Higher Desolvation Energy Reduces Molecular Recognition in Multi-Drug Resistant HIV-1 Protease

Abstract
:1. Introduction
2. Results and Discussion
2.1. The Four Clinical MDR HIV-1 Protease Isolates Are Resistant to All FDA-Approved HIV-1 Protease Inhibitors

{kind=link}
{kind=link}
{kind=link}
| HIV-1 Proteases | IC50 of HIV-1 protease inhibitors * in nM | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| DRV | ATV | LPV | TPV | NFV | APV | SQV | IDV | RTV | |
| NL4-3 | 0.26 | 0.19 | 0.28 | 0.24 | 1.6 | 0.43 | 0.50 | 0.47 | 0.34 |
| MDR 769 | 0.74 | 2.9 | 0.50 | 0.65 | 110 | 4.8 | 290 | 120 | 61 |
| MDR 807 | 2.0 | 7.6 | 1.2 | 0.63 | 210 | 2.9 | 850 | 280 | 14.6 |
| MDR 1385 | 3.0 | 4.6 | 2.3 | 2.8 | 230 | 3.1 | 29 | 140 | 8.0 |
| MDR 3761 | 0.89 | 2.2 | 0.39 | 0.63 | 430 | 4.1 | 110 | 210 | 21 |
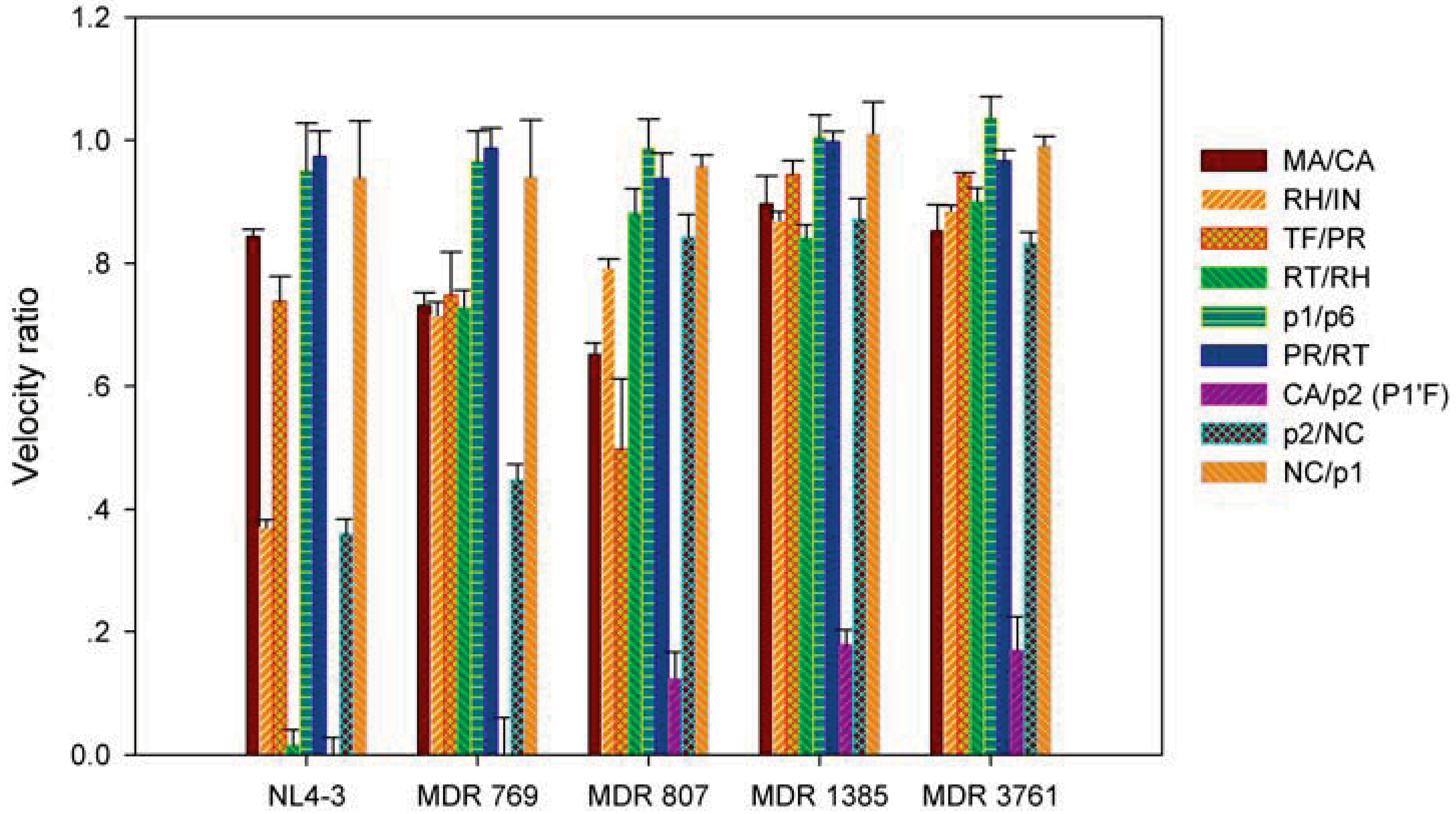
2.2. The MDR HIV-1 Protease Isolates Exhibited Different Substrate Binding Preference Relative to the WT Protease
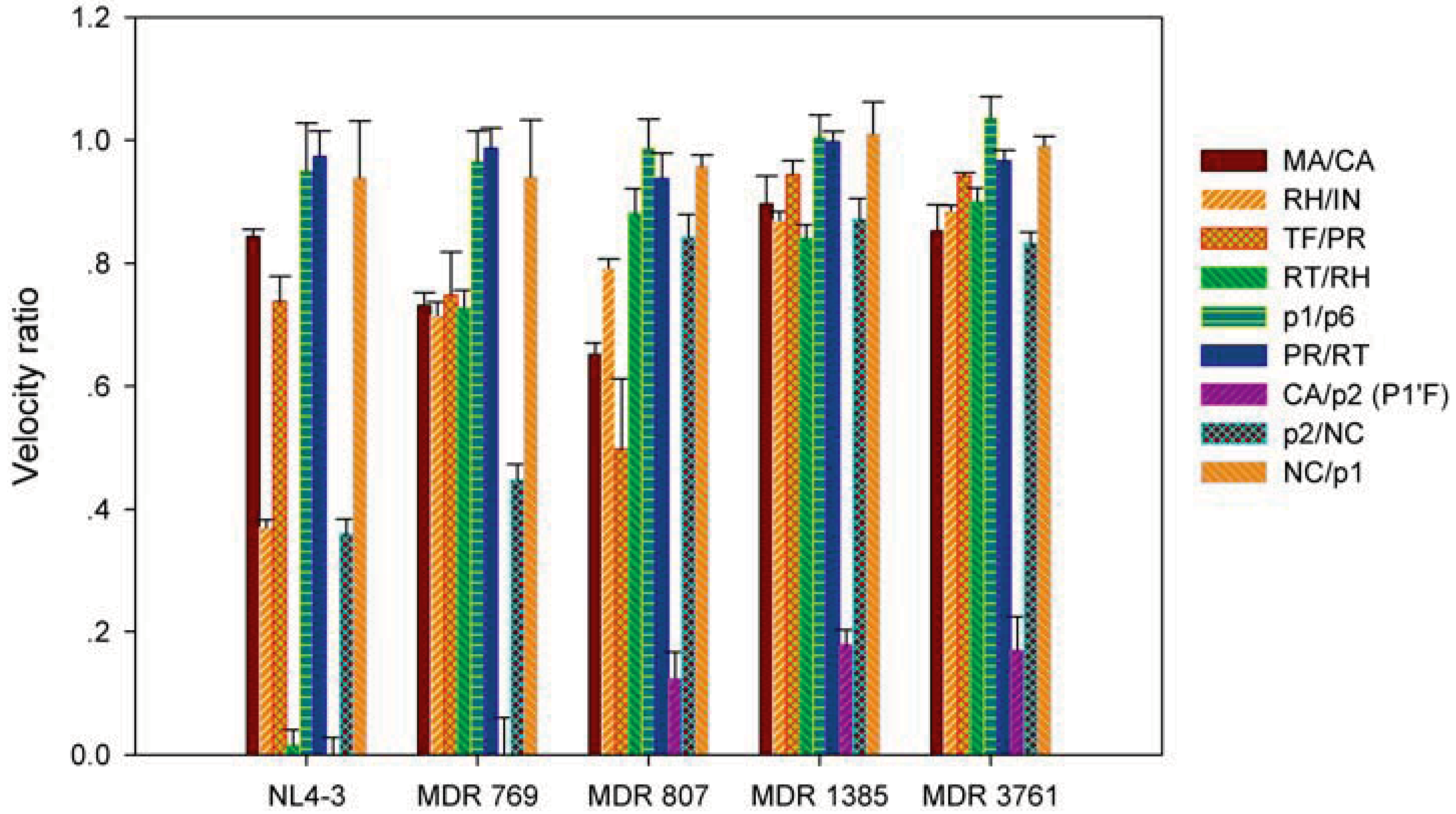
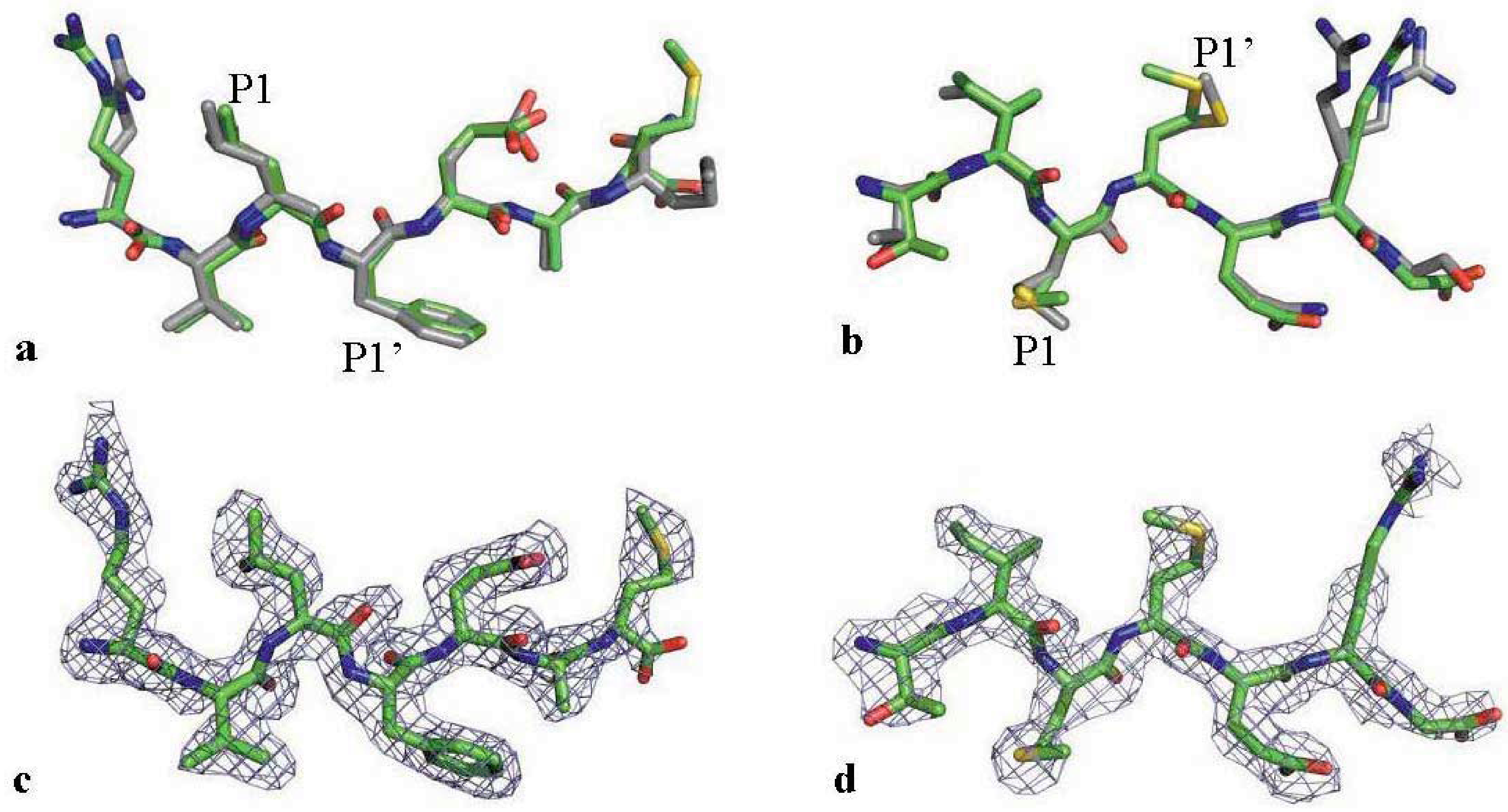
2.3. The MDR Protease-Substrate Co-Crystal Structures Were Insufficient to Explain the Different Substrate Binding Behaviors between the MDR and WT HIV-1 Proteases
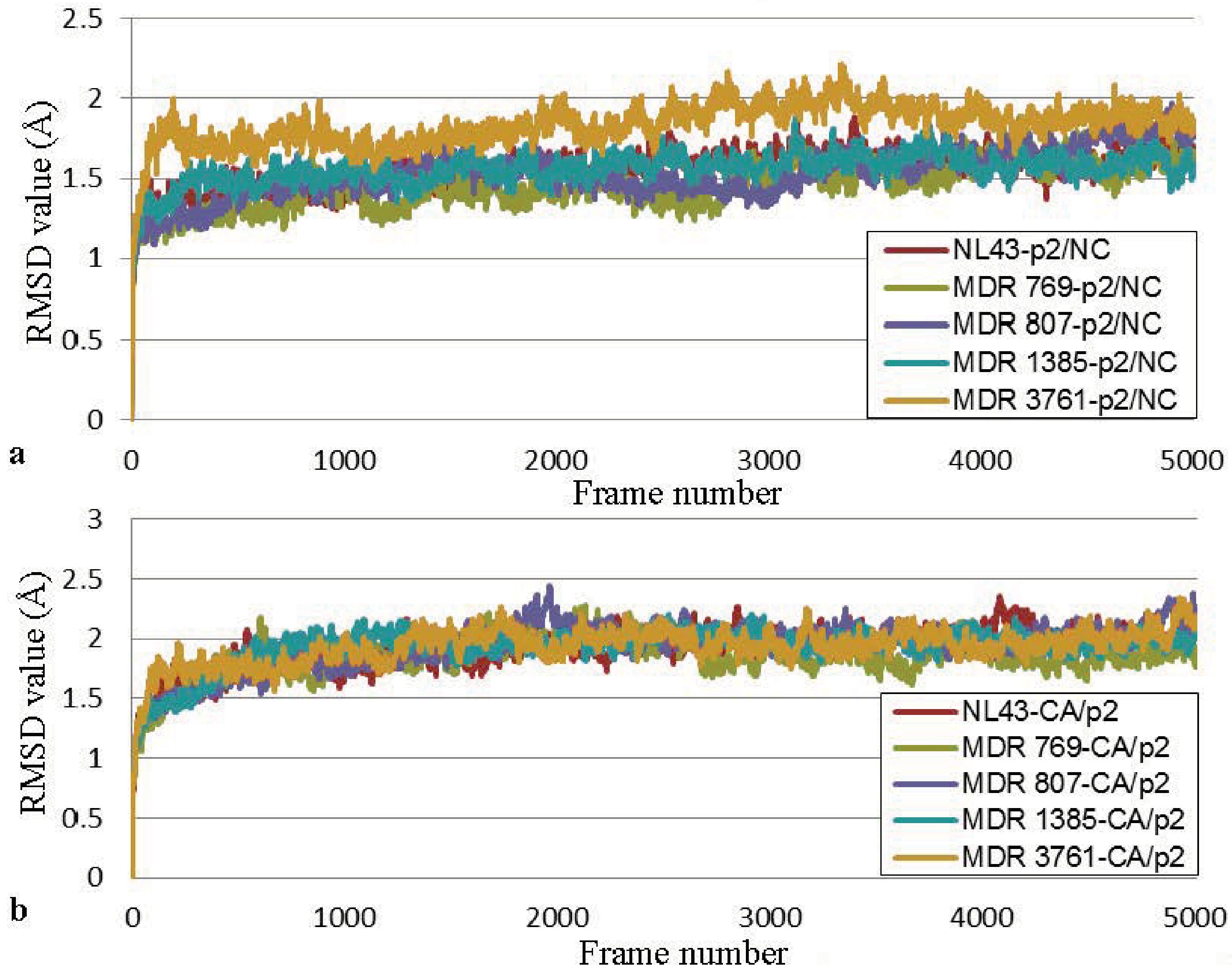
2.4. The Desolvation Energy Required by the MDR HIV-1 Protease Variants to Form Protease-Substrate Complexes Correlated with the Substrate Binding Assay
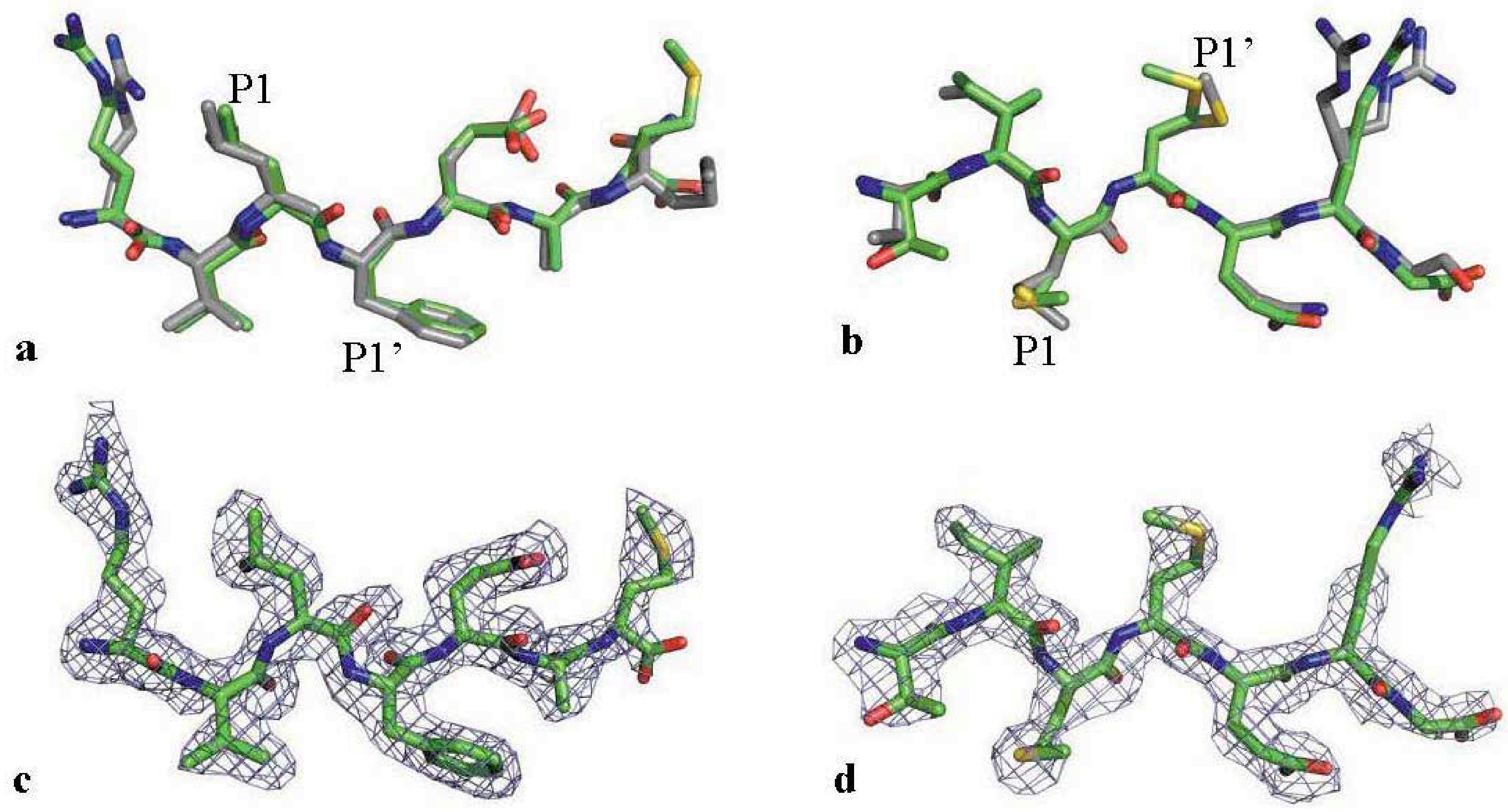
| Dataset | The MDR 769 in complex of substrate CA/p2 | The MDR 769 in complex of substrate p2/NC | |
|---|---|---|---|
| Data collection | |||
| Space group | P212121 | P212121 | |
| Wavelength (Å) | 0.979 | 0.979 | |
| Cell constants (Å) | a = 28.76 b = 65.38 c = 92.80 | a = 28.62 b = 63.85 c = 91.11 | |
| Resolution range (Å) | 30.00−2.10 (2.14−2.10) | 30.00−2.30 (2.38−2.30) | |
| Number of unique reflections | 10882 (507) | 7945 (787) | |
| Completeness (%) | 99.9 (99.0) | 98.8 (99.5) | |
| Redundancy | 7.6 (6.0) | 4.0 (4.0) | |
| Mean I/σ (I) | 13.2 (3.4) | 10.0 (2.4) | |
| Rmerge a | 0.162 (0.520) | 0.114 (0.451) | |
| Refinement | |||
| Rwork (%)b | 17.29 | 20.00 | |
| Rfree (%)b | 23.99 | 27.86 | |
| Number of atoms | |||
| Ligand | 60 | 56 | |
| Protease | 1529 | 1529 | |
| Solvent | 258 | 137 | |
| Average isotropic B factor (Å2) | |||
| Ligand | 20.28 | 35.32 | |
| Protease | 16.83 | 30.46 | |
| Solvent | 32.62 | 45.32 | |
| RMSD bond length (Å) | 0.008 | 0.009 | |
| RMSD bond angle (°) | 1.06 | 1.26 | |
| Ramachandran plot | |||
| Allowed/generous/disallowed (%) | 100/0/0 | 99.0/1.0/0 | |
| Terms of binding free energy (kcal/mol) | HIV-1 protease | ||||
|---|---|---|---|---|---|
| NL4-3 | MDR 769 | MDR 807 | MDR 1385 | MDR 3761 | |
| ΔGdesolvelec | 178 ± 10 | 188 ± 11 | 210 ± 16 | 228 ± 19 | 256 ± 10 |
| ΔGdesolvnonpolar | −7.6 ± 0.2 | −7.7 ± 0.2 | −7.9 ± 0.2 | −7.6 ± 0.2 | −7.7 ± 0.1 |
| ΔGdesolv | 170 | 180 | 202 | 220 | 248 |
| Terms of binding free energy (kcal/mol) | HIV-1 protease | ||||
|---|---|---|---|---|---|
| NL4-3 | MDR 769 | MDR 807 | MDR 1385 | MDR 3761 | |
| ΔGdesolvelec | 278 ± 22 | 283 ± 25 | 315 ± 18 | 302 ± 20 | 330 ± 18 |
| ΔGdesolvnonpolar | −7.5 ± 0.1 | −7.8 ± 0.2 | −7.6 ± 0.2 | −6.9 ± 0.1 | −7.3 ± 0.1 |
| ΔGdesolv | 271 | 275. | 307 | 295 | 322 |
3. Experimental Section
3.1. Protein Expression and Purification
3.2. Protease Inhibition and Substrate Interference Assays
| Residues | HIV-1 protease | Sequences * |
|---|---|---|
| 1–50 | NL4-3 | PQITLWKRPL VTIKIGGQLK EALLDTGADD TVLEEMNLPG RWKPKMIGGI |
| MDR 769 | PQITLWKRPI VTIKIGGQLK EALLDTGADD TVLEEVNLPG RWKPKLIGGI | |
| MDR 807 | PQITLWKRPI VTIKIGGQLK EALLDTGADD TVLEEMNLPG KWKPKIIVGI | |
| MDR 1385 | PQITLWKRPF VTIKIGGQLK EALLDTGADD TVLEEIDLPG RWKPKIIGGI | |
| MDR 3761 | PQITLWKRPI VAIKVGGQII EALLDTGADD TVLEEMNLPG RWKPKIIGGI | |
| 51–99 | NL4-3 | GGFIKVRQYD QILIEICGHK AIGTVLVGPT PVNIIGRNLL TQIGCTLNF |
| MDR 769 | GGFVKVRQYD QVPIEICGHK VIGTVLVGPT PANVIGRNLM TQIGCTLNF | |
| MDR 807 | GGFTKVRQYD NVQIEICGHK VIGAVLIGPT PANIIGRNLL TQLGCTLNF | |
| MDR 1385 | GGFIKVKQYD QIPIEICGHK VIGTVLVGPT PTNIIGRNMM TQLGCTLNF | |
| MDR 3761 | GGFIKVRQYD QIPVEICGHK IITTVLVGST PVNVIGRNLM TQLGCTLNF |
3.3. Crystallization, Data Collection, and Structure Refinement
| substrate | P3 | P2 | P1 | P1’ | P2’ | P3’ | P4’ |
|---|---|---|---|---|---|---|---|
| MA/CA | Gln | Asn | Tyr | Pro | Ile | Val | Gln |
| CA/p2* | Arg | Val | Leu | Phe | Glu | Ala | Met |
| p2/NC | Thr | Ile | Met | Met | Gln | Arg | Gly |
| NC/p1 | Gln | Ala | Asn | Phe | Leu | Gly | Lys |
| p1/p6 | Gly | Asn | Phe | Leu | Gln | Ser | Arg |
| TF/PR | Phe | Asn | Phe | Pro | Gln | Ile | Thr |
| PR/RT | Leu | Asn | Phe | Pro | Ile | Ser | Pro |
| RT/RH | Glu | Thr | Phe | Tyr | Val | Asp | Gly |
| RH/IN | Lys | Ile | Leu | Phe | Leu | Asp | Gly |
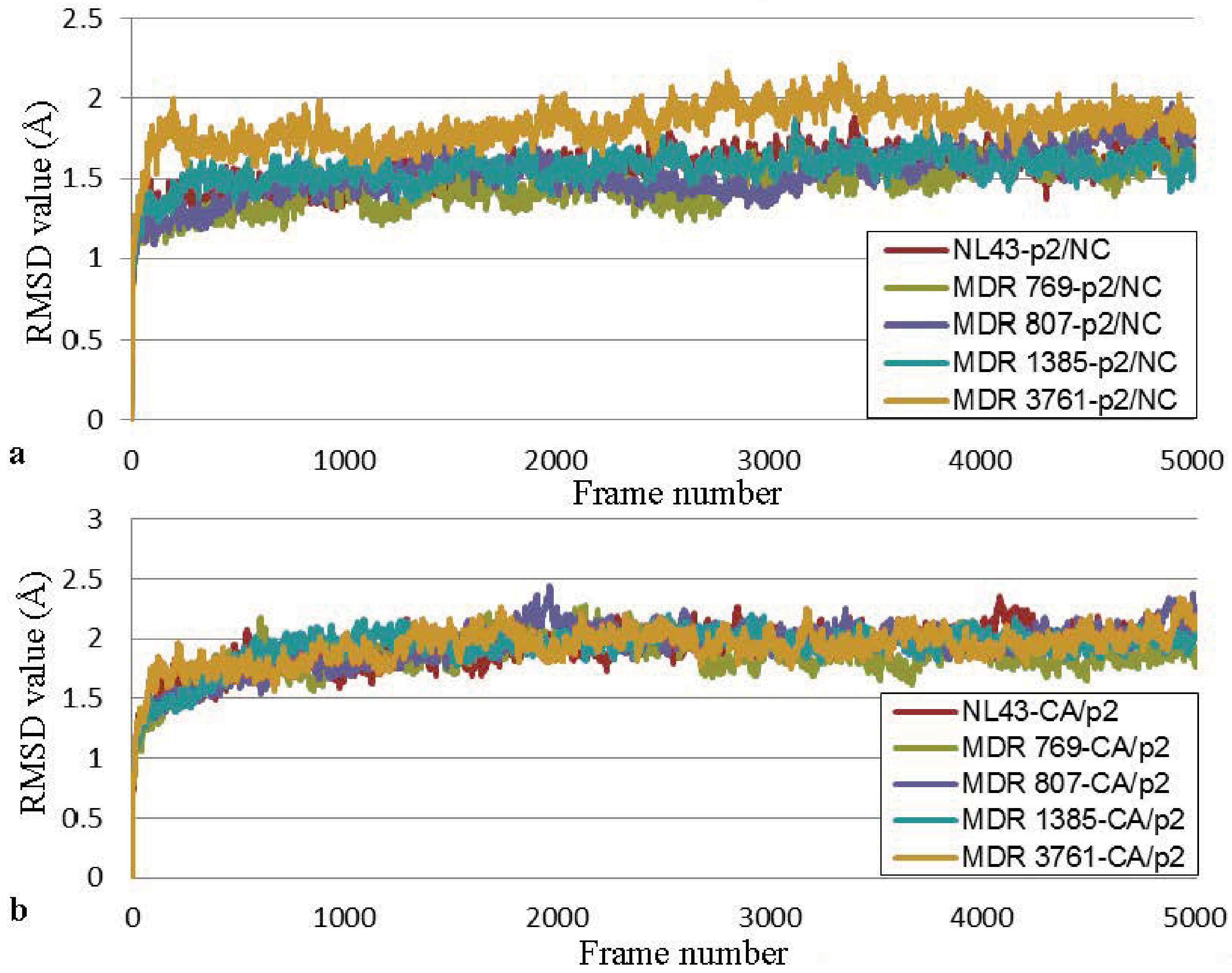
3.4. Molecular Dynamics Simulation
3.5. Energy Calculations
4. Conclusions
References
- Nalam, M.N.L.; Ali, A.; Altman, M.D.; Reddy, G.S.K.K.; Chellappan, S.; Kairys, V.; Ozen, A.; Cao, H.; Gilson, M.K.; Tidor, B.; Rana, T.M.; Schiffer, C.A. Evaluating the substrate-envelope hypothesis: Structural analysis of novel HIV-1 protease inhibitors designed to be robust against drug resistance. J. Virol. 2010, 84, 5368–5378. [Google Scholar]
- Prabu-Jeyabalan, M.; Schiffer, C.A.; Nalivaika, E. Substrate shape determines specificity of recognition for HIV-1 protease: Analysis of crystal structures of six substrate complexes. Structure 2002, 10, 369–381. [Google Scholar] [CrossRef]
- Ozen, A.; Haliloglu, T.; Schiffer, C.A. Dynamics of preferential substrate recognition in HIV-1 protease: Redefining the substrate envelope. J. Mol. Biol. 2011, 410, 726–744. [Google Scholar] [CrossRef]
- Luque, I.; Todd, M.J.; Gomez, J.; Semo, N.; Freire, E. Molecular basis of resistance to HIV-1 protease inhibition: A plausible hypothesis. Biochemistry 1998, 37, 5791–5797. [Google Scholar]
- Tie, Y.; Boross, P.I.; Wang, Y.F.; Gaddis, L.; Liu, F.; Chen, X.; Tozser, J.; Harrison, R.W.; Weber, I.T. Molecular basis for substrate recognition and drug resistance from 1.1 to 1.6 angstroms resolution crystal structures of HIV-1 protease mutants with substrate analogs. FEBS J. 2005, 272, 5265–5277. [Google Scholar]
- Ohtaka, H.; Freire, E. Adaptive inhibitors of the HIV-1 protease. Prog. Biophys. Mol. Biol. 2005, 88, 193–208. [Google Scholar] [CrossRef]
- Palmer, S.; Shafer, R.W.; Merigan, T.C. Highly drug-resistant HIV-1 clinical isolates are cross-resistant to many antiretroviral compounds in current clinical development. AIDS 1999, 13, 661–667. [Google Scholar]
- Wang, Y.; Liu, Z.; Brunzelle, J.S.; Kovari, I.A.; Dewdney, T.G.; Reiter, S.J.; Kovari, L.C. The higher barrier of darunavir and tipranavir resistance for HIV-1 protease. Biochem. Biophys. Res. Commun. 2011, 412, 737–742. [Google Scholar] [CrossRef]
- Logsdon, B.C.; Vickrey, J.F.; Martin, P.; Proteasa, G.; Koepke, J.I.; Terlecky, S.R.; Wawrzak, Z.; Winters, M.A.; Merigan, T.C.; Kovari, L.C. Crystal structures of a multidrug-resistant human immunodeficiency virus type 1 protease reveal an expanded active-site cavity. J. Virol. 2004, 78, 3123–3132. [Google Scholar]
- Vondrasek, J.; Wlodawer, A. HIVdb: A database of the structures of human immunodeficiency virus protease. Proteins 2002, 49, 429–431. [Google Scholar] [CrossRef]
- HHS, Panel on Antiretroviral Guidelines for Adults and Adolescents, Guidelines for the Use of Antiretroviral Agents in HIV-1-infected Adults and Adolescents; Department of Health and Human Services: Washington, DC, USA, 2011.
- Ozer, N.; Schiffer, C.A.; Haliloglu, T. Rationale for more diverse inhibitors in competition with substrates in HIV-1 protease. Biophys. J. 2010, 99, 1650–1659. [Google Scholar] [CrossRef]
- Szeltner, Z.; Polgar, L. Rate-determining steps in HIV-1 protease catalysis - The hydrolysis of the most specific substrate. J. Bio.l Chem. 1996, 271, 32180–32184. [Google Scholar] [CrossRef]
- Pettit, S.C.; Moody, M.D.; Wehbie, R.S.; Kaplan, A.H.; Nantermet, P.V.; Klein, C.A.; Swanstrom, R. The P2 domain of human-immunodeficiency-virus Type-1 gag regulates sequential proteolytic processing and is required to produce fully infectious virions. J. Virol. 1994, 68, 8017–8027. [Google Scholar]
- Vickrey, J.F.; Logsdon, B.C.; Proteasa, G.; Palmer, S.; Winters, M.A.; Merigan, T.C.; Kovari, L.C. HIV-1 protease variants from 100-fold drug resistant clinical isolates: Expression, purification, and crystallization. Protein Expr. Purif. 2003, 28, 165–172. [Google Scholar] [CrossRef]
- Galiano, L.; Ding, F.; Veloro, A.M.; Blackburn, M.E.; Simmerling, C.; Fanucci, G.E. Drug pressure selected mutations in HIV-1 protease alter flap conformations. J. Am. Chem. Soc. 2009, 131, 430–431. [Google Scholar]
- Otwinowski, Z.; Minor, W. Processing of X-ray Diffraction Data Collected in Oscillation Mode. In Methods in Enzymology; Carter, C.W., Sweet, R.M., Eds.; Academic Press, Inc.: New York, NY, USA, 1997; Volume 276, pp. 307–326. [Google Scholar]
- Collaborative Computational Project Number 4. The CCP4 suite: Programs for protein crystallography. Acta Crystallogr. D Biol. Crystallogr. 1994, 50, 760–763. [CrossRef]
- Arnold, K.; Bordoli, L.; Kopp, J.; Schwede, T. The SWISS-MODEL workspace: A web-based environment for protein structure homology modelling. Bioinformatics 2006, 22, 195–201. [Google Scholar] [CrossRef]
- Phillips, J.C.; Braun, R.; Wang, W.; Gumbart, J.; Tajkhorshid, E.; Villa, E.; Chipot, C.; Skeel, R.D.; Kale, L.; Schulten, K. Scalable molecular dynamics with NAMD. J. Comput. Chem. 2005, 26, 1781–1802. [Google Scholar] [CrossRef]
- Darden, T.; York, D.; Pedersen, L. Particle mesh ewald - an N.Log(N) method for ewald sums in large systems. J. Chem. Phys. 1993, 98, 10089–10092. [Google Scholar] [CrossRef]
- Hazuda, D.J.; Felock, P.; Witmer, M.; Wolfe, A.; Stillmock, K.; Grobler, J.A.; Espeseth, A.; Gabryelski, L.; Schleif, W.; Blau, C.; Miller, M.D. Inhibitors of strand transfer that prevent integration and inhibit HIV-1 replication in cells. Science 2000, 287, 646–650. [Google Scholar]
- Baker, N.A.; Sept, D.; Joseph, S.; Holst, M.J.; McCammon, J.A. Electrostatics of nanosystems: Application to microtubules and the ribosome. Proc. Natl. Acad. Sci. USA 2001, 98, 10037–10041. [Google Scholar]
- Dolinsky, T.J.; Czodrowski, P.; Li, H.; Nielsen, J.E.; Jensen, J.H.; Klebe, G.; Baker, N.A. PDB2PQR: Expanding and upgrading automated preparation of biomolecular structures for molecular simulations. Nucleic Acids Res. 2007, 35, W522–W525. [Google Scholar]
- Sitkoff, D.; Sharp, K.A.; Honig, B. Accurate calculation of hydration free-energies using macroscopic solvent models. J. Phys. Chem. 1994, 98, 1978–1988. [Google Scholar]
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Wang, Y.; Dewdney, T.G.; Liu, Z.; Reiter, S.J.; Brunzelle, J.S.; Kovari, I.A.; Kovari, L.C. Higher Desolvation Energy Reduces Molecular Recognition in Multi-Drug Resistant HIV-1 Protease. Biology 2012, 1, 81-93. https://doi.org/10.3390/biology1010081
Wang Y, Dewdney TG, Liu Z, Reiter SJ, Brunzelle JS, Kovari IA, Kovari LC. Higher Desolvation Energy Reduces Molecular Recognition in Multi-Drug Resistant HIV-1 Protease. Biology. 2012; 1(1):81-93. https://doi.org/10.3390/biology1010081
Chicago/Turabian StyleWang, Yong, Tamaria G. Dewdney, Zhigang Liu, Samuel J. Reiter, Joseph S. Brunzelle, Iulia A. Kovari, and Ladislau C. Kovari. 2012. "Higher Desolvation Energy Reduces Molecular Recognition in Multi-Drug Resistant HIV-1 Protease" Biology 1, no. 1: 81-93. https://doi.org/10.3390/biology1010081
APA StyleWang, Y., Dewdney, T. G., Liu, Z., Reiter, S. J., Brunzelle, J. S., Kovari, I. A., & Kovari, L. C. (2012). Higher Desolvation Energy Reduces Molecular Recognition in Multi-Drug Resistant HIV-1 Protease. Biology, 1(1), 81-93. https://doi.org/10.3390/biology1010081