A Comparison of Two Single-Stranded DNA Binding Models by Mutational Analysis of APOBEC3G

Abstract
:1. Introduction
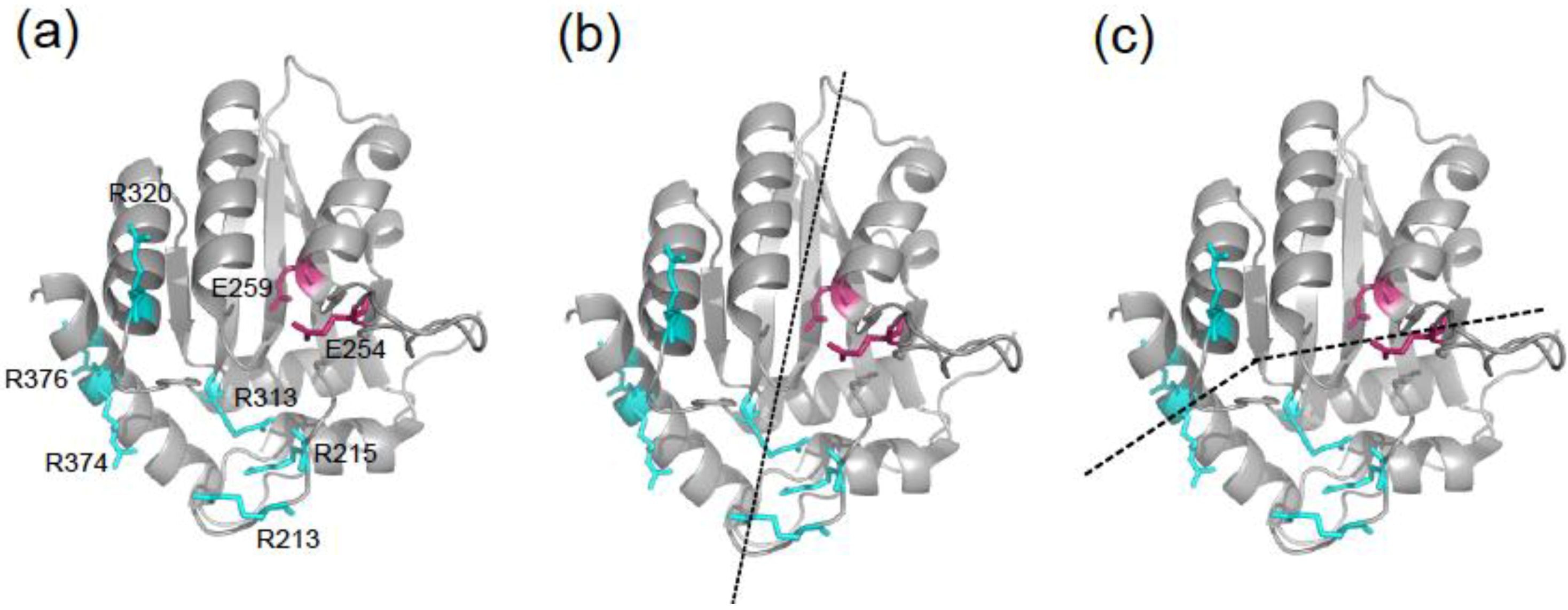
2. Results and Discussion
2.1. DNA Deaminase Activity Experiments
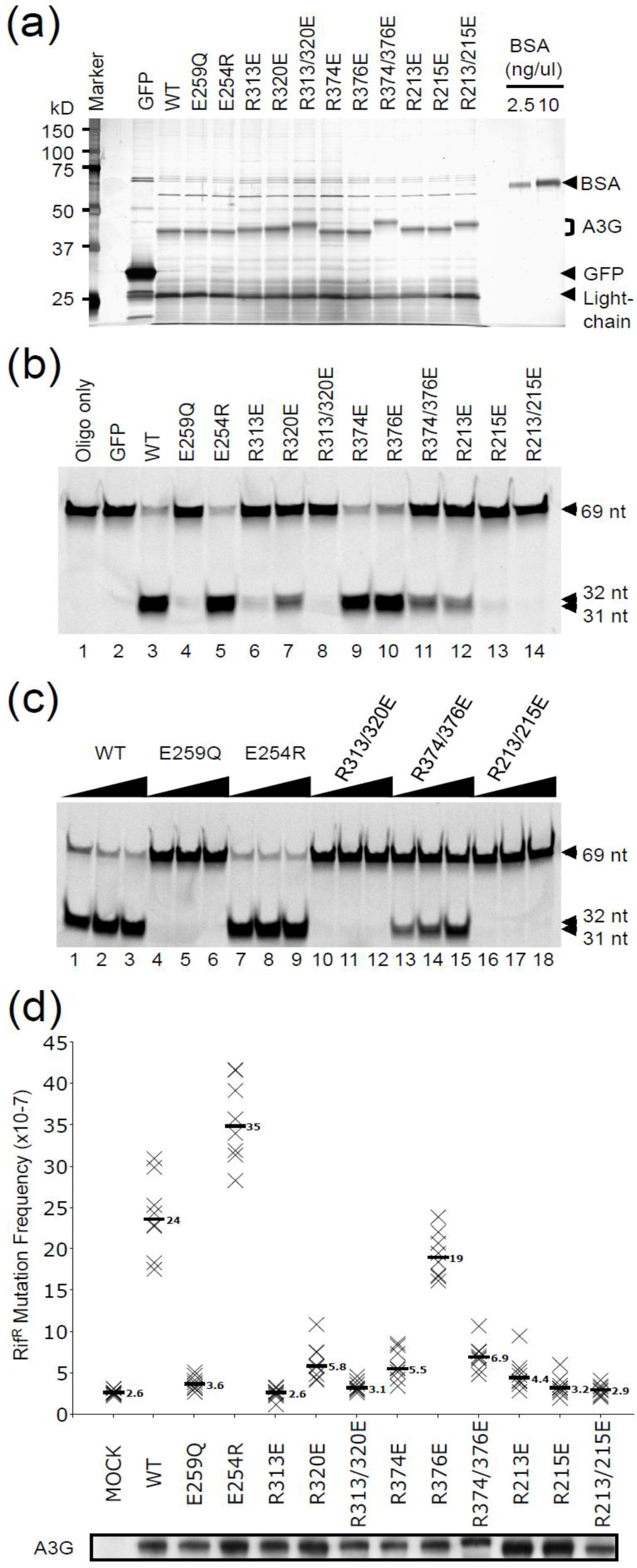
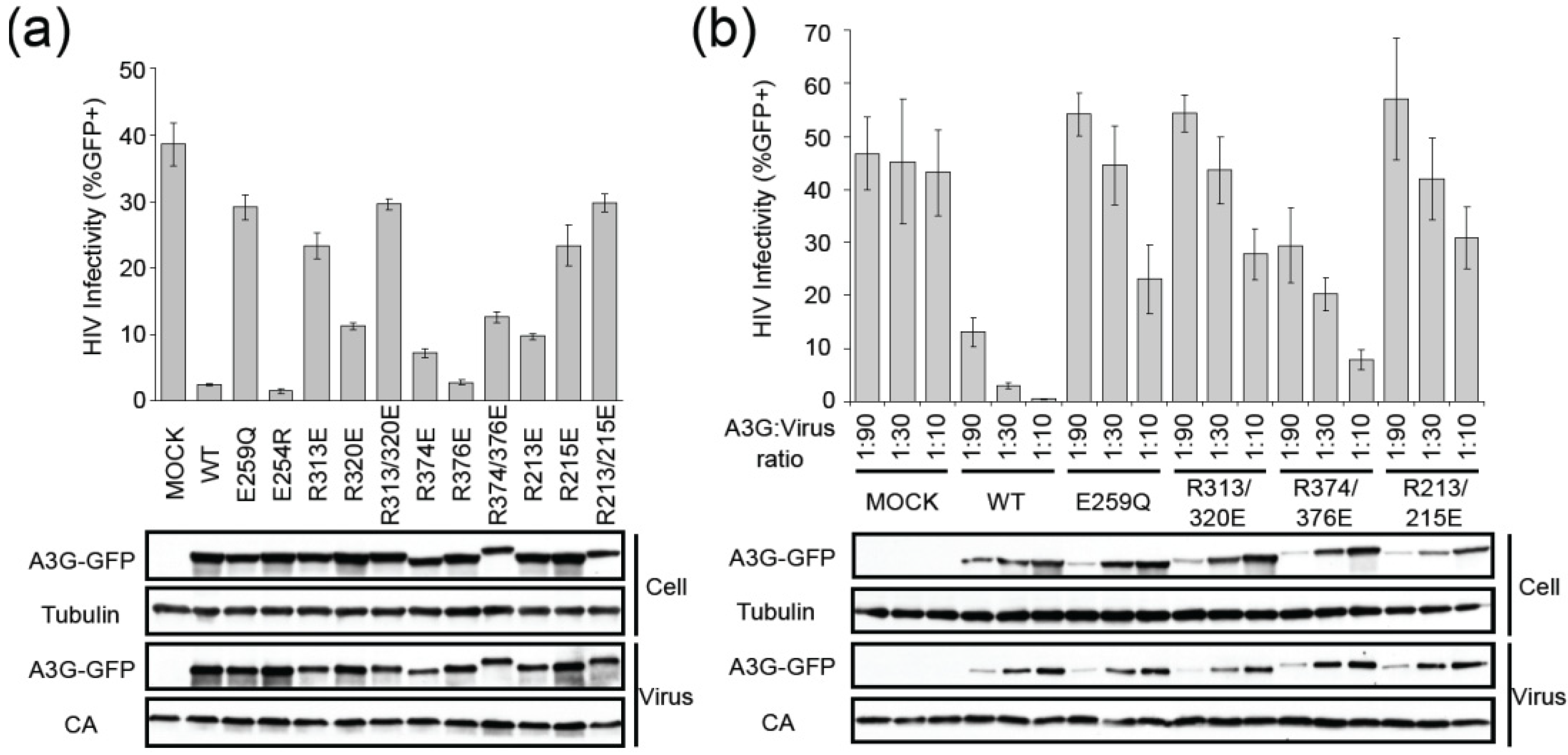
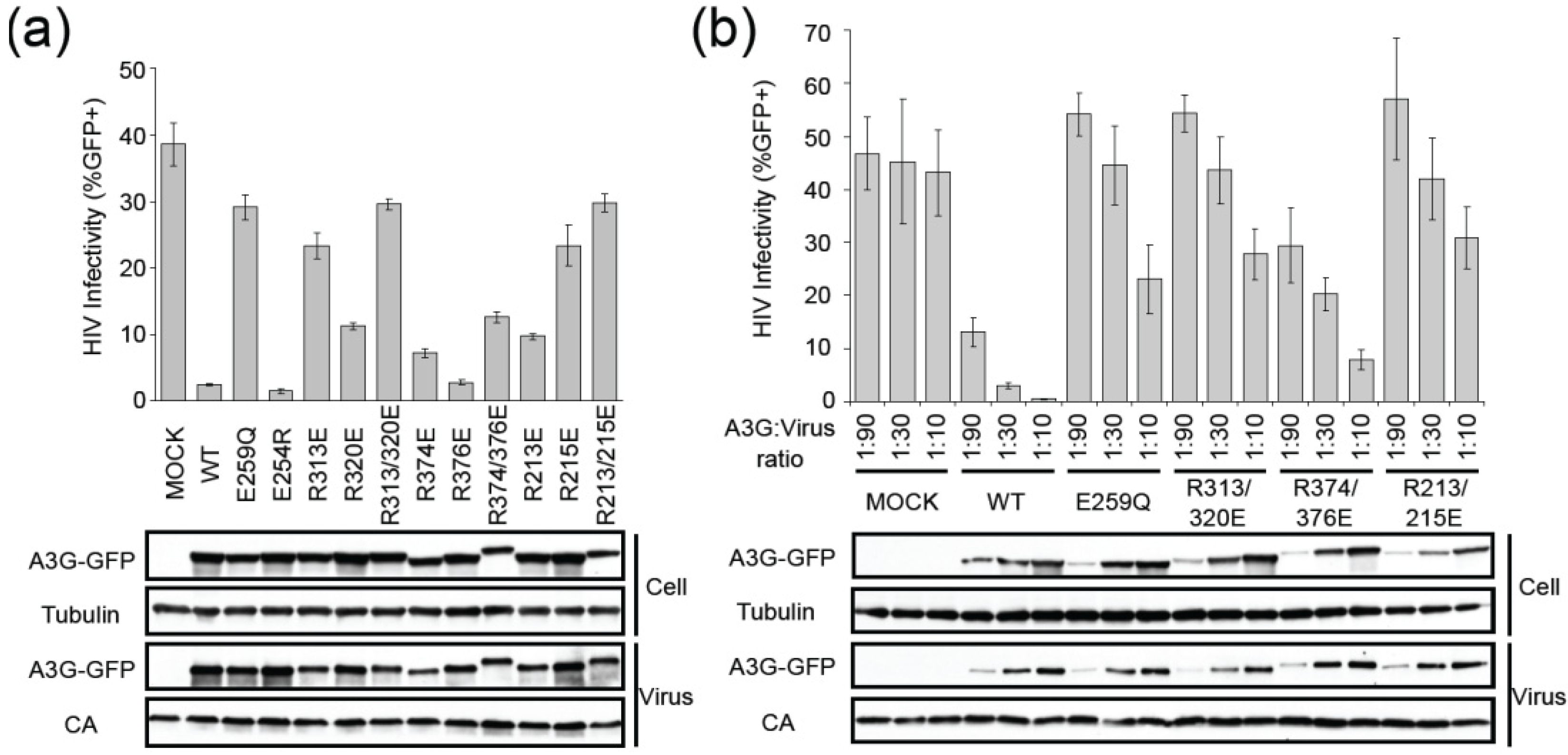
2.2. HIV Restriction Experiments
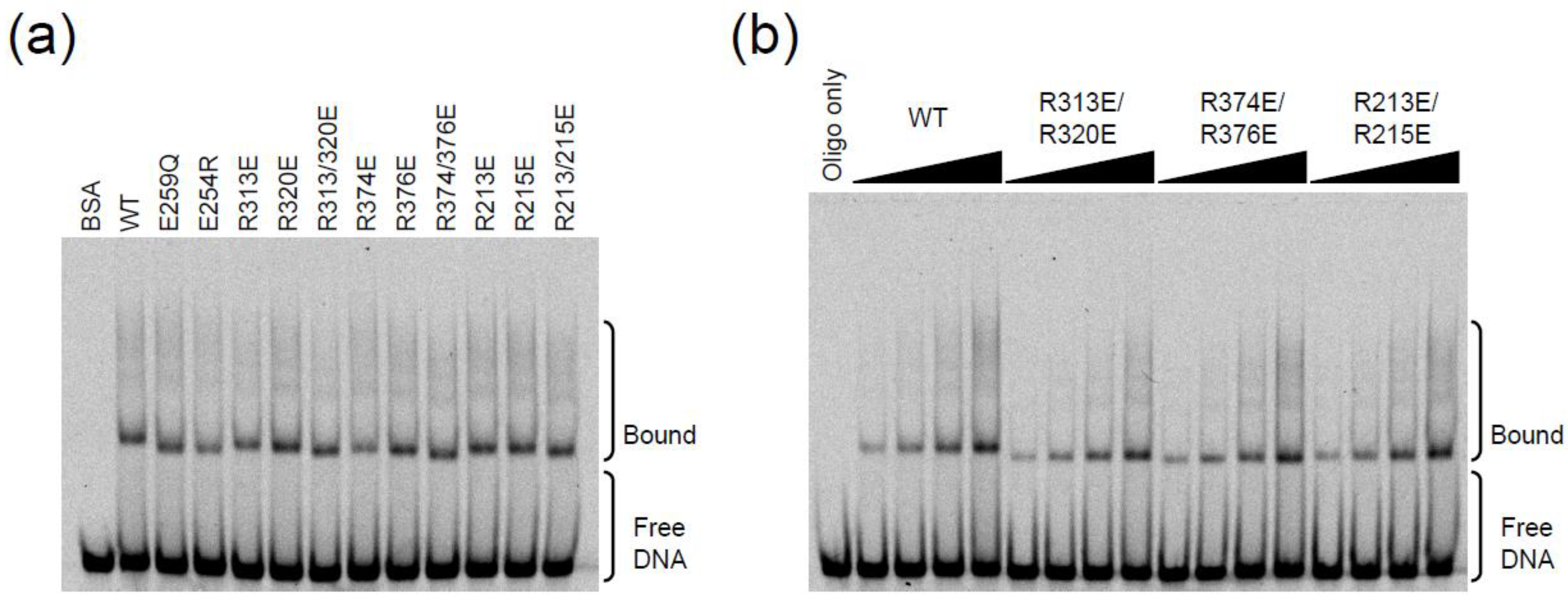
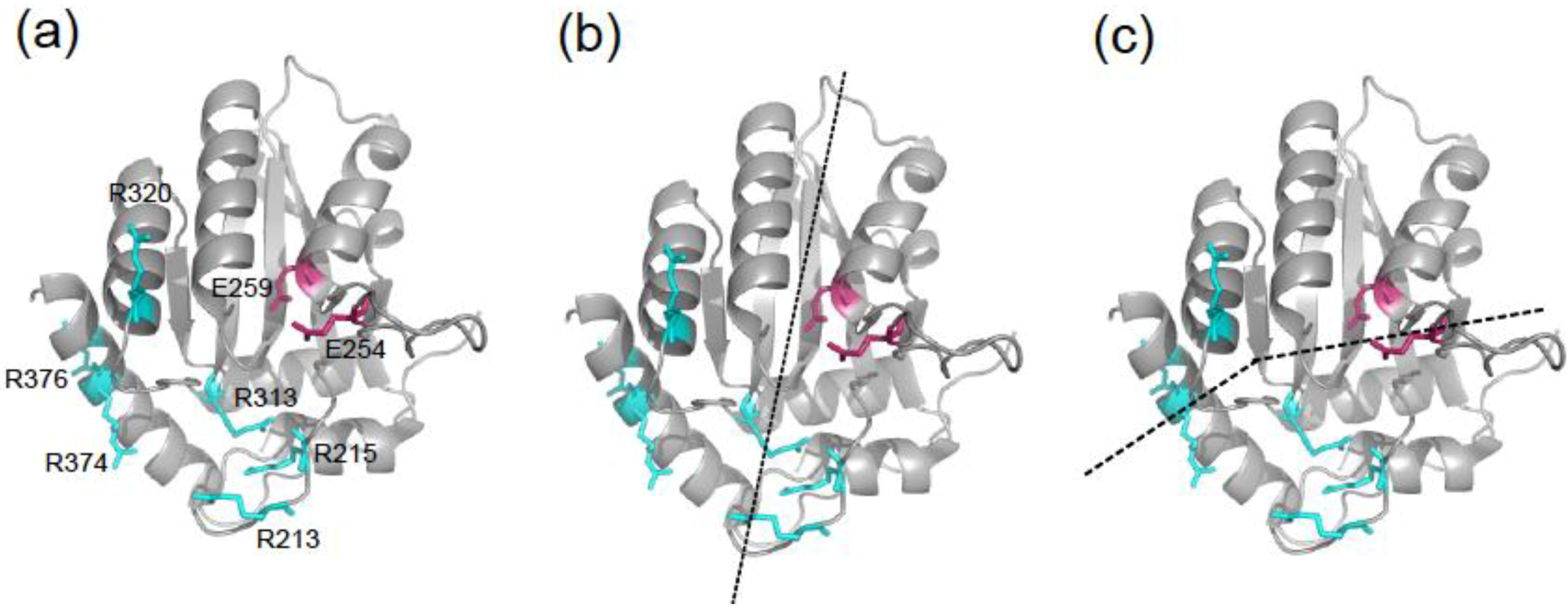
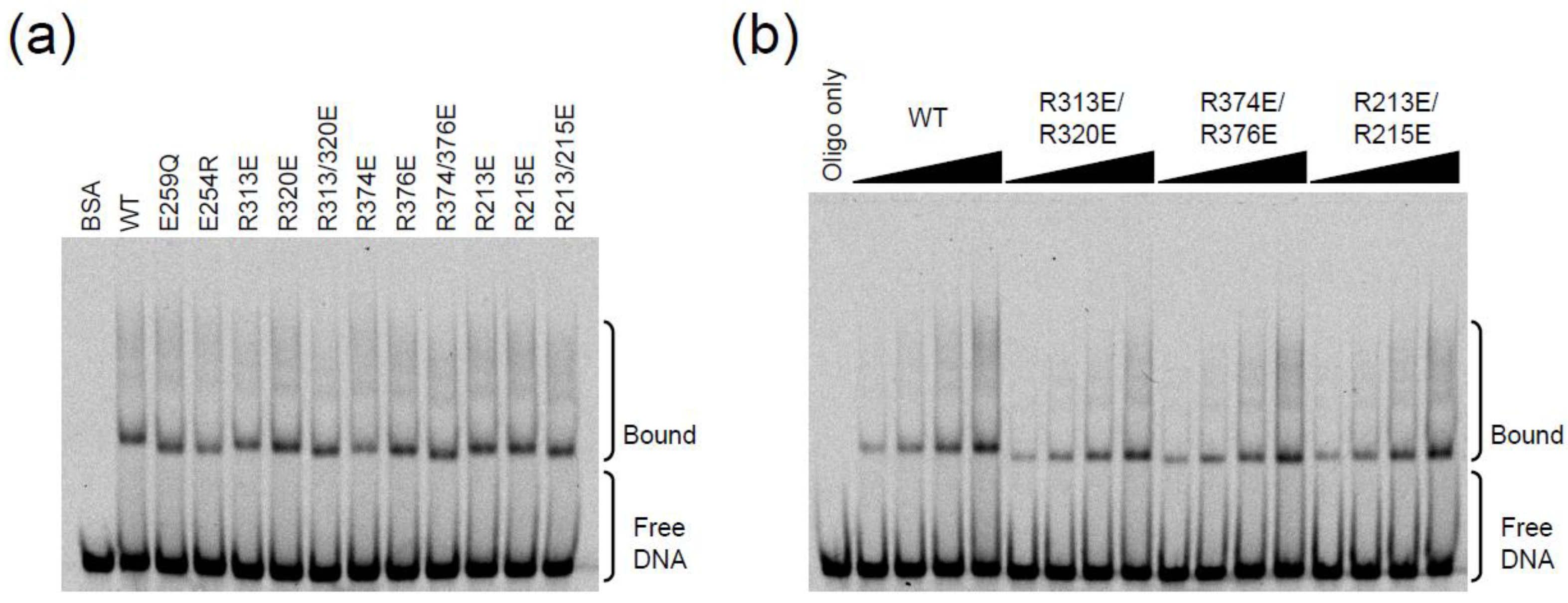
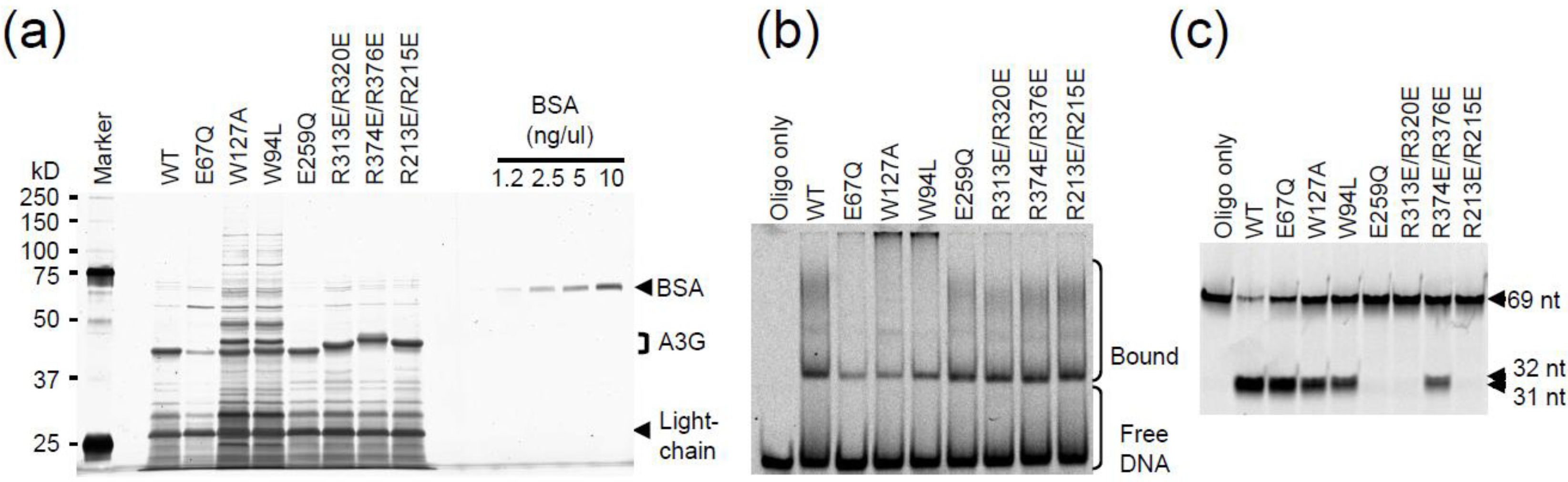
2.3. DNA Binding Experiments
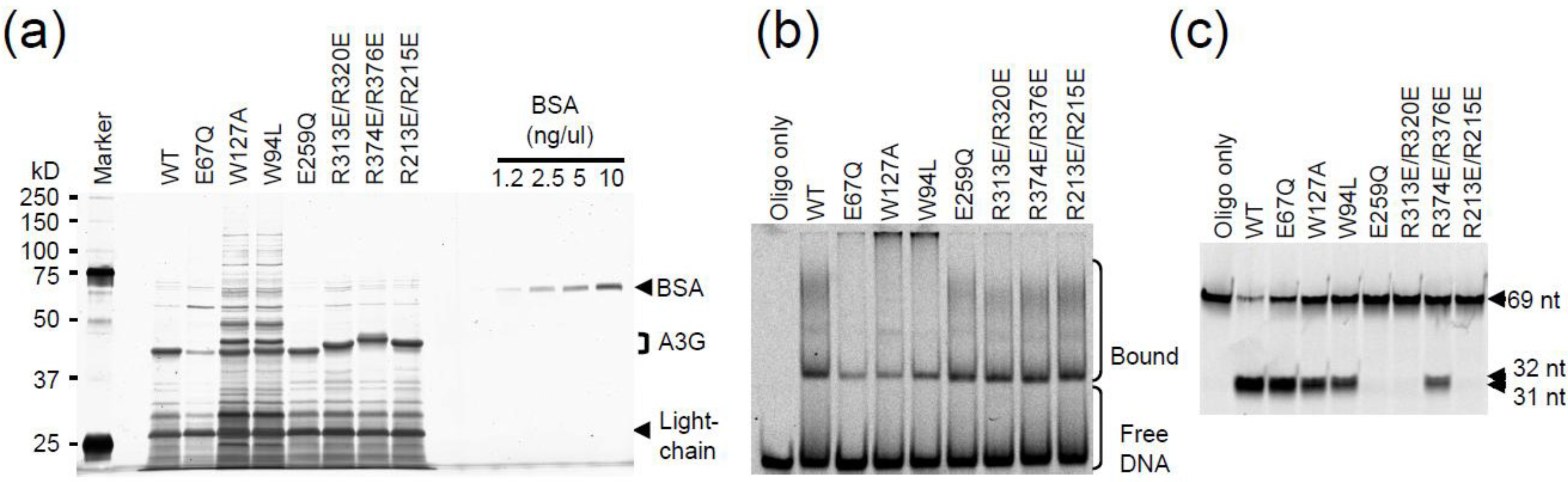
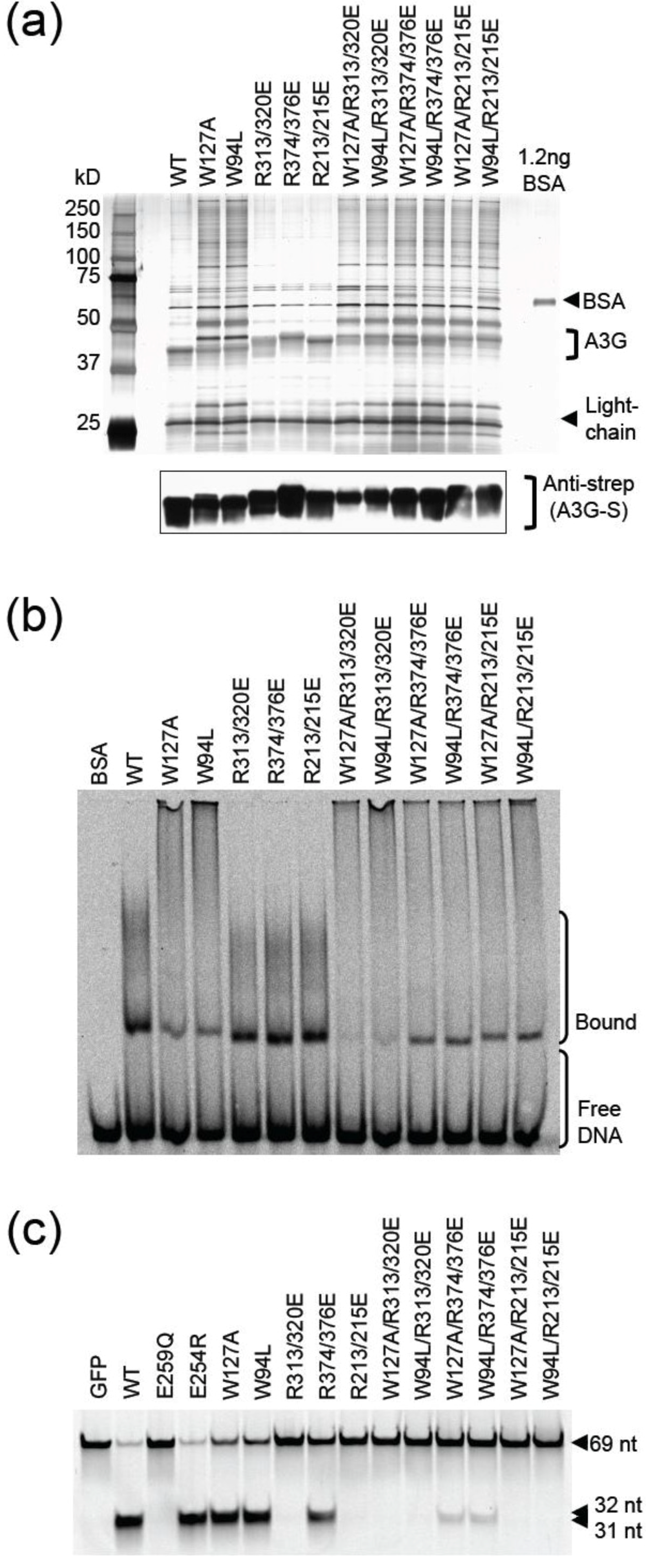
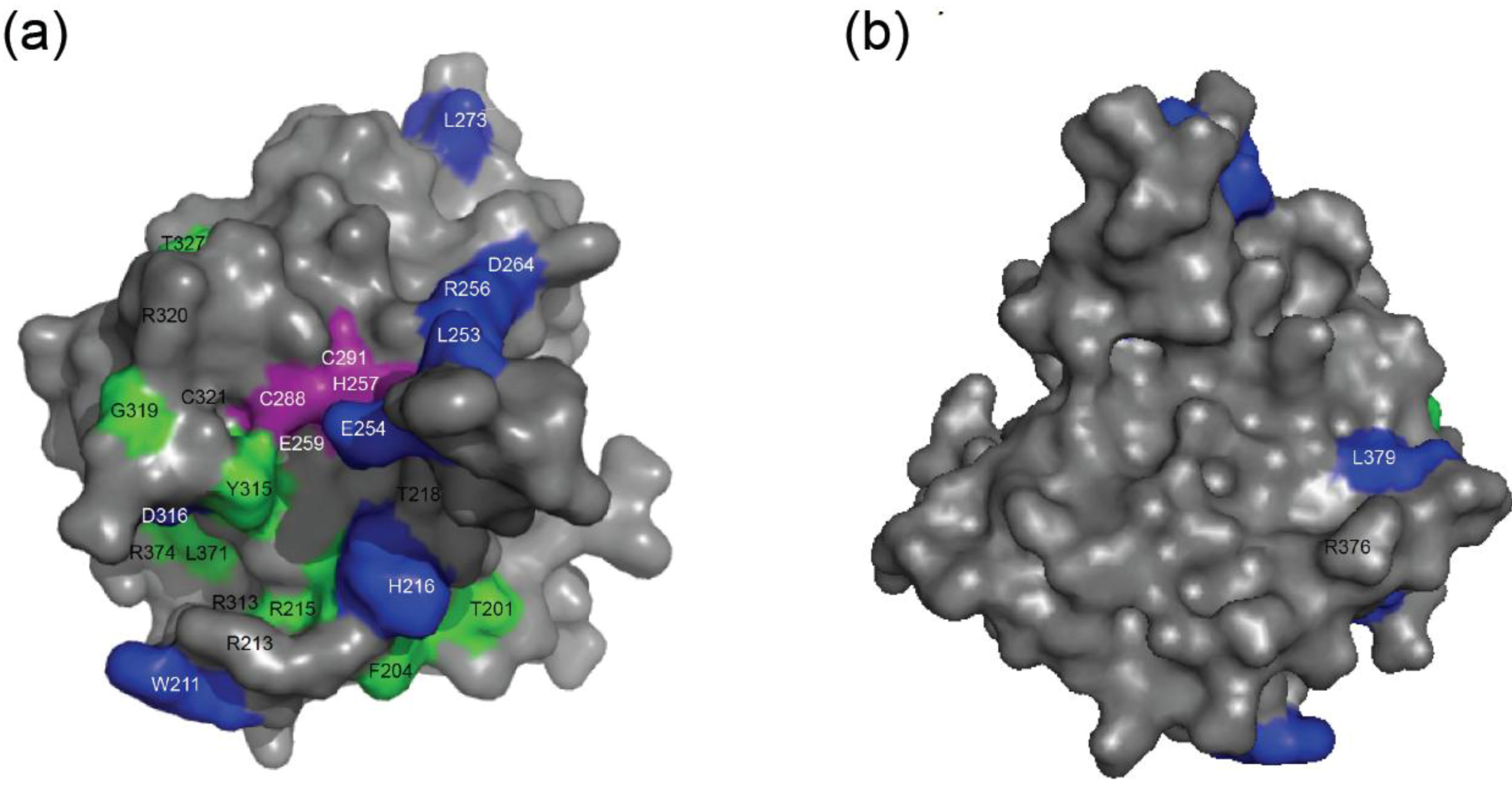
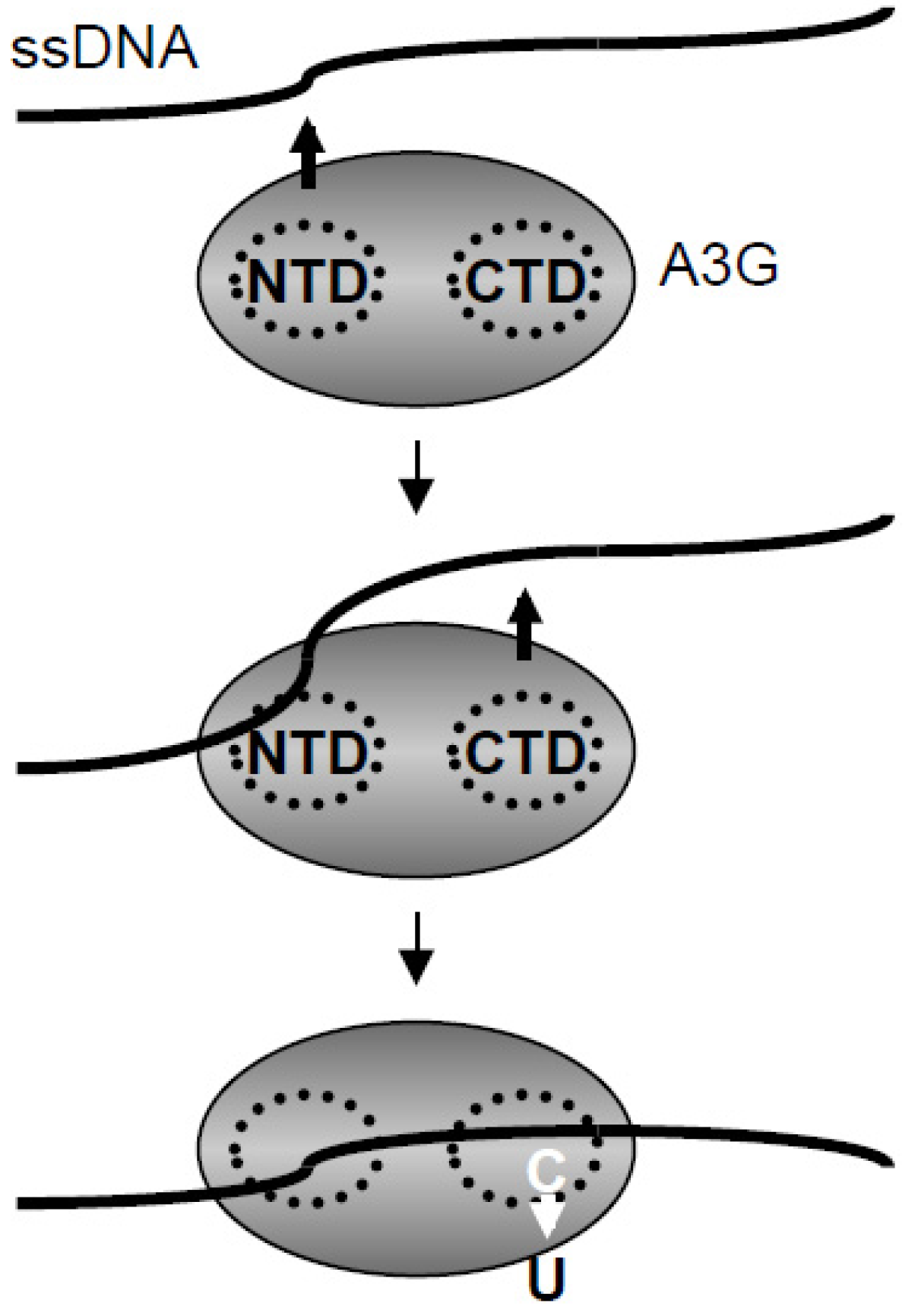
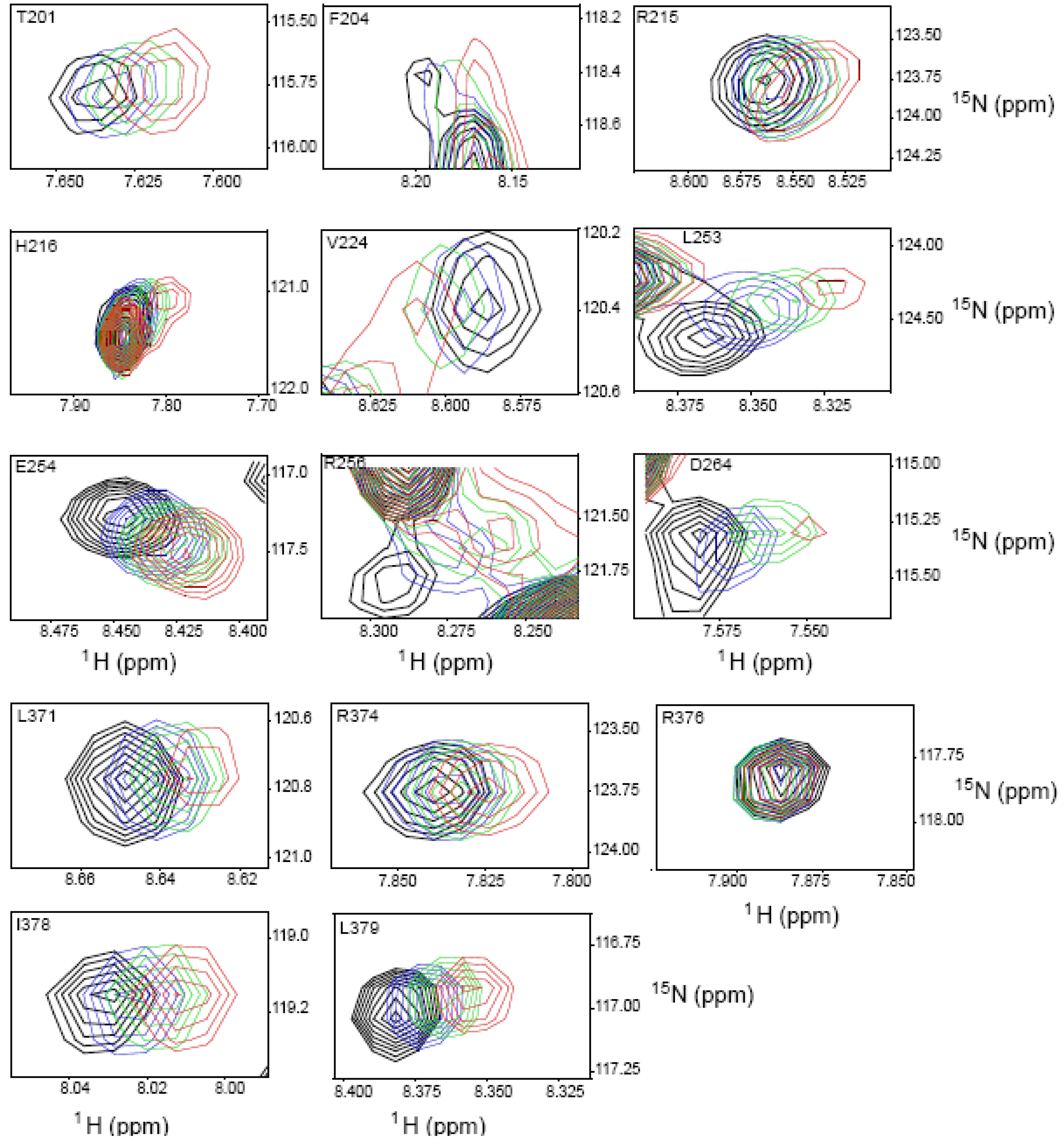
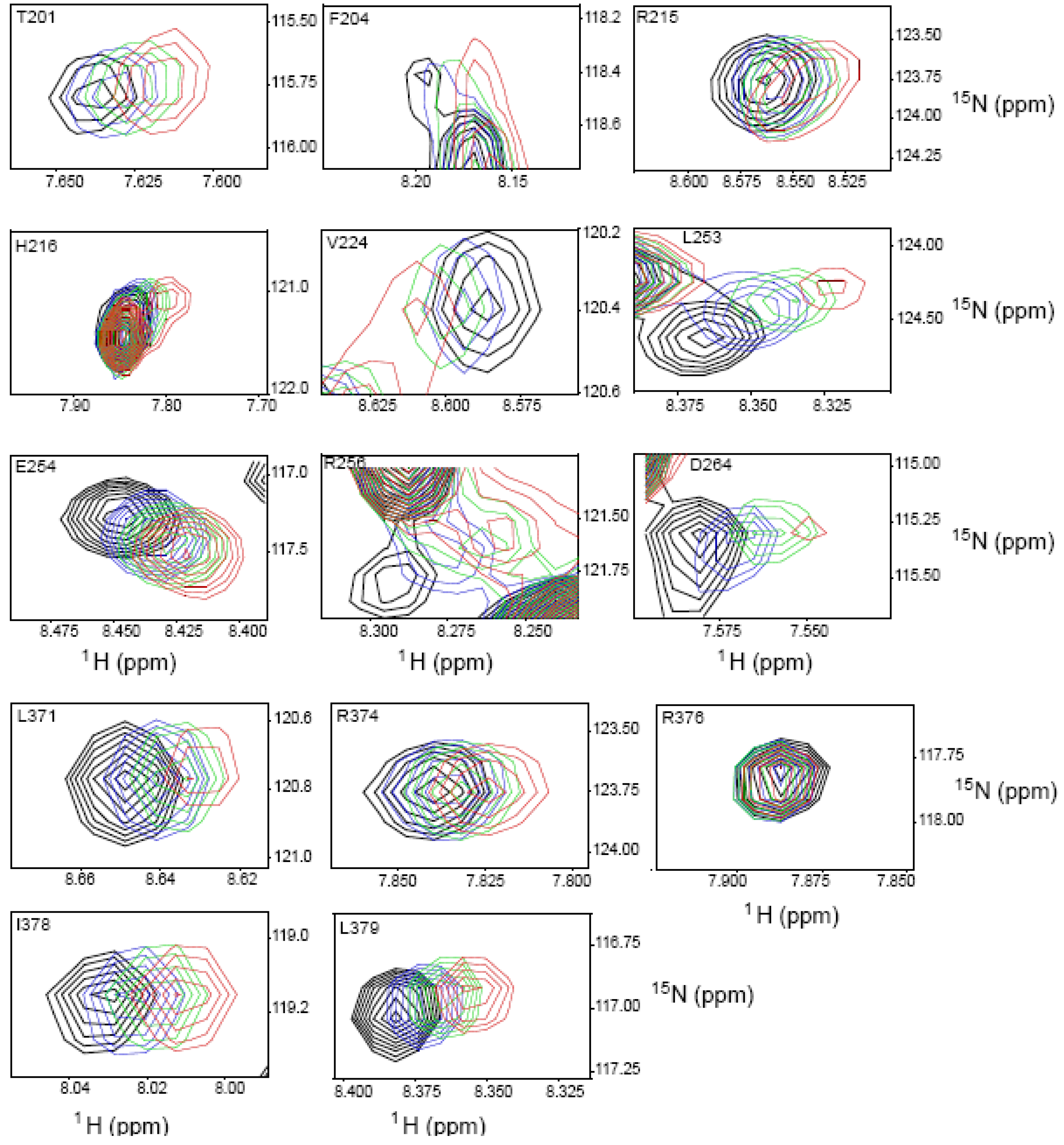
2.4. NMR Chemical Shift Perturbations

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Amino acid number | Shift in 1H (ppm) | Shift in 15N (ppm) | Combined Shift (ppm) |
|---|---|---|---|
| 201 | 0.012 | 0.025 | 0.013 |
| 204 | 0.014 | 0.051 | 0.017 |
| 211 side chain | 0.038 | 0.137 | 0.047 |
| 215 | 0.012 | 0.009 | 0.012 |
| 216 | 0.023 | 0.137 | 0.035 |
| 224 | 0.013 | 0.00 | 0.013 |
| 253 | 0.028 | 0.256 | 0.059 |
| 254 | 0.019 | 0.137 | 0.033 |
| 256 | 0.028 | 0.155 | 0.042 |
| 264 | 0.024 | 0.00 | 0.024 |
| 273 | 0.012 | 0.222 | 0.046 |
| 275 | 0.024 | 0.093 | 0.03 |
| 276 | 0.016 | 0.144 | 0.033 |
| 284 | 0.008 | 0.00 | 0.008 |
| 313 | 0.008 | 0.004 | 0.01 |
| 315 | 0.016 | 0.007 | 0.016 |
| 316 | 0.051 | 0.128 | 0.057 |
| 319 | 0.016 | 0.00 | 0.016 |
| 327 | 0.011 | 0.00 | 0.011 |
| 371 | 0.015 | 0.009 | 0.015 |
| 374 | 0.007 | 0.00 | 0.007 |
| 376 | 0.00 | 0.00 | 0.00 |
| 378 | 0.012 | 0.00 | 0.012 |
| 379 | 0.017 | 0.11 | 0.028 |
2.5. Discussion
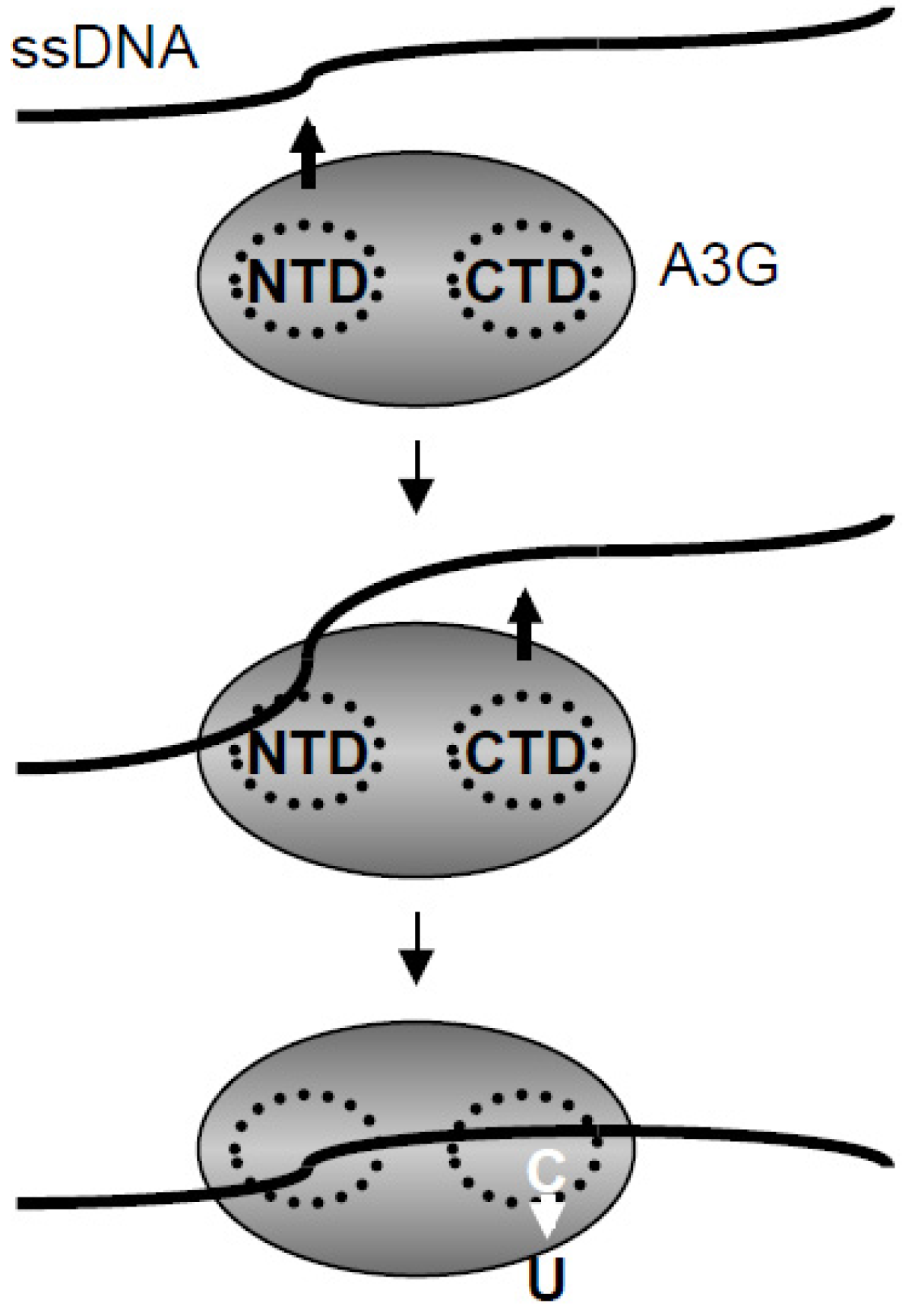
3. Methods
3.1. Plasmid Construction
3.2. E. coli-Based DNA Deamination Assays
3.3. HIV-Restriction Assays and Immunoblots
3.4. Protein Expression and Purification
3.5. EMSAs
3.6. In Vitro DNA Deamination Assays
3.7. NMR Chemical Shift Perturbation Experiments
Acknowledgements
References
- Conticello, S.G. The AID/APOBEC family of nucleic acid mutators. Genome Biol. 2008, 9, 229. [Google Scholar] [CrossRef]
- LaRue, R.S.; Andresdottir, V.; Blanchard, Y.; Conticello, S.G.; Derse, D.; Emerman, M.; Greene, W.C.; Jonsson, S.R.; Landau, N.R.; Lochelt, M.; et al. Guidelines for naming nonprimate APOBEC3 genes and proteins. J. Virol. 2009, 83, 494–497. [Google Scholar]
- Albin, J.S.; Harris, R.S. Interactions of host APOBEC3 restriction factors with HIV-1 in vivo: implications for therapeutics. Expert. Rev. Mol. Med. 2010, 12, e4. [Google Scholar] [CrossRef]
- Malim, M.H.; Emerman, M. HIV-1 accessory proteins—Ensuring viral survival in a hostile environment. Cell Host Microbe 2008, 3, 388–398. [Google Scholar] [CrossRef]
- Smith, J.L.; Bu, W.; Burdick, R.C.; Pathak, V.K. Multiple ways of targeting APOBEC3-virion infectivity factor interactions for anti-HIV-1 drug development. Trends. Pharmacol. Sci. 2009, 30, 638–646. [Google Scholar] [CrossRef]
- Hultquist, J.F.; Lengyel, J.A.; Refsland, E.W.; LaRue, R.S.; Lackey, L.; Brown, W.L.; Harris, R.S. Human and rhesus APOBEC3D, APOBEC3F, APOBEC3G, and APOBEC3H demonstrate a conserved capacity to restrict Vif-deficient HIV-1. J. Virol. 2011, 85, 11220–11234. [Google Scholar] [CrossRef]
- Refsland, E.W.; Hultquist, J.F.; Harris, R.S. Endogenous origins of HIV-1 G-to-A hypermutation and restriction in the nonpermissive T cell line CEM2n. PLoS Pathog. 2012, in press. [Google Scholar]
- Harris, R.S.; Petersen-Mahrt, S.K.; Neuberger, M.S. RNA editing enzyme APOBEC1 and some of its homologs can act as DNA mutators. Mol. Cell 2002, 10, 1247–1253. [Google Scholar] [CrossRef]
- Jarmuz, A.; Chester, A.; Bayliss, J.; Gisbourne, J.; Dunham, I.; Scott, J.; Navaratnam, N. An anthropoid-specific locus of orphan C to U RNA-editing enzymes on chromosome 22. Genomics 2002, 79, 285–296. [Google Scholar]
- Sheehy, A.M.; Gaddis, N.C.; Choi, J.D.; Malim, M.H. Isolation of a human gene that inhibits HIV-1 infection and is suppressed by the viral Vif protein. Nature 2002, 418, 646–650. [Google Scholar] [CrossRef]
- Chelico, L.; Pham, P.; Calabrese, P.; Goodman, M.F. APOBEC3G DNA deaminase acts processively 3' --> 5' on single-stranded DNA. Nat. Struct. Mol. Biol. 2006, 13, 392–399. [Google Scholar] [CrossRef]
- Holden, L.G.; Prochnow, C.; Chang, Y.P.; Bransteitter, R.; Chelico, L.; Sen, U.; Stevens, R.C.; Goodman, M.F.; Chen, X.S. Crystal structure of the anti-viral APOBEC3G catalytic domain and functional implications. Nature 2008, 456, 121–124. [Google Scholar]
- Furukawa, A.; Nagata, T.; Matsugami, A.; Habu, Y.; Sugiyama, R.; Hayashi, F.; Kobayashi, N.; Yokoyama, S.; Takaku, H.; Katahira, M. Structure, interaction and real-time monitoring of the enzymatic reaction of wild-type APOBEC3G. EMBO J. 2009, 28, 440–451. [Google Scholar] [CrossRef]
- Iwatani, Y.; Takeuchi, H.; Strebel, K.; Levin, J.G. Biochemical activities of highly purified, catalytically active human APOBEC3G: Correlation with antiviral effect. J. Virol. 2006, 80, 5992–6002. [Google Scholar] [CrossRef]
- Harris, R.S.; Bishop, K.N.; Sheehy, A.M.; Craig, H.M.; Petersen-Mahrt, S.K.; Watt, I.N.; Neuberger, M.S.; Malim, M.H. DNA deamination mediates innate immunity to retroviral infection. Cell 2003, 113, 803–809. [Google Scholar] [CrossRef]
- Chen, K.M.; Harjes, E.; Gross, P.J.; Fahmy, A.; Lu, Y.; Shindo, K.; Harris, R.S.; Matsuo, H. Structure of the DNA deaminase domain of the HIV-1 restriction factor APOBEC3G. Nature 2008, 452, 116–119. [Google Scholar]
- Shandilya, S.M.D.; Nalam, M.N.L.; Nalivaika, E.A.; Gross, P.J.; Valesano, J.C.; Shindo, K.; Li, M.; Munson, M.; Royer, W.E.; Harjes, E.; et al. Crystal structure of the APOBEC3G catalytic domain reveals potential oligomerization interfaces. Structure 2010, 18, 28–38. [Google Scholar] [CrossRef]
- Stenglein, M.D.; Matsuo, H.; Harris, R.S. Two regions within the amino-terminal half of APOBEC3G cooperate to determine cytoplasmic localization. J. Virol. 2008, 82, 9591–9599. [Google Scholar] [CrossRef]
- Xu, H.; Svarovskaia, E.S.; Barr, R.; Zhang, Y.; Khan, M.A.; Strebel, K.; Pathak, V.K. A single amino acid substitution in human APOBEC3G antiretroviral enzyme confers resistance to HIV-1 virion infectivity factor-induced depletion. Proc. Natl. Acad. Sci. U. S. A. 2004, 101, 5652–5657. [Google Scholar]
- Navarro, F.; Bollman, B.; Chen, H.; Konig, R.; Yu, Q.; Chiles, K.; Landau, N.R. Complementary function of the two catalytic domains of APOBEC3G. Virology 2005, 333, 374–386. [Google Scholar] [CrossRef]
- Bennett, R.P.; Presnyak, V.; Wedekind, J.E.; Smith, H.C. Nuclear Exclusion of the HIV-1 host defense factor APOBEC3G requires a novel cytoplasmic retention signal and is not dependent on RNA binding. J. Biol. Chem. 2008, 283, 7320–7327. [Google Scholar]
- Haché, G.; Liddament, M.T.; Harris, R.S. The retroviral hypermutation specificity of APOBEC3F and APOBEC3G is governed by the C-terminal DNA cytosine deaminase domain. J. Biol. Chem. 2005, 280, 10920–10924. [Google Scholar]
- Iwatani, Y.; Chan, D.S.; Liu, L.; Yoshii, H.; Shibata, J.; Yamamoto, N.; Levin, J.G.; Gronenborn, A.M.; Sugiura, W. HIV-1 Vif-mediated ubiquitination/degradation of APOBEC3G involves four critical lysine residues in its C-terminal domain. Proc. Natl. Acad. Sci. U. S. A. 2009, 106, 19539–19544. [Google Scholar]
- Kohli, R.M.; Abrams, S.R.; Gajula, K.S.; Maul, R.W.; Gearhart, P.J.; Stivers, J.T. A portable hot spot recognition loop transfers sequence preferences from APOBEC family members to activation-induced cytidine deaminase. J. Biol. Chem. 2009, 284, 22898–22904. [Google Scholar]
- Carpenter, M.A.; Rajagurubandara, E.; Wijesinghe, P.; Bhagwat, A.S. Determinants of sequence-specificity within human AID and APOBEC3G. DNA Repair (Amst) 2010, 9, 579–587. [Google Scholar] [CrossRef]
- Schumacher, A.J.; Haché, G.; MacDuff, D.A.; Brown, W.L.; Harris, R.S. The DNA deaminase activity of human APOBEC3G is required for Ty1, MusD, and human immunodeficiency virus type 1 restriction. J. Virol. 2008, 82, 2652–2660. [Google Scholar] [CrossRef]
- Harjes, E.; Gross, P.J.; Chen, K.M.; Lu, Y.; Shindo, K.; Nowarski, R.; Gross, J.D.; Kotler, M.; Harris, R.S.; Matsuo, H. An extended structure of the APOBEC3G catalytic domain suggests a unique holoenzyme model. J. Mol. Biol. 2009, 389, 819–832. [Google Scholar] [CrossRef]
- Li, M.; Shandilya, S.M.; Carpenter, M.A.; Rathore, A.; Brown, W.L.; Perkins, A.L.; Harki, D.A.; Solberg, J.; Hook, D.J.; Pandey, K.K.; et al. First-in-class small molecule inhibitors of the single-strand DNA cytosine deaminase APOBEC3G. ACS Chem. Biol. 2012, 7, 506–517. [Google Scholar] [CrossRef]
- Newman, E.N.; Holmes, R.K.; Craig, H.M.; Klein, K.C.; Lingappa, J.R.; Malim, M.H.; Sheehy, A.M. Antiviral function of APOBEC3G can be dissociated from cytidine deaminase activity. Curr. Biol. 2005, 15, 166–170. [Google Scholar]
- Mangeat, B.; Turelli, P.; Caron, G.; Friedli, M.; Perrin, L.; Trono, D. Broad antiretroviral defence by human APOBEC3G through lethal editing of nascent reverse transcripts. Nature 2003, 424, 99–103. [Google Scholar]
- Shindo, K.; Takaori-Kondo, A.; Kobayashi, M.; Abudu, A.; Fukunaga, K.; Uchiyama, T. The enzymatic activity of CEM15/APOBEC3G is essential for the regulation of the infectivity of HIV-1 virion but not a sole determinant of its antiviral activity. J. Biol. Chem. 2003, 278, 44412–44416. [Google Scholar]
- Haché, G.; Shindo, K.; Albin, J.S.; Harris, R.S. Evolution of HIV-1 isolates that use a novel Vif-independent mechanism to resist restriction by human APOBEC3G. Curr. Biol. 2008, 18, 819–824. [Google Scholar] [CrossRef]
- Browne, E.P.; Allers, C.; Landau, N.R. Restriction of HIV-1 by APOBEC3G is cytidine deaminase-dependent. Virology 2009, 387, 313–321. [Google Scholar] [CrossRef]
- Miyagi, E.; Opi, S.; Takeuchi, H.; Khan, M.; Goila-Gaur, R.; Kao, S.; Strebel, K. Enzymatically active APOBEC3G is required for efficient inhibition of human immunodeficiency virus type 1. J. Virol. 2007, 81, 13346–13353. [Google Scholar] [CrossRef]
- Holmes, R.K.; Koning, F.A.; Bishop, K.N.; Malim, M.H. APOBEC3F can inhibit the accumulation of HIV-1 reverse transcription products in the absence of hypermutation. Comparisons with APOBEC3G. J. Biol. Chem. 2007, 282, 2587–2595. [Google Scholar]
- Iwatani, Y.; Chan, D.S.; Wang, F.; Maynard, K.S.; Sugiura, W.; Gronenborn, A.M.; Rouzina, I.; Williams, M.C.; Musier-Forsyth, K.; Levin, J.G. Deaminase-independent inhibition of HIV-1 reverse transcription by APOBEC3G. Nucleic Acids Res. 2007, 35, 7096–7108. [Google Scholar] [CrossRef]
- Bishop, K.N.; Verma, M.; Kim, E.Y.; Wolinsky, S.M.; Malim, M.H. APOBEC3G inhibits elongation of HIV-1 reverse transcripts. PLoS Pathog. 2008, 4, e1000231. [Google Scholar] [CrossRef]
- Yu, Q.; Konig, R.; Pillai, S.; Chiles, K.; Kearney, M.; Palmer, S.; Richman, D.; Coffin, J.M.; Landau, N.R. Single-strand specificity of APOBEC3G accounts for minus-strand deamination of the HIV genome. Nat. Struct. Mol. Biol. 2004, 11, 435–442. [Google Scholar]
- McDougall, W.M.; Okany, C.; Smith, H.C. Deaminase activity on single-stranded DNA (ssDNA) occurs in vitro when APOBEC3G cytidine deaminase forms homotetramers and higher-order complexes. J. Biol. Chem. 2011, 286, 30655–30661. [Google Scholar]
- Shlyakhtenko, L.S.; Lushnikov, A.Y.; Li, M.; Lackey, L.; Harris, R.S.; Lyubchenko, Y.L. Atomic force microscopy studies provide direct evidence for dimerization of the HIV restriction factor APOBEC3G. J. Biol. Chem. 2011, 286, 3387–3395. [Google Scholar]
- Zhang, K.L.; Mangeat, B.; Ortiz, M.; Zoete, V.; Trono, D.; Telenti, A.; Michielin, O. Model structure of human APOBEC3G. PLoS One 2007, 2, e378. [Google Scholar] [CrossRef]
- Huthoff, H.; Autore, F.; Gallois-Montbrun, S.; Fraternali, F.; Malim, M.H. RNA-dependent oligomerization of APOBEC3G is required for restriction of HIV-1. PLoS Pathog. 2009, 5, e1000330. [Google Scholar] [CrossRef]
- Huthoff, H.; Malim, M.H. Cytidine deamination and resistance to retroviral infection: Towards a structural understanding of the APOBEC proteins. Virology 2005, 334, 147–153. [Google Scholar]
- Friew, Y.N.; Boyko, V.; Hu, W.S.; Pathak, V.K. Intracellular interactions between APOBEC3G, RNA, and HIV-1 Gag: APOBEC3G multimerization is dependent on its association with RNA. Retrovirology 2009, 6, 56. [Google Scholar] [CrossRef]
- Opi, S.; Takeuchi, H.; Kao, S.; Khan, M.A.; Miyagi, E.; Goila-Gaur, R.; Iwatani, Y.; Levin, J.G.; Strebel, K. Monomeric APOBEC3G is catalytically active and has antiviral activity. J. Virol. 2006, 80, 4673–4682. [Google Scholar] [CrossRef]
- Stenglein, M.D.; Harris, R.S. APOBEC3B and APOBEC3F inhibit L1 retrotransposition by a DNA deamination-independent mechanism. J. Biol. Chem. 2006, 281, 16837–16841. [Google Scholar] [CrossRef]
- Liddament, M.T.; Brown, W.L.; Schumacher, A.J.; Harris, R.S. APOBEC3F properties and hypermutation preferences indicate activity against HIV-1 in vivo. Curr. Biol. 2004, 14, 1385–1391. [Google Scholar] [CrossRef]
- Simon, J.H.; Fouchier, R.A.; Southerling, T.E.; Guerra, C.B.; Grant, C.K.; Malim, M.H. The Vif and Gag proteins of human immunodeficiency virus type 1 colocalize in infected human T cells. J. Virol. 1997, 71, 5259–5267. [Google Scholar]
Supplementary Materials
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Shindo, K.; Li, M.; Gross, P.J.; Brown, W.L.; Harjes, E.; Lu, Y.; Matsuo, H.; Harris, R.S. A Comparison of Two Single-Stranded DNA Binding Models by Mutational Analysis of APOBEC3G. Biology 2012, 1, 260-276. https://doi.org/10.3390/biology1020260
Shindo K, Li M, Gross PJ, Brown WL, Harjes E, Lu Y, Matsuo H, Harris RS. A Comparison of Two Single-Stranded DNA Binding Models by Mutational Analysis of APOBEC3G. Biology. 2012; 1(2):260-276. https://doi.org/10.3390/biology1020260
Chicago/Turabian StyleShindo, Keisuke, Ming Li, Phillip J. Gross, William L. Brown, Elena Harjes, Yongjian Lu, Hiroshi Matsuo, and Reuben S. Harris. 2012. "A Comparison of Two Single-Stranded DNA Binding Models by Mutational Analysis of APOBEC3G" Biology 1, no. 2: 260-276. https://doi.org/10.3390/biology1020260