
Chromosome-Level Genome Assembly of Protosalanx chinensis and Response to Air Exposure Stress

1
College of Fisheries, Key Lab of Freshwater Animal Breeding, Ministry of Agriculture and Rural Affair, Engineering Research Center of Green Development for Conventional Aquatic Biological Industry in the Yangtze River Economic Belt, Ministry of Education, Huazhong Agricultural University, Wuhan 430070, China
2
Key Laboratory of Freshwater Fisheries and Germplasm Resources Utilization, Ministry of Agriculture and Rural Affairs, Freshwater Fisheries Research Center, Chinese Academy of Fishery Sciences, Wuxi 214081, China
3
Wuxi Fisheries College, Nanjing Agricultural University, Wuxi 214081, China
*
Author to whom correspondence should be addressed.
Biology 2023, 12(9), 1266; https://doi.org/10.3390/biology12091266
Submission received: 2 June 2023
/
Revised: 5 September 2023
/
Accepted: 11 September 2023
/
Published: 21 September 2023
(This article belongs to the Section Genetics and Genomics)
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
In the present study, we assembled a high-quality chromosome-level genome of Protosalanx chinensis, which is the first chromosome-level genome for Salangidae. These genomic data provide a fundamental resource for ecological and adaptation studies of Protosalanx chinensis, and offer a deeper understanding of the response to air exposure stress and species conservation.
Abstract
Protosalanx chinensis is a suitable particular species for genetic studies on nearly scaleless skin, transparency and high sensitivity to hypoxia stress. Here, we generated a high-quality chromosome-level de novo assembly of P. chinensis. The final de novo assembly yielded 379.47 Mb with 28 pseudo-chromosomes and a scaffold N50 length of 14.52 Mb. In total, 21,074 protein-coding genes were predicted. P. chinensis, Esox lucius and Hypomesus transpacificus had formed a clade, which diverged about 115.5 million years ago. In the air exposure stress experiment, we found that some genes play an essential role during P. chinensis hypoxia, such as bhlh, Cry1, Clock, Arntl and Rorb in the circadian rhythm pathway. These genomic data offer a crucial foundation for P. chinensis ecology and adaptation studies, as well as a deeper understanding of the response to air exposure stress.
1. Introduction
Protosalanx chinensis (family Salangidae, order Salmoniformes, Figure 1) is a small annual cold-temperature fish endemic to East Asia and has some specific morphological characteristics, including transparency and scaleless skin [1,2,3,4]. P. chinensis exhibits strong ecological plasticity, with populations found in both freshwater and seawater habitats, including the Yangtze River Basin and its associated lakes (Taihu Lake, Hongze Lake, etc.), as well as the offshore waters of the Yellow Sea, Bohai Sea and East China Sea [1,5,6]. Due to the important economic value of P. chinensis, it was widely translocated to many lakes and reservoirs in northern China in the mid-1980s, and gradually formed a stable population [7]. This deliberate act of artificial translocation has engendered a remarkable shift, resulting in a notable proliferation of P. chinensis across diverse aquatic habitats, effectively amplifying its presence [7]. Studies of the genomic and physiological characteristics of P. chinensis have helped us to better understand the environmental adaptations of P. chinensis.
P. chinensis is a valuable model for studying the molecular mechanisms underlying the evolution of hypoxia. P. chinensis is difficult to obtain alive and can die quickly when stressed by hypoxia during net fishing [1]. Previous studies on the genome of P. chinensis have focused only on phylogeny, sexual differentiation, and skeletal development [2,3]. A basic understanding of stress and the corresponding physiological state of P. chinensis is still lacking. An air exposure stress experiment is an effective experimental tool with which to understand the stress state. An air exposure experiment on gilthead seabream showed that stress-induced hormonal changes affected the liver’s metabolic organization and highlighted the crucial role of vasotocinergic and isotocinergic pathway [8]. A study of rainbow trout demonstrated changes in miRNAs in fish blood during air exposure and identified several miRNA markers [9]. As an economically valuable fish, P. chinensis has not yet been fully cultured in captivity and transported live, so there is potential value in studying its response to hypoxic stress.
With the development of genomic (particularly long-reads) sequencing, two draft genome assemblies of P. chinensis had recently been reported with assembly quality with a contig N50 of 17.2 Kb [2], and a contig N50 of 103 Kb [3], respectively (Protosalanx hyalocranius and Protosalanx chinensis were the same species). Although these two genome drafts provide preliminary genetic information of P. chinensis, these genomes are deficient due to the limitation of sequencing technology. Therefore, chromosome-level genomes and comparative genomics resource are essential to understanding ecological and evolutionary research, translocation adaptation, and genetic improvement.
Here, we generated a high-quality chromosome-level de novo assembly of P. chinensis. A set of protein-coding genes was annotated, and the evolutionary history of P. chinensis was analyzed. In the air exposure stress experiment, the expression pattern of differentially expressed genes (DEGs) was investigated. These genomic data offer a crucial foundation for P. chinensis ecology and adaptation studies, as well as a deeper understanding of the response to air exposure stress and species conservation.
2. Materials and Methods
2.1. Sample Collection and DNA and RNA Sequencing
We collected muscle samples from an adult P. chinensis individual in the Hongze Lake at Jiangsu, China, for sequencing (Figure 1). After the muscle samples were collected, they were rapidly frozen in liquid nitrogen and stored at −80 °C until DNA extraction. DNA was extracted from muscle tissue. RNA was extracted from the larvae. DNA was extracted following the phenol/chloroform DNA extraction method. After the extraction of the DNA and RNA, corresponding quality control was conducted according to different library construction types. The quality control included assessing the concentration, purity, and fragment integrity of the samples.
With the BGI MGISEQ platform, a short insert WGS library was generated according to the manufacturer’s recommendations. A PacBio HiFi library was constructed using a QIAGEN Blood & Cell Culture DNA Midi Kit following the manufacturer’s instructions (QIAGEN, Hilden, Germany) and then sequenced on the PacBio Sequel II system. A Hi-C library was generated using the Mbo I restriction enzyme and sequenced on the BGI MGISEQ platform. We constructed one PacBio HiFi library with an insert fragment size of approximately 15 kb, and one Hi-C library with an insert fragment size of approximately 300 bp.
Fifteen RNA libraries were constructed using the TRIzol Total RNA Isolation Kit (Takara, San Jose, CA, USA) after which the concentration and purity of the extracted RNA were measured to ensure quality. Subsequently, the RNA was fragmented into appropriate lengths using digestion enzymes. This fragmented RNA was then reverse-transcribed to synthesize cDNA, which underwent end repair, addition of specific adapter sequences, and PCR amplification for library construction. Finally, the constructed RNA library was sequenced on the BGI MGISEQ platform.
2.2. Sequencing QC and Genome Assembly
We used the SOAPnuke v2.1.7 [10] pipeline to filter out the low-quality and adaptor reads. After that, we calculated the K-mer (k = 21) frequency distribution with Jellyfish v2.2.6 [11] and analyzed the result using GenomeScope v1.0 [12]. HiFi reads with about 62× coverage were sequenced using the PacBio Sequel platform and cleaned with SMRTLink v8. The contig assembly was carried out using Hifiasm v0.16.1-r375 [13], followed by a removal of the redundant sequences with the Purge-Haplotigs [14] program. Subsequently, the contigs were further connected to the chromosome level using the Juicer v1.5 [15] and 3D-DNA v180922 [16] pipelines. The BUSCO completeness score of the P. chinensis genome was calculated using BUSCO v5.2.2 [17] based on the actinopterygii (odb10) dataset.
2.3. Identification of Repetitive Sequences
We identified the repetitive sequences using a combination of de novo and homolog-based methods. For de novo annotation, we used RepeatModeler v1.0.4 (http://www.repeatmasker.org/RepeatModeler/, accessed date: 23 February 2023) and LTR-FINDER v1.0.7 [18] software to construct a primary library. This customed library was used to screen repeat sequences via the program RepeatMasker v4.0.7 [19]. For the homolog-based prediction, we utilized RepeatMasker v4.0.7 [19], RepeatProteinMasker v4.0.7 [19] and Tandem Repeat Finder v4.10.0 [20] based on the Repbase database.
2.4. Genome Annotation
Gene prediction was conducted through a combination of homology-based prediction, ab initio prediction and transcriptome-based prediction methods. Next, 96.4 Gb RNA-seq data were directly mapped to P. chinensis assembly with Hisat2 v2.1.0 [21] to identify putative exon regions and splice junctions. StringTie v1.3.5 [22] was then used to assemble the mapped reads into gene models and validated using PASA v2.5.2 [23]. Finally, we identified the candidate coding regions by employing TransDecoder v5.5.0 (https://github.com/TransDecoder/TransDecoder, accessed date: 23 February 2023). For homology-based annotation, we downloaded the assemblies and gene annotation files of four actinopterygii species (Danio rerio, Oryzias. latipes, P. hyalocranius and Salmo salar) from the NCBI database. Combined with the above RNA-seq and homolog data, we predicted the homology-like coding sequences using GeMoMa v1.8 [24]. A total of 1200 high-quality coding genes were used to train the predictors using August v3.2.1 [25] and SNAP v2006-07-28 [26] (Korf, 2004) and then ab initio prediction was performed. Lastly, we integrated all the protein-coding genes predicted using the above three strategies with the EVidenceModeler (EVM) pipeline v1.1.1 [23].
2.5. Phylogenetic and Gene Family Analysis
We used OrthoFinder v2.3.11 [27] to cluster protein-coding genes. Single-copy orthologous genes (1:1:1) were aligned using MAFFT v7.310 [28]. Referring to the methods used in previous studies on P. chinensis [3], we used PhyML v3.3 [29] with the HKY85 model to construct a maximum-likelihood phylogenetic tree with 100 pseudoreplicates. All branches had 100/100 bootstrap support, showing phylogeny consistent with a previous study [3]. We estimated the species divergence time using MCMCTREE in PAML v4.9 [30]. Four divergence time points from TimeTree (http://timetree.org.cn, accessed date: 23 February 2023) were used to calibrate the divergence times: (a) Callorhinchus milii and Latimeria chalumnae (421.5–461.6 MYA), (b) L. chalumnae and Lepisosteus oculatus (416.4–422.2 MYA), (c) L. oculatus and Anguilla Anguilla (372.4–383.4 MYA) and (d) Xiphophorus maculatus and Oryzias latipes (122.3–138.4 MYA).
Based on the core-orthologous gene sets, we used MCscanX v1.5.2 [31] to define syntenic blocks between P. chinensis and H. transpacificus, P. chinensis and E. Lucius. The core-orthologous gene sets were identified using Blast (v2.0.14) with an E-value threshold of 1 × 10−5 (at least 20 syntenic genes allowed), and visualized using NGenomeSyn v1.41 (https://github.com/hewm2008/NGenomeSyn, accessed date: 23 February 2023). We used CAFE v2.1 [32] to calculate the overall P-value of each branch and node with the Viterbi method.
2.6. Transcriptome under Air Exposure Stress
During the harvesting of P. chinensis, hypoxia frequently arises when they are removed from water, leading to significant mortality due to oxygen deprivation. Based on a preliminary experiment, exposure to air for more than 10 min resulted in massive mortality of the larvae. We designed an experiment to simulate the hypoxia process with five groups (the larvae fish re-entered the water after 10 min out of the water, and they either died immediately (DIC), died 15 min later (DIF), died half an hour later (DHH), died an hour later (DOH), or remained alive an hour later (UOH)). Subsequently, the clean reads were aligned onto the CDS of P. chinensis using Bowtie2 v 2.4.1 [33]. Gene expression levels were measured using a software package named RSEM [34]. The fragments per kilobase of exon per million mapped reads (FPKM) [35] method was used to calculate the expression level. In total, 17,299 genes were detected in at least one of the samples (FPKM ≥ 1). Additionally, a total of 1921 DEGs were detected via pairwise comparison with R package DESeq2 [36]. Subsequently, trend analysis was performed using the OmicShare tools (https://www.omicshare.com/tools, accessed date: 23 February 2023).
3. Results
3.1. Chromosome-Scale Genome Assembly
After sequencing, we obtained 45 Gb short-insert-size data for the P. chinensis genome survey (Table S1). The genome size of P. chinensis was estimated to be ~392.80 Mb with 0.68% heterozygosity (Figure S1 and Table S2). A total size of 378.20 Mb genome assembly with a contig N50 of 0.53 Mb was acquired (Table S3). Notably, the size of contig N50 was 5-fold longer than P. chinensis [3] and 25-fold longer than P. hyalocranius [2] (Table S3). Finally, we constructed a better P. chinensis genome assembly with 379.47 Mb genome size with a scaffold N50 size of 14.52 Mb, and 98.35% of the assembly sequences were assigned to 28 pseudochromosomes (Figure 2 and Figure 3 and Table S4).
3.2. Repetitive Sequences
In total, we identified 35.35% (134.14 Mb) repeat sequences of the P. chinensis genome assembly, of which the highest proportion was DNA transposons (16.42%), followed by long interspersed nuclear elements (LINEs, 11.73%), and long terminal repeats (LTRs, 8.17%) (Tables S5 and S6, Figure S2).
3.3. Genome Annotation
In total, we have identified 21,074 protein-coding genes with an average gene length of 8017 bp and average coding sequence (CDS) length of 1695 bp. The average exon number per gene was 10 with an average exon length of 175 bp, and an average intron length of 727 bp (Table S7). To evaluate the credibility of gene annotation, we calculated the overlap ratio between the P. chinensis gene models and the prediction results from de novo, homolog-based, and RNA-seq prediction. 99.66% of P. chinensis gene models were supported by at least one piece of evidence at the level of greater than 80% CDS overlap ratio (Table S8). Notably, the length of gene models had a similar distribution trend at the genes, CDS, exons and introns level, compared to D. rerio, O. latipes, P. hyalocranius and S. salar (Figure S3). The Benchmarking Universal Single-Copy Orthologs (BUSCO) test, referencing the 3640 actinopterygii protein set, identified 88.10% of gene sets as complete, 83.40% of the complete actinopterygii (odb10) genes were found (Table S9).
In addition, we aligned 95.7% of coding proteins into seven functional databases, including the non-redundant (NR) protein database of NCBI (95.25%), SwissProt [37] (87.64%), Kyoto Encyclopedia of Genes and Genomes (KEGG) [38] (86.15%), KOG (76.36%), Translation of European Molecular Biology Laboratory (Trembl) (95.33%), InterPro (91.59%), and Gene Ontology (GO) databases (68.66%) (Table S10 and Figure S4).
3.4. Phylogenetic and Gene Family Analysis
Finally, 357,384 protein-coding genes were clustered into 29,939 gene families in 15 Osteichthyes and 1 Chondrichthyes (Figure S5). Of these, 347,307 (97.18%) genes were identified as 19,862 orthologous groups (Table S11). Our analysis indicated that P. chinensis and H. transpacificus diverged about 115.5 MYA, and P. chinensis and Esox lucius diverged about 256.2 MYA (Figure S6). P. chinensis and H. transpacificus had a highly similar synteny to P. chinensis vs. E. Lucius (Figure 4 and Table S12).
Compared to the last common ancestor, P. chinensis genome had a total of 509 expanded gene families and 3701 contracted gene families (Figure 5). Of these, 103 expanded gene families (including 1373 genes) and 86 contracted gene families (including 81 genes) were calculated to be markedly changed with a p-value less than 0.01 (Figure 5). Through KEGG enrichment analysis, we observed that the significant expansion of gene families was mainly enriched into pathways like the pentose phosphate pathway, galactose metabolism, fructose and mannose metabolism, and so on (Figure S7 and Table S13), while the significant extraction gene families were mainly clustered into pathways including ascorbate and aldarate metabolism (Figure S8, Tables S14 and S15).
3.5. Gene Expression under Air Exposure Stress
Three biological replicates were included for each group, which yielded approximately 96.4 Gb clean reads in total (Table S16). In total, 17,299 genes were detected in at least one of the samples (FPKM ≥ 1). Furthermore, 1921 DEGs were detected. We observed that the expression pattern of 35 DEGs had an upward trend, whereas the expression profile of 71 DEGs demonstrated a downward trend (Figure 6). Upward trending DEGs were significantly enriched in circadian rhythm, glycosaminoglycan biosynthesis—keratan sulfate, lysosome and phagosome. Downward trending DEGs were significantly enriched in circadian rhythm—fly, circadian rhythm, arginine and proline metabolism, dopaminergic synapse, IL-17 signaling pathway, and oxytocin signaling pathway (Tables S17 and S18). In the circadian rhythm pathway, bhlh gene and cry1 gene were consistently upregulated, while clock gene, arntl gene, and rorb gene were consistently downregulated.
4. Discussion
The high-quality fish genome serves as a transformative key into the intricate world of aquatic life, revealing the evolutionary history, environmental adaptations, and potential applications for aquaculture [39,40]. The P. chinensis has attracted the attention of researchers due to its unique biological properties and economic value [2,3]. With the development of genomics technology, researchers have been able to study the genome of P. chinensis. In terms of genome quality, our genome exceeds that of previous genomes. The use of PacBio HIFI sequencing for library construction provides higher accuracy and longer read-continuity compared to previous PacBio CLR and WGS sequencing [2,3]. As a result, the genome assembly generated from HiFi data has improved contiguity, as indicated by an increased contig N50. The size of contig N50 was 5-fold longer than P. chinensis [3] and 25-fold longer than P. hyalocranius [2] (Tables S3 and S4). The size of scaffold N50 was 2.8-fold longer than P. chinensis [3] and 12-fold longer than P. hyalocranius [2] (Tables S3 and S4). And we assembled 28 pseudochromosomes, with 98.35% of the assembled sequences attributed to 28 chromosomes. We believe that our study can provide more detailed basic information for the study of the germplasm resources of P. chinensis.
By constructing a phylogenetic tree of 16 fish species, including P. chinensis, our results equally support previous studies that Osmeriformes (P. chinensis) acts as a sister order to Esociformes (E. lucius) [3]. This revelation underscores the close evolutionary affinity between these two orders, elucidating the shared ancestry and evolutionary trajectories that have shaped their distinct yet interconnected genetic landscapes. Delving deeper into the annals of evolutionary history within the Osmeriformes order, we directed our attention towards an intriguing comparison of divergence times between Hypomesus (H. transpacificus) and Protosalanx (P. chinensis). The time of divergence between the two genera is estimated to be about 115.5 MYA, i.e., the Cretaceous period.
As with the genomes of most fish, the repetitive elements of P. chinensis form a large part of the genome. Our comprehensive analysis of the P. chinensis genome assembly unveiled a rich tapestry of repeat sequences, amounting to a total of 35.35% (134.14 Mb) of the genome. Within this repetitive landscape, we observed the dominance of DNA transposons, constituting the largest proportion at 16.42%. Following closely behind were long interspersed nuclear elements (LINEs) at 11.73% and long terminal repeats (LTRs) at 8.17% (Tables S5 and S6, Figure S2). These findings shed light on the intricate composition and dynamics of repetitive elements within the P. chinensis genome, paving the way for further investigations into their functional significance and evolutionary implications.
Previous studies on the feeding habits of P. chinensis have shown that there is a shift in feeding habits from phytoplankton to carnivorous after the juvenile stage of P. chinensis [41]. Analysis of gene families showed that 103 gene families including 1373 genes had been expanded. Interestingly, these genes are significantly enriched in metabolic pathways such as galactose metabolism, fructose and mannose metabolism, biosynthesis of amino acids, carbon metabolism, and metabolic pathways. Although the expansion or contraction of gene families may be the result of random and natural selection and difficult to prove [42], the potential association between the expansion of these genes and the feeding habits of P. chinensis provides an important reference for future studies.
Air exposure stress is a useful method for studying the physiological responses of aquatic economic animals under environmental stress. In commerce, fish and shrimps encounter environmental stresses of hypoxia during capture, loading and transport. And studying the physiological characteristics and performance of aquatic economic animals under hypoxic conditions can help improve their survival rates and reduce losses [43,44]. In evolutionary terms, air exposure stress also helps to explore potential mechanisms of aquatic to terrestrial evolution [45]. Due to the difficult availability and high mortality rate of adult P. chinensis, we selected available larvae at early developmental stages as experimental subjects to analyse physiological trends through their transcriptional expression.
In the enrichment pathway of the upregulation trend, lysosomal and phagosome responses are important predictors of environmental stress in aquatic animals [46]. Lysosome and phagosome pathway related genes indicated that the organism of P. chinensis exhibited a continuous environmental stress response under the air exposure stress. In addition, IL-17 signaling showed a downward trend. The IL-17 signaling pathway is thought to be important for the maintenance of health under physiological stress [47]. Studies have shown that IL-17 plays a role in the resistance of fish to bacterial infection and is involved in the immune response. Differential expression of IL-17 family genes in skin tissues occurred in turbot after infection with Vibrio anguillarum [48]. The persistent decline of IL-17-related genes during air exposure stress in P. chinensis may indicate that air exposure stress affects the immune function of P. chinensis fingerlings to some extent.
Interestingly, pathway enrichment analysis showed that these two clusters of genes were significantly enriched in circadian rhythm pathways. Circadian rhythm-related genes showed both significant and consistent upregulation and consistent downregulation. Some studies have shown that the circadian rhythm is associated with a stress response in fish [49]. Significant circadian rhythm changes in heart rate in rainbow trout have been observed after stress from transport, etc. [50]. A study on spiny ginseng suggests that circadian rhythms are involved in the response to heat, hypoxia, and thermo-hypoxic stress through DNA methylation [51]. Although the specific function and role of circadian rhythm-related genes during stress in P. chinensis could not be clarified in this study, the specific mechanism of action deserves further investigation.
Genes associated with circadian rhythms are affected in fish following environmental stresses such as temperature, light, and hypoxia [52,53,54]. A hypoxia exposure study in Phoxinus lagowskii showed that sustained hypoxia exposure resulted in significant upregulation of cry1b gene. Under diel-cycling hypoxia exposure, the expression of three of the eight clock genes was increased, including per1a, clocka, and cry1b [53]. cry1a and cry1b expression is increased by heat shock in zebrafish ZEM-2S cells under a light–dark cycle [52]. Similarly, cold shock leads to dysregulation of the expression of genes such as zebrafish circadian rhythms, phototransduction and the IL-17 signing pathway and causes inflammation [54]. In this study, we showed that the expression of clock genes was changed in the larvae of P. chinensis under air exposure stress, indicating that hypoxic stress induced dysregulation of physiological rhythms in P. chinensis.
Future studies on the genome of P. chinensis should enhance the assembly of the “Telomere to Telomere” (T2T) genome by increasing long-read ONT sequencing [55]. It is also essential to validate the assembled genome’s accuracy using methods like PCR or Sanger sequencing. An in-depth exploration of comparative and functional genomics can uncover more biological significance. By incorporating these improvements, the study’s methodological robustness and reliability can be improved, resulting in more detailed and persuasive findings.
These genomic datasets aimed at unraveling the ecological dynamics and adaptive mechanisms of P. chinensis. Furthermore, they provided invaluable insights into the species’ ability to cope with air exposure stress, shedding light on the molecular underpinnings of its resilience. Beyond these immediate implications, our findings have far-reaching implications for the conservation and preservation of this remarkable species, elevating our understanding of its unique biological makeup and enabling more effective conservation strategies to safeguard its future.
5. Conclusions
In summary, a better P. chinensis genome assembly with 379.47 Mb genome size was reported with a scaffold N50 size of 14.52 Mb, and 98.35% of the assembly sequences was assigned to 28 pseudochromosomes. The expansion of gene families related to galactose metabolism, fructose and mannose metabolism, biosynthesis of amino acids, and carbon metabolism pathways in the genome of P. chinensis provides novel insights. Based on the intricate patterns of gene expression, our findings unveil the remarkable physiological activities and molecular responses exhibited by P. chinensis in hypoxic conditions. The circadian rhythm pathway is likely to be critical in the process of air exposure stress in P. chinensis, and its role cannot be ignored. These insights serve as a crucial stepping-stone towards unraveling the intricate mechanisms governing the species’ survival and adaptation strategies, further expanding our understanding of the complex interplay between genetics, physiology, and environmental stressors.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/biology12091266/s1, Figure S1: 21-mers analysis for estimating the genome size of P. chinensis; Figure S2: Divergence distribution of repetitive elements in P. chinensis genome; Figure S3: Distribution of gene, coding sequence, exon, and intron lengths, and exon number in P. chinensis and other four genomes; Figure S4: Gene function annotation results in the five databases of NR, InterPro, KEGG, SwissProt and KOG statistics Venn diagram; Figure S5: Phylogenetic tree of 16 species based on maximum-likelihood using 2152 single-copy orthologs; Figure S6: Estimation of divergence times of 16 species; Figure S7: Functional enrichment results of expansion gene families in P. chinensis genome, Terms with p < 0.01 was selected; Figure S8: Functional enrichment results of extraction gene families in P. chinensis genome, Terms with p < 0.01 was selected; Table S1: Sequencing data used for the genome P. chinensis assembly; Table S2: The information of P. chinensis genome survey analysis; Table S3: The statistics of length and number for the de novo assembled Protosalanx genomes; Table S4: Statistics of chromosomal length of P. chinensis genome; Table S5: Repetitive sequences in P. chinensis genome; Table S6: Transposable elements in P. chinensis genome; Table S7: Gene predictions in P. chinensis genome; Table S8: The evidence supporting the gene models of P. chinese genome; Table S9: BUSCO analysis result of P. chinensis genome; Table S10: Functional annotations of P. chinensis genes; Table S11: Gene family clustered; Table S12: The statics of Syntenic Blocks; Table S13: Top 20 pathway resulted from KEGG. KEGG enrichment of the markable expanded gene family in P. chinensis genome; Table S14: Top 20 pathway resulted from KEGG. KEGG enrichment of the markable extracted gene family in P. chinensis genome; Table S15: Data for analysis in this study; Table S16: RNA map ratio; Table S17: KEGG Pathway enrichment analysis for upward trend DEGs; Table S18: Pathway enrichment analysis for downward trend DEGs.
Author Contributions
Conceptualization, Y.Z. (Yanfeng Zhou); methodology, Y.Z. (Yanfeng Zhou); formal analysis, X.Z. and Y.D.; investigation, X.T. and Y.Z. (Yifan Zhou); writing—original draft preparation, Y.Z. (Yanfeng Zhou) and X.Z.; writing—review and editing, H.L. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by the grant from the Key R&D project of Hubei Province (2022BBA0050) and Basic scientific research project of the Chinese Academy of Fishery Sciences (2020TD61).
Institutional Review Board Statement
The animal study protocol was approved by the Ethics Committee of Freshwater Fisheries Research Center (FFRC) of the Chinese Academy of Fishery Sciences (CAFS) (FEH20200807, 2020/08/07).
Informed Consent Statement
Not applicable.
Data Availability Statement
The raw genome sequencing data for P. chinensis were deposited in the NCBI Sequence Read Archive (SRA) database under Accession the BioProjectID PRJNA915822. The genome assembly, genome annotation, coding sequences, protein sequences, repeat annotation and functional annotation files were deposited in Figshare: https://doi.org/10.6084/m9.figshare.22144694.v1 (accessed date: 23 February 2023).
Conflicts of Interest
The authors declare no conflict of interest.
References
- Tang, F.; Gao, W.; Li, H.; Liu, W. Biology and fishery ecology of Protosalanx chinensis: A review. J. Fish. China 2020, 44, 2100–2111. [Google Scholar]
- Liu, K.; Xu, D.; Li, J.; Bian, C.; Duan, J.; Zhou, Y.; Zhang, M.; You, X.; You, Y.; Chen, J.; et al. Whole Genome Sequencing of Chinese Clearhead Icefish, Protosalanx hyalocranius. Gigascience 2017, 6, giw012. [Google Scholar] [CrossRef]
- Zhang, J.; Qi, J.; Shi, F.; Pan, H.; Liu, M.; Tian, R.; Geng, Y.; Li, H.; Qu, Y.; Chen, J.; et al. Insights into the Evolution of Neoteny from the Genome of the Asian Icefish Protosalanx chinensis. iScience 2020, 23, 101267. [Google Scholar] [CrossRef]
- Wang, Z.; Fu, C.; Lei, G. Biodiversity of Chinese Icefishes (Salangidae) and Their Conserving Strategies. Biodivers. Sci. 2002, 10, 416–424. [Google Scholar] [CrossRef]
- Zhang, Y.; Dong, S.; Wang, Q.; Sun, Z. The isozyme genetic structures in large icefish (Protosalanx hyalocranius) and Taihu Lake icefish (Neosalanx taihuensis). J. Dalian Fish. Coll. 2005, 20, 111–115. [Google Scholar]
- Jian, Y.; Xun, X.; HongBo, L. Bioaccumulation of elements in icefish Protosalanx hyalocranius from the Taihu Lake and Hongze Lake. Oceanol. Et Limnol. Sin. Hai Yang Yu Hu Chao 2009, 40, 201–207. [Google Scholar]
- Kang, B.; Deng, J.; Wang, Z.; Zhang, J. Transplantation of Icefish (Salangidae) in China: Glory or Disaster? Rev. Aquac. 2015, 7, 13–27. [Google Scholar] [CrossRef]
- Skrzynska, A.K.; Maiorano, E.; Bastaroli, M.; Naderi, F.; Míguez, J.M.; Martínez-Rodríguez, G.; Mancera, J.M.; Martos-Sitcha, J.A. Impact of Air Exposure on Vasotocinergic and Isotocinergic Systems in Gilthead Sea Bream (Sparus aurata): New Insights on Fish Stress Response. Front. Physiol. 2018, 9, 96. [Google Scholar] [CrossRef]
- Ikert, H.; Lynch, M.D.J.; Doxey, A.C.; Giesy, J.P.; Servos, M.R.; Katzenback, B.A.; Craig, P.M. High Throughput Sequencing of MicroRNA in Rainbow Trout Plasma, Mucus, and Surrounding Water Following Acute Stress. Front. Physiol. 2021, 11, 588313. [Google Scholar] [CrossRef]
- Chen, Y.; Chen, Y.; Shi, C.; Huang, Z.; Zhang, Y.; Li, S.; Li, Y.; Ye, J.; Yu, C.; Li, Z.; et al. SOAPnuke: A MapReduce Acceleration-Supported Software for Integrated Quality Control and Preprocessing of High-Throughput Sequencing Data. Gigascience 2018, 7, gix120. [Google Scholar] [CrossRef]
- Marçais, G.; Kingsford, C. A Fast, Lock-Free Approach for Efficient Parallel Counting of Occurrences of k-Mers. Bioinformatics 2011, 27, 764–770. [Google Scholar] [CrossRef]
- Vurture, G.W.; Sedlazeck, F.J.; Nattestad, M.; Underwood, C.J.; Fang, H.; Gurtowski, J.; Schatz, M.C. GenomeScope: Fast Reference-Free Genome Profiling from Short Reads. Bioinformatics 2017, 33, 2202–2204. [Google Scholar] [CrossRef]
- Cheng, H.; Concepcion, G.T.; Feng, X.; Zhang, H.; Li, H. Haplotype-Resolved de Novo Assembly Using Phased Assembly Graphs with Hifiasm. Nat. Methods 2021, 18, 170–175. [Google Scholar] [CrossRef] [PubMed]
- Roach, M.J.; Schmidt, S.A.; Borneman, A.R. Purge Haplotigs: Allelic Contig Reassignment for Third-Gen Diploid Genome Assemblies. BMC Bioinform. 2018, 19, 460. [Google Scholar] [CrossRef] [PubMed]
- Durand, N.C.; Shamim, M.S.; Machol, I.; Rao, S.S.; Huntley, M.H.; Lander, E.S.; Aiden, E.L. Juicer Provides a One-Click System for Analyzing Loop-Resolution Hi-C Experiments. Cell Syst. 2016, 3, 95–98. [Google Scholar] [CrossRef]
- Dudchenko, O.; Batra, S.S.; Omer, A.D.; Nyquist, S.K.; Hoeger, M.; Durand, N.C.; Shamim, M.S.; Machol, I.; Lander, E.S.; Aiden, A.P.; et al. De Novo Assembly of the Aedes aegypti Genome Using Hi-C Yields Chromosome-Length Scaffolds. Science 2017, 356, 92–95. [Google Scholar] [CrossRef] [PubMed]
- Simão, F.A.; Waterhouse, R.M.; Ioannidis, P.; Kriventseva, E.V.; Zdobnov, E.M. BUSCO: Assessing Genome Assembly and Annotation Completeness with Single-Copy Orthologs. Bioinformatics 2015, 31, 3210–3212. [Google Scholar] [CrossRef]
- Xu, Z.; Wang, H. LTR_FINDER: An Efficient Tool for the Prediction of Full-Length LTR Retrotransposons. Nucleic Acids Res. 2007, 35, W265–W268. [Google Scholar] [CrossRef] [PubMed]
- Chen, N. Using Repeat Masker to Identify Repetitive Elements in Genomic Sequences. Curr. Protoc. Bioinform. 2004, 5, 4–10. [Google Scholar] [CrossRef] [PubMed]
- Benson, G. Tandem Repeats Finder: A Program to Analyze DNA Sequences. Nucleic Acids Res. 1999, 27, 573–580. [Google Scholar] [CrossRef]
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: A Fast Spliced Aligner with Low Memory Requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef] [PubMed]
- Kovaka, S.; Zimin, A.V.; Pertea, G.M.; Razaghi, R.; Salzberg, S.L.; Pertea, M. Transcriptome Assembly from Long-Read RNA-Seq Alignments with StringTie2. Genome Biol. 2019, 20, 278. [Google Scholar] [CrossRef] [PubMed]
- Haas, B.J.; Salzberg, S.L.; Zhu, W.; Pertea, M.; Allen, J.E.; Orvis, J.; White, O.; Buell, C.R.; Wortman, J.R. Automated Eukaryotic Gene Structure Annotation Using EVidenceModeler and the Program to Assemble Spliced Alignments. Genome Biol. 2008, 9, R7. [Google Scholar] [CrossRef]
- Keilwagen, J.; Hartung, F.; Grau, J. GeMoMa: Homology-Based Gene Prediction Utilizing Intron Position Conservation and RNA-Seq Data. Gene Predict. Methods Protoc. 2019, 1962, 161–177. [Google Scholar]
- Stanke, M.; Waack, S. Gene Prediction with a Hidden Markov Model and a New Intron Submodel. Bioinformatics 2003, 19, ii215–ii225. [Google Scholar] [CrossRef]
- Korf, I. Gene Finding in Novel Genomes. BMC Bioinformatics 2004, 5, 59. [Google Scholar] [CrossRef] [PubMed]
- Emms, D.M.; Kelly, S. OrthoFinder: Phylogenetic Orthology Inference for Comparative Genomics. Genome Biol. 2019, 20, 1–14. [Google Scholar] [CrossRef]
- Nakamura, T.; Yamada, K.D.; Tomii, K.; Katoh, K. Parallelization of MAFFT for Large-Scale Multiple Sequence Alignments. Bioinformatics 2018, 34, 2490–2492. [Google Scholar] [CrossRef]
- Guindon, S.; Dufayard, J.-F.; Lefort, V.; Anisimova, M.; Hordijk, W.; Gascuel, O. New Algorithms and Methods to Estimate Maximum-Likelihood Phylogenies: Assessing the Performance of PhyML 3.0. Syst. Biol. 2010, 59, 307–321. [Google Scholar] [CrossRef]
- Yang, Z. PAML 4: Phylogenetic Analysis by Maximum Likelihood. Mol. Biol. Evol. 2007, 24, 1586–1591. [Google Scholar] [CrossRef]
- Wang, Y.; Tang, H.; Debarry, J.D.; Tan, X.; Li, J.; Wang, X.; Lee, T.; Jin, H.; Marler, B.; Guo, H.; et al. MCScanX: A Toolkit for Detection and Evolutionary Analysis of Gene Synteny and Collinearity. Nucleic Acids Res. 2012, 40, e49. [Google Scholar] [CrossRef] [PubMed]
- De Bie, T.; Cristianini, N.; Demuth, J.P.; Hahn, M.W. CAFE: A Computational Tool for the Study of Gene Family Evolution. Bioinformatics 2006, 22, 1269–1271. [Google Scholar] [CrossRef] [PubMed]
- Langmead, B.; Salzberg, S.L. Fast Gapped-Read Alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef]
- Li, B.; Dewey, C.N. RSEM: Accurate Transcript Quantification from RNA-Seq Data with or without a Reference Genome. BMC Bioinform. 2011, 12, 323. [Google Scholar] [CrossRef] [PubMed]
- Roberts, A.; Trapnell, C.; Donaghey, J.; Rinn, J.L.; Pachter, L. Improving RNA-Seq Expression Estimates by Correcting for Fragment Bias. Genome Biol. 2011, 12, R22. [Google Scholar] [CrossRef] [PubMed]
- Love, M.I.; Huber, W.; Anders, S. Moderated Estimation of Fold Change and Dispersion for RNA-Seq Data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef]
- Bairoch, A.; Apweiler, R. The SWISS-PROT Protein Sequence Data Bank and Its Supplement TrEMBL in 1999. Nucleic Acids Res. 1999, 27, 49–54. [Google Scholar] [CrossRef]
- Kanehisa, M.; Goto, S. KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef]
- Gui, J.-F.; Zhou, L.; Li, X.-Y. Rethinking Fish Biology and Biotechnologies in the Challenge Era for Burgeoning Genome Resources and Strengthening Food Security. Water Biol. Secur. 2022, 1, 100002. [Google Scholar] [CrossRef]
- Lu, G.; Luo, M. Genomes of Major Fishes in World Fisheries and Aquaculture: Status, Application and Perspective. Aquac. Fish. 2020, 5, 163–173. [Google Scholar] [CrossRef]
- Tang, F.-J.; Liu, W.; Wang, J.-L.; Li, Z.; Xie, S.-G. Diet Composition and Transition of Clearhead Icefish (Protosalanx hyalocranius) in Lake Xingkai; Kunming Institute of Zoology, Chinese Academy of Sciences: Kunming, China, 2013; Volume 34, pp. 493–498. [Google Scholar] [PubMed]
- Harris, R.M.; Hofmann, H.A. Seeing Is Believing: Dynamic Evolution of Gene Families. Proc. Natl. Acad. Sci. USA 2015, 112, 1252–1253. [Google Scholar] [CrossRef] [PubMed]
- Mu, Y.; Li, W.; Wu, B.; Chen, J.; Chen, X. Transcriptome Analysis Reveals New Insights into Immune Response to Hypoxia Challenge of Large Yellow Croaker (Larimichthys crocea). Fish Shellfish Immunol. 2020, 98, 738–747. [Google Scholar] [CrossRef]
- Lu, Y.-P.; Zheng, P.-H.; Zhang, X.-X.; Li, J.-T.; Zhang, Z.-L.; Xu, J.-R.; Meng, Y.-Q.; Li, J.-J.; Xian, J.-A.; Wang, A.-L. New Insights into the Regulation Mechanism of Red Claw Crayfish (Cherax quadricarinatus) Hepatopancreas under Air Exposure Using Transcriptome Analysis. Fish Shellfish Immunol. 2023, 132, 108505. [Google Scholar] [CrossRef]
- Wu, L.; Tang, D.; Shen, C.; Bai, Y.; Jiang, K.; Yu, Q.; Wang, Z. Comparative Transcriptome Analysis of the Gills of Cardisoma armatum Provides Novel Insights into the Terrestrial Adaptive Related Mechanism of Air Exposure Stress. Genomics 2021, 113, 1193–1202. [Google Scholar] [CrossRef] [PubMed]
- Moore, M.N.; Allen, J.I.; McVeigh, A.; Shaw, J. Lysosomal and Autophagic Reactions as Predictive Indicators of Environmental Impact in Aquatic Animals. Autophagy 2006, 2, 217–220. [Google Scholar] [CrossRef]
- McGeachy, M.J.; Cua, D.J.; Gaffen, S.L. The IL-17 Family of Cytokines in Health and Disease. Immunity 2019, 50, 892–906. [Google Scholar] [CrossRef]
- Xue, T.; Liu, Y.; Cao, M.; Zhang, X.; Fu, Q.; Yang, N.; Li, C. Genome-Wide Identification of Interleukin-17 (IL-17)/Interleukin-17 Receptor (IL- 17R) in Turbot (Scophthalmus maximus) and Expression Pattern Analysis after Vibrio anguillarum Infection. Dev. Comp. Immunol. 2021, 121, 104070. [Google Scholar] [CrossRef]
- Liu, L.; Zhang, R.; Wang, X.; Zhu, H.; Tian, Z. Transcriptome Analysis Reveals Molecular Mechanisms Responsive to Acute Cold Stress in the Tropical Stenothermal Fish Tiger Barb (Puntius tetrazona). BMC Genom. 2020, 21, 737. [Google Scholar] [CrossRef]
- Brijs, J.; Sandblom, E.; Axelsson, M.; Sundell, K.; Sundh, H.; Huyben, D.; Broström, R.; Kiessling, A.; Berg, C.; Gräns, A. The Final Countdown: Continuous Physiological Welfare Evaluation of Farmed Fish during Common Aquaculture Practices before and during Harvest. Aquaculture 2018, 495, 903–911. [Google Scholar] [CrossRef]
- Wu, J.; Zhang, W.; Li, C. Heat and Hypoxia Exposure Mediates Circadian Rhythms Response via Methylation Modification in Apostichopus Japonicas. Front. Mar. Sci. 2021, 8, 721465. [Google Scholar] [CrossRef]
- Jerônimo, R.; Moraes, M.N.; de Assis, L.V.M.; Ramos, B.C.; Rocha, T.; Castrucci, A.M.d.L. Thermal Stress in Danio Rerio: A Link between Temperature, Light, Thermo-TRP Channels, and Clock Genes. J. Therm. Biol. 2017, 68, 128–138. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Yang, Y.; Wang, Z.; Xu, K.; Xiao, X.; Mu, W. Comparison of Effects in Sustained and Diel-Cycling Hypoxia on Hypoxia Tolerance, Histology, Physiology and Expression of Clock Genes in High Latitude Fish Phoxinus lagowskii. Comp. Biochem. Physiol. Part A Mol. Integr. Physiol. 2021, 260, 111020. [Google Scholar] [CrossRef]
- Peng, L.-B.; Wang, D.; Han, T.; Wen, Z.; Cheng, X.; Zhu, Q.-L.; Zheng, J.-L.; Wang, P. Histological, Antioxidant, Apoptotic and Transcriptomic Responses under Cold Stress and the Mitigation of Blue Wavelength Light of Zebrafish Eyes. Aquac. Rep. 2022, 26, 101291. [Google Scholar] [CrossRef]
- Mao, Y.; Zhang, G. A Complete, Telomere-to-Telomere Human Genome Sequence Presents New Opportunities for Evolutionary Genomics. Nat. Methods 2022, 19, 635–638. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Illustration of P. chinensis.

Figure 2.
Genome characteristics of P. chinensis.
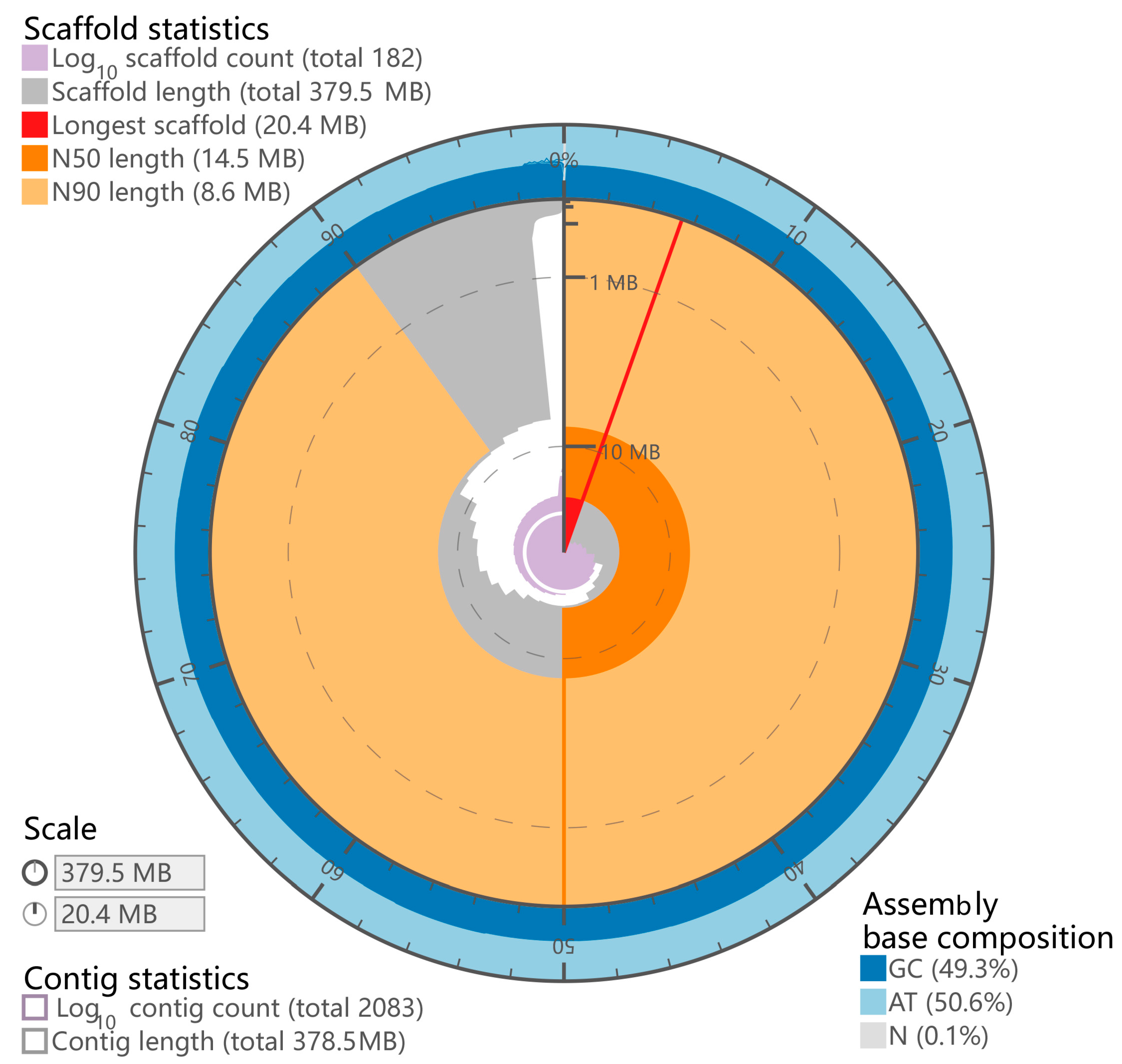
Figure 3.
Genome-wide Hi-C heatmap of P. chinensis.
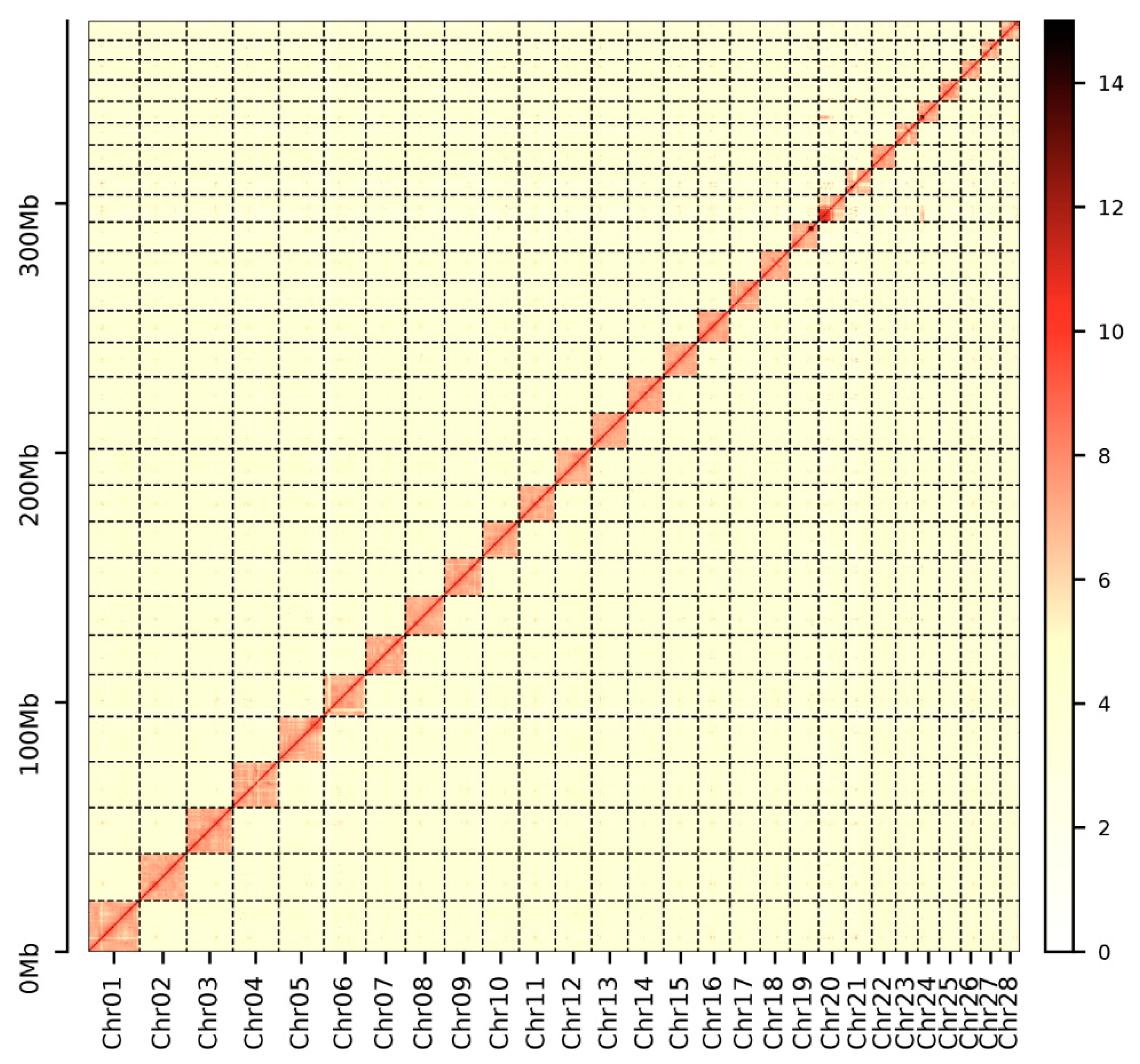
Figure 4.
Genome synteny between P. chinensis and H. transpacificus, P. chinensis, and E. lucius.

Figure 5.
Number of expanded and contracted gene families in P. chinensis.
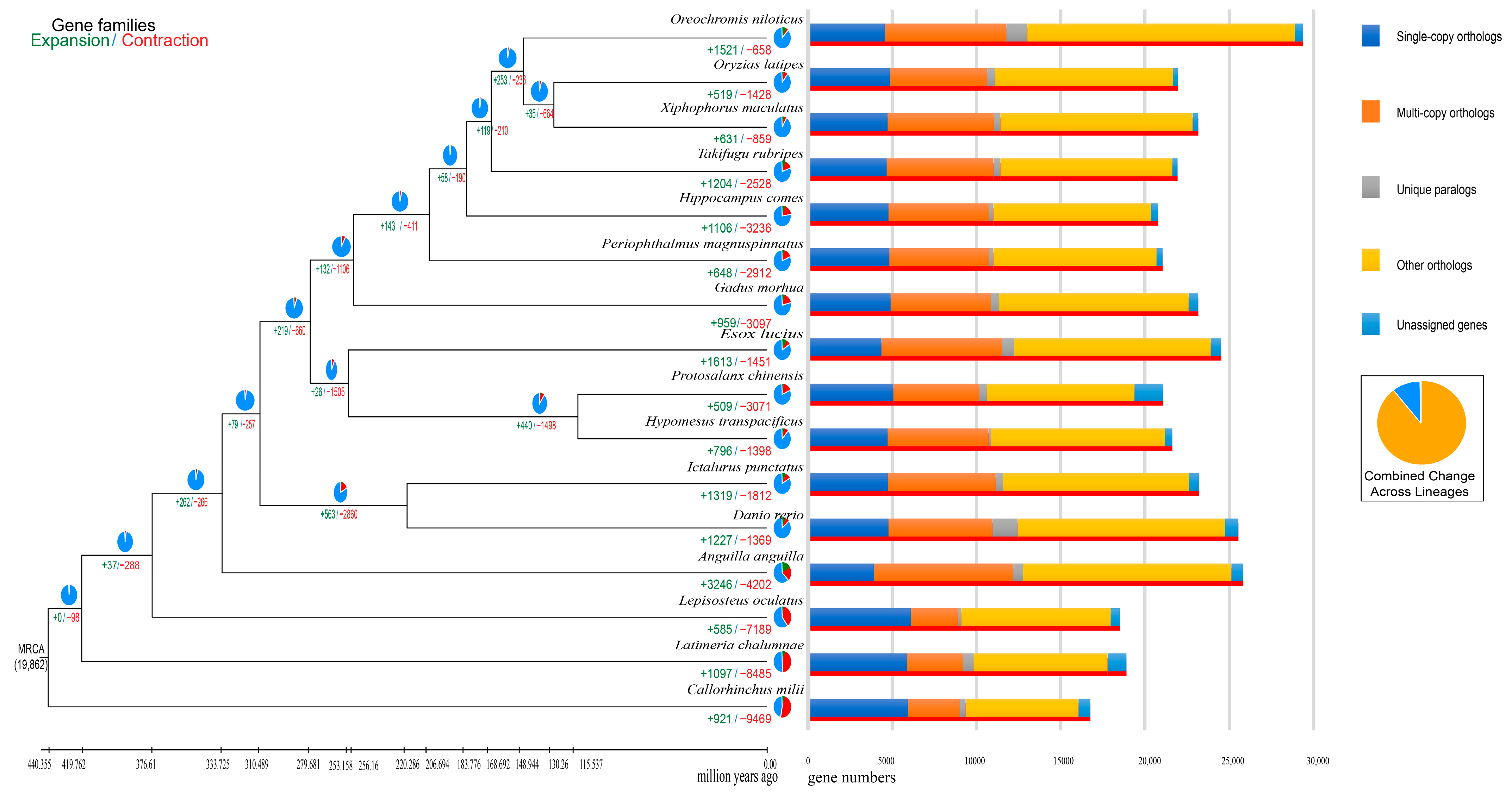
Figure 6.
DGEs analysis. (a) UpSetR plots depicting the number of unique and shared DEGs. (b) Trend analysis of DEGs expression profiles.
Figure 6.
DGEs analysis. (a) UpSetR plots depicting the number of unique and shared DEGs. (b) Trend analysis of DEGs expression profiles.

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Zhou, Y.; Zhang, X.; Tang, X.; Zhou, Y.; Ding, Y.; Liu, H. Chromosome-Level Genome Assembly of Protosalanx chinensis and Response to Air Exposure Stress. Biology 2023, 12, 1266. https://doi.org/10.3390/biology12091266
AMA Style
Zhou Y, Zhang X, Tang X, Zhou Y, Ding Y, Liu H. Chromosome-Level Genome Assembly of Protosalanx chinensis and Response to Air Exposure Stress. Biology. 2023; 12(9):1266. https://doi.org/10.3390/biology12091266
Chicago/Turabian StyleZhou, Yanfeng, Xizhao Zhang, Xuemei Tang, Yifan Zhou, Yuting Ding, and Hong Liu. 2023. "Chromosome-Level Genome Assembly of Protosalanx chinensis and Response to Air Exposure Stress" Biology 12, no. 9: 1266. https://doi.org/10.3390/biology12091266
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.