
Bulked Segregant Analysis and Association Analysis Identified the Polymorphisms Related to the Intermuscular Bones in Common Carp (Cyprinus carpio)

 , ,
, ,
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Ethics Statement
2.2. Counting and Classifying the IBs
2.3. Genome Resequencing and Bulk Segregant Analysis
2.4. Amplification, SNP Calling, and Genotyping
2.5. Examining the Genetic Diversities of the Examined SNPs
2.6. Identifying the SNPs Associated with the Numbers of IBs
2.7. Joint Effects of Significant SNPs on the IB Numbers
3. Results
3.1. Comparison of the IB Numbers in Different Strains
3.2. Identification of Segregant SNPs by BSA
3.3. Genetic Diversity of Three Sequenced BSA-SNP Regions
3.4. SNPs Associated with the IB Numbers
3.5. Joint Effects of Significant SNPs on the IB Numbers
4. Discussions
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Yang, K.; Jiang, W.; Wang, X.; Zhang, Y.; Pan, X.; Yang, J. Evolution of the intermuscular bones in the Cyprinidae (Pisces) from a phylogenetic perspective. Ecol. Evol. 2019, 9, 8555–8566. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, B.; Zhang, Y.W.; Liu, X.; Ma, L.; Yang, J.X. Molecular mechanisms of intermuscular bone development in fish: A review. Zool. Res. 2021, 42, 362–376. [Google Scholar] [CrossRef] [PubMed]
- Nie, C.-H.; Hilsdorf, A.W.S.; Wan, S.-M.; Gao, Z.-X. Understanding the development of intermuscular bones in teleost: Status and future directions for aquaculture. Rev. Aquac. 2020, 12, 759–772. [Google Scholar] [CrossRef]
- Li, L.; Zhong, Z.; Zeng, M.; Liu, S.; Zhou, Y.; Xiao, J.; Wang, J.; Liu, Y. Comparative analysis of intermuscular bones in fish of different ploidies. Sci. China Life Sci. 2013, 56, 341–350. [Google Scholar] [CrossRef] [Green Version]
- Wan, S.-M.; Xiong, X.-M.; Tomljanović, T.; Chen, Y.-L.; Liu, H.; Treer, T.; Gao, Z.-X. Identification and mapping of SNPs associated with number of intermuscular bone in blunt snout bream. Aquaculture 2019, 507, 75–82. [Google Scholar] [CrossRef]
- Wan, S.M.; Yi, S.K.; Zhong, J.; Nie, C.H.; Guan, N.N.; Chen, B.X.; Gao, Z.X. Identification of MicroRNA for Intermuscular Bone Development in Blunt Snout Bream (Megalobrama amblycephala). Int. J. Mol. Sci. 2015, 16, 10686–10703. [Google Scholar] [CrossRef] [Green Version]
- Wan, S.M.; Yi, S.K.; Zhong, J.; Nie, C.H.; Guan, N.N.; Zhang, W.Z.; Gao, Z.X. Dynamic mRNA and miRNA expression analysis in response to intermuscular bone development of blunt snout bream (Megalobrama amblycephala). Sci. Rep. 2016, 6, 31050. [Google Scholar] [CrossRef] [Green Version]
- Sun, X.; Zhang, R.; Liu, M.; Chen, H.; Chen, L.; Luo, F.; Zhang, D.; Huang, J.; Li, F.; Ni, Z.; et al. Rmrp Mutation Disrupts Chondrogenesis and Bone Ossification in Zebrafish Model of Cartilage-Hair Hypoplasia via Enhanced Wnt/β-Catenin Signaling. J. Bone Miner. Res. 2019, 34, 2101–2116. [Google Scholar] [CrossRef]
- Nunes, J.R.S.; Pértille, F.; Andrade, S.C.S.; Perazza, C.A.; Villela, P.M.S.; Almeida-Val, V.M.F.; Gao, Z.X.; Coutinho, L.L.; Hilsdorf, A.W.S. Genome-wide association study reveals genes associated with the absence of intermuscular bones in tambaqui (Colossoma macropomum). Anim. Genet. 2020, 51, 899–909. [Google Scholar] [CrossRef]
- Yang, G.; Qin, Z.; Kou, H.; Liang, R.; Zhao, L.; Jiang, S.; Lin, L.; Zhang, K. A Comparative Genomic and Transcriptional Survey Providing Novel Insights into Bone Morphogenetic Protein 2 (bmp2) in Fishes. Int. J. Mol. Sci. 2019, 20, 6137. [Google Scholar] [CrossRef] [Green Version]
- Zhong, Z.; Niu, P.; Wang, M.; Huang, G.; Xu, S.; Sun, Y.; Xu, X.; Hou, Y.; Sun, X.; Yan, Y.; et al. Targeted disruption of sp7 and myostatin with CRISPR-Cas9 results in severe bone defects and more muscular cells in common carp. Sci. Rep. 2016, 6, 22953. [Google Scholar] [CrossRef] [Green Version]
- Nie, C.; Wan, S.; Chen, Y.; Zhu, D.; Wang, X.; Dong, X.; Gao, Z.-X. Loss of scleraxis leads to distinct reduction of mineralized intermuscular bone in zebrafish. Aquac. Fish. 2021, 6, 169–177. [Google Scholar] [CrossRef]
- Kague, E.; Hughes, S.M.; Lawrence, E.A.; Cross, S.; Martin-Silverstone, E.; Hammond, C.L.; Hinits, Y. Scleraxis genes are required for normal musculoskeletal development and for rib growth and mineralization in zebrafish. FASEB J. 2019, 33, 9116–9130. [Google Scholar] [CrossRef] [Green Version]
- Xiong, X.-M.; Robinson, N.A.; Zhou, J.-J.; Chen, Y.-L.; Wang, W.; Wang, X.-B.; Gao, Z.-X. Genetic parameter estimates for intermuscular bone in blunt snout bream (Megalobrama amblycephala) based on a microsatellite-based pedigree. Aquaculture 2019, 502, 371–377. [Google Scholar] [CrossRef]
- Li, J.-T.; Wang, Q.; Huang Yang, M.-D.; Li, Q.-S.; Cui, M.-S.; Dong, Z.-J.; Wang, H.-W.; Yu, J.-H.; Zhao, Y.-J.; Yang, C.-R.; et al. Parallel subgenome structure and divergent expression evolution of allo-tetraploid common carp and goldfish. Nat. Genet. 2021, 53, 1493–1503. [Google Scholar] [CrossRef]
- Yang, M.H.; Wang, Q.; Zhao, R.; Li, Q.S.; Cui, M.S.; Zhang, Y.; Li, J.T. Cyprinus carpio (common carp). Trends Genet. 2022, 38, 305–306. [Google Scholar] [CrossRef]
- Tang, G.; Lv, W.; Sun, Z.; Cao, D.; Zheng, X.; Tong, G.; Wang, H.; Zhang, X.; Kuang, Y. Heritability and quantitative trait locus analyses of intermuscular bones in mirror carp (Cyprinus carpio). Aquaculture 2020, 515, 734601. [Google Scholar] [CrossRef]
- Peng, J.; Zeng, D.; He, P.; Wei, P.; Hui, W.; Wu, T.; Zhuo, X.; Lin, Y. mRNA and microRNA transcriptomics analyses in intermuscular bones of two carp species, rice flower carp (Cyprinus carpio var. Quanzhounensis) and Jian carp (Cyprinus carpio var. Jian). Comp. Biochem. Physiol. Part D Genom. Proteom. 2019, 30, 71–80. [Google Scholar]
- Xu, P.; Zhang, X.; Wang, X.; Li, J.; Liu, G.; Kuang, Y.; Xu, J.; Zheng, X.; Ren, L.; Wang, G.; et al. Genome sequence and genetic diversity of the common carp, Cyprinus carpio. Nat. Genet. 2014, 46, 1212–1219. [Google Scholar] [CrossRef] [Green Version]
- Lv, Y.P.; Bao, B.L.; Jiang, Y.; Yang, L.L.; Li, J.L. Comparative analysis of intermuscular bones in lower teleosts. J. Fish. China 2007, 31, 661–668. [Google Scholar]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [Green Version]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M.; et al. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef] [Green Version]
- Tribhuvan, K.U.; Kumar, K.; Sevanthi, A.M.; Gaikwad, K. MutMap: A versatile tool for identification of mutant loci and mapping of genes. Indian J. Plant Physiol. 2018, 23, 612–621. [Google Scholar] [CrossRef]
- Wang, K.; Li, M.; Hakonarson, H. ANNOVAR: Functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010, 38, e164. [Google Scholar] [CrossRef]
- Chen, C.; Chen, H.; Zhang, Y.; Thomas, H.R.; Frank, M.H.; He, Y.; Xia, R. TBtools: An Integrative Toolkit Developed for Interactive Analyses of Big Biological Data. Mol. Plant 2020, 13, 1194–1202. [Google Scholar] [CrossRef]
- Weckx, S.; Del-Favero, J.; Rademakers, R.; Claes, L.; Cruts, M.; De Jonghe, P.; Van Broeckhoven, C.; De Rijk, P. novoSNP, a novel computational tool for sequence variation discovery. Genome Res. 2005, 15, 436–442. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Sun, X.Q.; Ye, Y.Q.; Wang, Q.; Li, Q.S.; Zhao, R.; Wang, H.W.; Li, J.T. Association between the Polymorphisms of fads2a and fads2b and Poly-Unsaturated Fatty Acids in Common Carp (Cyprinus carpio). Animals 2021, 11, 1780. [Google Scholar] [CrossRef]
- Bradbury, P.J.; Zhang, Z.; Kroon, D.E.; Casstevens, T.M.; Ramdoss, Y.; Buckler, E.S. TASSEL: Software for association mapping of complex traits in diverse samples. Bioinformatics 2007, 23, 2633–2635. [Google Scholar] [CrossRef]
- Alexander, D.H.; Novembre, J.; Lange, K. Fast model-based estimation of ancestry in unrelated individuals. Genome Res. 2009, 19, 1655–1664. [Google Scholar] [CrossRef] [Green Version]
- Francis, R.M. pophelper: An R package and web app to analyse and visualize population structure. Mol. Ecol. Resour. 2017, 17, 27–32. [Google Scholar] [CrossRef] [Green Version]
- Raymond, M.; Rousset, F. GENEPOP (Version 1.2): Population Genetics Software for Exact Tests and Ecumenicism. J. Hered. 1995, 86, 248–249. [Google Scholar] [CrossRef]
- Feng, X.; Yu, X.; Tong, J. Novel Single Nucleotide Polymorphisms of the Insulin-Like Growth Factor-I Gene and Their Associations with Growth Traits in Common Carp (Cyprinus carpio L.). Int. J. Mol. Sci. 2014, 15, 22471–22482. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, Y.; Li, H.; Li, J.; Yu, F.; Yu, J. Association study between single nucleotide polymorphisms in leptin and growth traits in Cyprinus carpio var. Jian. Genet. Mol. Res. GMR 2016, 15, gmr.15037635. [Google Scholar] [CrossRef]
- Wang, X.; Yu, X.; Tong, J. Molecular Characterization and Growth Association of Two Apolipoprotein A-Ib Genes in Common Carp (Cyprinus carpio). Int. J. Mol. Sci. 2016, 17, 1569. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, Y.; Yu, X.; Tong, J. Polymorphisms in Myostatin Gene and associations with growth traits in the common carp (Cyprinus carpio L.). Int. J. Mol. Sci. 2012, 13, 14956–14961. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Knüppel, S.; Meidtner, K.; Arregui, M.; Holzhütter, H.G.; Boeing, H. Joint effect of unlinked genotypes: Application to type 2 diabetes in the EPIC-Potsdam case-cohort study. Ann. Hum. Genet. 2015, 79, 253–263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilkinson, G.N.; Rogers, C.E. Symbolic Description of Factorial Models for Analysis of Variance. J. R. Stat. Soc. Ser. C 1973, 22, 392–399. [Google Scholar] [CrossRef]
- Della Ragione, F.; Takabayashi, K.; Mastropietro, S.; Mercurio, C.; Oliva, A.; Russo, G.L.; Della Pietra, V.; Borriello, A.; Nobori, T.; Carson, D.A.; et al. Purification and characterization of recombinant human 5′-methylthioadenosine phosphorylase: Definite identification of coding cDNA. Biochem. Biophys. Res. Commun. 1996, 223, 514–519. [Google Scholar] [CrossRef]
- Peters, A.E.; Knöpper, K.; Grafen, A.; Kastenmüller, W. A multifunctional mouse model to study the role of Samd3. Eur. J. Immunol. 2021, 52, 328–337. [Google Scholar] [CrossRef]
- Hagihara, M.; Endo, M.; Hata, K.; Higuchi, C.; Takaoka, K.; Yoshikawa, H.; Yamashita, T. Neogenin, a Receptor for Bone Morphogenetic Proteins. J. Biol. Chem. 2011, 286, 5157–5165. [Google Scholar] [CrossRef] [Green Version]
- Havens, B.A.; Velonis, D.; Kronenberg, M.S.; Lichtler, A.C.; Oliver, B.; Mina, M. Roles of FGFR3 during morphogenesis of Meckel’s cartilage and mandibular bones. Dev. Biol. 2008, 316, 336–349. [Google Scholar] [CrossRef] [Green Version]
- Nakashima, K.; de Crombrugghe, B. Transcriptional mechanisms in osteoblast differentiation and bone formation. Trends Genet. 2003, 19, 458–466. [Google Scholar] [CrossRef]
- Perrien, D.S.; Brown, E.C.; Aronson, J.; Skinner, R.A.; Montague, D.C.; Badger, T.M.; Lumpkin, C.K., Jr. Immunohistochemical study of osteopontin expression during distraction osteogenesis in the rat. J. Histochem. Cytochem. 2002, 50, 567–574. [Google Scholar] [CrossRef] [Green Version]
- Su, S.; Dong, Z. Comparative expression analyses of bone morphogenetic protein 4 (BMP4) expressions in muscles of tilapia and common carp indicate that BMP4 plays a role in the intermuscular bone distribution in a dose-dependent manner. Gene Expr. Patterns 2018, 27, 106–113. [Google Scholar] [CrossRef]
- Chinipardaz, Z.; Liu, M.; Graves, D.T.; Yang, S. Role of Primary Cilia in Bone and Cartilage. J. Dent. Res. 2021, 220345211046606. [Google Scholar] [CrossRef]
- Shi, W.; Ma, Z.; Zhang, G.; Wang, C.; Jiao, Z. Novel functions of the primary cilium in bone disease and cancer. Cytoskeleton 2019, 76, 233–242. [Google Scholar] [CrossRef] [Green Version]
- Camacho-Vanegas, O.; Camacho, S.C.; Till, J.; Miranda-Lorenzo, I.; Terzo, E.; Ramirez, M.C.; Schramm, V.; Cordovano, G.; Watts, G.; Mehta, S.; et al. Primate Genome Gain and Loss: A Bone Dysplasia, Muscular Dystrophy, and Bone Cancer Syndrome Resulting from Mutated Retroviral-Derived MTAP Transcripts. Am. J. Hum. Genet. 2012, 90, 614–627. [Google Scholar] [CrossRef] [Green Version]
- Topaz, O.; Indelman, M.; Chefetz, I.; Geiger, D.; Metzker, A.; Altschuler, Y.; Choder, M.; Bercovich, D.; Uitto, J.; Bergman, R.; et al. A Deleterious Mutation in SAMD9 Causes Normophosphatemic Familial Tumoral Calcinosis. Am. J. Hum. Genet. 2006, 79, 759–764. [Google Scholar] [CrossRef] [Green Version]
- Cheng, F.; Sun, R.; Hou, X.; Zheng, H.; Zhang, F.; Zhang, Y.; Liu, B.; Liang, J.; Zhuang, M.; Liu, Y.; et al. Subgenome parallel selection is associated with morphotype diversification and convergent crop domestication in Brassica rapa and Brassica oleracea. Nat. Genet. 2016, 48, 1218–1224. [Google Scholar] [CrossRef]
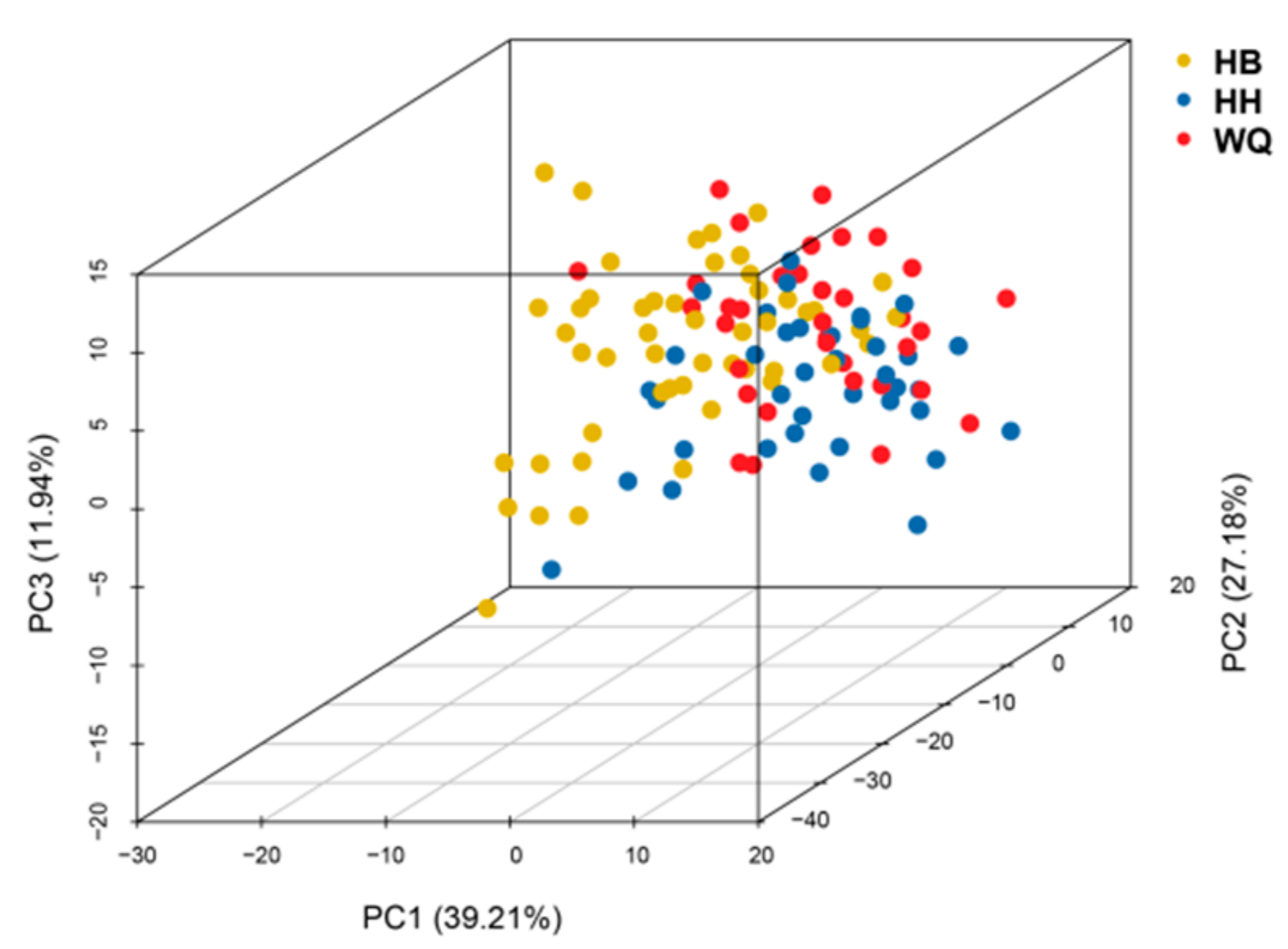
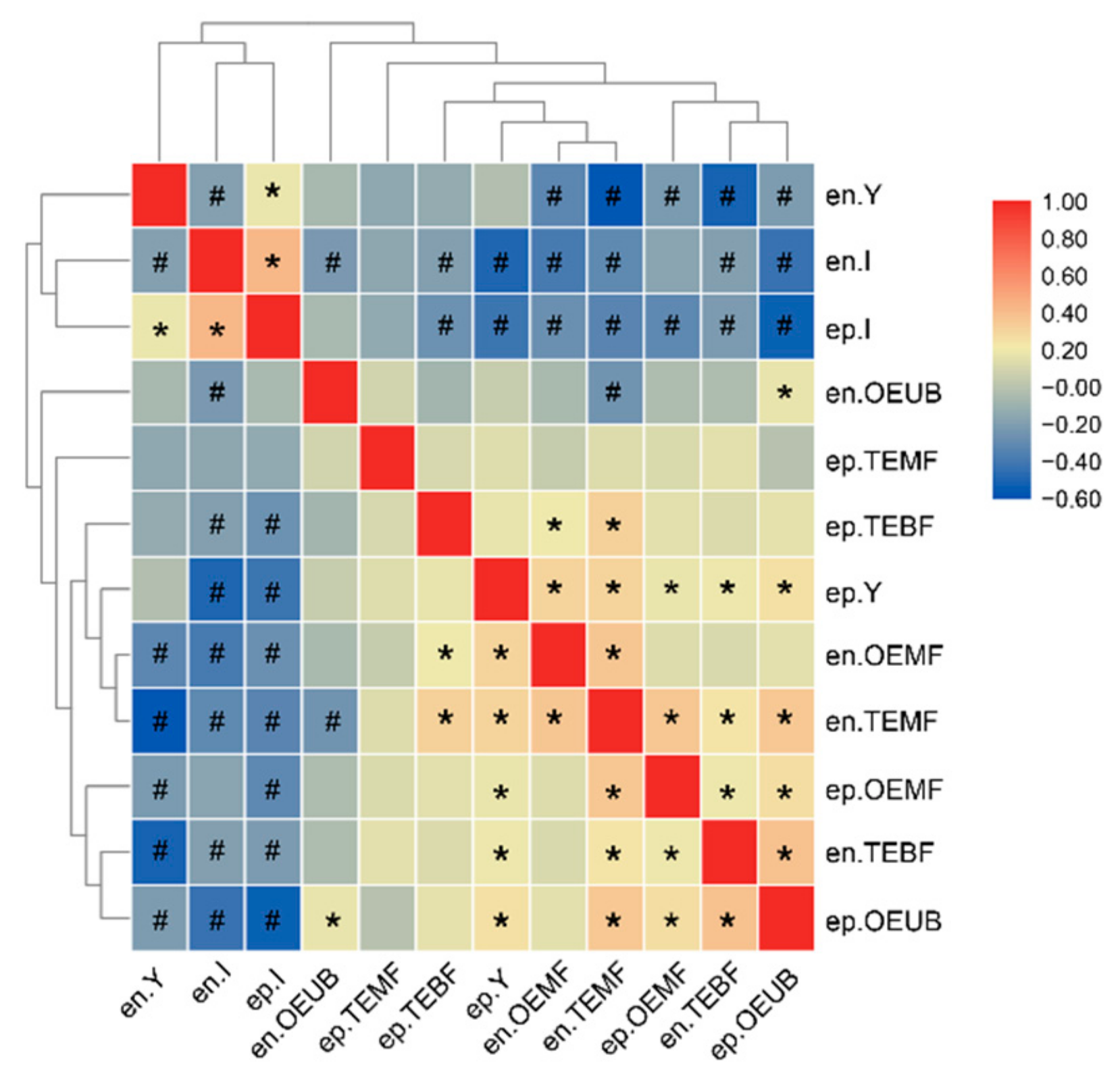

{kind=link}
{kind=link}
| Sample | Q20 (%) | Clean Read Pairs | Depth | Mapping Ratio (%) |
|---|---|---|---|---|
| HBF | 96.83 | 205,678,671 | 37.24 | 99.20 |
| HBM | 96.77 | 211,458,150 | 38.28 | 98.88 |
| WQF | 96.90 | 206,924,782 | 37.46 | 99.29 |
| WQM | 96.70 | 198,379,639 | 35.92 | 99.20 |
| HHF | 97.02 | 231,095,221 | 41.84 | 99.31 |
| HHM | 97.01 | 228,265,661 | 41.33 | 99.27 |
| Genomic Location | Number | |
|---|---|---|
| downstream | 3 | |
| exonic | nonsynonymous | 4 |
| synonymous | 4 | |
| intergenic | 108 | |
| intronic | 85 | |
| upstream | 4 |
| Trait | SNP | GLM | ANOVA | |||
|---|---|---|---|---|---|---|
| p Value | Marker R2 (%) # | MM | Mm | p Value | ||
| IB | A1.3023796 | 0.0487 | 4.72 | 96.0 ± 5.35 | 90.2 ± 8.79 | 0.0236 |
| B25.3800532 | 0.0224 | 5.90 | 96.1 ± 5.41 | 90.1 ± 5.87 | 0.0059 | |
| en-IB | B25.3800532 | 0.0228 | 5.57 | 66.1 ± 3.05 | 62.6 ± 2.64 | 0.0039 |
| en-I | A1.3023490 | 0.0167 | 6.76 | 20.5 ± 5.45 | 27.2 ± 12.80 | 0.0022 |
| B25.3800904 | 0.0369 | 5.49 | 21.4 ± 6.22 | 15.9 ± 4.26 | 0.0213 | |
| A1.3023694 | 0.0382 | 5.47 | 20.6 ± 5.90 | 22.2 ± 6.95 | 0.0208 | |
| B25.3800710 * | 0.0498 | 5.04 | 20.6 ± 5.46 | 27.3 ± 12.58 | 0.0055 | |
| en-OEMF | B25.3800526 | 0.0019 | 7.51 | 3.1 ± 2.94 | 6.00 ± 3.44 | 0.0014 |
| B25.3800904 | 0.00001 | 16.39 | 3.1 ± 2.80 | 8.6 ± 2.07 | 0.00002 | |
| B25.3800999 | 0.0059 | 8.02 | 2.9 ± 2.40 | 4.5 ± 3.72 | 0.0342 | |
| A16.2710728 | 0.0297 | 5.65 | 4.6 ± 3.50 | 3.0 ± 2.85 | 0.0418 | |
| en-OEUB | A1.3023466 | 0.0221 | 6.26 | 11.4 ± 4.26 | 8.3 ± 2.80 | 0.0126 |
| A16.2710488 | 0.0450 | 5.13 | 9.9 ± 3.15 | 11.9 ± 4.57 | 0.0301 | |
| en-TEBF | A1.3023753 * | 0.0333 | 4.80 | 7.9 ± 3.89 | 7.7 ± 3.87 | 0.0005 |
| A1.3023814 | 0.0414 | 2.96 | 9.7 ± 4.57 | 6.9 ± 3.78 | 0.0014 | |
| B25.3800710 * | 0.0425 | 4.47 | 8.7 ± 4.39 | 5.1 ± 5.79 | 0.0433 | |
| B25.3800929 | 0.0206 | 3.79 | 8.7 ± 4.50 | 13.0 ± 4.12 | 0.0401 | |
| A1.3023730 | 0.0358 | 4.66 | 9.0 ± 4.35 | 5.3 ± 3.13 | 0.0469 | |
| B25.3801033 | 0.0268 | 5.06 | 8.5 ± 4.49 | 8.7 ± 3.95 | 0.0148 | |
| en-TEMF | A1.3023814 | 0.0138 | 4.64 | 6.9 ± 6.57 | 3.2 ± 3.68 | 0.0013 |
| A16.2710488 | 0.0183 | 6.09 | 4.6 ± 5.79 | 7.7 ± 6.37 | 0.0198 | |
| B25.3800685 | 0.0276 | 5.48 | 5.8 ± 5.99 | 3.0 ± 4.07 | 0.0488 | |
| B25.3800710 * | 0.0349 | 5.14 | 5.6 ± 5.85 | 1.00 ± 1.91 | 0.0414 | |
| en-Y | A1.3023447 | 0.00001 | 15.84 | 14.8 ± 6.67 | 19.2 ± 7.38 | 0.0013 |
| A1.3023694 | 0.0155 | 6.02 | 14.7 ± 6.92 | 16.4 ± 7.49 | 0.0395 | |
| B25.3801016 | 0.0330 | 6.32 | 15.5 ± 6.63 | 19.8 ± 7.42 | 0.0137 | |
| ep-I | A1.3023730 | 0.0302 | 5.22 | 19.2 ± 3.81 | 22.0 ± 2.14 | 0.0177 |
| ep-OEMF | B25.3801033 | 0.0165 | 4.65 | 0.4 ± 0.72 | 0.9 ± 1.19 | 0.0357 |
| ep-TEMF | B25.3800904 | 0.0149 | 7.02 | 0.2 ± 0.53 | 0.9 ± 1.07 | 0.0047 |
| B25.3800911 | 0.0380 | 3.64 | 0.2 ± 0.40 | 0.5 ± 0.76 | 0.0118 | |
| ep-Y | B25.3801016 | 0.0399 | 4.87 | 3.6 ± 1.62 | 2.1 ± 1.37 | 0.0073 |
| B25.3801028 | 0.0306 | 3.55 | 3.7 ± 1.47 | 1.9 ± 1.37 | 0.0019 | |
| Trait | SNP | Each SNP Marker R2 (%) | Marker R2 of Genotype Combination (%) |
|---|---|---|---|
| IB | A1.3023796 | 4.72 | 10 |
| B25.3800532 | 5.90 | ||
| en-I | A1.3023490 | 6.76 | 15.9 |
| B25.3800904 | 5.49 | ||
| A1.3023694 | 5.47 | ||
| B25.3800710 * | 5.04 | ||
| en-OEMF | B25.3800526 | 7.51 | 34.3 |
| B25.3800904 | 16.39 | ||
| B25.3800999 | 8.02 | ||
| A16.2710728 | 5.65 | ||
| en-OEUB | A1.3023466 | 6.26 | 7.44 |
| A16.2710488 | 5.13 | ||
| en-TEBF | A1.3023753 * | 4.80 | 42.07 |
| A1.3023814 | 2.96 | ||
| B25.3800710 * | 4.47 | ||
| B25.3800929 | 3.79 | ||
| A1.3023730 | 4.66 | ||
| B25.3801033 | 5.06 | ||
| en-TEMF | A1.3023814 | 4.64 | 17.57 |
| A16.2710488 | 6.09 | ||
| B25.3800685 | 5.48 | ||
| B25.3800710 * | 5.14 | ||
| en-Y | A1.3023447 | 15.84 | 9.99 |
| A1.3023694 | 6.02 | ||
| B25.3801016 | 6.32 | ||
| ep-TEMF | B25.3800904 | 7.02 | 7.95 |
| B25.3800911 | 3.64 | ||
| ep-Y | B25.3801016 | 4.87 | 18.21 |
| B25.3801028 | 3.55 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cui, M.-S.; Zhao, R.; Wang, Q.; Zhang, Y.; Li, Q.-S.; Huang Yang, M.-D.; Sun, X.-Q.; Li, J.-T. Bulked Segregant Analysis and Association Analysis Identified the Polymorphisms Related to the Intermuscular Bones in Common Carp (Cyprinus carpio). Biology 2022, 11, 477. https://doi.org/10.3390/biology11030477
Cui M-S, Zhao R, Wang Q, Zhang Y, Li Q-S, Huang Yang M-D, Sun X-Q, Li J-T. Bulked Segregant Analysis and Association Analysis Identified the Polymorphisms Related to the Intermuscular Bones in Common Carp (Cyprinus carpio). Biology. 2022; 11(3):477. https://doi.org/10.3390/biology11030477
Chicago/Turabian StyleCui, Ming-Shu, Ran Zhao, Qi Wang, Yan Zhang, Qing-Song Li, Mei-Di Huang Yang, Xiao-Qing Sun, and Jiong-Tang Li. 2022. "Bulked Segregant Analysis and Association Analysis Identified the Polymorphisms Related to the Intermuscular Bones in Common Carp (Cyprinus carpio)" Biology 11, no. 3: 477. https://doi.org/10.3390/biology11030477