Transcriptional Gene Silencing (TGS) via the RNAi Machinery in HIV-1 Infections


{kind=link}
{kind=link}
Abstract
:1. Introduction
2. MicroRNA and the RNAi Molecular Machinery
2.1. MicroRNA Biogenesis and the RNAi Machinery
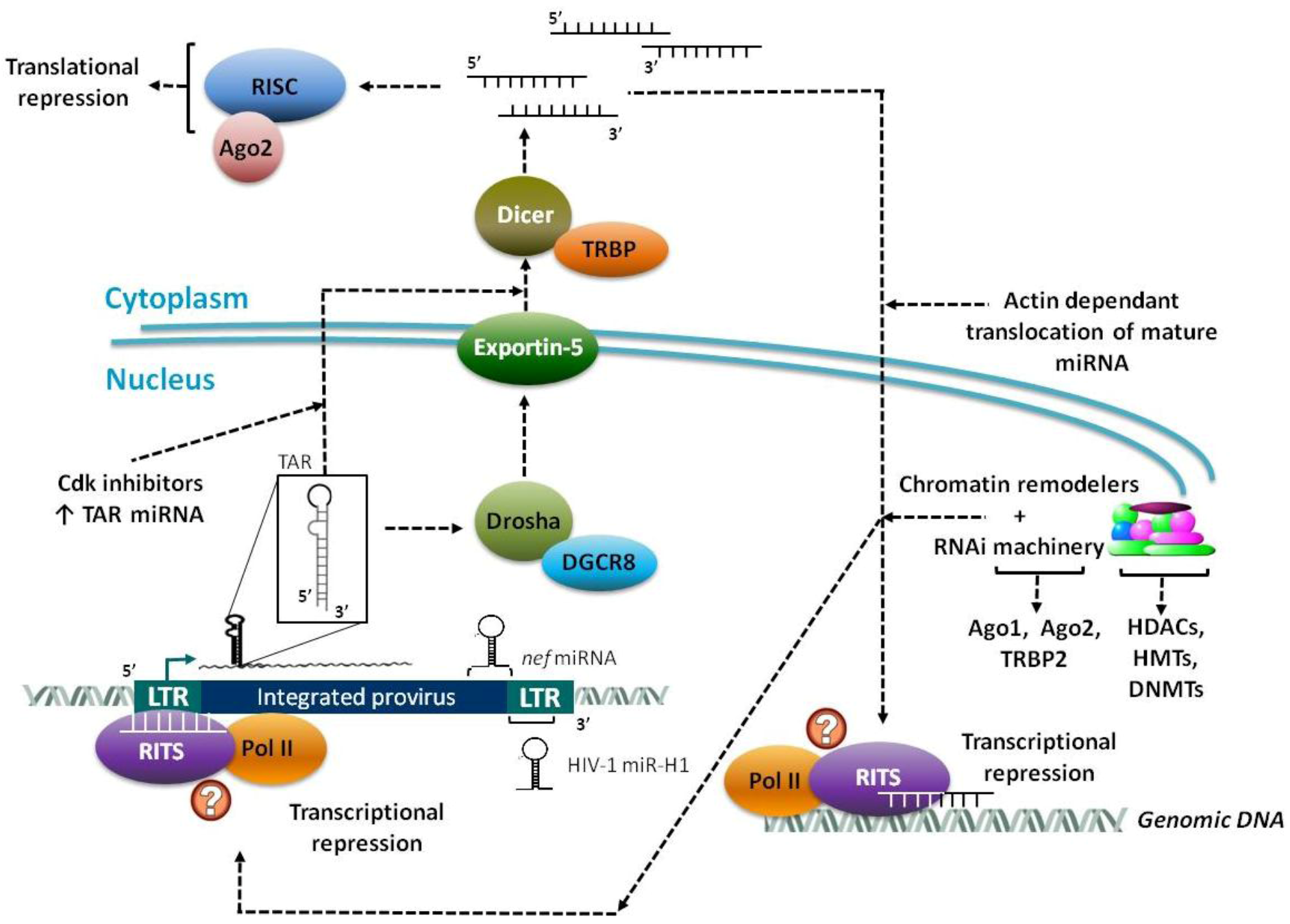
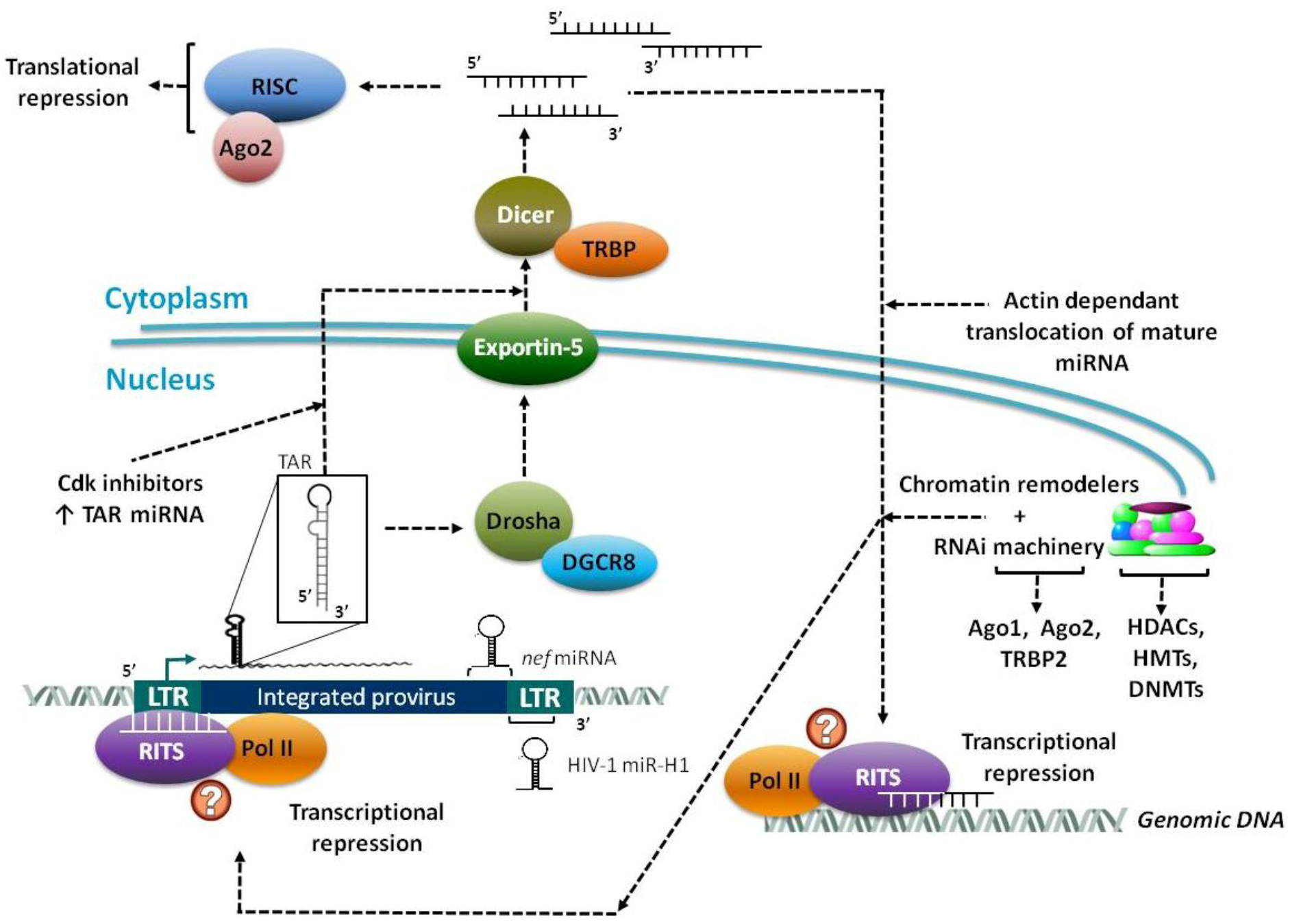
2.2. Virally Encoded miRNAs
2.2.1. Primary Functions of Viral miRNAs
2.2.2. MicroRNAs from DNA and RNA Viruses
2.2.3. HIV-1 Derived miRNAs
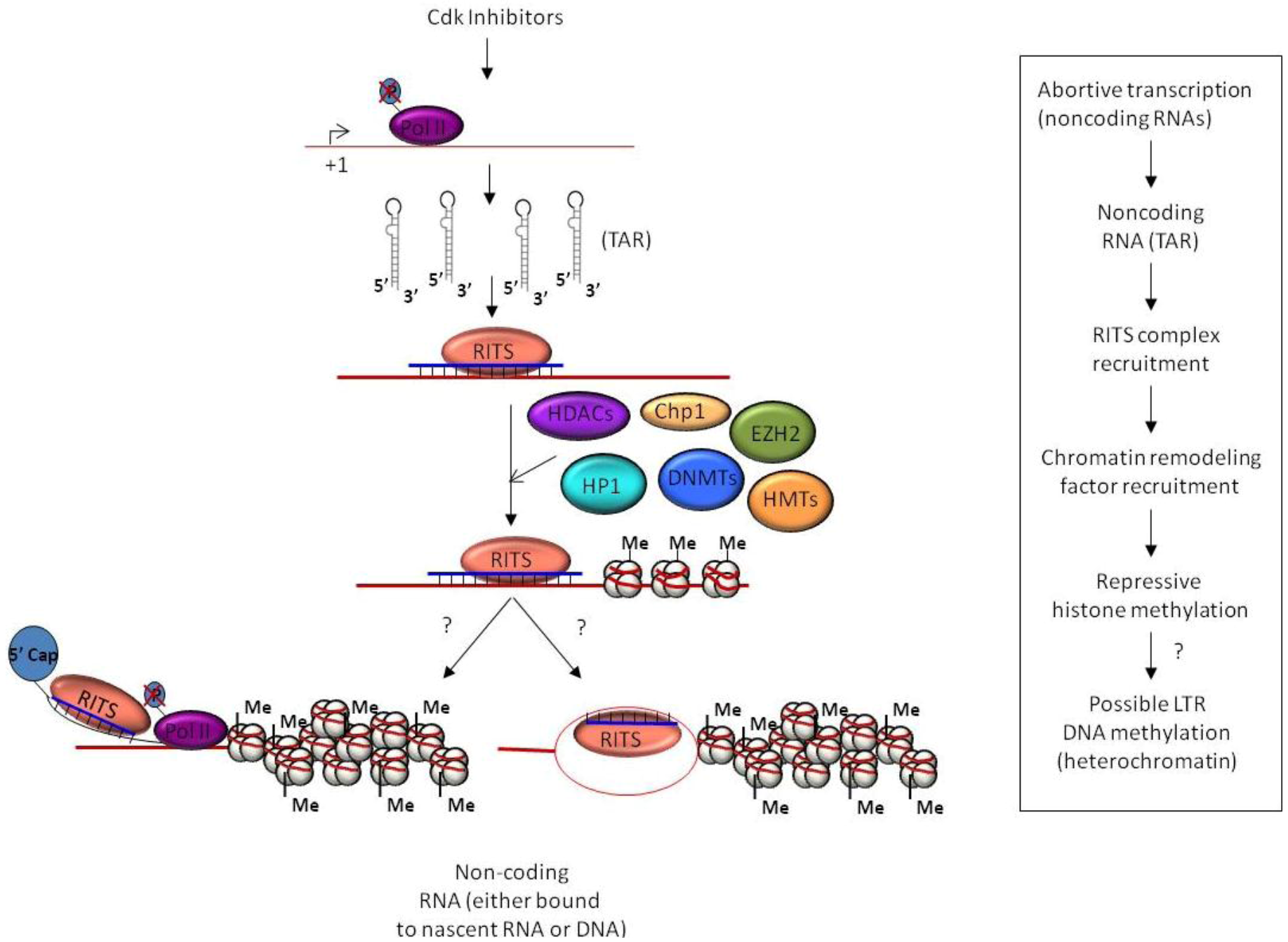
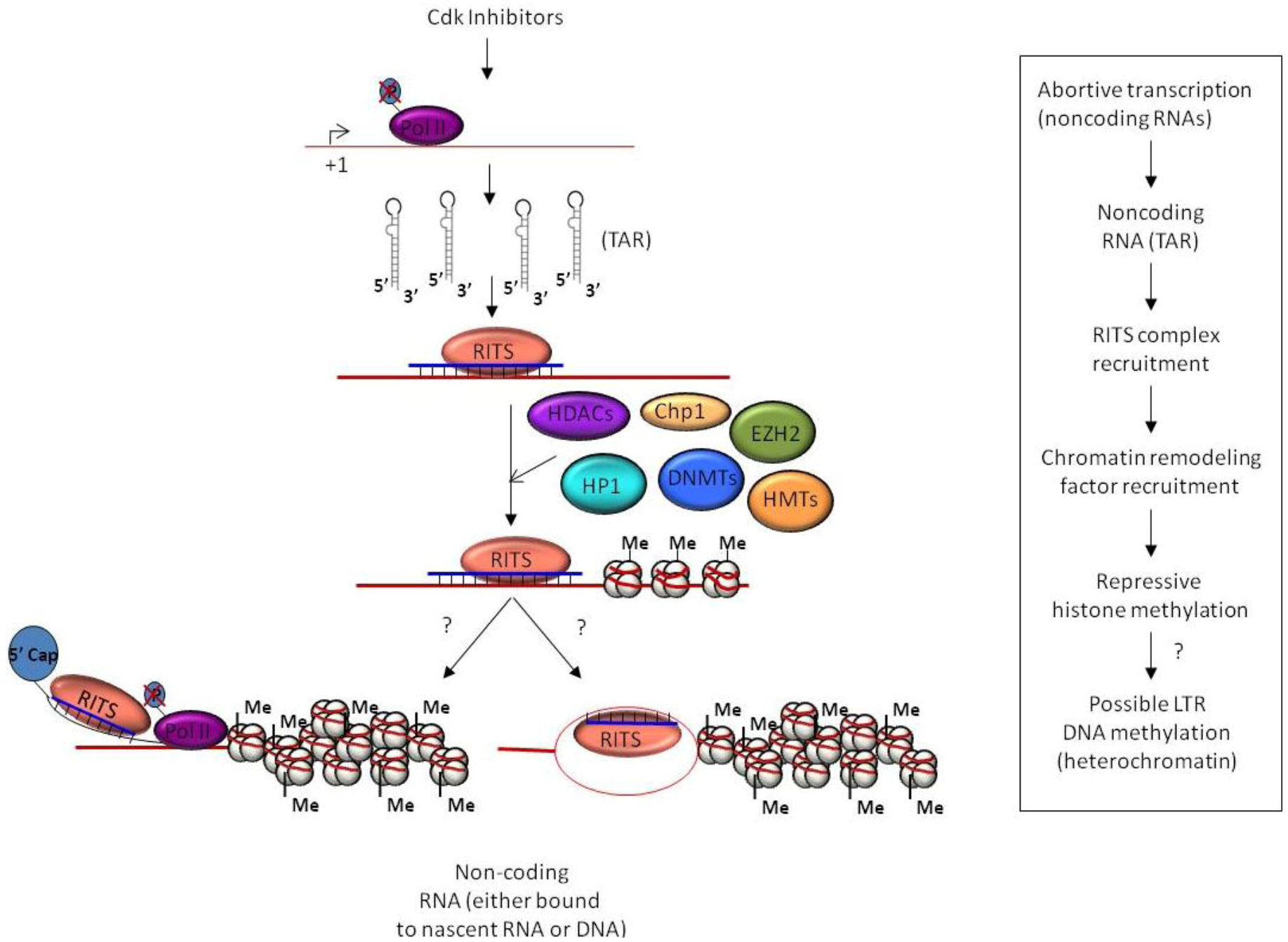
3. Epigenetic Regulation and Chromatin Remodeling
3.1. Histone Acetyltransferases
3.2. Histone Deacetylases
3.3. Histone Methyltransferases and Histone Demethyltransferases
3.4. DNA Methyltransferases
3.5. ATP-Dependent Chromatin Remodelers
4. TGS in Mammalians
4.1. TGS and HIV-1 Infection
5. Conclusions
Acknowledgements
References
- UNAIDS, Global Report: Unaids Report on the Global Aids Epidemic 2010; Joint United Nations Programme on HIV/AIDS (UNAIDS): Geneva, Switzerland, 2010.
- Nachega, J.B.; Marconi, V.C.; van Zyl, G.U.; Gardner, E.M.; Preiser, W.; Hong, S.Y.; Mills, E.J.; Gross, R. Hiv treatment adherence, drug resistance, virologic failure: Evolving concepts. Infect. Disord. Drug Targets 2011, 11, 167–174. [Google Scholar]
- Schweighardt, B.; Ortiz, G.M.; Grant, R.M.; Wellons, M.; Miralles, G.D.; Kostrikis, L.G.; Bartlett, J.A.; Nixon, D.F. Emergence of drug-resistant hiv-1 variants in patients undergoing structured treatment interruptions. Aids 2002, 16, 2342–2344. [Google Scholar]
- Mette, M.F.; Aufsatz, W.; van der Winden, J.; Matzke, M.A.; Matzke, A.J. Transcriptional silencing and promoter methylation triggered by double-stranded rna. EMBO J. 2000, 19, 5194–5201. [Google Scholar]
- Verdel, A.; Jia, S.; Gerber, S.; Sugiyama, T.; Gygi, S.; Grewal, S.I.; Moazed, D. Rnai-mediated targeting of heterochromatin by the rits complex. Science 2004, 303, 672–676. [Google Scholar]
- Noma, K.; Sugiyama, T.; Cam, H.; Verdel, A.; Zofall, M.; Jia, S.; Moazed, D.; Grewal, S.I. Rits acts in cis to promote rna interference-mediated transcriptional and post-transcriptional silencing. Nat. Genet. 2004, 36, 1174–1180. [Google Scholar]
- Morris, K.V.; Chan, S.W.; Jacobsen, S.E.; Looney, D.J. Small interfering rna-induced transcriptional gene silencing in human cells. Science 2004, 305, 1289–1292. [Google Scholar]
- Omoto, S.; Ito, M.; Tsutsumi, Y.; Ichikawa, Y.; Okuyama, H.; Brisibe, E.A.; Saksena, N.K.; Fujii, Y.R. Hiv-1 nef suppression by virally encoded microRNA. Retrovirology 2004, 1. [Google Scholar] [CrossRef] [Green Version]
- Chua, J.H.; Armugam, A.; Jeyaseelan, K. Micrornas: Biogenesis, function and applications. Curr. Opin. Mol. Ther. 2009, 11, 189–199. [Google Scholar]
- Ying, S.Y.; Lin, S.L. Intron-mediated rna interference and microrna biogenesis. Methods Mol. Biol. 2009, 487, 387–413. [Google Scholar]
- Perron, M.P.; Provost, P. Protein components of the microrna pathway and human diseases. Methods Mol. Biol. 2009, 487, 369–385. [Google Scholar]
- Winter, J.; Jung, S.; Keller, S.; Gregory, R.I.; Diederichs, S. Many roads to maturity: Microrna biogenesis pathways and their regulation. Nat. Cell Biol. 2009, 11, 228–234. [Google Scholar]
- Faller, M.; Guo, F. Microrna biogenesis: There’s more than one way to skin a cat. Biochim. Biophys. Acta 2008, 1779, 663–667. [Google Scholar]
- Van Wynsberghe, P.M.; Chan, S.P.; Slack, F.J.; Pasquinelli, A.E. Analysis of microrna expression and function. Methods Cell Biol. 2011, 106, 219–252. [Google Scholar]
- Yang, J.S.; Lai, E.C. Alternative mirna biogenesis pathways and the interpretation of core mirna pathway mutants. Mol. Cell 2011, 43, 892–903. [Google Scholar]
- Perron, M.P.; Provost, P. Protein interactions and complexes in human microrna biogenesis and function. Frontiers Biosci. A J. Virtual Libr. 2008, 13, 2537–2547. [Google Scholar] [CrossRef]
- Cai, X.; Hagedorn, C.H.; Cullen, B.R. Human micrornas are processed from capped, polyadenylated transcripts that can also function as mrnas. RNA 2004, 10, 1957–1966. [Google Scholar]
- Lee, Y.; Ahn, C.; Han, J.; Choi, H.; Kim, J.; Yim, J.; Lee, J.; Provost, P.; Radmark, O.; Kim, S.; et al. The nuclear rnase iii drosha initiates microrna processing. Nature 2003, 425, 415–419. [Google Scholar]
- Denli, A.M.; Tops, B.B.; Plasterk, R.H.; Ketting, R.F.; Hannon, G.J. Processing of primary micrornas by the microprocessor complex. Nature 2004, 432, 231–235. [Google Scholar]
- Han, J.; Lee, Y.; Yeom, K.H.; Kim, Y.K.; Jin, H.; Kim, V.N. The drosha-dgcr8 complex in primary microrna processing. Genes Dev. 2004, 18, 3016–3027. [Google Scholar]
- Yi, R.; Qin, Y.; Macara, I.G.; Cullen, B.R. Exportin-5 mediates the nuclear export of pre-micrornas and short hairpin rnas. Genes Dev. 2003, 17, 3011–3016. [Google Scholar] [CrossRef]
- Kim, V.N. Microrna precursors in motion: Exportin-5 mediates their nuclear export. Trends Cell Biol. 2004, 14, 156–159. [Google Scholar] [CrossRef]
- Vermeulen, A.; Behlen, L.; Reynolds, A.; Wolfson, A.; Marshall, W.S.; Karpilow, J.; Khvorova, A. The contributions of dsrna structure to dicer specificity and efficiency. RNA 2005, 11, 674–682. [Google Scholar]
- Macrae, I.J.; Li, F.; Zhou, K.; Cande, W.Z.; Doudna, J.A. Structure of dicer and mechanistic implications for rnai. Cold Spring Harb. Symp Quant. Biol. 2006, 71, 73–80. [Google Scholar] [CrossRef]
- Zhang, X.; Zeng, Y. The terminal loop region controls microrna processing by drosha and dicer. Nucleic Acids Res. 2010, 38, 7689–7697. [Google Scholar] [CrossRef]
- Chendrimada, T.P.; Gregory, R.I.; Kumaraswamy, E.; Norman, J.; Cooch, N.; Nishikura, K.; Shiekhattar, R. Trbp recruits the dicer complex to ago2 for microrna processing and gene silencing. Nature 2005, 436, 740–744. [Google Scholar]
- Haase, A.D.; Jaskiewicz, L.; Zhang, H.; Laine, S.; Sack, R.; Gatignol, A.; Filipowicz, W. Trbp, a regulator of cellular pkr and hiv-1 virus expression, interacts with dicer and functions in rna silencing. EMBO Rep. 2005, 6, 961–967. [Google Scholar]
- Kawamata, T.; Tomari, Y. Making risc. Trends Biochem. Sci. 2010, 35, 368–376. [Google Scholar] [CrossRef]
- Easow, G.; Teleman, A.A.; Cohen, S.M. Isolation of microrna targets by mirnp immunopurification. RNA 2007, 13, 1198–1204. [Google Scholar]
- Lytle, J.R.; Yario, T.A.; Steitz, J.A. Target mrnas are repressed as efficiently by microrna-binding sites in the 5' utr as in the 3' utr. Proc. Natl. Acad. Sci. USA 2007, 104, 9667–9672. [Google Scholar]
- Orom, U.A.; Nielsen, F.C.; Lund, A.H. Microrna-10a binds the 5'utr of ribosomal protein mrnas and enhances their translation. Mol. Cell 2008, 30, 460–471. [Google Scholar] [CrossRef]
- Hafner, M.; Landthaler, M.; Burger, L.; Khorshid, M.; Hausser, J.; Berninger, P.; Rothballer, A.; Ascano, M., Jr.; Jungkamp, A.C.; Munschauer, M.; et al. Transcriptome-wide identification of rna-binding protein and microrna target sites by par-clip. Cell 2010, 141, 129–141. [Google Scholar]
- Rigoutsos, I. New tricks for animal micrornas: Targeting of amino acid coding regions at conserved and nonconserved sites. Cancer Res. 2009, 69, 3245–3248. [Google Scholar] [CrossRef]
- Parker, J.S.; Roe, S.M.; Barford, D. Molecular mechanism of target rna transcript recognition by argonaute-guide complexes. Cold Spring Harb. Symp. Quant. Biol. 2006, 71, 45–50. [Google Scholar] [CrossRef]
- Parker, J.S.; Parizotto, E.A.; Wang, M.; Roe, S.M.; Barford, D. Enhancement of the seed-target recognition step in rna silencing by a piwi/mid domain protein. Mol. Cell 2009, 33, 204–214. [Google Scholar] [CrossRef]
- Zofall, M.; Grewal, S.I. Rnai-mediated heterochromatin assembly in fission yeast. Cold Spring Harb. Symp. Quant. Biol. 2006, 71, 487–496. [Google Scholar] [CrossRef]
- Buhler, M.; Moazed, D. Transcription and rnai in heterochromatic gene silencing. Nat. Struct. Mol. Biol. 2007, 14, 1041–1048. [Google Scholar] [CrossRef]
- Li, L.C.; Okino, S.T.; Zhao, H.; Pookot, D.; Place, R.F.; Urakami, S.; Enokida, H.; Dahiya, R. Small dsrnas induce transcriptional activation in human cells. Proc. Natl. Acad Sci. USA 2006, 103, 17337–17342. [Google Scholar]
- Place, R.F.; Li, L.C.; Pookot, D.; Noonan, E.J.; Dahiya, R. Microrna-373 induces expression of genes with complementary promoter sequences. Proc. Natl. Acad. Sci. USA 2008, 105, 1608–1613. [Google Scholar]
- Morris, K.V.; Santoso, S.; Turner, A.M.; Pastori, C.; Hawkins, P.G. Bidirectional transcription directs both transcriptional gene activation and suppression in human cells. PLoS Genet. 2008, 4, e1000258. [Google Scholar]
- Umbach, J.L.; Cullen, B.R. The role of rnai and micrornas in animal virus replication and antiviral immunity. Genes Dev. 2009, 23, 1151–1164. [Google Scholar]
- Botos, I.; Liu, L.; Wang, Y.; Segal, D.M.; Davies, D.R. The toll-like receptor 3: Dsrna signaling complex. Biochim. Biophys. Acta 2009, 1789, 667–674. [Google Scholar]
- Kok, K.H.; Lei, T.; Jin, D.Y. Identification and validation of the cellular targets of virus-encoded micrornas. Methods Mol. Biol. 2010, 667, 319–326. [Google Scholar]
- Pfeffer, S.; Zavolan, M.; Grasser, F.A.; Chien, M.; Russo, J.J.; Ju, J.; John, B.; Enright, A.J.; Marks, D.; Sander, C.; et al. Identification of virus-encoded micrornas. Science 2004, 304, 734–736. [Google Scholar]
- Griffiths-Jones, S.; Grocock, R.J.; van Dongen, S.; Bateman, A.; Enright, A.J. Mirbase: Microrna sequences, targets and gene nomenclature. Nucleic Acids Res. 2006, 34, D140–D144. [Google Scholar]
- Griffiths-Jones, S.; Saini, H.K.; van Dongen, S.; Enright, A.J. Mirbase: Tools for microrna genomics. Nucleic Acids Res. 2008, 36, D154–D158. [Google Scholar]
- Sullivan, C.S.; Ganem, D. Micrornas and viral infection. Mol. Cell 2005, 20, 3–7. [Google Scholar] [CrossRef]
- Barth, S.; Pfuhl, T.; Mamiani, A.; Ehses, C.; Roemer, K.; Kremmer, E.; Jaker, C.; Hock, J.; Meister, G.; Grasser, F.A. Epstein-barr virus-encoded microrna mir-bart2 down-regulates the viral DNA polymerase balf5. Nucleic Acids Res. 2008, 36, 666–675. [Google Scholar]
- Schopman, N.C.; Willemsen, M.; Liu, Y.P.; Bradley, T.; van Kampen, A.; Baas, F.; Berkhout, B.; Haasnoot, J. Deep sequencing of virus-infected cells reveals hiv-encoded small rnas. Nucleic Acids Res. 2012, 40, 414–427. [Google Scholar]
- Riley, K.J.; Rabinowitz, G.S.; Yario, T.A.; Luna, J.M.; Darnell, R.B.; Steitz, J.A. Ebv and human micrornas co-target oncogenic and apoptotic viral and human genes during latency. EMBO J. 2012. [Google Scholar] [CrossRef]
- Gupta, A.; Gartner, J.J.; Sethupathy, P.; Hatzigeorgiou, A.G.; Fraser, N.W. Anti-apoptotic function of a microrna encoded by the hsv-1 latency-associated transcript. Nature 2006, 442, 82–85. [Google Scholar]
- Bennasser, Y.; Le, S.Y.; Yeung, M.L.; Jeang, K.T. Hiv-1 encoded candidate micro-rnas and their cellular targets. Retrovirology 2004, 1. [Google Scholar] [CrossRef] [Green Version]
- Grey, F.; Meyers, H.; White, E.A.; Spector, D.H.; Nelson, J. A human cytomegalovirus-encoded microrna regulates expression of multiple viral genes involved in replication. PLoS Pathog. 2007, 3, e163. [Google Scholar]
- Lo, A.K.; To, K.F.; Lo, K.W.; Lung, R.W.; Hui, J.W.; Liao, G.; Hayward, S.D. Modulation of lmp1 protein expression by ebv-encoded micrornas. Proc. Natl. Acad Sci. USA 2007, 104, 16164–16169. [Google Scholar]
- Murphy, E.; Vanicek, J.; Robins, H.; Shenk, T.; Levine, A.J. Suppression of immediate-early viral gene expression by herpesvirus-coded micrornas: Implications for latency. Proc. Natl. Acad Sci. USA 2008, 105, 5453–5458. [Google Scholar]
- Hall, I.M.; Shankaranarayana, G.D.; Noma, K.; Ayoub, N.; Cohen, A.; Grewal, S.I. Establishment and maintenance of a heterochromatin domain. Science 2002, 297, 2232–2237. [Google Scholar]
- Jenuwein, T. Molecular biology. An rna-guided pathway for the epigenome. Science 2002, 297, 2215–2218. [Google Scholar] [CrossRef]
- Matzke, M.A.; Birchler, J.A. Rnai-mediated pathways in the nucleus. Nat. Rev. Genet. 2005, 6, 24–35. [Google Scholar] [CrossRef]
- Volpe, T.A.; Kidner, C.; Hall, I.M.; Teng, G.; Grewal, S.I.; Martienssen, R.A. Regulation of heterochromatic silencing and histone h3 lysine-9 methylation by rnai. Science 2002, 297, 1833–1837. [Google Scholar]
- Klase, Z.; Kale, P.; Winograd, R.; Gupta, M.V.; Heydarian, M.; Berro, R.; McCaffrey, T.; Kashanchi, F. Hiv-1 tar element is processed by dicer to yield a viral micro-rna involved in chromatin remodeling of the viral ltr. BMC Mol. Biol. 2007, 8. [Google Scholar] [CrossRef]
- Cantalupo, P.; Doering, A.; Sullivan, C.S.; Pal, A.; Peden, K.W.; Lewis, A.M.; Pipas, J.M. Complete nucleotide sequence of polyomavirus sa12. J. Virol. 2005, 79, 13094–13104. [Google Scholar]
- Hussain, M.; Taft, R.J.; Asgari, S. An insect virus-encoded microrna regulates viral replication. J. Virol. 2008, 82, 9164–9170. [Google Scholar]
- Aparicio, O.; Razquin, N.; Zaratiegui, M.; Narvaiza, I.; Fortes, P. Adenovirus virus-associated rna is processed to functional interfering rnas involved in virus production. J. Virol. 2006, 80, 1376–1384. [Google Scholar] [CrossRef]
- Grundhoff, A.; Sullivan, C.S. Virus-encoded micrornas. Virology 2011, 411, 325–343. [Google Scholar] [CrossRef]
- Zhu, J.Y.; Pfuhl, T.; Motsch, N.; Barth, S.; Nicholls, J.; Grasser, F.; Meister, G. Identification of novel epstein-barr virus microrna genes from nasopharyngeal carcinomas. J. Virol. 2009, 83, 3333–3341. [Google Scholar]
- Grundhoff, A.; Sullivan, C.S.; Ganem, D. A combined computational and microarray-based approach identifies novel micrornas encoded by human gamma-herpesviruses. RNA 2006, 12, 733–750. [Google Scholar]
- Sullivan, C.S.; Sung, C.K.; Pack, C.D.; Grundhoff, A.; Lukacher, A.E.; Benjamin, T.L.; Ganem, D. Murine polyomavirus encodes a microrna that cleaves early rna transcripts but is not essential for experimental infection. Virology 2009, 387, 157–167. [Google Scholar] [CrossRef]
- Singh, J.; Singh, C.P.; Bhavani, A.; Nagaraju, J. Discovering micrornas from bombyx mori nucleopolyhedrosis virus. Virology 2010, 407, 120–128. [Google Scholar] [CrossRef]
- Seo, G.J.; Chen, C.J.; Sullivan, C.S. Merkel cell polyomavirus encodes a microrna with the ability to autoregulate viral gene expression. Virology 2009, 383, 183–187. [Google Scholar] [CrossRef]
- Narayanan, A.; Kehn-Hall, K.; Bailey, C.; Kashanchi, F. Analysis of the roles of hiv-derived micrornas. Expert Opin. Biol. Ther. 2011, 11, 17–29. [Google Scholar] [CrossRef]
- Lin, J.; Cullen, B.R. Analysis of the interaction of primate retroviruses with the human rna interference machinery. J. Virol. 2007, 81, 12218–12226. [Google Scholar] [CrossRef]
- Klase, Z.; Winograd, R.; Davis, J.; Carpio, L.; Hildreth, R.; Heydarian, M.; Fu, S.; McCaffrey, T.; Meiri, E.; Ayash-Rashkovsky, M.; et al. Hiv-1 tar mirna protects against apoptosis by altering cellular gene expression. Retrovirology 2009, 6. [Google Scholar] [CrossRef]
- Kincaid, R.P.; Burke, J.M.; Sullivan, C.S. Rna virus microrna that mimics a b-cell oncomir. Proc. Natl. Acad. Sci. USA 2012, 109, 3077–3082. [Google Scholar]
- Ouellet, D.L.; Plante, I.; Landry, P.; Barat, C.; Janelle, M.E.; Flamand, L.; Tremblay, M.J.; Provost, P. Identification of functional micrornas released through asymmetrical processing of hiv-1 tar element. Nucleic Acids Res. 2008, 36, 2353–2365. [Google Scholar] [CrossRef]
- Perez, J.T.; Varble, A.; Sachidanandam, R.; Zlatev, I.; Manoharan, M.; Garcia-Sastre, A.; tenOever, B.R. Influenza a virus-generated small rnas regulate the switch from transcription to replication. Proc. Natl. Acad. Sci. USA 2010, 107, 11525–11530. [Google Scholar]
- Althaus, C.F.; Vongrad, V.; Niederost, B.; Joos, B.; di Giallonardo, F.; Rieder, P.; Pavlovic, J.; Trkola, A.; Gunthard, H.F.; Metzner, K.J.; et al. Tailored enrichment strategy detects low abundant small noncoding rnas in hiv-1 infected cells. Retrovirology 2012, 9. [Google Scholar] [CrossRef] [Green Version]
- Yeung, M.L.; Bennasser, Y.; Watashi, K.; Le, S.Y.; Houzet, L.; Jeang, K.T. Pyrosequencing of small non-coding rnas in hiv-1 infected cells: Evidence for the processing of a viral-cellular double-stranded rna hybrid. Nucleic Acids Res. 2009, 37, 6575–6586. [Google Scholar]
- Gatignol, A.; Buckler-White, A.; Berkhout, B.; Jeang, K.T. Characterization of a human tar rna-binding protein that activates the hiv-1 ltr. Science 1991, 251, 1597–1600. [Google Scholar]
- Feng, S.; Holland, E.C. Hiv-1 tat trans-activation requires the loop sequence within tar. Nature 1988, 334, 165–167. [Google Scholar]
- Berkhout, B.; Jeang, K.T. Detailed mutational analysis of tar rna: Critical spacing between the bulge and loop recognition domains. Nucleic Acids Res. 1991, 19, 6169–6176. [Google Scholar] [CrossRef]
- Rana, T.M.; Jeang, K.T. Biochemical and functional interactions between hiv-1 tat protein and tar rna. Arch. Biochem. Biophys. 1999, 365, 175–185. [Google Scholar] [CrossRef]
- Bennasser, Y.; Yeung, M.L.; Jeang, K.T. Hiv-1 tar rna subverts rna interference in transfected cells through sequestration of tar rna-binding protein, trbp. J. Biol. Chem. 2006, 281, 27674–27678. [Google Scholar] [CrossRef]
- Cummings, M.; Higginbottom, K.; McGurk, C.J.; Wong, O.G.; Koberle, B.; Oliver, R.T.; Masters, J.R. Xpa versus ercc1 as chemosensitising agents to cisplatin and mitomycin c in prostate cancer cells: Role of ercc1 in homologous recombination repair. Biochem. Pharmacol. 2006, 72, 166–175. [Google Scholar]
- Chang, I.Y.; Kim, M.H.; Kim, H.B.; Lee, D.Y.; Kim, S.H.; Kim, H.Y.; You, H.J. Small interfering rna-induced suppression of ercc1 enhances sensitivity of human cancer cells to cisplatin. Biochem. Biophys. Res. Commun. 2005, 327, 225–233. [Google Scholar]
- Imbeault, M.; Ouellet, M.; Tremblay, M.J. Microarray study reveals that hiv-1 induces rapid type-i interferon-dependent p53 mrna up-regulation in human primary cd4+ t cells. Retrovirology 2009, 6. [Google Scholar] [CrossRef]
- Rotger, M.; Dang, K.K.; Fellay, J.; Heinzen, E.L.; Feng, S.; Descombes, P.; Shianna, K.V.; Ge, D.; Gunthard, H.F.; Goldstein, D.B.; et al. Genome-wide mrna expression correlates of viral control in cd4+ t-cells from hiv-1-infected individuals. PLoS Pathog. 2010, 6, e1000781. [Google Scholar]
- Kaul, D.; Ahlawat, A.; Gupta, S.D. Hiv-1 genome-encoded hiv1-mir-h1 impairs cellular responses to infection. Mol. Cell. Biochem. 2009, 323, 143–148. [Google Scholar] [CrossRef]
- Arhel, N.J.; Kirchhoff, F. Implications of nef: Host cell interactions in viral persistence and progression to aids. Curr. Top. Microbiol. Immunol. 2009, 339, 147–175. [Google Scholar]
- Omoto, S.; Fujii, Y.R. Regulation of human immunodeficiency virus 1 transcription by nef microrna. J. Gen. Virol. 2005, 86, 751–755. [Google Scholar] [CrossRef]
- Workman, J.L.; Kingston, R.E. Alteration of nucleosome structure as a mechanism of transcriptional regulation. Annu. Rev. Biochem. 1998, 67, 545–579. [Google Scholar] [CrossRef]
- Armstrong, J.A.; Emerson, B.M. Transcription of chromatin: These are complex times. Curr. Opin. Genet. Dev. 1998, 8, 165–172. [Google Scholar] [CrossRef]
- Kadonaga, J.T. Eukaryotic transcription: An interlaced network of transcription factors and chromatin-modifying machines. Cell 1998, 92, 307–313. [Google Scholar] [CrossRef]
- Kingston, R.E.; Narlikar, G.J. Atp-dependent remodeling and acetylation as regulators of chromatin fluidity. Genes Dev. 1999, 13, 2339–2352. [Google Scholar] [CrossRef]
- Lyko, F.; Paro, R. Chromosomal elements conferring epigenetic inheritance. Bioessays 1999, 21, 824–832. [Google Scholar] [CrossRef]
- Rohlf, T.; Steiner, L.; Przybilla, J.; Prohaska, S.; Binder, H.; Galle, J. Modeling the dynamic epigenome: From histone modifications towards self-organizing chromatin. Epigenomics 2012, 4, 205–219. [Google Scholar] [CrossRef]
- Weng, N.P.; Araki, Y.; Subedi, K. The molecular basis of the memory t cell response: Differential gene expression and its epigenetic regulation. Nat. Rev. Immunol. 2012, 12, 306–315. [Google Scholar]
- Tripathy, M.K.; Abbas, W.; Herbein, G. Epigenetic regulation of hiv-1 transcription. Epigenomics 2011, 3, 487–502. [Google Scholar] [CrossRef]
- Van Duyne, R.; Guendel, I.; Narayanan, A.; Gregg, E.; Shafagati, N.; Tyagi, M.; Easley, R.; Klase, Z.; Nekhai, S.; Kehn-Hall, K.; et al. Varying modulation of hiv-1 ltr activity by baf complexes. J. Mol. Biol. 2011, 411, 581–596. [Google Scholar] [CrossRef]
- Hakre, S.; Chavez, L.; Shirakawa, K.; Verdin, E. Epigenetic regulation of hiv latency. Curr. Opin. HIV AIDS 2011, 6, 19–24. [Google Scholar] [CrossRef]
- El Kharroubi, A.; Piras, G.; Zensen, R.; Martin, M.A. Transcriptional activation of the integrated chromatin-associated human immunodeficiency virus type 1 promoter. Mol. Cell Biol. 1998, 18, 2535–2544. [Google Scholar]
- Ravindra, K.C.; Narayan, V.; Lushington, G.H.; Peterson, B.R.; Prabhu, K.S. Targeting of histone acetyltransferase p300 by cyclopentenone prostaglandin delta(12)-pgj(2) through covalent binding to cys(1438). Chem. Res. Toxicol. 2012, 25, 337–347. [Google Scholar]
- Sterner, D.E.; Berger, S.L. Acetylation of histones and transcription-related factors. Microbiol. Mol. Biol. Rev. MMBR 2000, 64, 435–459. [Google Scholar] [CrossRef]
- Di Fenza, A.; Rocchia, W.; Tozzini, V. Complexes of hiv-1 integrase with hat proteins: Multiscale models, dynamics, and hypotheses on allosteric sites of inhibition. Proteins 2009, 76, 946–958. [Google Scholar] [CrossRef]
- Easley, R.; van Duyne, R.; Coley, W.; Guendel, I.; Dadgar, S.; Kehn-Hall, K.; Kashanchi, F. Chromatin dynamics associated with hiv-1 tat-activated transcription. Biochim. Biophys. Acta 2010, 1799, 275–285. [Google Scholar]
- Voss, A.K.; Thomas, T. Myst family histone acetyltransferases take center stage in stem cells and development. BioEssays News Rev. Mol. Cell. Dev. Biol. 2009, 31, 1050–1061. [Google Scholar]
- Avvakumov, N.; Cote, J. The myst family of histone acetyltransferases and their intimate links to cancer. Oncogene 2007, 26, 5395–5407. [Google Scholar] [CrossRef]
- Rekowski, M.W.; Giannis, A. Histone acetylation modulation by small molecules: A chemical approach. Biochim. Biophys. Acta 2010, 1799, 760–767. [Google Scholar]
- Ogryzko, V.V.; Schiltz, R.L.; Russanova, V.; Howard, B.H.; Nakatani, Y. The transcriptional coactivators p300 and cbp are histone acetyltransferases. Cell 1996, 87, 953–959. [Google Scholar]
- Deng, L.; de la Fuente, C.; Fu, P.; Wang, L.; Donnelly, R.; Wade, J.D.; Lambert, P.; Li, H.; Lee, C.G.; Kashanchi, F. Acetylation of hiv-1 tat by cbp/p300 increases transcription of integrated hiv-1 genome and enhances binding to core histones. Virology 2000, 277, 278–295. [Google Scholar] [CrossRef]
- Cereseto, A.; Manganaro, L.; Gutierrez, M.I.; Terreni, M.; Fittipaldi, A.; Lusic, M.; Marcello, A.; Giacca, M. Acetylation of hiv-1 integrase by p300 regulates viral integration. EMBO J. 2005, 24, 3070–3081. [Google Scholar]
- Harrod, R.; Nacsa, J.; van Lint, C.; Hansen, J.; Karpova, T.; McNally, J.; Franchini, G. Human immunodeficiency virus type-1 tat/co-activator acetyltransferase interactions inhibit p53lys-320 acetylation and p53-responsive transcription. J. Biol. Chem. 2003, 278, 12310–12318. [Google Scholar]
- Imai, K.; Ochiai, K. Role of histone modification on transcriptional regulation and hiv-1 gene expression: Possible mechanisms of periodontal diseases in aids progression. J. Oral Sci. 2011, 53, 1–13. [Google Scholar] [CrossRef]
- Col, E.; Caron, C.; Seigneurin-Berny, D.; Gracia, J.; Favier, A.; Khochbin, S. The histone acetyltransferase, hgcn5, interacts with and acetylates the hiv transactivator, tat. J. Biol. Chem. 2001, 276, 28179–28184. [Google Scholar]
- Kiernan, R.E.; Vanhulle, C.; Schiltz, L.; Adam, E.; Xiao, H.; Maudoux, F.; Calomme, C.; Burny, A.; Nakatani, Y.; Jeang, K.T.; et al. Hiv-1 tat transcriptional activity is regulated by acetylation. EMBO J. 1999, 18, 6106–6118. [Google Scholar] [CrossRef]
- D’Orso, I.; Frankel, A.D. Tat acetylation modulates assembly of a viral-host rna-protein transcription complex. Proc. Natl. Acad. Sci. USA 2009, 106, 3101–3106. [Google Scholar]
- Weissman, J.D.; Brown, J.A.; Howcroft, T.K.; Hwang, J.; Chawla, A.; Roche, P.A.; Schiltz, L.; Nakatani, Y.; Singer, D.S. Hiv-1 tat binds tafii250 and represses tafii250-dependent transcription of major histocompatibility class i genes. Proc. Natl. Acad. Sci. USA 1998, 95, 11601–11606. [Google Scholar]
- Triboulet, R.; Mari, B.; Lin, Y.L.; Chable-Bessia, C.; Bennasser, Y.; Lebrigand, K.; Cardinaud, B.; Maurin, T.; Barbry, P.; Baillat, V.; et al. Suppression of microrna-silencing pathway by hiv-1 during virus replication. Science 2007, 315, 1579–1582. [Google Scholar] [CrossRef]
- Balasubramanyam, K.; Varier, R.A.; Altaf, M.; Swaminathan, V.; Siddappa, N.B.; Ranga, U.; Kundu, T.K. Curcumin, a novel p300/creb-binding protein-specific inhibitor of acetyltransferase, represses the acetylation of histone/nonhistone proteins and histone acetyltransferase-dependent chromatin transcription. J. Biol. Chem. 2004, 279, 51163–51171. [Google Scholar]
- Dorr, A.; Kiermer, V.; Pedal, A.; Rackwitz, H.R.; Henklein, P.; Schubert, U.; Zhou, M.M.; Verdin, E.; Ott, M. Transcriptional synergy between tat and pcaf is dependent on the binding of acetylated tat to the pcaf bromodomain. EMBO J. 2002, 21, 2715–2723. [Google Scholar]
- Mantelingu, K.; Reddy, B.A.; Swaminathan, V.; Kishore, A.H.; Siddappa, N.B.; Kumar, G.V.; Nagashankar, G.; Natesh, N.; Roy, S.; Sadhale, P.P.; et al. Specific inhibition of p300-hat alters global gene expression and represses hiv replication. Chem. Biol. 2007, 14, 645–657. [Google Scholar] [CrossRef]
- Gregoretti, I.V.; Lee, Y.M.; Goodson, H.V. Molecular evolution of the histone deacetylase family: Functional implications of phylogenetic analysis. J. Mol. Biol. 2004, 338, 17–31. [Google Scholar] [CrossRef]
- De Ruijter, A.J.; van Gennip, A.H.; Caron, H.N.; Kemp, S.; van Kuilenburg, A.B. Histone deacetylases (hdacs): Characterization of the classical hdac family. Biochem. J. 2003, 370, 737–749. [Google Scholar] [CrossRef]
- Keedy, K.S.; Archin, N.M.; Gates, A.T.; Espeseth, A.; Hazuda, D.J.; Margolis, D.M. A limited group of class i histone deacetylases acts to repress human immunodeficiency virus type 1 expression. J. Virol. 2009, 83, 4749–4756. [Google Scholar]
- Coull, J.J.; Romerio, F.; Sun, J.M.; Volker, J.L.; Galvin, K.M.; Davie, J.R.; Shi, Y.; Hansen, U.; Margolis, D.M. The human factors yy1 and lsf repress the human immunodeficiency virus type 1 long terminal repeat via recruitment of histone deacetylase 1. J. Virol. 2000, 74, 6790–6799. [Google Scholar] [CrossRef]
- Williams, S.A.; Chen, L.F.; Kwon, H.; Ruiz-Jarabo, C.M.; Verdin, E.; Greene, W.C. Nf-kappab p50 promotes hiv latency through hdac recruitment and repression of transcriptional initiation. EMBO J. 2006, 25, 139–149. [Google Scholar]
- Kutsch, O.; Benveniste, E.N.; Shaw, G.M.; Levy, D.N. Direct and quantitative single-cell analysis of human immunodeficiency virus type 1 reactivation from latency. J. Virol. 2002, 76, 8776–8786. [Google Scholar]
- Huber, K.; Doyon, G.; Plaks, J.; Fyne, E.; Mellors, J.W.; Sluis-Cremer, N. Inhibitors of histone deacetylases: Correlation between isoform specificity and reactivation of hiv type 1 (hiv-1) from latently infected cells. J. Biol. Chem. 2011, 286, 22211–22218. [Google Scholar]
- Kim, D.H.; Villeneuve, L.M.; Morris, K.V.; Rossi, J.J. Argonaute-1 directs sirna-mediated transcriptional gene silencing in human cells. Nat. Struct. Mol. Biol. 2006, 13, 793–797. [Google Scholar]
- Suzuki, K.; Juelich, T.; Lim, H.; Ishida, T.; Watanebe, T.; Cooper, D.A.; Rao, S.; Kelleher, A.D. Closed chromatin architecture is induced by an rna duplex targeting the hiv-1 promoter region. J. Biol. Chem. 2008, 283, 23353–23363. [Google Scholar]
- Huo, L.; Li, D.; Sun, X.; Shi, X.; Karna, P.; Yang, W.; Liu, M.; Qiao, W.; Aneja, R.; Zhou, J. Regulation of tat acetylation and transactivation activity by the microtubule-associated deacetylase hdac6. J. Biol. Chem. 2011, 286, 9280–9286. [Google Scholar]
- Valenzuela-Fernandez, A.; Alvarez, S.; Gordon-Alonso, M.; Barrero, M.; Ursa, A.; Cabrero, J.R.; Fernandez, G.; Naranjo-Suarez, S.; Yanez-Mo, M.; Serrador, J.M.; et al. Histone deacetylase 6 regulates human immunodeficiency virus type 1 infection. Mol. Biol. Cell 2005, 16, 5445–5454. [Google Scholar]
- Van Duyne, R.; Easley, R.; Wu, W.; Berro, R.; Pedati, C.; Klase, Z.; Kehn-Hall, K.; Flynn, E.K.; Symer, D.E.; Kashanchi, F. Lysine methylation of hiv-1 tat regulates transcriptional activity of the viral ltr. Retrovirology 2008, 5. [Google Scholar] [CrossRef]
- Dillon, S.C.; Zhang, X.; Trievel, R.C.; Cheng, X. The set-domain protein superfamily: Protein lysine methyltransferases. Genome Biol. 2005, 6. [Google Scholar] [CrossRef]
- Luo, M. Current chemical biology approaches to interrogate protein methyltransferases. ACS Chem. Biol. 2012, 7, 443–463. [Google Scholar] [CrossRef]
- Zhang, L.; Eugeni, E.E.; Parthun, M.R.; Freitas, M.A. Identification of novel histone post-translational modifications by peptide mass fingerprinting. Chromosoma 2003, 112, 77–86. [Google Scholar]
- Feng, Q.; Wang, H.; Ng, H.H.; Erdjument-Bromage, H.; Tempst, P.; Struhl, K.; Zhang, Y. Methylation of h3-lysine 79 is mediated by a new family of hmtases without a set domain. Curr. Biol. 2002, 12, 1052–1058. [Google Scholar]
- Ng, H.H.; Feng, Q.; Wang, H.; Erdjument-Bromage, H.; Tempst, P.; Zhang, Y.; Struhl, K. Lysine methylation within the globular domain of histone h3 by dot1 is important for telomeric silencing and sir protein association. Genes Dev. 2002, 16, 1518–1527. [Google Scholar] [CrossRef]
- Lachner, M.; O'Carroll, D.; Rea, S.; Mechtler, K.; Jenuwein, T. Methylation of histone h3 lysine 9 creates a binding site for hp1 proteins. Nature 2001, 410, 116–120. [Google Scholar] [CrossRef]
- Schneider, R.; Bannister, A.J.; Myers, F.A.; Thorne, A.W.; Crane-Robinson, C.; Kouzarides, T. Histone h3 lysine 4 methylation patterns in higher eukaryotic genes. Nat. Cell Biol. 2004, 6, 73–77. [Google Scholar] [CrossRef]
- Ng, H.H.; Robert, F.; Young, R.A.; Struhl, K. Targeted recruitment of set1 histone methylase by elongating pol ii provides a localized mark and memory of recent transcriptional activity. Mol. Cell 2003, 11, 709–719. [Google Scholar]
- Rao, B.; Shibata, Y.; Strahl, B.D.; Lieb, J.D. Dimethylation of histone h3 at lysine 36 demarcates regulatory and nonregulatory chromatin genome-wide. Mol. Cell Biol. 2005, 25, 9447–9459. [Google Scholar] [CrossRef]
- Schotta, G.; Lachner, M.; Sarma, K.; Ebert, A.; Sengupta, R.; Reuter, G.; Reinberg, D.; Jenuwein, T. A silencing pathway to induce h3-k9 and h4-k20 trimethylation at constitutive heterochromatin. Genes Dev. 2004, 18, 1251–1262. [Google Scholar] [CrossRef]
- Le Douce, V.; Colin, L.; Redel, L.; Cherrier, T.; Herbein, G.; Aunis, D.; Rohr, O.; van Lint, C.; Schwartz, C. Lsd1 cooperates with ctip2 to promote hiv-1 transcriptional silencing. Nucleic Acids Res. 2012, 40, 1904–1915. [Google Scholar] [CrossRef]
- Li, F.; Huarte, M.; Zaratiegui, M.; Vaughn, M.W.; Shi, Y.; Martienssen, R.; Cande, W.Z. Lid2 is required for coordinating h3k4 and h3k9 methylation of heterochromatin and euchromatin. Cell 2008, 135, 272–283. [Google Scholar] [CrossRef]
- Wysocka, J.; Allis, C.D.; Coonrod, S. Histone arginine methylation and its dynamic regulation. Frontiers Biosci. A J. Virtual Libr. 2006, 11, 344–355. [Google Scholar] [CrossRef]
- Bachand, F. Protein arginine methyltransferases: From unicellular eukaryotes to humans. Eukaryot. Cell 2007, 6, 889–898. [Google Scholar] [CrossRef]
- Cuthbert, G.L.; Daujat, S.; Snowden, A.W.; Erdjument-Bromage, H.; Hagiwara, T.; Yamada, M.; Schneider, R.; Gregory, P.D.; Tempst, P.; Bannister, A.J.; et al. Histone deimination antagonizes arginine methylation. Cell 2004, 118, 545–553. [Google Scholar] [CrossRef]
- Willemsen, N.M.; Hitchen, E.M.; Bodetti, T.J.; Apolloni, A.; Warrilow, D.; Piller, S.C.; Harrich, D. Protein methylation is required to maintain optimal hiv-1 infectivity. Retrovirology 2006, 3. [Google Scholar] [CrossRef]
- Kwak, Y.T.; Guo, J.; Prajapati, S.; Park, K.J.; Surabhi, R.M.; Miller, B.; Gehrig, P.; Gaynor, R.B. Methylation of spt5 regulates its interaction with rna polymerase ii and transcriptional elongation properties. Mol. Cell 2003, 11, 1055–1066. [Google Scholar] [CrossRef]
- Xie, B.; Invernizzi, C.F.; Richard, S.; Wainberg, M.A. Arginine methylation of the human immunodeficiency virus type 1 tat protein by prmt6 negatively affects tat interactions with both cyclin t1 and the tat transactivation region. J. Virol. 2007, 81, 4226–4234. [Google Scholar]
- Boulanger, M.C.; Liang, C.; Russell, R.S.; Lin, R.; Bedford, M.T.; Wainberg, M.A.; Richard, S. Methylation of tat by prmt6 regulates human immunodeficiency virus type 1 gene expression. J. Virol. 2005, 79, 124–131. [Google Scholar]
- Friedman, J.; Cho, W.K.; Chu, C.K.; Keedy, K.S.; Archin, N.M.; Margolis, D.M.; Karn, J. Epigenetic silencing of hiv-1 by the histone h3 lysine 27 methyltransferase enhancer of zeste 2. J. Virol. 2011, 85, 9078–9089. [Google Scholar] [CrossRef]
- Weinberg, M.S.; Villeneuve, L.M.; Ehsani, A.; Amarzguioui, M.; Aagaard, L.; Chen, Z.X.; Riggs, A.D.; Rossi, J.J.; Morris, K.V. The antisense strand of small interfering rnas directs histone methylation and transcriptional gene silencing in human cells. RNA 2006, 12, 256–262. [Google Scholar]
- Meissner, A.; Mikkelsen, T.S.; Gu, H.; Wernig, M.; Hanna, J.; Sivachenko, A.; Zhang, X.; Bernstein, B.E.; Nusbaum, C.; Jaffe, D.B.; et al. Genome-scale DNA methylation maps of pluripotent and differentiated cells. Nature 2008, 454, 766–770. [Google Scholar]
- Fan, S.; Zhang, M.Q.; Zhang, X. Histone methylation marks play important roles in predicting the methylation status of cpg islands. Biochem. Biophys. Res. Commun. 2008, 374, 559–564. [Google Scholar]
- Cedar, H.; Bergman, Y. Linking DNA methylation and histone modification: Patterns and paradigms. Nat. Rev. Genet. 2009, 10, 295–304. [Google Scholar] [CrossRef]
- Ooi, S.K.; Qiu, C.; Bernstein, E.; Li, K.; Jia, D.; Yang, Z.; Erdjument-Bromage, H.; Tempst, P.; Lin, S.P.; Allis, C.D.; et al. Dnmt3l connects unmethylated lysine 4 of histone h3 to de novo methylation of DNA. Nature 2007, 448, 714–717. [Google Scholar]
- Jia, D.; Jurkowska, R.Z.; Zhang, X.; Jeltsch, A.; Cheng, X. Structure of dnmt3a bound to dnmt3l suggests a model for de novo DNA methylation. Nature 2007, 449, 248–251. [Google Scholar]
- Ishida, T.; Hamano, A.; Koiwa, T.; Watanabe, T. 5' long terminal repeat (ltr)-selective methylation of latently infected hiv-1 provirus that is demethylated by reactivation signals. Retrovirology 2006, 3. [Google Scholar] [CrossRef]
- Blazkova, J.; Murray, D.; Justement, J.S.; Funk, E.K.; Nelson, A.K.; Moir, S.; Chun, T.W.; Fauci, A.S. Paucity of hiv DNA methylation in latently infected, resting cd4+ t cells from infected individuals receiving antiretroviral therapy. J. Virol. 2012, 86, 5390–5392. [Google Scholar]
- Ting, A.H.; Schuebel, K.E.; Herman, J.G.; Baylin, S.B. Short double-stranded rna induces transcriptional gene silencing in human cancer cells in the absence of DNA methylation. Nat. Genet. 2005, 37, 906–910. [Google Scholar]
- Ansari, K.I.; Mishra, B.P.; Mandal, S.S. Mll histone methylases in gene expression, hormone signaling and cell cycle. Front. Biosci. 2009, 14, 3483–3495. [Google Scholar]
- Tsukiyama, T.; Wu, C. Chromatin remodeling and transcription. Curr. Opin. Genet. Dev. 1997, 7, 182–191. [Google Scholar] [CrossRef]
- Reyes, J.C.; Barra, J.; Muchardt, C.; Camus, A.; Babinet, C.; Yaniv, M. Altered control of cellular proliferation in the absence of mammalian brahma (snf2alpha). EMBO J. 1998, 17, 6979–6991. [Google Scholar]
- Chiba, H.; Muramatsu, M.; Nomoto, A.; Kato, H. Two human homologues of saccharomyces cerevisiae swi2/snf2 and drosophila brahma are transcriptional coactivators cooperating with the estrogen receptor and the retinoic acid receptor. Nucleic Acids Res. 1994, 22, 1815–1820. [Google Scholar]
- De la Serna, I.L.; Ohkawa, Y.; Imbalzano, A.N. Chromatin remodelling in mammalian differentiation: Lessons from atp-dependent remodellers. Nat. Rev. Genet. 2006, 7, 461–473. [Google Scholar]
- Wang, W.; Xue, Y.; Zhou, S.; Kuo, A.; Cairns, B.R.; Crabtree, G.R. Diversity and specialization of mammalian swi/snf complexes. Genes Dev. 1996, 10, 2117–2130. [Google Scholar] [CrossRef]
- Sudarsanam, P.; Winston, F. The swi/snf family nucleosome-remodeling complexes and transcriptional control. Trends Genet. 2000, 16, 345–351. [Google Scholar] [CrossRef]
- Narlikar, G.J.; Fan, H.Y.; Kingston, R.E. Cooperation between complexes that regulate chromatin structure and transcription. Cell 2002, 108, 475–487. [Google Scholar]
- Sims, R.J., 3rd; Mandal, S.S.; Reinberg, D. Recent highlights of rna-polymerase-ii-mediated transcription. Curr. Opin. Cell Biol. 2004, 16, 263–271. [Google Scholar]
- Fan, H.Y.; Narlikar, G.J.; Kingston, R.E. Noncovalent modification of chromatin: Different remodeled products with different atpase domains. Cold Spring Harb. Symp. Quant. Biol. 2004, 69, 183–192. [Google Scholar] [CrossRef]
- Liu, N.; Balliano, A.; Hayes, J.J. Mechanism(s) of swi/snf-induced nucleosome mobilization. Chembiochem 2011, 12, 196–204. [Google Scholar] [CrossRef]
- Castanotto, D.; Tommasi, S.; Li, M.; Li, H.; Yanow, S.; Pfeifer, G.P.; Rossi, J.J. Short hairpin rna-directed cytosine (cpg) methylation of the rassf1a gene promoter in hela cells. Mol. Ther. 2005, 12, 179–183. [Google Scholar] [CrossRef]
- Ahlenstiel, C.L.; Lim, H.G.; Cooper, D.A.; Ishida, T.; Kelleher, A.D.; Suzuki, K. Direct evidence of nuclear argonaute distribution during transcriptional silencing links the actin cytoskeleton to nuclear rnai machinery in human cells. Nucleic Acids Res. 2012, 40, 1579–1595. [Google Scholar]
- Janowski, B.A.; Huffman, K.E.; Schwartz, J.C.; Ram, R.; Nordsell, R.; Shames, D.S.; Minna, J.D.; Corey, D.R. Involvement of ago1 and ago2 in mammalian transcriptional silencing. Nat. Struct. Mol. Biol. 2006, 13, 787–792. [Google Scholar] [CrossRef]
- Vire, E.; Brenner, C.; Deplus, R.; Blanchon, L.; Fraga, M.; Didelot, C.; Morey, L.; van Eynde, A.; Bernard, D.; Vanderwinden, J.M.; et al. The polycomb group protein ezh2 directly controls DNA methylation. Nature 2006, 439, 871–874. [Google Scholar]
- Han, J.; Kim, D.; Morris, K.V. Promoter-associated rna is required for rna-directed transcriptional gene silencing in human cells. Proc. Natl. Acad. Sci. USA 2007, 104, 12422–12427. [Google Scholar]
- Janowski, B.A.; Huffman, K.E.; Schwartz, J.C.; Ram, R.; Hardy, D.; Shames, D.S.; Minna, J.D.; Corey, D.R. Inhibiting gene expression at transcription start sites in chromosomal DNA with antigene rnas. Nat. Chem. Biol. 2005, 1, 216–222. [Google Scholar]
- Hawkins, P.G.; Santoso, S.; Adams, C.; Anest, V.; Morris, K.V. Promoter targeted small rnas induce long-term transcriptional gene silencing in human cells. Nucleic Acids Res. 2009, 37, 2984–2995. [Google Scholar]
- Clerc, I.; Laverdure, S.; Torresilla, C.; Landry, S.; Borel, S.; Vargas, A.; Arpin-Andre, C.; Gay, B.; Briant, L.; Gross, A.; et al. Polarized expression of the membrane asp protein derived from hiv-1 antisense transcription in t cells. Retrovirology 2011, 8. [Google Scholar] [CrossRef]
- Lefebvre, G.; Desfarges, S.; Uyttebroeck, F.; Munoz, M.; Beerenwinkel, N.; Rougemont, J.; Telenti, A.; Ciuffi, A. Analysis of hiv-1 expression level and sense of transcription by high-throughput sequencing of the infected cell. J. Virol. 2011, 85, 6205–6211. [Google Scholar] [CrossRef]
- Bansal, A.; Carlson, J.; Yan, J.; Akinsiku, O.T.; Schaefer, M.; Sabbaj, S.; Bet, A.; Levy, D.N.; Heath, S.; Tang, J.; et al. Cd8 t cell response and evolutionary pressure to hiv-1 cryptic epitopes derived from antisense transcription. J. Exp. Med. 2010, 207, 51–59. [Google Scholar]
- Bentley, K.; Deacon, N.; Sonza, S.; Zeichner, S.; Churchill, M. Mutational analysis of the hiv-1 ltr as a promoter of negative sense transcription. Arch. Virol. 2004, 149, 2277–2294. [Google Scholar]
- Briquet, S.; Vaquero, C. Immunolocalization studies of an antisense protein in hiv-1-infected cells and viral particles. Virology 2002, 292, 177–184. [Google Scholar] [CrossRef]
- Tagieva, N.E.; Vaquero, C. Expression of naturally occurring antisense rna inhibits human immunodeficiency virus type 1 heterologous strain replication. J. Gen. Virol. 1997, 78(Pt. 10), 2503–2511. [Google Scholar]
- Peeters, A.; Lambert, P.F.; Deacon, N.J. A fourth sp1 site in the human immunodeficiency virus type 1 long terminal repeat is essential for negative-sense transcription. J. Virol. 1996, 70, 6665–6672. [Google Scholar]
- Landry, S.; Halin, M.; Lefort, S.; Audet, B.; Vaquero, C.; Mesnard, J.M.; Barbeau, B. Detection, characterization and regulation of antisense transcripts in hiv-1. Retrovirology 2007, 4. [Google Scholar] [CrossRef]
- Ludwig, L.B.; Ambrus, J.L., Jr.; Krawczyk, K.A.; Sharma, S.; Brooks, S.; Hsiao, C.B.; Schwartz, S.A. Human immunodeficiency virus-type 1 ltr DNA contains an intrinsic gene producing antisense rna and protein products. Retrovirology 2006, 3. [Google Scholar] [CrossRef]
- Vanhee-Brossollet, C.; Thoreau, H.; Serpente, N.; D’Auriol, L.; Levy, J.P.; Vaquero, C. A natural antisense rna derived from the hiv-1 env gene encodes a protein which is recognized by circulating antibodies of hiv+ individuals. Virology 1995, 206, 196–202. [Google Scholar]
- Michael, N.L.; Vahey, M.T.; d’Arcy, L.; Ehrenberg, P.K.; Mosca, J.D.; Rappaport, J.; Redfield, R.R. Negative-strand rna transcripts are produced in human immunodeficiency virus type 1-infected cells and patients by a novel promoter downregulated by tat. J. Virol. 1994, 68, 979–987. [Google Scholar]
- Miller, R.H. Human immunodeficiency virus may encode a novel protein on the genomic DNA plus strand. Science 1988, 239, 1420–1422. [Google Scholar]
- Kobayashi-Ishihara, M.; Yamagishi, M.; Hara, T.; Matsuda, Y.; Takahashi, R.; Miyake, A.; Nakano, K.; Yamochi, T.; Ishida, T.; Watanabe, T. Hiv-1-encoded antisense rna suppresses viral replication for a prolonged period. Retrovirology 2012, 9. [Google Scholar] [CrossRef]
- Suzuki, K.; Shijuuku, T.; Fukamachi, T.; Zaunders, J.; Guillemin, G.; Cooper, D.; Kelleher, A. Prolonged transcriptional silencing and cpg methylation induced by sirnas targeted to the hiv-1 promoter region. J. RNAi Gene Silenc. 2005, 1, 66–78. [Google Scholar]
- Suzuki, K.; Ishida, T.; Yamagishi, M.; Ahlenstiel, C.; Swaminathan, S.; Marks, K.; Murray, D.; McCartney, E.M.; Beard, M.R.; Alexander, M.; et al. Transcriptional gene silencing of hiv-1 through promoter targeted rna is highly specific. RNA Biol. 2011, 8, 1035–1046. [Google Scholar] [CrossRef]
- Carpio, L.; Klase, Z.; Coley, W.; Guendel, I.; Choi, S.; Van Duyne, R.; Narayanan, A.; Kehn-Hall, K.; Meijer, L.; Kashanchi, F. Microrna machinery is an integral component of drug-induced transcription inhibition in hiv-1 infection. J RNAi Gene Silenc. 2010, 6, 386–400. [Google Scholar]
- Coley, W.; Van Duyne, R.; Carpio, L.; Guendel, I.; Kehn-Hall, K.; Chevalier, S.; Narayanan, A.; Luu, T.; Lee, N.; Klase, Z.; et al. Absence of dicer in monocytes and its regulation by hiv-1. J. Biol. Chem. 2010, 285, 31930–31943. [Google Scholar]
- Morris, K.V. Therapeutic potential of sirna-mediated transcriptional gene silencing. Biotechniques 2006, 40, S7–S13. [Google Scholar]
- Turner, A.M.; Ackley, A.M.; Matrone, M.A.; Morris, K.V. Characterization of an hiv-targeted transcriptional gene-silencing rna in primary cells. Hum. Gene Ther. 2012, 23, 473–483. [Google Scholar]
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Sampey, G.C.; Guendel, I.; Das, R.; Jaworski, E.; Klase, Z.; Narayanan, A.; Kehn-Hall, K.; Kashanchi, F. Transcriptional Gene Silencing (TGS) via the RNAi Machinery in HIV-1 Infections. Biology 2012, 1, 339-369. https://doi.org/10.3390/biology1020339
Sampey GC, Guendel I, Das R, Jaworski E, Klase Z, Narayanan A, Kehn-Hall K, Kashanchi F. Transcriptional Gene Silencing (TGS) via the RNAi Machinery in HIV-1 Infections. Biology. 2012; 1(2):339-369. https://doi.org/10.3390/biology1020339
Chicago/Turabian StyleSampey, Gavin C., Irene Guendel, Ravi Das, Elizabeth Jaworski, Zachary Klase, Aarthi Narayanan, Kylene Kehn-Hall, and Fatah Kashanchi. 2012. "Transcriptional Gene Silencing (TGS) via the RNAi Machinery in HIV-1 Infections" Biology 1, no. 2: 339-369. https://doi.org/10.3390/biology1020339