
PoxiPred: An Artificial-Intelligence-Based Method for the Prediction of Potential Antigens and Epitopes to Accelerate Vaccine Development Efforts against Poxviruses
 , and
, and 
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Retrieval of Known T-Cell Epitopes
2.2. Retrieval of Proteomes
2.3. Data Preparation
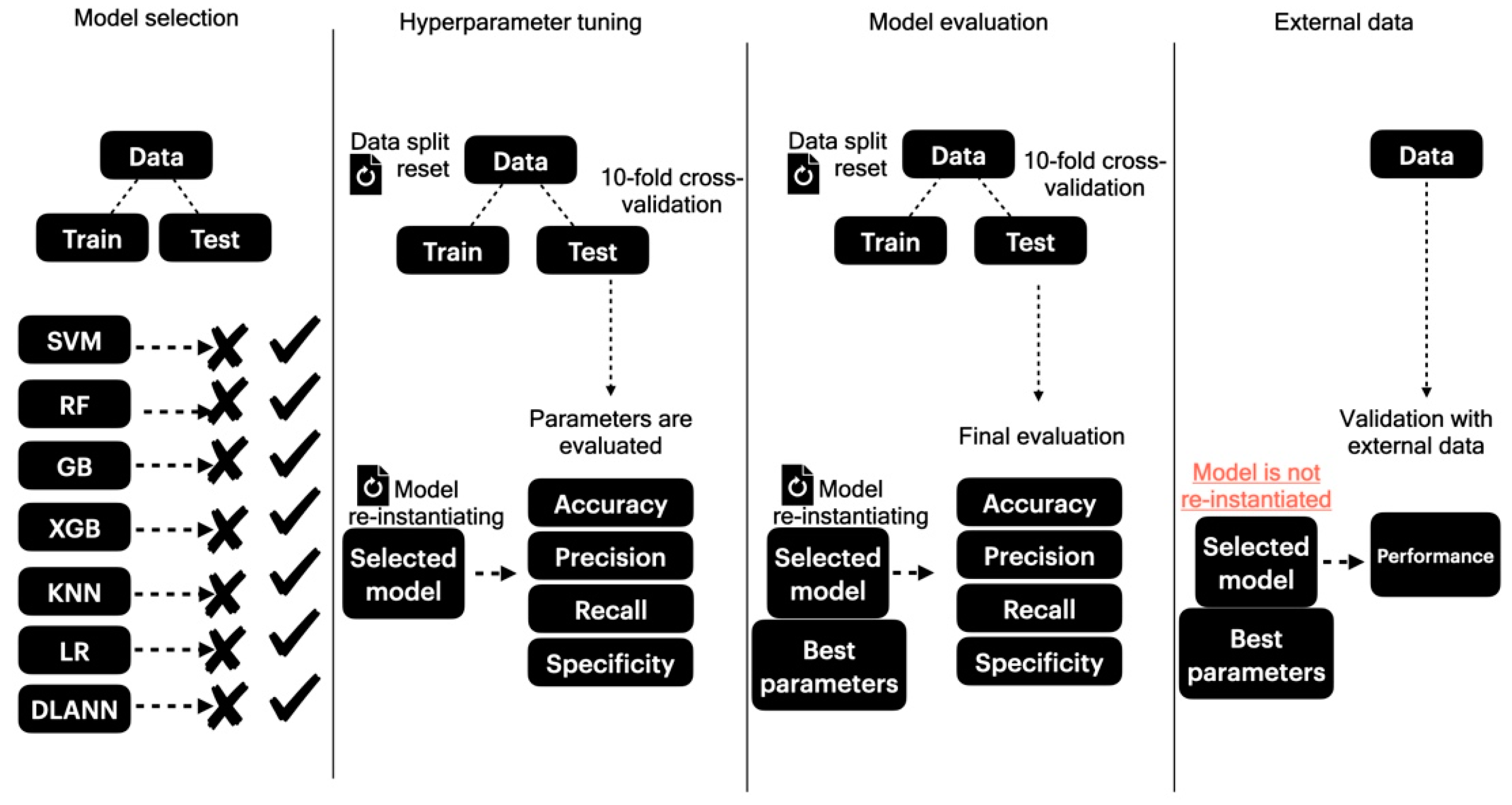
2.4. Classification Routines
3. Results
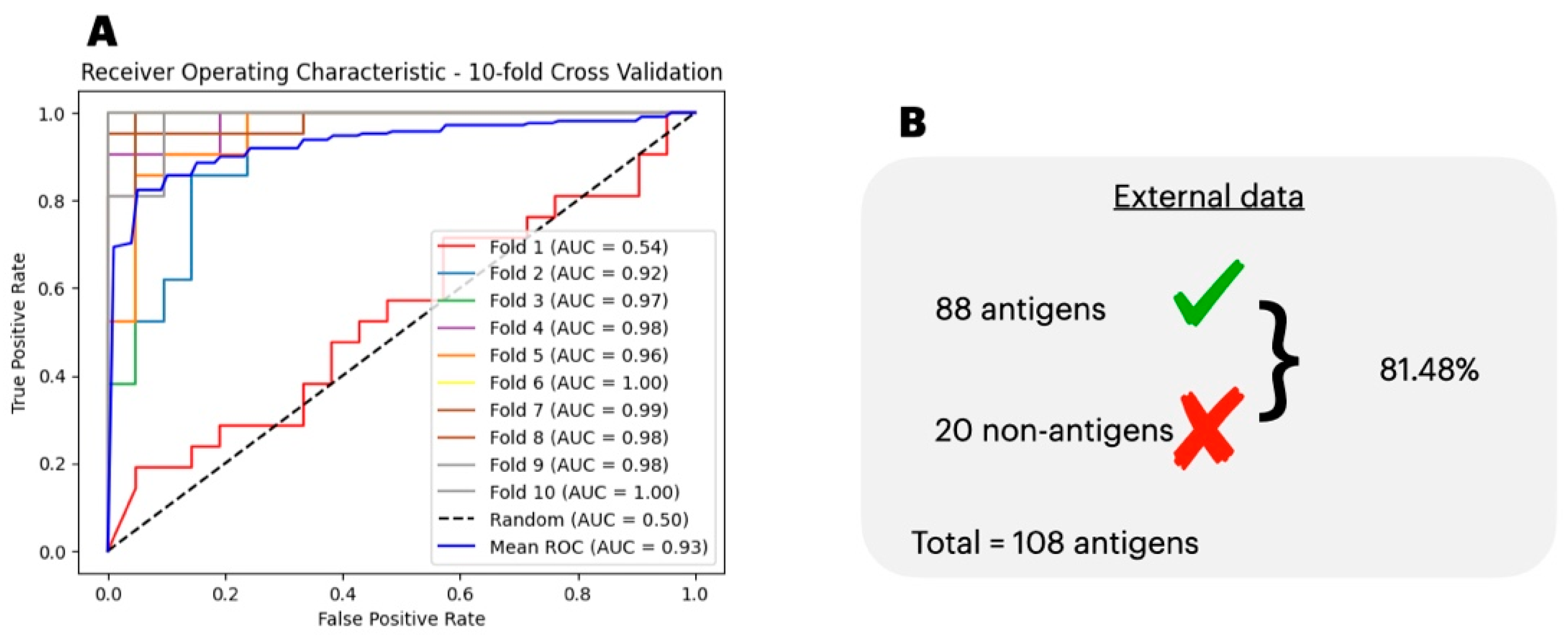
3.1. Antigenicity Classification
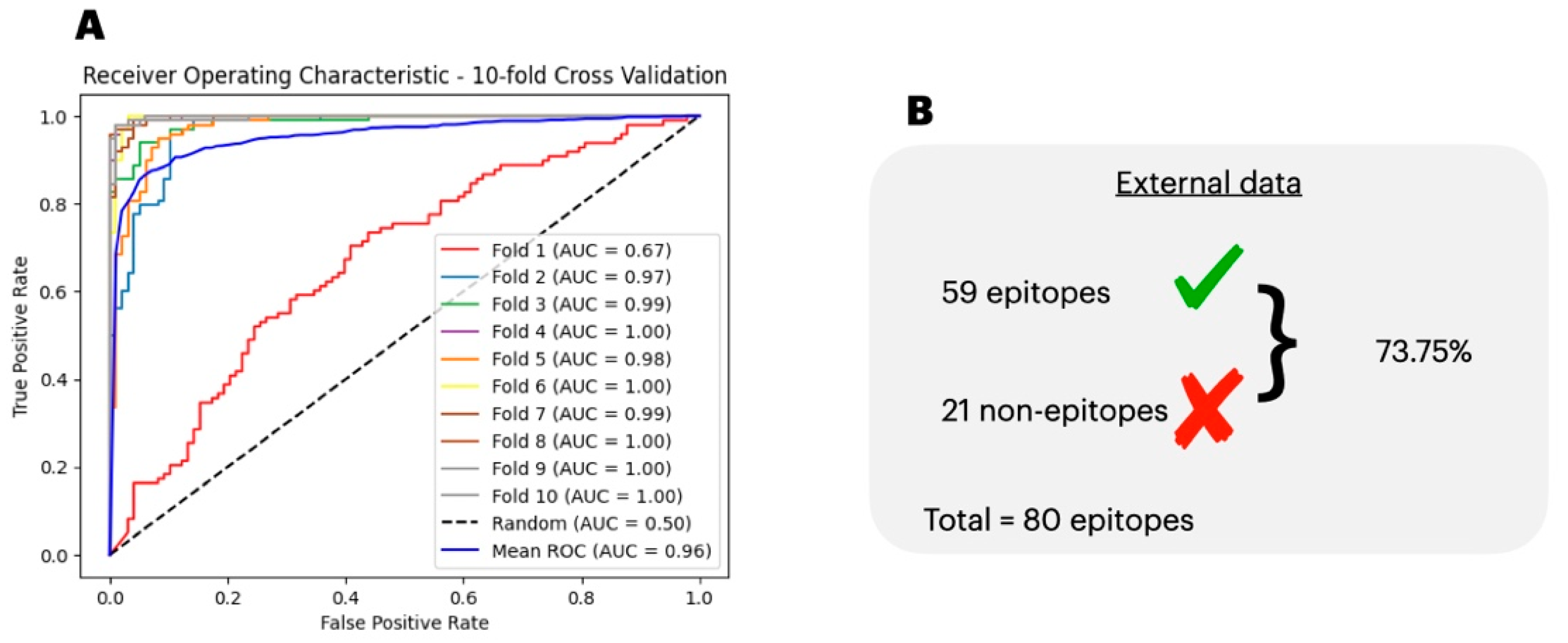
3.2. Epitope Classification
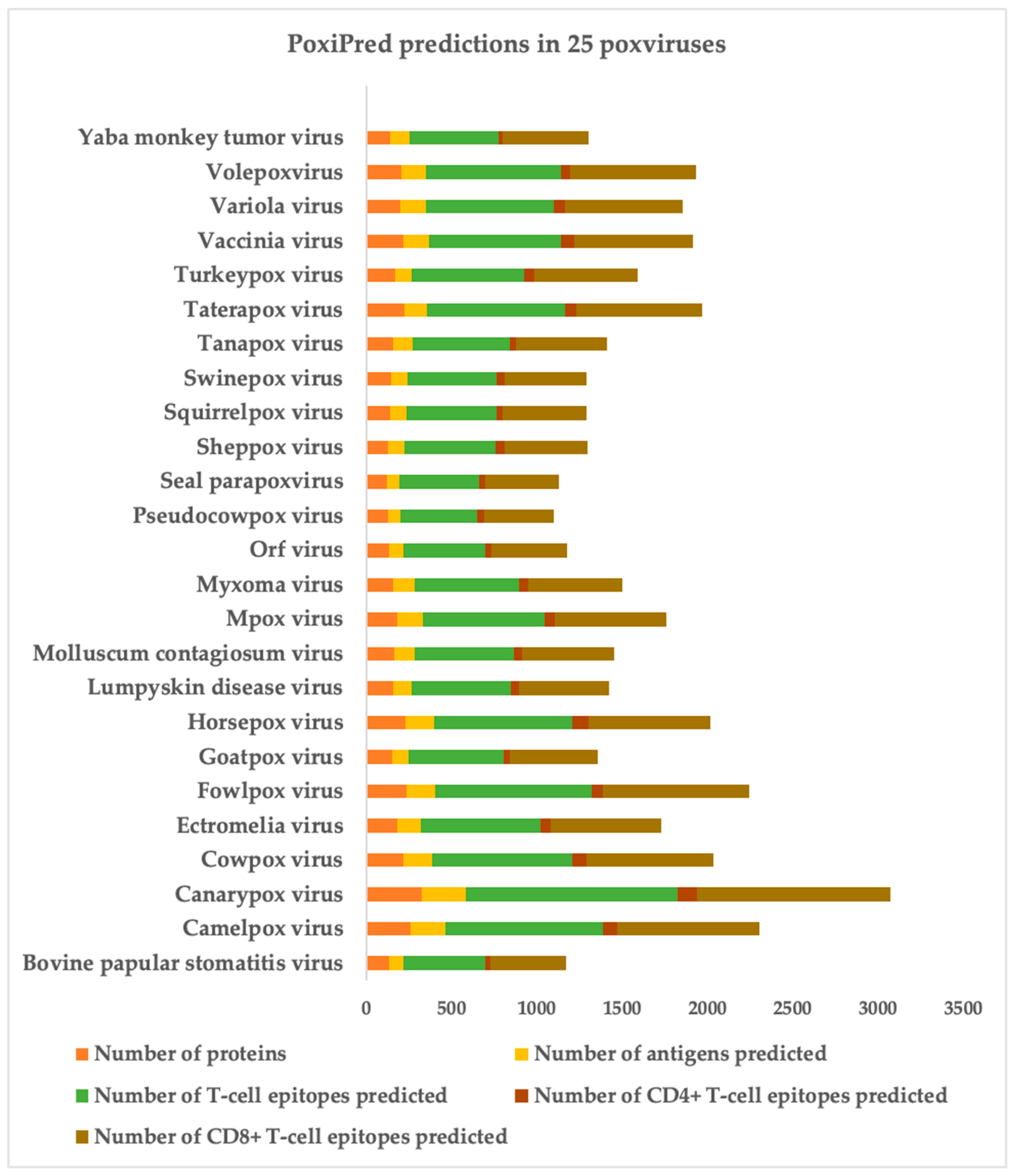
3.3. Antigen and Epitope Prediction in the Proteome Files of 25 Poxviruses
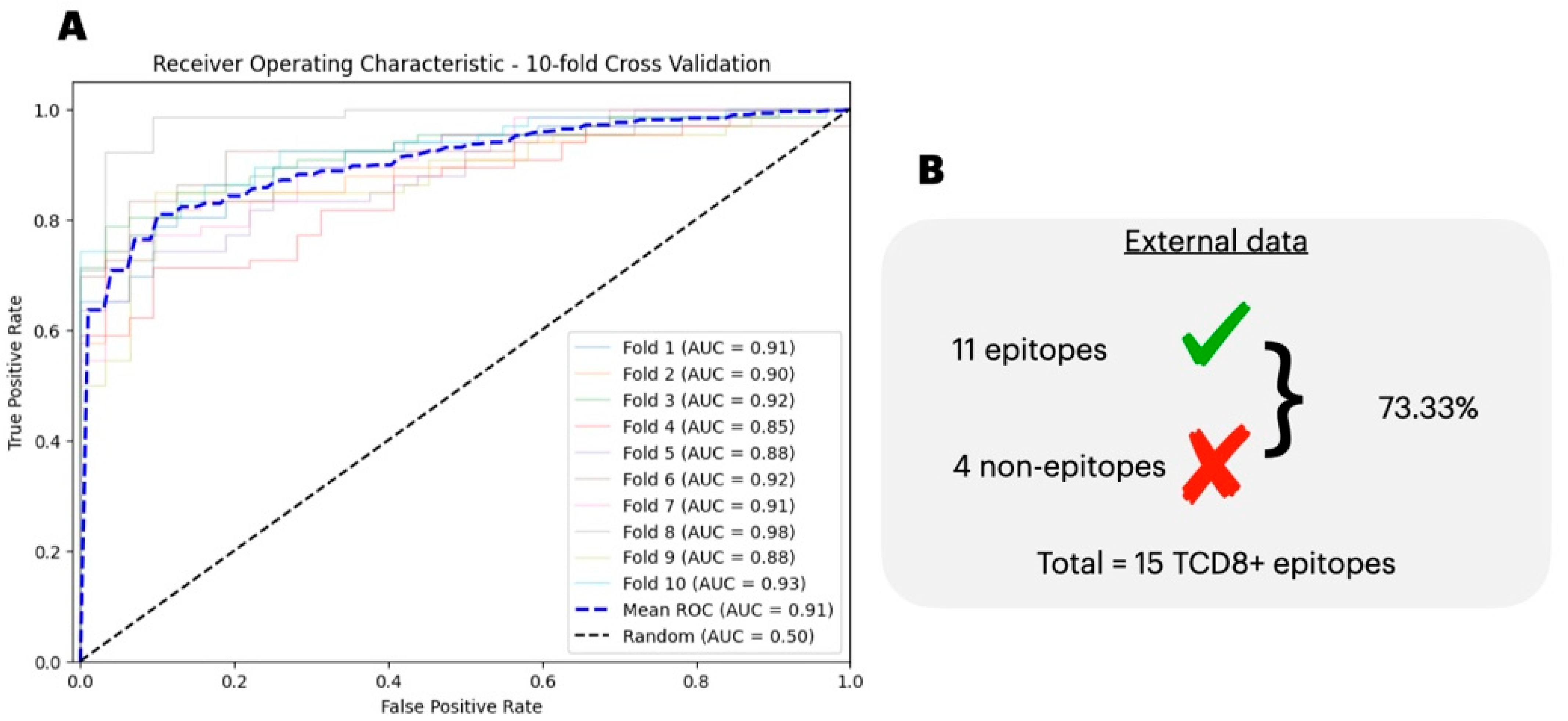
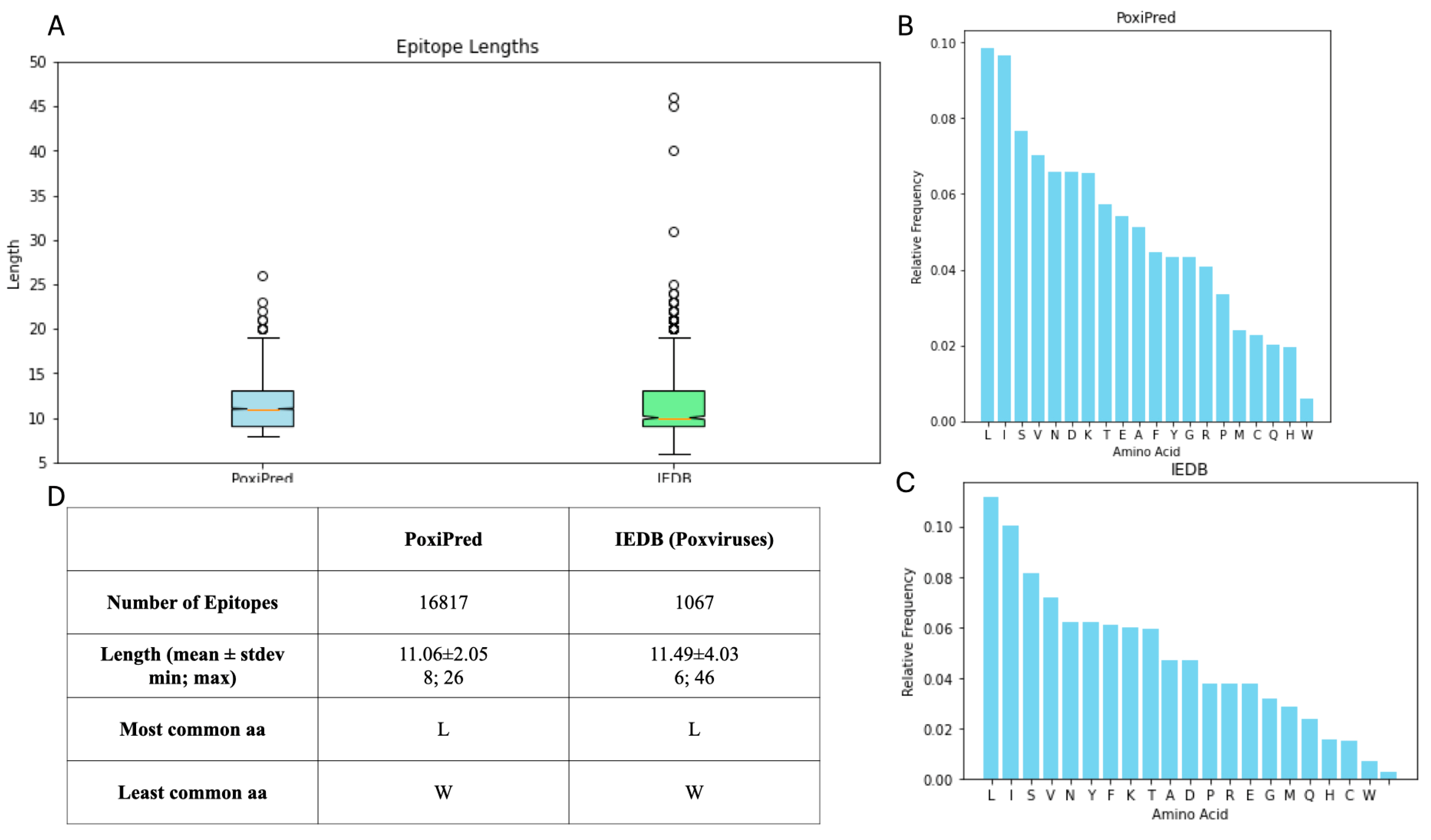
3.4. Comparison of the Predicted with Experimentally Verified Epitopes
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Günther, T.; Haas, L.; Alawi, M.; Wohlsein, P.; Marks, J.; Grundhoff, A.; Becher, P.; Fischer, N. Recovery of the first full-length genome sequence of a parapoxvirus directly from a clinical sample. Sci. Rep. 2017, 7, 3734. [Google Scholar] [CrossRef] [PubMed]
- Gjessing, M.C.; Yutin, N.; Tengs, T.; Senkevich, T.; Koonin, E.; Rønning, H.P.; Alarcon, M.; Ylving, S.; Lie, K.-I.; Saure, B.; et al. Salmon Gill Poxvirus, the Deepest Representative of the Chordopoxvirinae. J. Virol. 2015, 89, 9348–9367. [Google Scholar] [CrossRef] [PubMed]
- Buller, R.M.; Palumbo, G.J. Poxvirus pathogenesis. Microbiol. Rev. 1991, 55, 80–122. [Google Scholar] [CrossRef] [PubMed]
- Upton, C.; Slack, S.; Hunter, A.L.; Ehlers, A.; Roper, R.L. Poxvirus Orthologous Clusters: Toward Defining the Minimum Essential Poxvirus Genome. J. Virol. 2003, 77, 7590–7600. [Google Scholar] [CrossRef] [PubMed]
- Hughes, A.L.; Irausquin, S.; Friedman, R. The Evolutionary Biology of Poxviruses. Infect. Genet. Evol. 2010, 10, 50–59. [Google Scholar] [CrossRef]
- Kaler, J.; Hussain, A.; Flores, G.; Kheiri, S.; Desrosiers, D. Monkeypox: A Comprehensive Review of Transmission, Pathogenesis, and Manifestation. Cureus 2022, 14, e26531. [Google Scholar] [CrossRef]
- McInnes, C.J.; Damon, I.K.; Smith, G.L.; McFadden, G.; Isaacs, S.N.; Roper, R.L.; Evans, D.H.; Damaso, C.R.; Carulei, O.; Wise, L.M.; et al. ICTV Virus Taxonomy Profile: Poxviridae 2023. J. Gen. Virol. 2023, 104, 001849. [Google Scholar] [CrossRef]
- Dutt, M.; Kumar, A.; Rout, M.; Dehury, B.; Martinez, G.; Ndishimye, P.; Kelvin, A.A.; Kelvin, D.J. Drug repurposing for Mpox: Discovery of small molecules as potential inhibitors against DNA-dependent RNA polymerase using molecular modeling approach. J. Cell. Biochem. 2023, 124, 701–715. [Google Scholar] [CrossRef]
- Meyer, H.; Ehmann, R.; Smith, G.L. Smallpox in the Post-Eradication Era. Viruses 2020, 12, 138. [Google Scholar] [CrossRef] [PubMed]
- Isidro, J.; Borges, V.; Pinto, M.; Sobral, D.; Santos, J.D.; Nunes, A.; Mixão, V.; Ferreira, R.; Santos, D.; Duarte, S.; et al. Phylogenomic characterization and signs of microevolution in the 2022 multi-country outbreak of monkeypox virus. Nat. Med. 2022, 28, 1569–1572. [Google Scholar] [CrossRef] [PubMed]
- 2022-23 Mpox Outbreak: Global Trends. World Health Organization: Geneva, Switzerland, 2023. Available online: https://worldhealthorg.shinyapps.io/mpx_global/ (accessed on 7 September 2023).
- Eom, H.J.; Lee, E.S.; Yoo, H.S. Lumpy skin disease as an emerging infectious disease. J. Vet. Sci. 2023, 24, e42. [Google Scholar] [CrossRef]
- Liang, Z.; Yao, K.; Wang, S.; Yin, J.; Ma, X.; Yin, X.; Wang, X.; Sun, Y. Understanding the research advances on lumpy skin disease: A comprehensive literature review of experimental evidence. Front. Microbiol. 2022, 13, 1065894. [Google Scholar] [CrossRef] [PubMed]
- Tsai, K.J.; Tu, Y.C.; Wu, C.H.; Huang, C.W.; Ting, L.J.; Huang, Y.L.; Pan, C.H.; Chang, C.Y.; Deng, M.C.; Lee, F. First detection and phylogenetic analysis of lumpy skin disease virus from Kinmen Island, Taiwan in 2020. J. Vet. Med. Sci. 2022, 84, 1093–1100. [Google Scholar] [CrossRef] [PubMed]
- Crandell, R.A.; Casey, H.W.; Brumlow, W.B. Studies of a Newly Recognized Poxvirus of Monkeys. J. Infect. Dis. 1969, 119, 80–88. [Google Scholar] [CrossRef] [PubMed]
- Downie, A.W. The epidemiology of tanapox and Yaba virus infections. J. Med. Microbiol. 1972, 5, 14. [Google Scholar]
- Espana, C. Review of some outbreaks of viral disease in captive nonhuman primates. Lab. Anim. Sci. 1971, 21, 1023–1031. [Google Scholar] [PubMed]
- Kupper, J.L.; Casey, H.W.; Johnson, D.K. Experimental Yaba and benign epidermal monkey pox in rhesus monkeys. Lab. Anim. Care 1970, 20, 979–988. [Google Scholar] [PubMed]
- Yang, X.; Zhao, L.; Wei, F.; Li, J. DeepNetBim: Deep learning model for predicting HLA-epitope interactions based on network analysis by harnessing binding and immunogenicity information. BMC Bioinform. 2020, 22, 231. [Google Scholar] [CrossRef] [PubMed]
- Yurina, V.; Adianingsih, O.R. Predicting epitopes for vaccine development using bioinformatics tools. Ther. Adv. Vaccines Immunother. 2022, 10, 25151355221100218. [Google Scholar] [CrossRef]
- Oli, A.N.; Obialor, W.O.; Ifeanyichukwu, M.O.; Odimegwu, D.C.; Okoyeh, J.N.; Emechebe, G.O.; Adejumo, S.A.; Ibeanu, G.C. Immunoinformatics and Vaccine Development: An Overview. ImmunoTargets Ther. 2020, 9, 13–30. [Google Scholar] [CrossRef]
- Parvizpour, S.; Pourseif, M.M.; Razmara, J.; Rafi, M.A.; Omidi, Y. Epitope-based vaccine design: A comprehensive overview of bioinformatics approaches. Drug Discov. Today 2020, 25, 1034–1042. [Google Scholar] [CrossRef]
- Sanchez-Trincado, J.L.; Gomez-Perosanz, M.; Reche, P.A. Fundamentals and Methods for T- and B-Cell Epitope Prediction. J. Immunol. Res. 2017, 2017, 2680160. [Google Scholar] [CrossRef]
- Hellberg, S.; Sjöström, M.; Skagerberg, B.; Wold, S. Peptide Quantitative Structure-Activity Relationships, a Multivariate Approach. J. Med. Chem. 1987, 30, 1126–1135. [Google Scholar] [CrossRef]
- Doytchinova, I.A.; Flower, D.R. VaxiJen: A server for prediction of protective antigens, tumour antigens and subunit vaccines. BMC Bioinform. 2006, 8, 4. [Google Scholar] [CrossRef] [PubMed]
- Grifoni, A.; Zhang, Y.; Tarke, A.; Sidney, J.; Rubiro, P.; Reina-Campos, M.; Filaci, G.; Dan, J.M.; Scheuermann, R.H.; Sette, A. Defining antigen targets to dissect vaccinia virus and monkeypox virus-specific T cell responses in humans. Cell Host Microbe 2022, 30. [Google Scholar] [CrossRef]
- Wold, S.; Jonsson, J.; Sjörström, M.; Sandberg, M.; Rännar, S. DNA and peptide sequences and chemical processes multivariately modelled by principal component analysis and partial least-squares projections to latent structures. Anal. Chim. Acta 1993, 277, 239–253. [Google Scholar] [CrossRef]
- Peters, B.; Nielsen, M.; Sette, A. T Cell Epitope Predictions. Annu. Rev. Immunol. 2020, 38, 123–145. [Google Scholar] [CrossRef] [PubMed]
- Desai, D.V.; Kulkarni-Kale, U. T-cell epitope prediction methods: An overview. Methods Mol. Biol. 2014, 1184, 333–364. [Google Scholar] [CrossRef] [PubMed]
- Vujović, M.; Borch, A.; Hadrup, S.R.; Marcatili, P. T Cell Epitope Prediction and Its Application to Immunotherapy. Front. Immunol. 2021, 12, 712488. [Google Scholar] [CrossRef]
- Whang, S.E.; Roh, Y.; Song, H.; Lee, J.G. Data collection and quality challenges in deep learning: A data-centric AI perspective. VLDB J. 2023, 32, 791–813. [Google Scholar] [CrossRef]
- Yang, Z.; Bogdan, P.; Nazarian, S. An in silico deep learning approach to multi-epitope vaccine design: A SARS-CoV-2 case study. Sci. Rep. 2021, 11, 3238. [Google Scholar] [CrossRef] [PubMed]
- Cihan, P.; Ozger, Z.B. A new approach for determining SARS-CoV-2 epitopes using machine learning-based in silico methods. Comput. Biol. Chem. 2022, 98, 107688. [Google Scholar] [CrossRef] [PubMed]
- Choong, A.C.H.; Lee, N.K. Evaluation of convolutionary neural networks modeling of DNA sequences using ordinal versus one-hot encoding method. In Proceedings of the 1st International Conference on Computer and Drone Applications: Ethical Integration of Computer and Drone Technology for Humanity Sustainability, IConDA, Kuching, Malaysia, 9–11 November 2017. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Hidden Layers | Neurons | Accuracy | Specificity | Recall | Precision | Loss | Epochs | |
|---|---|---|---|---|---|---|---|---|
| Train | 1 | 10 | 0.542857 | 1 | 0.085714 | 1 | 0.27382 | 500 |
| 2 | 0.697884 | 1 | 0.395767 | 1 | 0.065661 | |||
| 3 | 0.85873 | 1 | 0.71746 | 1 | 0.009175 | |||
| 1 | 25 | 0.661111 | 1 | 0.322222 | 1 | 0.059489 | ||
| 2 | 0.933598 | 1 | 0.867196 | 1 | 0.000419 | |||
| 3 | 0.983069 | 1 | 0.966138 | 1 | 0.000034 | |||
| 1 | 50 | 0.747354 | 1 | 0.494709 | 1 | 0.014109 | ||
| 2 | 0.953704 | 1 | 0.907407 | 1 | 0.000072 | |||
| 3 | 1 | 1 | 1 | 1 | 0.000001 | |||
| Test | 1 | 10 | 0.533333 | 0.995238 | 0.071429 | 0.4875 | 0.616637 | 500 |
| 2 | 0.680952 | 0.97619 | 0.385714 | 0.871985 | 0.986844 | |||
| 3 | 0.788095 | 0.966667 | 0.609524 | 0.932477 | 1.191654 | |||
| 1 | 25 | 0.65 | 0.995238 | 0.304762 | 0.885714 | 0.490774 | ||
| 2 | 0.861905 | 0.985714 | 0.738095 | 0.9625 | 0.364647 | |||
| 3 | 0.840476 | 0.97619 | 0.704762 | 0.958974 | 0.832513 | |||
| 1 | 50 | 0.707143 | 0.990476 | 0.42381 | 0.933333 | 0.30011 | ||
| 2 | 0.883333 | 0.985714 | 0.780952 | 0.957143 | 0.370044 | |||
| 3 | 0.959524 | 0.995238 | 0.92381 | 0.995 | 0.066186 |
| Hidden Layers | Neurons | Accuracy | Specificity | Recall | Precision | Loss | Epochs | |
|---|---|---|---|---|---|---|---|---|
| Train | 1 | 10 | 0.500057 | 1 | 0.000114 | 0.1 | 0.492214 | 100 |
| 2 | 0.506767 | 1 | 0.013529 | 0.7 | 0.406405 | |||
| 3 | 0.534196 | 1 | 0.046385 | 0.9 | 0.368531 | |||
| 1 | 25 | 0.505401 | 1 | 0.0108 | 0.7 | 0.353444 | ||
| 2 | 0.700928 | 1 | 0.401827 | 1 | 0.073519 | |||
| 3 | 0.80755 | 0.999886 | 0.615183 | 0.999833 | 0.028656 | |||
| 1 | 50 | 0.56396 | 1 | 0.127903 | 0.9 | 0.163809 | ||
| 2 | 0.900867 | 1 | 0.801725 | 0.00165 | 0.827208 | |||
| 3 | 0.999943 | 1 | 0.999886 | 1 | 0.000021 | |||
| Test | 1 | 10 | 0.5 | 1 | 0 | 0 | 0.621593 | 100 |
| 2 | 0.5082 | 0.998969 | 0.017473 | 0.06 | 0.587974 | |||
| 3 | 0.519487 | 0.99898 | 0.040112 | 0.591667 | 0.580427 | |||
| 1 | 25 | 0.503074 | 0.99898 | 0.007185 | 0.55 | 0.569142 | ||
| 2 | 0.688061 | 0.998969 | 0.377341 | 0.9975 | 0.395682 | |||
| 3 | 0.765824 | 0.988776 | 0.543194 | 0.940396 | 0.408892 | |||
| 1 | 50 | 0.568184 | 0.998969 | 0.137513 | 0.895455 | 0.41917 | ||
| 2 | 0.827208 | 0.981633 | 0.672996 | 0.960404 | 0.32027 | |||
| 3 | 0.93145 | 0.996928 | 0.86599 | 0.996532 | 0.090237 |
| Accuracy | Precision | Recall | F1 Score | |
|---|---|---|---|---|
| Random Forest | 0.76 | 0.78 | 0.92 | 0.84 |
| Support Vector Machines | 0.69 | 0.69 | 1 | 0.82 |
| Logistic Regression | 0.65 | 0.68 | 0.93 | 0.79 |
| Gradient Boosting | 0.80 | 0.82 | 0.81 | 0.86 |
| Extreme Gradient Boosting | 0.82 | 0.84 | 0.90 | 0.87 |
| K-Nearest Neighbors | 0.61 | 0.84 | 0.54 | 0.66 |
| Fold n. | F1 Score | Balanced Accuracy | Geometric Mean |
|---|---|---|---|
| 1 | 0.89 | 0.72 | 0.82 |
| 2 | 0.85 | 0.74 | 0.72 |
| 3 | 0.88 | 0.84 | 0.84 |
| 4 | 0.82 | 0.71 | 0.70 |
| 5 | 0.82 | 0.78 | 0.78 |
| 6 | 0.90 | 0.87 | 0.87 |
| 7 | 0.84 | 0.79 | 0.79 |
| 8 | 0.94 | 0.93 | 0.93 |
| 9 | 0.89 | 0.87 | 0.87 |
| 10 | 0.88 | 0.84 | 0.83 |
| Mean | 0.87 | 0.82 | 0.82 |
| Standard deviation | 0.04 | 0.06 | 0.07 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Martinez, G.S.; Dutt, M.; Kelvin, D.J.; Kumar, A. PoxiPred: An Artificial-Intelligence-Based Method for the Prediction of Potential Antigens and Epitopes to Accelerate Vaccine Development Efforts against Poxviruses. Biology 2024, 13, 125. https://doi.org/10.3390/biology13020125
Martinez GS, Dutt M, Kelvin DJ, Kumar A. PoxiPred: An Artificial-Intelligence-Based Method for the Prediction of Potential Antigens and Epitopes to Accelerate Vaccine Development Efforts against Poxviruses. Biology. 2024; 13(2):125. https://doi.org/10.3390/biology13020125
Chicago/Turabian StyleMartinez, Gustavo Sganzerla, Mansi Dutt, David J. Kelvin, and Anuj Kumar. 2024. "PoxiPred: An Artificial-Intelligence-Based Method for the Prediction of Potential Antigens and Epitopes to Accelerate Vaccine Development Efforts against Poxviruses" Biology 13, no. 2: 125. https://doi.org/10.3390/biology13020125