1. Introduction
The development of a multicellular organism involves the generation of many different cell types from a single fertilized egg cell. Understanding the molecular mechanisms by which cells become differentiated from each other is a fundamental goal of developmental biology. Our goal is to study how exposures to ionizing radiation (IR) alter or modify these cellular mechanisms.
The nematode
Caenorhabditis elegans (
C. elegans) stands as an ideal model organism for dissecting cellular intricacies at the single-cell level. The developmental trajectory of
C. elegans, encompassing cell divisions, migrations, and differentiations, has been meticulously documented [
1,
2,
3,
4]. The pursuit of this comprehensive understanding has been aided by three key attributes intrinsic to
C. elegans. Firstly, its little size (1 mm for adults) and ease of cultivation on nonpathogenic
Escherichia coli (
E. coli) in laboratory settings render it prolific, with a mere three-day life cycle yielding a multitude of progeny from a single individual. Secondly, the organism’s transparency facilitates in situ cell observation. Its consistent cellular lineage, meticulously tracked through direct microscopic observation, remains largely invariant.
C. elegans have a fixed number of only 558 larval and 959 adult somatic cells, which constitute organs such as muscles, intestine, and neurons. The germ line constitutes a substantial portion of the body, featuring germ line stem cells that sequentially produce sperm and oocytes that will undergo fertilization in the spermatheca and advance to early cleavage stages before emergence via the vulva. Lastly, the prevalence of self-fertile hermaphroditism among
C. elegans greatly facilitates the generation of inbred lines and genetic analyses. Numerous investigations have delved into the spectrum of cell-to-cell interactions using increasingly sophisticated strategies and phenotypic evaluations facilitated by green fluorescent protein (GFP)-tagged strains to facilitate phenotype detection with a diverse range of documented mutants and transgenic strains (a vast collection which is available at the
Caenorhabditis Genome Center, University of Minnesota).
A strategy for investigating cellular functionality is to eliminate individual cells, subsequently observing the resultant developmental or behavioral abnormalities. This method is commonly executed through the precise killing of specific cells or cell groups using a laser microbeam, a technique known as laser ablation. Notably, laser ablation has been extensively used to uncover the roles of diverse mature cell types, encompassing neurons engaged in locomotion, feeding, mechano-sensation, and chemo-sensation [
5,
6,
7,
8,
9]. In addition, the elimination of distal tip cells within the somatic gonad triggers premature germ line differentiation, thus revealing that the somatic gonad is essential for sustaining an undifferentiated state within the germ line [
4,
10,
11]. Laser ablation has also proven particularly insightful for comprehending postembryonic cell interactions during gonad and vulva development, yielding substantial insights into these intricate processes [
4,
10,
11,
12]. Extending this approach to charged-particle microbeams could help to characterize the specific radiation-induced biological responses. Charged-particle microbeams provide the possibility of selectively irradiating living cells with a precise dose and dose delivery timing [
13,
14,
15,
16,
17,
18]. Initially developed to target adherent mammalian cells, the use of charged-particle microbeams have progressively broadened to encompass the irradiation of three-dimensional tissue models and small in vivo multicellular system [
19,
20,
21,
22]. Amongst the well-characterized biological models, the nematode
C. elegans is particularly suited to the specific constraints of targeted irradiations [
16,
23,
24,
25]. And several reports have detailed the use of
C. elegans at charged-particle microbeam end stations [
16,
17,
23,
24,
26,
27,
28].
The
C. elegans vulva serves as an exemplary model to unravel the intricate mechanisms governing pattern formation and morphogenesis [
29]. The cell divisions of vulval development occur over a period of only five hours and can be readily and directly analyzed in living animals using genetic and molecular methods. It has been demonstrated that cell signaling is central to vulval development. The vulva is induced by the gonad from the epithelium that envelops the animal. Inductive signaling seems to function by preventing the action of another intercellular signal that otherwise inhibits vulval development. Moreover, the responding vulval precursor cells interact to help to establish the precise spatial pattern of expression of vulval cell fates. Morphogenesis of the vulva culminates in a process called vulval eversion, whereby a passageway is made from the uterus through the vulva to the outside of the animals. Mutations causing abnormal eversion of the vulva (
evl mutants) have been identified. Using laser ablation, it has been shown that the uterus and anchor cell are important for correct vulval eversion. It has been also described that
evl mutations may influence vulval eversion by primarily affecting the development of the somatic gonad, while others affect the developments of both structures [
30].
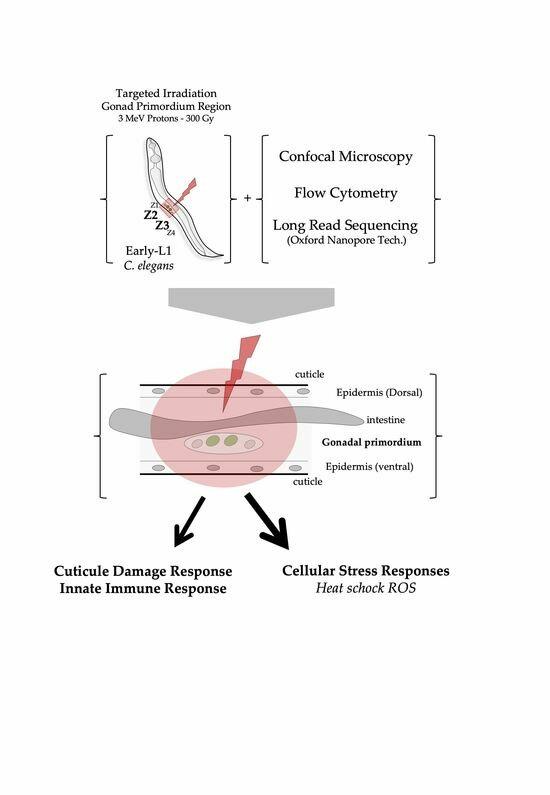
In the present study, we have applied charged-particle microbeams to selectively irradiate the stem progenitor gonad region (identified by the position of the progenitor gonad stem cells named Z2-Z3) in C. elegans at a defined developmental stage (synchronized population of L1 larvae). This approach provides a way to investigate the radiation-induced effects in a living organism in development using a well-defined and well-characterized organogenesis system: gonads and vulva. In addition, this work aimed to combine the use of selective and targeted irradiation and the use of a high-throughput nematode fluorescent analysis device (Complex Object Parametric Analysis and Sorting—COPAS, Union Biometrica) and third generation sequencing (Oxford Nanopore technologies) with a limited number of individuals (<300).
2. Materials and Methods
2.1. Worm Strains and Culture
C. elegans worm strains were cultivated on nematode growth medium (NGM) agar plates and provided unrestricted access to
Escherichia coli strain (
E. coli) OP50 at 19 °C, following established protocols [
31,
32]. We used the transgenic GZ264 strain carrying an appropriate fluorescent marker (GZ264 (isIs17[pGZ265:pie-1::GFP-pcn-1(W0D2.4)]), specifically expressed at the L1 stage in worms, onto the Z2-Z3 progenitor gonad stem cells. The
Caenorhabditis Genetics Center (CGC, University of Minnesota, Minneapolis, MN, USA) provided these
C. elegans strains and the
E. coli OP50.
2.2. Preparation of Large Population of Synchronized C. elegans (L1 Stage)
The bleaching technique was used for synchronizing C. elegans cultures at the first larval stage (L1). Populations of young gravid hermaphrodites from standard, well-fed culture stocks were collected with M9 buffer (3 g·L−1 KH2PO4 (Sigma-Aldrich, Saint-Quentin Fallavier, France, cat. no. P9791-500G), 6 g·L−1 Na2HPO4(Sigma-Aldrich, Saint-Quentin Fallavier, France, cat. no. S5136-500G), 5 g·L−1 NaCl (Sigma-Aldrich, Saint-Quentin Fallavier, France, cat. no. S3014-500G), and 1 mM MgSO4 (Sigma-Aldrich, Saint-Quentin Fallavier, France, cat. no. M2643-500G)) and washed three times with sterile water to remove bacteria. Then, worms pelleted via centrifugation (2 min, 2000 rpms, room temperature) were treated with a freshly prepared alkaline hypochlorite solution (1.5% (v/v) NaOCl (Merck KGaA, Darmstadt, Germany, cat. no. K50436114829) and 1 M NaOH (Sigma-Aldrich, Saint-Quentin Fallavier, France, cat. no. S5881-500G-M). The suspension was swirled every 2 min with vortex-mixing (~6 min). The released embryos were pelleted via centrifugation (2000 rpms; 2 min; 4 °C). The supernatant was carefully discarded, and embryos were washed three times with M9 buffer followed by centrifugation. The pelleted embryos were suspended in M9 and plated on an NGM agar plate without bacteria. The elapsed time between hatching and irradiation was reduced in order to favor healthy conditions (<20 h).
2.3. Samples Preparation and Mounting for Irradiation
We adapted the sample preparation conditions to use our custom-made support dish for micro-irradiation and live imaging. The day of irradiation, L1 larvae were collected by centrifugation (2000 rpms; 2 min; 4 °C) in the cold M9 medium and resuspended in the mounting medium (M9 supplemented with 0.25 mM Tetramisole hydrochloride (Sigma-Aldrich, Saint-Quentin Fallavier, France, cat. no. L9756-10G), with 30% (v/v) Poloxamer-407 (Sigma-Aldrich, Saint-Quentin Fallavier, France, cat. no. 16758-500G). L1 were stored at 4 °C to avoid their immobilization by the polymerization of the mounting medium at 20 °C. The number of worms was estimated to adjust the dilution volume to obtain ~100 worms per µL. 30 min before irradiation, an aliquot ~2 µL was directly deposited on a sterile 4 μm thick polypropylene (Goodfellow Cambridge Ltd., Huntingdon, UK, cat. no. PP301040) and immediately covered with an afresh agar pad (3% (w/v), Sigma-Aldrich, Saint-Quentin Fallavier, France, cat. no. A1296-1KG) in M9 to maintain worm immobilized in a thin layer of medium. To prevent desiccation and contamination, the dish was closed with a glass side coverslip. The elapsed time between mounting and irradiation was limited to 1 h.
2.4. Beam Line Characteristics and Irradiation Procedures
Worms were irradiated with 3 MeV protons delivered on the micro-irradiation setup installed on the AIFIRA facility (Applications Interdisciplinaires des Faisceaux d’Ions en Région Aquitaine, Bordeaux, France) [
14,
15]. The accelerator (Singletron
TM, High Voltage Engineering Europa, Amersfoort, The Netherlands) delivers light ion beams with energies up to 3 MeV [
33]. To target Z2-Z3 cells, the beam was strongly collimated to reduce the particle flux to a few thousand protons per second on the target and was focused using a triplet of magnetic quadrupoles. Before hitting the target, the protons pass through a 200 nm thick Si
3N
4 membrane (Silson Ltd., Southam, UK) and a 700 µm thick residual air layer, leading to a beam spot size of about 4 µm. The beam was targeted to Z2-Z3 thanks to the use of the GZ264 transgenic strain. Indeed, at the first larval stage (L1), of the whole organism, only these two cells expressed fluorescence based on GFP-tagged protein position (GFP::PCN-1). The beam spot raster scanned on a 16 × 16 µm
2 area to ensure a homogenous dose distribution on the gonad stem cells while preserving the worm body from irradiation. Assuming a constant LET of the protons (12 keV·µm
−1) when traversing the worms, and for a given fluence F, the mean dose was calculated as follows:
For each replicate, we irradiated a minimum of 3 independent worm populations, consisting of 300 to 450 worms in each population. This adds up to a total of 900 to 1200 irradiated worms per condition for every replicate [
33,
34]. The irradiation end station consists of a motorized inverted fluorescence microscope (Carl Zeiss Micro-Imaging S.A.S, Rueil-Malmaison, France, cat. no. AxioObserver Z1) equipped with a 14 bits Rolera EM-C
2TM Camera (Teledyne Photometrics, Tucson, AZ, USA, cat. no. QImaging) which is positioned horizontally at the end of the beam line. It is equipped with 63× objective (Carl Zeiss Micro-Imaging S.A.S, Rueil-Malmaison, France, cat. no. LD Plan-Neofluar 20×/0.4). Fluorescence light is provided by a light emitting diode (LED) illuminating system (Carl Zeiss Micro-Imaging S.A.S, Rueil-Malmaison, France, cat. no. Colibri2
TM) with negligible heat production. The image acquisition is performed using the Micromanager software [
35].
2.5. Sample Preparation for Confocal Imaging
Worm populations were immediately fixed in cold 4% (
w/
v) paraformaldehyde (Sigma-Aldrich, Saint-Quentin Fallavier, France, cat. no. 158127-500G) for 15 min. Then, worms were pelleted via centrifugation (2 min., 2000 rpms, RT) and paraformaldehyde was replaced by cold acetone (Sigma-Aldrich, Saint-Quentin Fallavier, France, cat. no. 650501-1L) for permeabilization (2 min, −20 °C) and finally washed twice in M9. After fixation and washing, M9 was removed and replaced by a freshly prepared solution of DyLight 594 Phalloidin (1:80 (
v/
v), Cell Signaling Technologies, Ozyme, Saint-Cyr-L’École, France, cat. no. 12877) and Hoecsht
33342 (2:4000 (
v/
v), Thermo Fischer Scientific, Illkirch, France, cat. no. H3570). Worms were incubated overnight at RT under gentle agitation and then washed via two series of centrifugation (2 min, 2000 rpms) with M9. The supernatant was discarded, and the pelleted worms were suspended in 2–3 drops of Prolong Gold Antifade reagent (Thermo Fischer Scientific, Illkirch, France, cat. no. P36934) and transferred via pipettes for mounting between the glass slides. Three-dimensional images were acquired with a Leica DMRE TCS SP2 AOBS confocal microscope (oil-immersion objective × 63, 1.4 NA), assembled, and reconstructed using Image J software (Rasband, W.S., ImageJ, U.S. National Institutes of Health, Bethesda, MD, USA,
https://imagej.nih.gov/ij/, accessed on 20 September 2023, 1997–2018).
2.6. Nematode Population Analysis and Gene Reporter Assay Using COPAS
Proper larval development is essential for the normal function of
C. elegans in adult life stages. Delayed growth in
C. elegans can be indicative of dysfunction of a number of developmental processes and can be measured in a high-throughput growth assay using the Complex Object Parameter Analysis Sorter (COPAS Biosort, Union Biometrica, Aalst, Belgium) [
36]. The COPAS Biosort is a flow cytometer designed specifically for large object analysis. It records worm axial length (time of flight, TOF), optical density (EXT), and longitudinal fluorescence variation in worms.
COPAS Biosort calibration and settings were defined analyzing a non-synchronized GZ264 population. A 488 nm laser source is used to excite GFP in worms as its passes through the flow cell. GFP intensity is then recorded using a combination of a dichroic mirror reflecting emitted fluorescence (510/523 nm) and a photomultiplier tube (PMT). PMT scale, gain, and threshold were kept constant during all acquisitions.
After irradiation and recovery time, populations were carefully collected by rinsing the Petri dish with ultrapure water. Suspended worms were then analyzed using the COPAS device. The dilution of worms was adjusted to keep the counting rate in the range of 10–50 per second. No gating of experimental parameters (TOF, EXT, and PhGreen (Peak Height Green)) was applied during acquisition. A dedicated python library was developed for data analysis. For each sample, it provides (i) the number of worms, (ii) their size (body length evaluated by TOF), and (iii) the expression level of the gene reporter per individual, along with its longitudinal profile.
Standard procedure to produce GFP longitudinal intensity profiles using our python library includes the following steps: reading raw data; filtering events, if any, corresponding to measure artefacts like bubbles; sorting individual profiles by length and filtering profiles with a length smaller than 80 in order to discard small objects (debris, eggs, or younger worms); aligning profiles from head to tail using a Pearson’s correlation test between adjacent individuals; and finally, merging worm profiles with the same length, producing mean profiles for each observed length.
2.7. RNA Collection
Efficient extraction to obtain a good amount and high quality of RNA remains critical in the success of transcription analysis. As our samples were not immediately processed for RNA extraction, they were collected by centrifugation in M9 buffer (3 h post-irradiation) and stored at −80 °C until analysis. Although C. elegans has proved to be a powerful tool for genetic analyses, its use has been stalled by the inability to efficiently and rapidly break its resistant cuticle that must be ruptured prior to RNA extraction. Thus, we adapted the protocol established by Qiagen (RNeasy Kit, Courtaboeuf, France), which allows efficient purification of total RNA from small amounts of starting material.
We need to consider that our primary goal is to minimize the time required to irradiate all the worms of a sample. Increasing the number of worms per sample presents several challenges. Worm density is a critical factor in our ability to accurately distinguish stem cells within their bodies, as overcrowding can lead to overlapping worms. Moreover, worms are sensitive to high-density populations, which can induce stress. The selected number of worms (>450) is the most suitable for our experimental conditions. Furthermore, even though we irradiated only 450 worms per sample, we irradiated several separate samples (at least 3). Since these samples were irradiated at different times, we need to perform distinct extractions at various intervals after the irradiation process.
After irradiation, L1 worms were cultured for 3 h on NGM plates seeding with an E. coli OP50 strain, washed in RNase-free water, and pelleted by centrifugation to remove bacteria. Samples need to be first lysed in RLT buffer and then homogenized and purified using a silica membrane via successive washing and centrifugation steps. We tested two methods to disrupt the cuticle of frozen worms using (i) bead beating with Precellys (Bertin Technologies) or (ii) Dounce homogenization combined with successive and alternate freeze-cracking steps. In the bead beating procedure, microscopic glass beads are used to mechanically disrupt the cuticle. Although this method is cheap and fast, variability in the RNA material recovery can be observed; it also requires a relatively large number of worms, and the RNA tends to degrade as the samples heat up. By contrast, Dounce homogenization combined with successive and alternate freeze-cracking steps resulted in an acceptable RNA quantity and reproducible quality from a small number of L1 populations (<450). In this procedure, worms were lysed via 20 cycles of freeze-cracking using a Dounce tissue homogenizer (Sigma-Aldrich, Saint-Quentin Fallavier, France, cat. no. 885301/885303) and total RNA was isolated with an RNeasy Mini kit according to manufacturer’s instructions (Qiagen SAS, Courtaboeuf, France, cat. no. 74104). Total RNA integrity was assessed with the Agilent high-sensitivity RNA system for TapeStation (Agilent Technologies, Les Ulis, France).
2.8. Library Preparation and Sequencing (Oxford Nanopore Technologies, PCR-cDNA Barcoding)
Whole transcriptome cDNA libraries were first constructed from extracted mRNA using a PCR-cDNA barcoding kit (Oxford Nanopore Technologies, Oxford, UK, cat. no. SQK-PCB109). Briefly, 50 ng total RNA (~1 ng (polyA)+ mRNA) from each condition was taken in RNase-free PCR tubes containing Maxima H Minus reverse transcriptase enzyme (Thermo Fischer Scientific, Illkirch, France, cat. no. EP0751), VN primer (2 µM, variant of oligo (dT) with complementary nucleotides for annealing of barcode primers), 10 mM dNTPs (New England Biolabs, Evry, France cat. no. N0447S), RNaseOUT (40 U/µL, Thermo Fischer Scientific, Illkirch, France, cat. no. 10777019), and strand-switching primer (10 µM, Oxford Nanopore Technologies Oxford, UK). Products were sequentially added and prepared as recommended by the manufacturers. Samples were incubated at +42 °C for 90 min, followed by enzyme inactivation at +85 °C for 5 min (one cycle) in a thermocycler for the generation of full-length cDNA from poly(A)+ messenger RNA. cDNA libraries from each sample then underwent full-length amplification and sample barcoding using 14 cycles of PCR. Each PCR reaction mix consisted of 25 µL 2× LongAmp Taq master mix (New England Biolabs, Evry, France, cat. no. M0287S), 1.5 µL barcoded primers (named BP01 to BP12), 18.5 µL nuclease-free water, and 5 µL (~0.25 ng) cDNA. The following PCR cycling conditions were used: initial denaturation at 95 °C for 30 s (1 cycle), denaturation at 95 °C for 15 s (14 cycles), annealing at 62 °C for 15 s (14 cycles), extension at 65 °C for 15 s (14 cycles), and final extension at 65 °C for 6 min (1 cycle). (The long extension time of 6 min was to selectively amplify cDNAs up to ~5 kb in length.) 1 µL Exonuclease 1 (New England Biolabs, Evry, France, cat. no. M0293S) was finally added in each PCR tube for incubation at +37 °C, followed by enzyme inactivation at +80 °C for 5 min (one cycle). To clean up the cDNA libraries, PCR reactions with the same barcode (from BP01 to BP12) were pooled in two 1.5 mL DNA tubes and primer dimers were removed using 0.8× volume equivalent Agencourt® AMPure® XP beads (Beckman Coulter, Villepinte, France, cat. no. A63880). Briefly, the beads (80 µL) were added to each pooled sample, incubated on a hula mixer for 5 min at room temperature, and spun and pelleted on a magnet. The supernatants were pipetted off and the resulting beads were washed with 70% (v/v) ethanol (200 µL, freshly prepared using nuclease-free water) without disturbing the pellet. The ethanol was removed using a pipette and the beads were washed again with ethanol; the pelleted beads were spun down and placed back on the magnet. Residual ethanol was pipetted off and the beads were briefly allowed to dry. While the beads still appeared glossy (with no cracking), they were resuspended in 12 µL elution buffer (provided with the PCR-cDNA barcoding kit; ONT) to recover the cDNA libraries. Prior to loading the barcoded cDNA libraries onto the flow cell for long-read sequencing, 100 ng of each cleaned up cDNA library was ligated with adapters. The barcoded libraries (100 fmol final) were pooled in 1.5 mL DNA, combined with rapid adapters (Oxford Nanopore Technologies, Oxford, UK, cat. no. SQK-PCB109), and incubated at room temperature for 5 min. The adapter-ligated pooled cDNA library was transferred to a fresh tube and combined with loading beads and sequencing buffer (SQK-PCB109 kit; ONT) to form the sequencing mix. The flow cell R9.4.1 (Oxford Nanopore Technologies, Oxford, UK, cat. no. FLO-MIN106D) was primed with pre-mixed flush buffer and flush tether (Oxford Nanopore Technologies, Oxford, UK, cat. no. Flow Cell Priming kit EXP-FLP002), then loaded with the sequencing mix and run for 24 h on a MinION Mk1C sequencing device (Oxford Nanopore Technologies, Oxford, UK).
2.9. Transcriptome Analysis
Upon completion, guppy 6.1.4 was used to perform super high accuracy base-calling and data demultiplexing (Raw fast5 files). Reads were mapped with minimap2 option ‘-ax map-ont’ to the
C. elegans WS287 transcriptome and pooled into an expression matrix via a dedicated Python script. Differential expression analysis was performed on R with edgeR and limma packages. Genes < 1 counts per million (cpm) were excluded, and then the data were normalized (limma-voom) and transformed into log2, a linear model which is adjusted to the data for each gene. The contrasts were then extracted and compared to the linear model according to the empirical Bayesian method, and the obtained
p-values were adjusted by the Bonferroni method [
37] to obtain the final differential expression results. A volcano plot of all of the genes’ status of differential expression was produced using Glimma using the foldchange as the X-axis and the negative log10 of the adjusted
p-value as Y-axis. A heatmap of compared relative expression between conditions of 136 genes, 36 differentially expressed genes, and 100 highest foldchange genes (50 up-regulated, 50 down-regulated) not classified as differentially expressed, was produced using ggplots. Enrichment analysis was performed with the R package gprofiler2 [
38], which queries public APIs from several databases and determines statistically significant results based on the number of genes impacted and the total number of genes attached for each ontology.
4. Discussion
Our main goal in targeted irradiation is to accurately cause damage in precise cellular areas within specific cellular regions in multicellular organisms. Laser ablation is the preferred method for achieving this goal due to its compatibility with microscopy, enabling simultaneous irradiation and observation. This technique has a long history of use in studying C. elegans cell lineage and development. Targeted irradiation using ionizing radiation in multicellular organisms faces limitations, including limited access to microbeam facilities and constraints on the number of irradiated animals due to technical challenges associated with sample manipulation and irradiation procedures. Using a charged-particle microbeam requires separate equipment for observation and irradiation, leading to increased complexity involving precise beam–microscope alignment, specialized sample preparation, and synchronization of time-lapse imaging. One significant limitation lies in the real-time monitoring of radiation-induced damage through time-lapse imaging, which generally restricts the study to a small number of individuals and is constrained by beam time availability. Nevertheless, charged-particle microbeams offer a unique advantage by enabling precise control over irradiation dosage down to a single particle, playing a crucial role in studying radiation-induced effects in biological models.
When conducting targeted irradiation within a multicellular organism, two key factors come into play: the precise identification of the target and the immobilization of the organism. We validated a methodology by focusing on the organogenesis of the vulva and gonad, two well-defined and extensively studied developmental systems in C. elegans. Our approach involved the precise manipulation of worm populations and targeted irradiation of stem progenitor gonad cells with three MeV protons during a specific developmental stage (L1). This required immobilization, identification, imaging, irradiation, and subsequent recovery of a synchronized population with minimal time and manipulation. To achieve this, we used a specialized mounting medium composed of M9 medium supplemented with Poloxamer-407, a unique non-ionic tri-block copolymer known for its property of reverse gelatinization. This property was instrumental in immobilizing the worms and facilitating their rapid recovery after irradiation. In this study, we chose to employ a high dose of irradiation as a proof of concept. Moving forward, we now have the flexibility to design new experiments with varying doses to explore the dose–response relationship and potentially identify threshold levels. The impact of micro-irradiation on the lifespan and different phenotypic traits could be studied in the future.
Additionally, we employed a
C. elegans strain expressing the PCN-1::GFP-tagged reporter under the control of the pie-1 promoter. This greatly aided in the identification of gonadal progenitor stem cells through fluorescence microscopy.
C. elegans is a unique multicellular model for conducting in vivo gene expression studies, thanks to its transparent body, highly predictable cell lineage, and numerous collections of GFP-tagged reporters, making it ideal for precise analyses of radiation-induced effects throughout development. Following irradiation and subsequent animal development, we conducted high-magnification fluorescence microscopy to assess gonadal and vulval development. While this analysis was time-consuming and labor-intensive, it provided valuable insights into the targeted organ development. Specifically, for targeted irradiation at 300 Gy, we observed not only organ absence (agenesis) but also anomalies such as vulvar eversion characterized by cells exhibiting tumor-like uncontrolled development, suggesting that targeted irradiation of the stem cell region induces alterations in tissue-specific cell signaling and cell development. Similar to other studies, we have identified the vulva as a somatic tissue exhibiting radiosensitivity, perhaps because the vulva is one of the few organs dividing post-embryonically [
42,
43].
To comprehensively analyze gonadal development post-irradiation at 300 Gy, we used COPAS to quantitatively evaluate developmental abnormalities in several hundred animals. GFP fluorescence measurements across a significant number of samples reaffirmed our initial observations made through confocal microscopy, confirming the absence of gonads in selective and targeted irradiated populations. Since GFP expression is specific to gonadal cells, we were able to measure GFP fluorescence across a significant number of samples. This approach provided us with a complementary dataset that reaffirmed the initial observations made through confocal microscopy, namely, the decrease in fluorescence, and consequently, the radiation-induced modifications in gonad development in irradiated populations.
Finally, we harnessed the latest advanced transcriptome analysis technologies, demonstrating the feasibility of conducting such experiments on a relatively small number of individuals. We identified a radiation-induced response through the expression of genes involved in pathways such as heat shock and oxidative stress responses (over-expression of the
hsp gene family,
hsp-16.1,
hsp-16.2 hsp-16.41,
hsp-16.48, and
hsp-16.49). Moreover, our findings revealed a response mediated by the cuticle defense response (
nlp-66) and by the innate immune response (
Scl-2), highlighting the significance of studying at the organism level, particularly in extensively characterized organisms like
C. elegans. Similar phenomena have been observed in instances of biomechanical damage caused by factors such as bacterial infections, mechanical abrasions, or laser injury. It would be interesting to test the
C. elegans strain expressing the inducible
pnlp-29::GFP reporter to demonstrate that
C. elegans responds to physical injury of the epidermis mediated by ionizing radiation. It has been shown that the activation of
nlp-29 via wounding suggests that in
C. elegans, tissue damage triggers an innate immune response [
53]. It would be interesting to test this hypothesis using targeted irradiation at different doses of exposure and at different developmental stages. Exploring the cuticle and epidermis of
C. elegans as a model for understanding radiation-induced responses in epithelial tissues holds significant promise. The data presented in our study open exciting opportunities to investigate the cellular and molecular mechanisms underlying radiodermatitis. Although the
C. elegans cuticle constitutes a highly specialized extracellular matrix (ECM) with intricate and unique features, its biogenesis shares fundamental molecules, mechanisms, and pathways with vertebrates. This commonality makes
C. elegans an invaluable experimental system for dissecting ECM formation. This research allows for the examination of alterations in gene expression, signaling pathways, and cellular responses within these tissues, leading to the identification of crucial molecular components and mechanisms (for example, the MAP kinase signaling pathways). These findings offer valuable insights into the molecular and cellular basis mechanism of radiodermatitis [
54,
55] in response to radiation exposure and hold significant promise for advancing future prospects in radiation therapy.

 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}





