Transcription sites are not correlated with chromosome territories in wheat nuclei. J. Cell Biol. 1998, 143, 5–12. [Google Scholar] [CrossRef]
- Rabl, C. Uber Zelltheilung. Morphol. Jahrb. 1885, 10, 214–330. [Google Scholar]
- Bannister, A.J.; Kouzarides, T. Reversing histone methylation. Nature 2005, 436, 1103–1106. [Google Scholar] [CrossRef]
- Santos-Rosa, H.; Schneider, R.; Bannister, A.J.; Sherriff, J.; Bernstein, B.E.; Emre, N.C.; Schreiber, S.L.; Mellor, J.; Kouzarides, T. Active genes are tri-methylated at K4 of histone H3. Nature 2002, 419, 407–411. [Google Scholar] [CrossRef]
- Fischle, W.; Tseng, B.S.; Dormann, H.L.; Ueberheide, B.M.; Garcia, B.A.; Shabanowitz, J.; Hunt, D.F.; Funabiki, H.; Allis, C.D. Regulation of HP1-chromatin binding by histone H3 methylation and phosphorylation. Nature 2005, 438, 1116–1122. [Google Scholar] [CrossRef]
- Turck, F.; Roudier, F.; Farrona, S.; Martin-Magniette, M.L.; Guillaume, E.; Buisine, N.; Gagnot, S.; Martienssen, R.A.; Coupland, G.; Colot, V. Arabidopsis TFL2/LHP1 specifically associates with genes marked by trimethylation of histone H3 lysine 27. PLoS Genet. 2007, 3, e86. [Google Scholar] [CrossRef]
- Zhang, X.; Germann, S.; Blus, B.J.; Khorasanizadeh, S.; Gaudin, V.; Jacobsen, S.E. The Arabidopsis LHP1 protein colocalizes with histone H3 Lys27 trimethylation. Nat. Struct. Mol. Biol. 2007, 14, 869–871. [Google Scholar] [CrossRef]
- Exner, V.; Aichinger, E.; Shu, H.; Wildhaber, T.; Alfarano, P.; Caflisch, A.; Gruissem, W.; Kohler, C.; Hennig, L. The chromodomain of LIKE HETEROCHROMATIN PROTEIN 1 is essential for H3K27me3 binding and function during Arabidopsis development. PLoS One 2009, 4, e5335. [Google Scholar] [CrossRef]
- Mylne, J.S.; Barrett, L.; Tessadori, F.; Mesnage, S.; Johnson, L.; Bernatavichute, Y.V.; Jacobsen, S.E.; Fransz, P.; Dean, C. LHP1, the Arabidopsis homologue of HETEROCHROMATIN PROTEIN1, is required for epigenetic silencing of FLC. Proc. Natl. Acad. Sci. USA 2006, 103, 5012–5017. [Google Scholar] [CrossRef]
- Bedford, M.T.; Clarke, S.G. Protein arginine methylation in mammals: Who, what, and why. Mol. Cell 2009, 33, 1–13. [Google Scholar] [CrossRef]
- Ahmad, K.; Henikoff, S. The histone variant H3.3 marks active chromatin by replication-independent nucleosome assembly. Mol. Cell 2002, 9, 1191–1200. [Google Scholar] [CrossRef]
- Talbert, P.B.; Henikoff, S. Histone variants—Ancient wrap artists of the epigenome. Nat. Rev. Mol. Cell Biol. 2010, 11, 264–275. [Google Scholar] [CrossRef]
- Malik, H.S.; Henikoff, S. Phylogenomics of the nucleosome. Nat. Struct. Biol. 2003, 10, 882–891. [Google Scholar] [CrossRef]
- Palmer, D.K.; O’Day, K.; Trong, H.L.; Charbonneau, H.; Margolis, R.L. Purification of the centromere-specific protein CENP-A and demonstration that it is a distinctive histone. Proc. Natl. Acad. Sci. USA 1991, 88, 3734–3738. [Google Scholar] [CrossRef]
- Buchwitz, B.J.; Ahmad, K.; Moore, L.L.; Roth, M.B.; Henikoff, S. A histone-H3-like protein in C. elegans. Nature 1999, 401, 547–548. [Google Scholar] [CrossRef]
- Henikoff, S.; Ahmad, K.; Platero, J.S.; van Steensel, B. Heterochromatic deposition of centromeric histone H3-like proteins. Proc. Natl. Acad. Sci. USA 2000, 97, 716–721. [Google Scholar]
- Meluh, P.B.; Yang, P.; Glowczewski, L.; Koshland, D.; Smith, M.M. Cse4p is a component of the core centromere of Saccharomyces cerevisiae. Cell 1998, 94, 607–613. [Google Scholar] [CrossRef]
- Henikoff, S.; Ahmad, K.; Malik, H.S. The centromere paradox: Stable inheritance with rapidly evolving DNA. Science 2001, 293, 1098–1102. [Google Scholar] [CrossRef]
- Wieland, G.; Orthaus, S.; Ohndorf, S.; Diekmann, S.; Hemmerich, P. Functional complementation of human centromere protein A (CENP-A) by Cse4p from Saccharomyces cerevisiae. Mol. Cell. Biol. 2004, 24, 6620–6630. [Google Scholar] [CrossRef]
- Fang, Y.; Spector, D.L. Centromere positioning and dynamics in living Arabidopsis plants. Mol. Biol. Cell 2005, 16, 5710–5718. [Google Scholar] [CrossRef]
- Dalal, Y.; Wang, H.; Lindsay, S.; Henikoff, S. Tetrameric structure of centromeric nucleosomes in interphase Drosophila cells. PLoS Biol. 2007, 5, e218. [Google Scholar] [CrossRef]
- Camahort, R.; Shivaraju, M.; Mattingly, M.; Li, B.; Nakanishi, S.; Zhu, D.X.; Shilatifard, A.; Workman, J.L.; Gerton, J.L. Cse4 is part of an octameric nucleosome in budding yeast. Mol. Cell 2009, 35, 794–805. [Google Scholar] [CrossRef]
- Furuyama, T.; Henikoff, S. Centromeric nucleosomes induce positive DNA supercoils. Cell 2009, 138, 104–113. [Google Scholar] [CrossRef]
- Tagami, H.; Ray-Gallet, D.; Almouzni, G.; Nakatani, Y. Histone H3.1 and H3.3 complexes mediate nucleosome assembly pathways dependent or independent of DNA synthesis. Cell 2004, 116, 51–61. [Google Scholar] [CrossRef]
- Hake, S.B.; Allis, C.D. Histone H3 variants and their potential role in indexing mammalian genomes: The “H3 barcode hypothesis”. Proc. Natl. Acad. Sci. USA 2006, 103, 6428–6435. [Google Scholar] [CrossRef]
- Chow, C.M.; Georgiou, A.; Szutorisz, H.; Maia e Silva, A.; Pombo, A.; Barahona, I.; Dargelos, E.; Canzonetta, C.; Dillon, N. Variant histone H3.3 marks promoters of transcriptionally active genes during mammalian cell division. EMBO Rep. 2005, 6, 354–360. [Google Scholar] [CrossRef]
- Mito, Y.; Henikoff, J.G.; Henikoff, S. Genome-scale profiling of histone H3.3 replacement patterns. Nat. Genet. 2005, 37, 1090–1097. [Google Scholar] [CrossRef]
- Wirbelauer, C.; Bell, O.; Schubeler, D. Variant histone H3.3 is deposited at sites of nucleosomal displacement throughout transcribed genes while active histone modifications show a promoter-proximal bias. Genes Dev. 2005, 19, 1761–1766. [Google Scholar] [CrossRef]
- Janicki, S.M.; Tsukamoto, T.; Salghetti, S.E.; Tansey, W.P.; Sachidanandam, R.; Prasanth, K.V.; Ried, T.; Shav-Tal, Y.; Bertrand, E.; Singer, R.H.; et al. From silencing to gene expression: Real-time analysis in single cells. Cell 2004, 116, 683–698. [Google Scholar] [CrossRef]
- Schwartz, B.E.; Ahmad, K. Transcriptional activation triggers deposition and removal of the histone variant H3.3. Genes Dev. 2005, 19, 804–814. [Google Scholar] [CrossRef]
- Jin, C.; Felsenfeld, G. Nucleosome stability mediated by histone variants H3.3 and H2A.Z. Genes Dev. 2007, 21, 1519–1529. [Google Scholar] [CrossRef]
- Van Daal, A.; White, E.M.; Elgin, S.C.; Gorovsky, M.A. Conservation of intron position indicates separation of major and variant H2As is an early event in the evolution of eukaryotes. J. Mol. Evol. 1990, 30, 449–455. [Google Scholar] [CrossRef]
- Thatcher, T.H.; Gorovsky, M.A. Phylogenetic analysis of the core histones H2A, H2B, H3, and H4. Nucleic Acids Res. 1994, 22, 174–179. [Google Scholar] [CrossRef]
- Clarkson, M.J.; Wells, J.R.; Gibson, F.; Saint, R.; Tremethick, D.J. Regions of variant histone His2AvD required for Drosophila development. Nature 1999, 399, 694–697. [Google Scholar] [CrossRef]
- Faast, R.; Thonglairoam, V.; Schulz, T.C.; Beall, J.; Wells, J.R.; Taylor, H.; Matthaei, K.; Rathjen, P.D.; Tremethick, D.J.; Lyons, I. Histone variant H2A.Z is required for early mammalian development. Curr. Biol. CB 2001, 11, 1183–1187. [Google Scholar] [CrossRef]
- Jackson, J.D.; Gorovsky, M.A. Histone H2A.Z has a conserved function that is distinct from that of the major H2A sequence variants. Nucleic Acids Res. 2000, 28, 3811–3816. [Google Scholar] [CrossRef]
- Eirin-Lopez, J.M.; Gonzalez-Romero, R.; Dryhurst, D.; Ishibashi, T.; Ausio, J. The evolutionary differentiation of two histone H2A.Z variants in chordates (H2A.Z-1 and H2A.Z-2) is mediated by a stepwise mutation process that affects three amino acid residues. BMC Evol. Biol. 2009, 9, 31. [Google Scholar]
- March-Diaz, R.; Garcia-Dominguez, M.; Lozano-Juste, J.; Leon, J.; Florencio, F.J.; Reyes, J.C. Histone H2A.Z and homologues of components of the SWR1 complex are required to control immunity in Arabidopsis. Plant J. 2008, 53, 475–487. [Google Scholar]
- Krogan, N.J.; Keogh, M.C.; Datta, N.; Sawa, C.; Ryan, O.W.; Ding, H.; Haw, R.A.; Pootoolal, J.; Tong, A.; Canadien, V.; et al. A Snf2 family ATPase complex required for recruitment of the histone H2A variant Htz1. Mol. Cell 2003, 12, 1565–1576. [Google Scholar] [CrossRef]
- Cai, Y.; Jin, J.; Florens, L.; Swanson, S.K.; Kusch, T.; Li, B.; Workman, J.L.; Washburn, M.P.; Conaway, R.C.; Conaway, J.W. The mammalian YL1 protein is a shared subunit of the TRRAP/TIP60 histone acetyltransferase and SRCAP complexes. J. Biol. Chem. 2005, 280, 13665–13670. [Google Scholar] [CrossRef]
- Choi, J.; Heo, K.; An, W. Cooperative action of TIP48 and TIP49 in H2A.Z exchange catalyzed by acetylation of nucleosomal H2A. Nucleic Acids Res. 2009, 37, 5993–6007. [Google Scholar] [CrossRef]
- Mizuguchi, G.; Shen, X.; Landry, J.; Wu, W.H.; Sen, S.; Wu, C. ATP-driven exchange of histone H2AZ variant catalyzed by SWR1 chromatin remodeling complex. Science 2004, 303, 343–348. [Google Scholar] [CrossRef]
- Ladurner, A.G.; Inouye, C.; Jain, R.; Tjian, R. Bromodomains mediate an acetyl-histone encoded antisilencing function at heterochromatin boundaries. Mol. Cell 2003, 11, 365–376. [Google Scholar] [CrossRef]
- Noh, Y.S.; Amasino, R.M. PIE1, an ISWI family gene, is required for FLC activation and floral repression in Arabidopsis. Plant Cell 2003, 15, 1671–1682. [Google Scholar] [CrossRef]
- March-Diaz, R.; Garcia-Dominguez, M.; Florencio, F.J.; Reyes, J.C. SEF, a new protein required for flowering repression in Arabidopsis, interacts with PIE1 and ARP6. Plant Physiol. 2007, 143, 893–901. [Google Scholar]
- Choi, K.; Kim, S.; Kim, S.Y.; Kim, M.; Hyun, Y.; Lee, H.; Choe, S.; Kim, S.G.; Michaels, S.; Lee, I. SUPPRESSOR OF FRIGIDA3 encodes a nuclear ACTIN-RELATED PROTEIN6 required for floral repression in Arabidopsis. Plant Cell 2005, 17, 2647–2660. [Google Scholar] [CrossRef]
- Deal, R.B.; Kandasamy, M.K.; McKinney, E.C.; Meagher, R.B. The nuclear actin-related protein ARP6 is a pleiotropic developmental regulator required for the maintenance of FLOWERING LOCUS C expression and repression of flowering in Arabidopsis. Plant Cell 2005, 17, 2633–2646. [Google Scholar] [CrossRef]
- Martin-Trillo, M.; Lazaro, A.; Poethig, R.S.; Gomez-Mena, C.; Pineiro, M.A.; Martinez-Zapater, J.M.; Jarillo, J.A. EARLY IN SHORT DAYS 1 (ESD1) encodes ACTIN-RELATED PROTEIN 6 (AtARP6), a putative component of chromatin remodelling complexes that positively regulates FLC accumulation in Arabidopsis. Development 2006, 133, 1241–1252. [Google Scholar] [CrossRef]
- Allis, C.D.; Glover, C.V.; Bowen, J.K.; Gorovsky, M.A. Histone variants specific to the transcriptionally active, amitotically dividing macronucleus of the unicellular eucaryote, Tetrahymena thermophila. Cell 1980, 20, 609–617. [Google Scholar] [CrossRef]
- Suto, R.K.; Clarkson, M.J.; Tremethick, D.J.; Luger, K. Crystal structure of a nucleosome core particle containing the variant histone H2A.Z. Nat. Struct. Biol. 2000, 7, 1121–1124. [Google Scholar]
- Abbott, D.W.; Ivanova, V.S.; Wang, X.; Bonner, W.M.; Ausio, J. Characterization of the stability and folding of H2A.Z chromatin particles: Implications for transcriptional activation. J. Biol. Chem. 2001, 276, 41945–41949. [Google Scholar]
- Fan, J.Y.; Gordon, F.; Luger, K.; Hansen, J.C.; Tremethick, D.J. The essential histone variant H2A.Z regulates the equilibrium between different chromatin conformational states. Nat. Struct. Biol. 2002, 9, 172–176. [Google Scholar]
- Park, Y.J.; Dyer, P.N.; Tremethick, D.J.; Luger, K. A new fluorescence resonance energy transfer approach demonstrates that the histone variant H2AZ stabilizes the histone octamer within the nucleosome. J. Biol. Chem. 2004, 279, 24274–24282. [Google Scholar]
- Dryhurst, D.; Thambirajah, A.A.; Ausio, J. New twists on H2A.Z: A histone variant with a controversial structural and functional past. Biochem. Cell Biol. 2004, 82, 490–497. [Google Scholar] [CrossRef]
- Guillemette, B.; Gaudreau, L. Reuniting the contrasting functions of H2A.Z. Biochem. Cell Biol. 2006, 84, 528–535. [Google Scholar]
- Raisner, R.M.; Hartley, P.D.; Meneghini, M.D.; Bao, M.Z.; Liu, C.L.; Schreiber, S.L.; Rando, O.J.; Madhani, H.D. Histone variant H2A.Z marks the 5' ends of both active and inactive genes in euchromatin. Cell 2005, 123, 233–248. [Google Scholar]
- Adam, M.; Robert, F.; Larochelle, M.; Gaudreau, L. H2A.Z is required for global chromatin integrity and for recruitment of RNA polymerase II under specific conditions. Mol. Cell. Biol. 2001, 21, 6270–6279. [Google Scholar] [CrossRef]
- Brickner, D.G.; Cajigas, I.; Fondufe-Mittendorf, Y.; Ahmed, S.; Lee, P.C.; Widom, J.; Brickner, J.H. Z-mediated localization of genes at the nuclear periphery confers epigenetic memory of previous transcriptional state. PLoS Biol. 2007, 5, e81. [Google Scholar] [CrossRef]
- Li, B.; Pattenden, S.G.; Lee, D.; Gutierrez, J.; Chen, J.; Seidel, C.; Gerton, J.; Workman, J.L. Preferential occupancy of histone variant H2AZ at inactive promoters influences local histone modifications and chromatin remodeling. Proc. Natl. Acad. Sci. USA 2005, 102, 18385–18390. [Google Scholar]
- Santisteban, M.S.; Kalashnikova, T.; Smith, M.M. Histone H2A.Z regulats transcription and is partially redundant with nucleosome remodeling complexes. Cell 2000, 103, 411–422. [Google Scholar] [CrossRef]
- Dhillon, N.; Oki, M.; Szyjka, S.J.; Aparicio, O.M.; Kamakaka, R.T. H2A.Z functions to regulate progression through the cell cycle. Mol. Cell. Biol. 2006, 26, 489–501. [Google Scholar] [CrossRef]
- Carr, A.M.; Dorrington, S.M.; Hindley, J.; Phear, G.A.; Aves, S.J.; Nurse, P. Analysis of a histone H2A variant from fission yeast: Evidence for a role in chromosome stability. Mol. Gen. Genet. 1994, 245, 628–635. [Google Scholar]
- Downs, J.A.; Allard, S.; Jobin-Robitaille, O.; Javaheri, A.; Auger, A.; Bouchard, N.; Kron, S.J.; Jackson, S.P.; Cote, J. Binding of chromatin-modifying activities to phosphorylated histone H2A at DNA damage sites. Mol. Cell 2004, 16, 979–990. [Google Scholar] [CrossRef]
- Meneghini, M.D.; Wu, M.; Madhani, H.D. Conserved histone variant H2A.Z protects euchromatin from the ectopic spread of silent heterochromatin. Cell 2003, 112, 725–736. [Google Scholar] [CrossRef]
- Zofall, M.; Fischer, T.; Zhang, K.; Zhou, M.; Cui, B.; Veenstra, T.D.; Grewal, S.I. Histone H2A.Z cooperates with RNAi and heterochromatin factors to suppress antisense RNAs. Nature 2009, 461, 419–422. [Google Scholar] [CrossRef]
- Creyghton, M.P.; Markoulaki, S.; Levine, S.S.; Hanna, J.; Lodato, M.A.; Sha, K.; Young, R.A.; Jaenisch, R.; Boyer, L.A. H2AZ is enriched at polycomb complex target genes in ES cells and is necessary for lineage commitment. Cell 2008, 135, 649–661. [Google Scholar] [CrossRef]
- Zilberman, D.; Coleman-Derr, D.; Ballinger, T.; Henikoff, S. Histone H2A.Z and DNA methylation are mutually antagonistic chromatin marks. Nature 2008, 456, 125–129. [Google Scholar]
- Sarcinella, E.; Zuzarte, P.C.; Lau, P.N.; Draker, R.; Cheung, P. Monoubiquitylation of H2A.Z distinguishes its association with euchromatin or facultative heterochromatin. Mol. Cell. Biol. 2007, 27, 6457–6468. [Google Scholar] [CrossRef]
- Rangasamy, D.; Berven, L.; Ridgway, P.; Tremethick, D.J. Pericentric heterochromatin becomes enriched with H2A.Z during early mammalian development. EMBO J. 2003, 22, 1599–1607. [Google Scholar] [CrossRef]
- Guillemette, B.; Bataille, A.R.; Gevry, N.; Adam, M.; Blanchette, M.; Robert, F.; Gaudreau, L. Variant histone H2A.Z is globally localized to the promoters of inactive yeast genes and regulates nucleosome positioning. PLoS Biol. 2005, 3, e384. [Google Scholar] [CrossRef]
- Raisner, R.M.; Madhani, H.D. Patterning chromatin: Form and function for H2A.Z variant nucleosomes. Curr. Opin. Genet. Dev. 2006, 16, 119–124. [Google Scholar]
- Zhang, H.; Roberts, D.N.; Cairns, B.R. Genome-wide dynamics of Htz1, a histone H2A variant that poises repressed/basal promoters for activation through histone loss. Cell 2005, 123, 219–231. [Google Scholar] [CrossRef]
- Barski, A.; Cuddapah, S.; Cui, K.; Roh, T.Y.; Schones, D.E.; Wang, Z.; Wei, G.; Chepelev, I.; Zhao, K. High-resolution profiling of histone methylations in the human genome. Cell 2007, 129, 823–837. [Google Scholar] [CrossRef]
- Draker, R.; Cheung, P. Transcriptional and epigenetic functions of histone variant H2A.Z. Biochem. Cell Biol. 2009, 87, 19–25. [Google Scholar]
- Gross, D.S.; Garrard, W.T. Nuclease hypersensitive sites in chromatin. Annu. Rev. Biochem. 1988, 57, 159–197. [Google Scholar] [CrossRef]
- Kumar, S.V.; Wigge, P.A. Z-containing nucleosomes mediate the thermosensory response in Arabidopsis. Cell 2010, 140, 136–147. [Google Scholar] [CrossRef]
- Coleman-Derr, D.; Zilberman, D. Deposition of histone variant H2A.Z within gene bodies regulates responsive genes. PLoS Genet. 2012, 8, e1002988. [Google Scholar] [CrossRef]
- Saha, A.; Wittmeyer, J.; Cairns, B.R. Chromatin remodelling: The industrial revolution of DNA around histones. Nat. Rev. Mol. Cell Biol. 2006, 7, 437–447. [Google Scholar]
- Langst, G.; Becker, P.B. Nucleosome remodeling: One mechanism, many phenomena? Biochim. Biophys. Acta 1677, 58–63. [Google Scholar]
- Kingston, R.E.; Narlikar, G.J. ATP-dependent remodeling and acetylation as regulators of chromatin fluidity. Genes Dev. 1999, 13, 2339–2352. [Google Scholar]
- Strohner, R.; Wachsmuth, M.; Dachauer, K.; Mazurkiewicz, J.; Hochstatter, J.; Rippe, K.; Langst, G. A ‘loop recapture’ mechanism for ACF-dependent nucleosome remodeling. Nat. Struct. Mol. Biol. 2005, 12, 683–690. [Google Scholar]
- Zofall, M.; Persinger, J.; Kassabov, S.R.; Bartholomew, B. Chromatin remodeling by ISW2 and SWI/SNF requires DNA translocation inside the nucleosome. Nat. Struct. Mol. Biol. 2006, 13, 339–346. [Google Scholar] [CrossRef]
- Fazzio, T.G.; Gelbart, M.E.; Tsukiyama, T. Two distinct mechanisms of chromatin interaction by the Isw2 chromatin remodeling complex in vivo. Mol. Cell. Biol. 2005, 25, 9165–9174. [Google Scholar] [CrossRef]
- Goldmark, J.P.; Fazzio, T.G.; Estep, P.W.; Church, G.M.; Tsukiyama, T. The Isw2 chromatin remodeling complex represses early meiotic genes upon recruitment by Ume6p. Cell 2000, 103, 423–433. [Google Scholar]
- Seo, S.; Herr, A.; Lim, J.W.; Richardson, G.A.; Richardson, H.; Kroll, K.L. Geminin regulates neuronal differentiation by antagonizing Brg1 activity. Genes Dev. 2005, 19, 1723–1734. [Google Scholar]
- Feng, Q.; Zhang, Y. The MeCP1 complex represses transcription through preferential binding, remodeling, and deacetylating methylated nucleosomes. Genes Dev. 2001, 15, 827–832. [Google Scholar]
- Mayer, C.; Schmitz, K.M.; Li, J.; Grummt, I.; Santoro, R. Intergenic transcripts regulate the epigenetic state of rRNA genes. Mol. Cell 2006, 22, 351–361. [Google Scholar] [CrossRef]
- Kobor, M.S.; Venkatasubrahmanyam, S.; Meneghini, M.D.; Gin, J.W.; Jennings, J.L.; Link, A.J.; Madhani, H.D.; Rine, J. A protein complex containing the conserved Swi2/Snf2-related ATPase Swr1p deposits histone variant H2A.Z into euchromatin. PLoS Biol. 2004, 2, E131. [Google Scholar]
- Bovery, T. Die Blastomerenkerne von Ascaris megalocephala und die Theorie der Chromosomenindividualitat. Arch. Zellforsch. 1909, 3, 181–286. [Google Scholar]
- Manuelidis, L. Individual interphase chromosome domains revealed by in situ hybridization. Hum. Genet. 1985, 71, 288–293. [Google Scholar]
- Schardin, M.; Cremer, T.; Hager, H.D.; Lang, M. Specific staining of human chromosomes in Chinese hamster x man hybrid cell lines demonstrates interphase chromosome territories. Hum. Genet. 1985, 71, 281–287. [Google Scholar] [CrossRef]
- Schwarzacher, T.; Leitch, A.R.; Bennett, M.D.; Heslop-Harrison, J.S. In situ localization of parental genomes in a wide hybrid. Ann. Bot. 1989, 64, 315–324. [Google Scholar]
- Lysak, M.A.; Fransz, P.F.; Ali, H.B.; Schubert, I. Chromosome painting in Arabidopsis thaliana. Plant J. 2001, 28, 689–697. [Google Scholar]
- Schubert, I.; Shaw, P. Organization and dynamics of plant interphase chromosomes. Trends Plant Sci. 2011, 16, 273–281. [Google Scholar] [CrossRef]
- Armstrong, S.J.; Franklin, F.C.; Jones, G.H. Nucleolus-associated telomere clustering and pairing precede meiotic chromosome synapsis in Arabidopsis thaliana. J. Cell Sci. 2001, 114, 4207–4217. [Google Scholar]
- Prieto, P.; Santos, A.P.; Moore, G.; Shaw, P. Chromosomes associate premeiotically and in xylem vessel cells via their telomeres and centromeres in diploid rice (Oryza sativa). Chromosoma 2004, 112, 300–307. [Google Scholar]
- Santos, A.P.; Ferreira, L.; Maroco, J.; Oliveira, M.M. Abiotic stress and induced DNA hypomethylation cause interphase chromatin structural changes in rice rDNA loci. Cytogenet. Genome Res. 2011, 132, 297–303. [Google Scholar] [CrossRef]
- Dietzel, S.; Schiebel, K.; Little, G.; Edelmann, P.; Rappold, G.A.; Eils, R.; Cremer, C.; Cremer, T. The 3D positioning of ANT2 and ANT3 genes within female X chromosome territories correlates with gene activity. Exp. Cell Res. 1999, 252, 363–375. [Google Scholar] [CrossRef]
- Chambeyron, S.; da Silva, N.R.; Lawson, K.A.; Bickmore, W.A. Nuclear re-organisation of the Hoxb complex during mouse embryonic development. Development 2005, 132, 2215–2223. [Google Scholar] [CrossRef]
- Volpi, E.; Mittendorfer, B.; Rasmussen, B.B.; Wolfe, R.R. The response of muscle protein anabolism to combined hyperaminoacidemia and glucose-induced hyperinsulinemia is impaired in the elderly. J. Clin. Endocrinol. Metab. 2000, 85, 4481–4490. [Google Scholar] [CrossRef]
- Wegel, E.; Koumproglou, R.; Shaw, P.; Osbourn, A. Cell type-specific chromatin decondensation of a metabolic gene cluster in oats. Plant Cell 2009, 21, 3926–3936. [Google Scholar] [CrossRef]
- Brown, K.E.; Guest, S.S.; Smale, S.T.; Hahm, K.; Merkenschlager, M.; Fisher, A.G. Association of transcriptionally silent genes with Ikaros complexes at centromeric heterochromatin. Cell 1997, 91, 845–854. [Google Scholar] [CrossRef]
- Brown, K.E.; Baxter, J.; Graf, D.; Merkenschlager, M.; Fisher, A.G. Dynamic repositioning of genes in the nucleus of lymphocytes preparing for cell division. Mol. Cell 1999, 3, 207–217. [Google Scholar] [CrossRef]
- Schubeler, D.; Francastel, C.; Cimbora, D.M.; Reik, A.; Martin, D.I.; Groudine, M. Nuclear localization and histone acetylation: A pathway for chromatin opening and transcriptional activation of the human beta-globin locus. Genes Dev. 2000, 14, 940–950. [Google Scholar]
- Kosak, S.T.; Skok, J.A.; Medina, K.L.; Riblet, R.; le Beau, M.M.; Fisher, A.G.; Singh, H. Subnuclear compartmentalization of immunoglobulin loci during lymphocyte development. Science 2002, 296, 158–162. [Google Scholar] [CrossRef]
- Hu, Y.; Kireev, I.; Plutz, M.; Ashourian, N.; Belmont, A.S. Large-scale chromatin structure of inducible genes: Transcription on a condensed, linear template. J. Cell Biol. 2009, 185, 87–100. [Google Scholar] [CrossRef]
- Costa, S.; Shaw, P. Chromatin organization and cell fate switch respond to positional information in Arabidopsis. Nature 2006, 439, 493–496. [Google Scholar] [CrossRef]
- Verschure, P.J.; van der Kraan, I.; Manders, E.M.; Hoogstraten, D.; Houtsmuller, A.B.; van Driel, R. Condensed chromatin domains in the mammalian nucleus are accessible to large macromolecules. EMBO Rep. 2003, 4, 861–866. [Google Scholar] [CrossRef]
- Chen, D.; Dundr, M.; Wang, C.; Leung, A.; Lamond, A.; Misteli, T.; Huang, S. Condensed mitotic chromatin is accessible to transcription factors and chromatin structural proteins. J. Cell Biol. 2005, 168, 41–54. [Google Scholar]
- Bancaud, A.; Huet, S.; Daigle, N.; Mozziconacci, J.; Beaudouin, J.; Ellenberg, J. Molecular crowding affects diffusion and binding of nuclear proteins in heterochromatin and reveals the fractal organization of chromatin. EMBO J. 2009, 28, 3785–3798. [Google Scholar] [CrossRef]
- Cook, P.R. The organization of replication and transcription. Science 1999, 284, 1790–1795. [Google Scholar] [CrossRef]
- Cook, P.R. A model for all genomes: The role of transcription factories. J. Mol. Biol. 2010, 395, 1–10. [Google Scholar] [CrossRef]
- Dekker, J. The three ‘C’ s of chromosome conformation capture: Controls, controls, controls. Nat. Methods 2006, 3, 17–21. [Google Scholar] [CrossRef]
- Simonis, M.; Kooren, J.; de Laat, W. An evaluation of 3C-based methods to capture DNA interactions. Nat. Methods 2007, 4, 895–901. [Google Scholar] [CrossRef]
- Sutherland, H.; Bickmore, W.A. Transcription factories: Gene expression in unions? Nat. Rev. Genet. 2009, 10, 457–466. [Google Scholar] [CrossRef]
- Simonis, M.; Klous, P.; Splinter, E.; Moshkin, Y.; Willemsen, R.; de Wit, E.; van Steensel, B.; de Laat, W. Nuclear organization of active and inactive chromatin domains uncovered by chromosome conformation capture-on-chip (4C). Nat. Genet. 2006, 38, 1348–1354. [Google Scholar] [CrossRef]
- Wurtele, H.; Chartrand, P. Genome-wide scanning of HoxB1-associated loci in mouse ES cells using an open-ended Chromosome Conformation Capture methodology. Chromosom. Res. 2006, 14, 477–495. [Google Scholar] [CrossRef]
- Dostie, J.; Richmond, T.A.; Arnaout, R.A.; Selzer, R.R.; Lee, W.L.; Honan, T.A.; Rubio, E.D.; Krumm, A.; Lamb, J.; Nusbaum, C.; et al. Chromosome Conformation Capture Carbon Copy (5C): A massively parallel solution for mapping interactions between genomic elements. Genome Res. 2006, 16, 1299–1309. [Google Scholar]
- Lieberman-Aiden, E.; van Berkum, N.L.; Williams, L.; Imakaev, M.; Ragoczy, T.; Telling, A.; Amit, I.; Lajoie, B.R.; Sabo, P.J.; Dorschner, M.O.; et al. Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science 2009, 326, 289–293. [Google Scholar] [CrossRef]
- Dekker, J.; Marti-Renom, M.A.; Mirny, L.A. Exploring the three-dimensional organization of genomes: Interpreting chromatin interaction data. Nat. Rev. Genet. 2013, 14, 390–403. [Google Scholar] [CrossRef]
- Shaw, P.J. Mapping chromatin conformation. F1000 Biol Rep. 2010, 2. [Google Scholar] [CrossRef]
- De Wit, E.; de Laat, W. A decade of 3C technologies: Insights into nuclear organization. Genes Dev. 2012, 26, 11–24. [Google Scholar] [CrossRef]
- Caron, F.; Thomas, J.O. Exchange of histone H1 between segments of chromatin. J. Mol. Biol. 1981, 146, 513–537. [Google Scholar] [CrossRef]
- Thomas, J.O.; Rees, C. Exchange of histones H1 and H5 between chromatin fragments. A preference of H5 for higher-order structures. Eur. J. Biochem. 1983, 134, 109–115. [Google Scholar] [CrossRef]
- Louters, L.; Chalkley, R. Exchange of histones H1, H2A, and H2B in vivo. Biochemistry 1985, 24, 3080–3085. [Google Scholar] [CrossRef]
- White, J.; Stelzer, E. Photobleaching GFP reveals protein dynamics inside live cells. Trends Cell Biol. 1999, 9, 61–65. [Google Scholar] [CrossRef]
- Phair, R.D.; Misteli, T. Kinetic modelling approaches to in vivo imaging. Nat. Rev. Mol. Cell Biol. 2001, 2, 898–907. [Google Scholar] [CrossRef]
- Misteli, T.; Gunjan, A.; Hock, R.; Bustin, M.; Brown, D.T. Dynamic binding of histone H1 to chromatin in living cells. Nature 2000, 408, 877–881. [Google Scholar] [CrossRef]
- Lever, M.A.; Th’ng, J.P.; Sun, X.; Hendzel, M.J. Rapid exchange of histone H1.1 on chromatin in living human cells. Nature 2000, 408, 873–876. [Google Scholar] [CrossRef]
- Phair, R.D.; Scaffidi, P.; Elbi, C.; Vecerova, J.; Dey, A.; Ozato, K.; Brown, D.T.; Hager, G.; Bustin, M.; Misteli, T. Global nature of dynamic protein-chromatin interactions in vivo: Three-dimensional genome scanning and dynamic interaction networks of chromatin proteins. Mol. Cell. Biol. 2004, 24, 6393–6402. [Google Scholar] [CrossRef]
- Kimura, H. Histone dynamics in living cells revealed by photobleaching. DNA Repair (Amst) 2005, 4, 939–950. [Google Scholar] [CrossRef]
- Kimura, H.; Cook, P.R. Kinetics of core histones in living human cells: Little exchange of H3 and H4 and some rapid exchange of H2B. J. Cell Biol. 2001, 153, 1341–1353. [Google Scholar] [CrossRef]
- Higashi, T.; Matsunaga, S.; Isobe, K.; Morimoto, A.; Shimada, T.; Kataoka, S.; Watanabe, W.; Uchiyama, S.; Itoh, K.; Fukui, K. Histone H2A mobility is regulated by its tails and acetylation of core histone tails. Biochem. Biophys. Res. Commun. 2007, 357, 627–632. [Google Scholar] [CrossRef]
- Ito, T.; Ikehara, T.; Nakagawa, T.; Kraus, W.L.; Muramatsu, M. p300-mediated acetylation facilitates the transfer of histone H2A-H2B dimers from nucleosomes to a histone chaperone. Genes Dev. 2000, 14, 1899–1907. [Google Scholar]
- Lippincott-Schwartz, J.; Altan-Bonnet, N.; Patterson, G.H. Photobleaching and photoactivation: Following protein dynamics in living cells. Nat. Cell Biol. 2003, Suppl:S7–14. [Google Scholar]
- Deal, R.B.; Henikoff, J.G.; Henikoff, S. Genome-wide kinetics of nucleosome turnover determined by metabolic labeling of histones. Science 2010, 328, 1161–1164. [Google Scholar] [CrossRef]
- Straight, A.F.; Belmont, A.S.; Robinett, C.C.; Murray, A.W. GFP tagging of budding yeast chromosomes reveals that protein-protein interactions can mediate sister chromatid cohesion. Curr. Biol. CB 1996, 6, 1599–1608. [Google Scholar] [CrossRef]
- Belmont, A.S. Visualizing chromosome dynamics with GFP. Trends Cell Biol. 2001, 11, 250–257. [Google Scholar] [CrossRef]
- Belmont, A.S.; Straight, A.F. In vivo visualization of chromosomes using lac operator-repressor binding. Trends Cell Biol. 1998, 8, 121–124. [Google Scholar] [CrossRef]
- Michaelis, C.; Ciosk, R.; Nasmyth, K. Cohesins: Chromosomal proteins that prevent premature separation of sister chromatids. Cell 1997, 91, 35–45. [Google Scholar] [CrossRef]
- Fuchs, J.; Lorenz, A.; Loidl, J. Chromosome associations in budding yeast caused by integrated tandemly repeated transgenes. J. Cell Sci. 2002, 115, 1213–1220. [Google Scholar]
- Belmont, A.S.; Li, G.; Sudlow, G.; Robinett, C. Visualization of large-scale chromatin structure and dynamics using the lac operator/lac repressor reporter system. Methods Cell Biol. 1999, 58, 203–222. [Google Scholar]
- Robinett, C.C.; Straight, A.; Li, G.; Willhelm, C.; Sudlow, G.; Murray, A.; Belmont, A.S. In vivo localization of DNA sequences and visualization of large-scale chromatin organization using lac operator/repressor recognition. J. Cell Biol. 1996, 135, 1685–1700. [Google Scholar] [CrossRef]
- Marshall, W.F.; Straight, A.; Marko, J.F.; Swedlow, J.; Dernburg, A.; Belmont, A.; Murray, A.W.; Agard, D.A.; Sedat, J.W. Interphase chromosomes undergo constrained diffusional motion in living cells. Curr. Biol. CB 1997, 7, 930–939. [Google Scholar] [CrossRef]
- Vazquez, J.; Belmont, A.S.; Sedat, J.W. Multiple regimes of constrained chromosome motion are regulated in the interphase Drosophila nucleus. Curr. Biol. CB 2001, 11, 1227–1239. [Google Scholar] [CrossRef]
- Chubb, J.R.; Boyle, S.; Perry, P.; Bickmore, W.A. Chromatin motion is constrained by association with nuclear compartments in human cells. Curr. Biol. CB 2002, 12, 439–445. [Google Scholar] [CrossRef]
- Rosa, A.; Maddocks, J.H.; Neumann, F.R.; Gasser, S.M.; Stasiak, A. Measuring limits of telomere movement on nuclear envelope. Biophys. J. 2006, 90, L24–L26. [Google Scholar] [CrossRef]
- Tumbar, T.; Belmont, A.S. Interphase movements of a DNA chromosome region modulated by VP16 transcriptional activator. Nat. Cell Biol. 2001, 3, 134–139. [Google Scholar] [CrossRef]
- Heun, P.; Laroche, T.; Shimada, K.; Furrer, P.; Gasser, S.M. Chromosome dynamics in the yeast interphase nucleus. Science 2001, 294, 2181–2186. [Google Scholar] [CrossRef]
- Kato, N.; Lam, E. Chromatin of endoreduplicated pavement cells has greater range of movement than that of diploid guard cells in Arabidopsis thaliana. J. Cell Sci. 2003, 116, 2195–2201. [Google Scholar] [CrossRef]
- Rosin, F.M.; Watanabe, N.; Cacas, J.L.; Kato, N.; Arroyo, J.M.; Fang, Y.; May, B.; Vaughn, M.; Simorowski, J.; Ramu, U.; et al. Genome-wide transposon tagging reveals location-dependent effects on transcription and chromatin organization in Arabidopsis. Plant J. 2008, 55, 514–525. [Google Scholar] [CrossRef]
- Matzke, A.J.; Huettel, B.; van der Winden, J.; Matzke, M. Use of two-color fluorescence-tagged transgenes to study interphase chromosomes in living plants. Plant Physiol. 2005, 139, 1586–1596. [Google Scholar] [CrossRef]
- Matzke, A.J.; Watanabe, K.; van der Winden, J.; Naumann, U.; Matzke, M. High frequency, cell type-specific visualization of fluorescent-tagged genomic sites in interphase and mitotic cells of living Arabidopsis plants. Plant Methods 2010, 6, 2. [Google Scholar] [CrossRef]
- Rosa, S.; de Lucia, F.; Mylne, J.S.; Zhu, D.; Ohmido, N.; Pendle, A.; Kato, N.; Shaw, P.; Dean, C. Physical clustering of FLC alleles during Polycomb-mediated epigenetic silencing in vernalization. Genes Dev. 2013, 27, 1845–1850. [Google Scholar] [CrossRef]
- Chuang, C.H.; Carpenter, A.E.; Fuchsova, B.; Johnson, T.; de Lanerolle, P.; Belmont, A.S. Long-range directional movement of an interphase chromosome site. Curr. Biol. CB 2006, 16, 825–831. [Google Scholar] [CrossRef]
- Hediger, F.; Taddei, A.; Neumann, F.R.; Gasser, S.M. Methods for visualizing chromatin dynamics in living yeast. Methods Enzymol. 2004, 375, 345–365. [Google Scholar]
- Svoboda, K.; Yasuda, R. Principles of two-photon excitation microscopy and its applications to neuroscience. Neuron 2006, 50, 823–839. [Google Scholar] [CrossRef]
- Levi, V.; Ruan, Q.; Gratton, E. 3-D particle tracking in a two-photon microscope: Application to the study of molecular dynamics in cells. Biophys. J. 2005, 88, 2919–2928. [Google Scholar] [CrossRef]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Rosa, S.; Shaw, P. Insights into Chromatin Structure and Dynamics in Plants. Biology 2013, 2, 1378-1410. https://doi.org/10.3390/biology2041378
Rosa S, Shaw P. Insights into Chromatin Structure and Dynamics in Plants. Biology. 2013; 2(4):1378-1410. https://doi.org/10.3390/biology2041378
Chicago/Turabian StyleRosa, Stefanie, and Peter Shaw. 2013. "Insights into Chromatin Structure and Dynamics in Plants" Biology 2, no. 4: 1378-1410. https://doi.org/10.3390/biology2041378
APA StyleRosa, S., & Shaw, P. (2013). Insights into Chromatin Structure and Dynamics in Plants. Biology, 2(4), 1378-1410. https://doi.org/10.3390/biology2041378