Microfluidic-Based Amplification-Free Bacterial DNA Detection by Dielectrophoretic Concentration and Fluorescent Resonance Energy Transfer Assisted in Situ Hybridization (FRET-ISH) †

Abstract
:1. Introduction

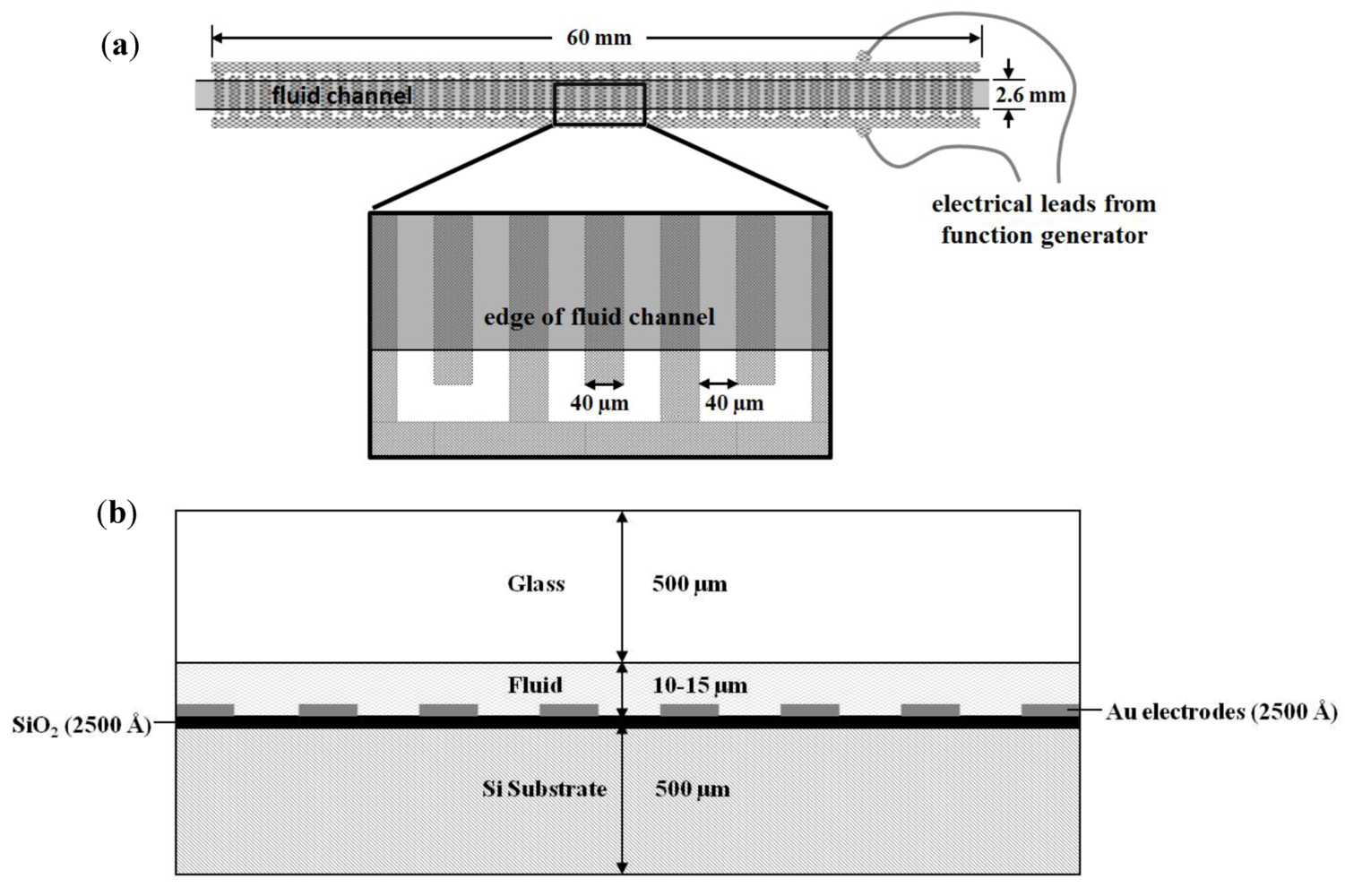{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Bacterial centrifugation and preparation | 6 min |
| Sample delivery to chip | 1 min |
| Dielectrophoretic capture and concentration | 1 min |
| Cell lysis, permeabilization and nucleic acid denaturation | 5 min |
| Nucleic acid hybridization | 5 min |
| Detection and data analysis | 5 min |
| Total Time | 23 min |
2. Experimental Section
2.1. Fluorescent Staining of Cells
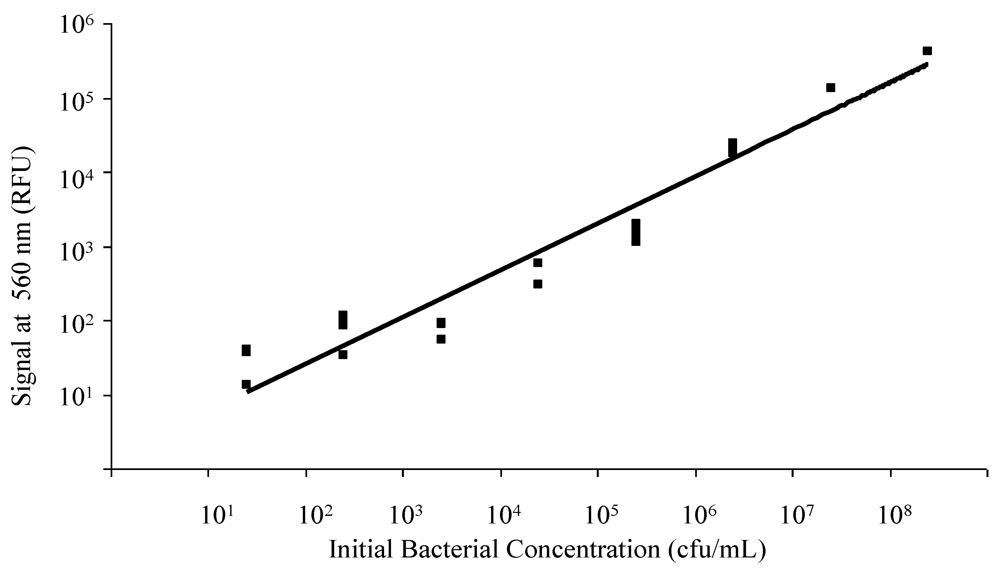
2.2. Spectrofluorometry for Confirmation of FRET-ISH Performance
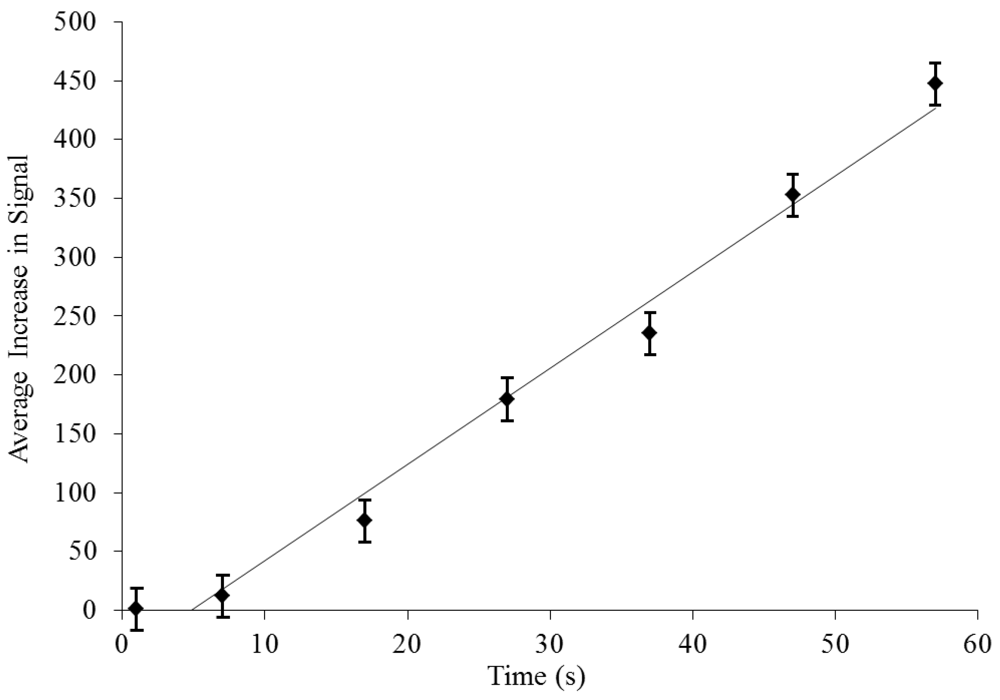
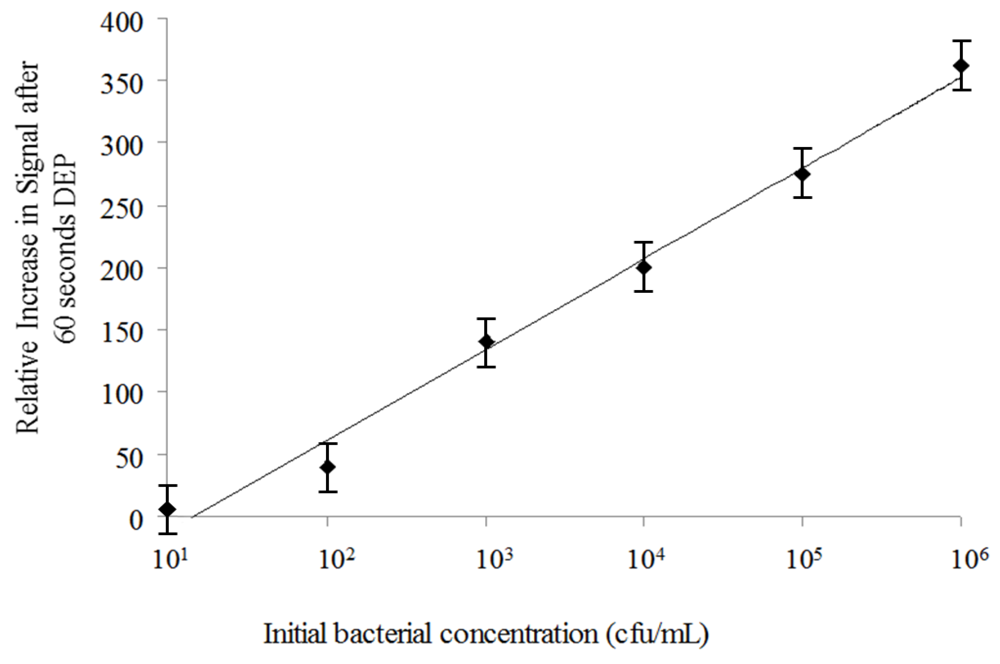
2.3. Dielectrophoretic Capture and Concentration of Cells
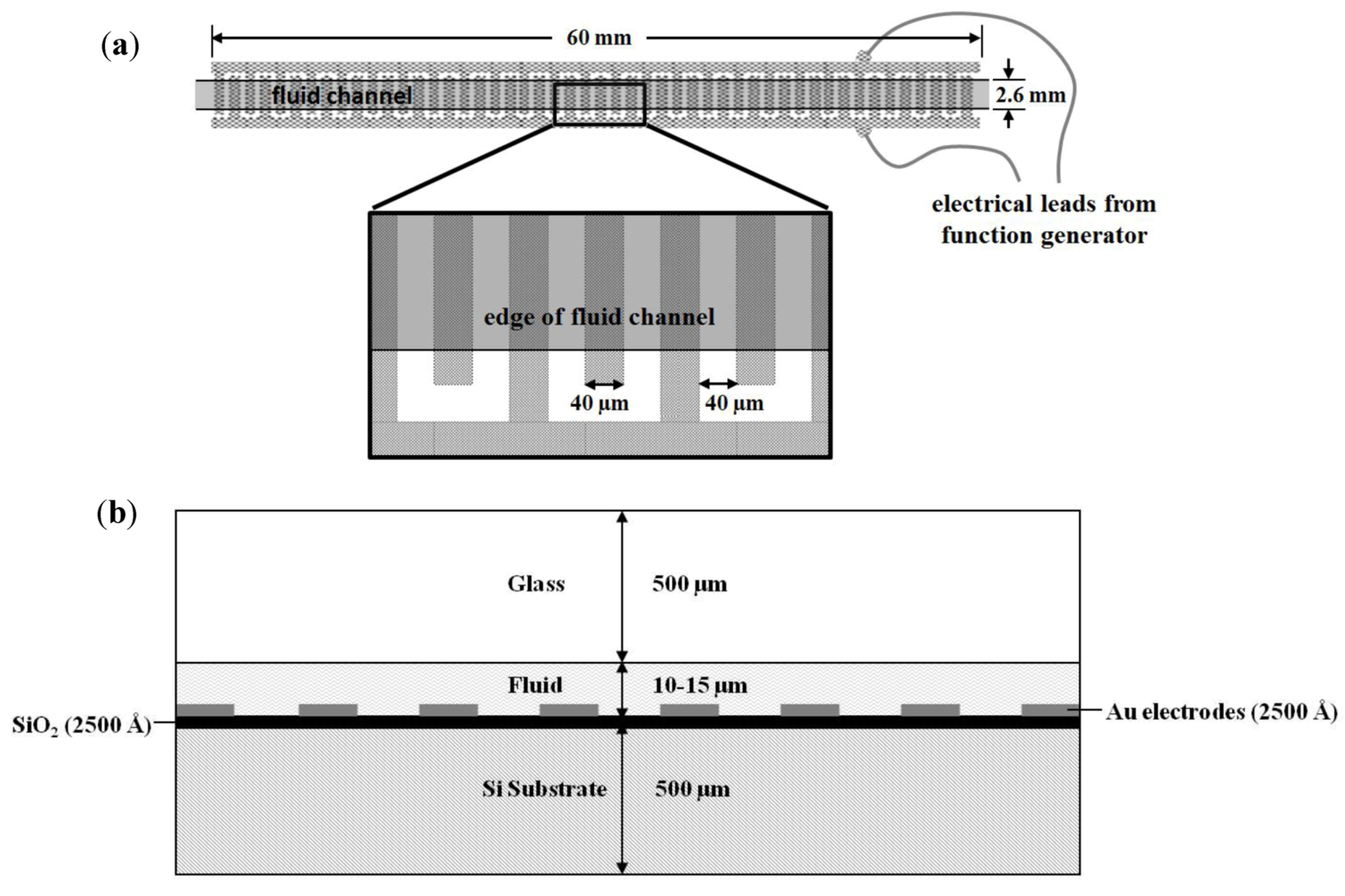
2.4. On-Chip Probe-Based Identification: FRET-ISH
2.5. On-Chip Thermal Lysis, Permeabilization and Nucleic Acid Denaturation and Hybridization
2.6. Imaging and Data Analysis
3. Results and Discussion
3.1. Spectrofluorometry for Confirmation of FRET-ISH Performance
3.2. Dielectrophoretic Capture and Concentration of Cells
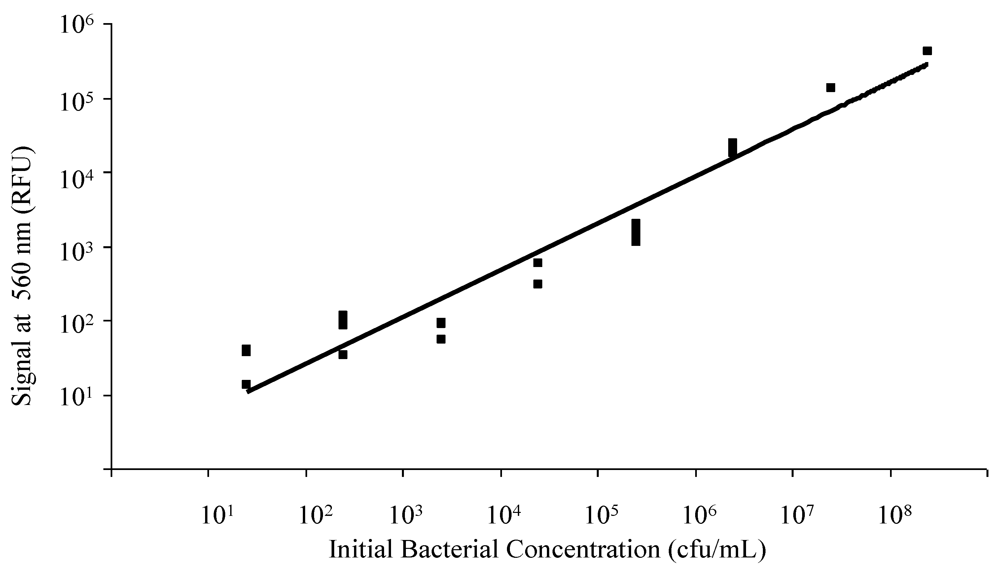

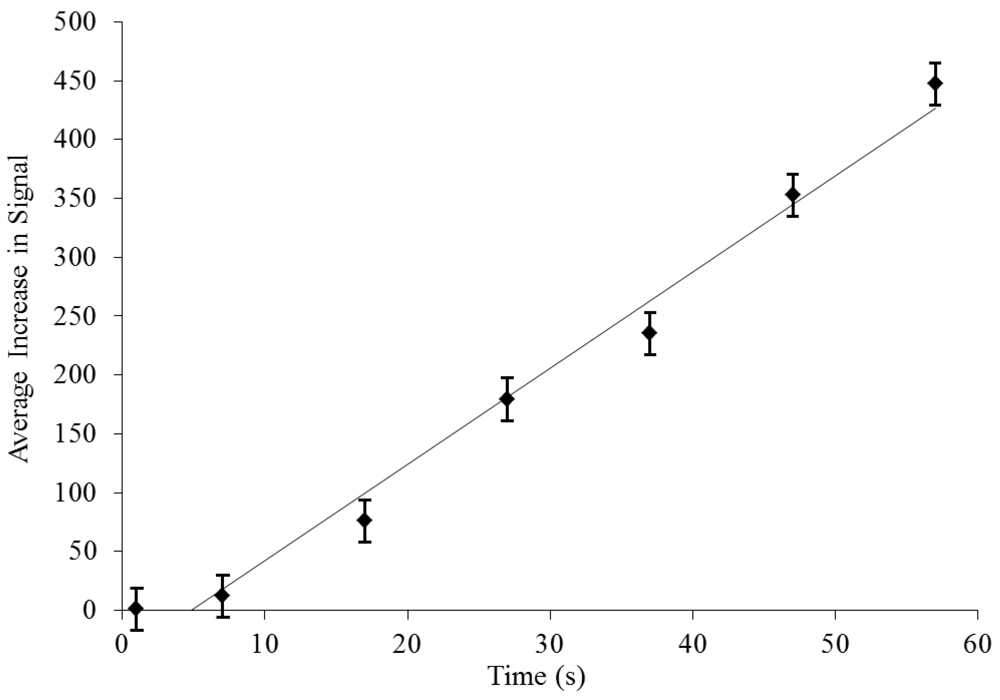
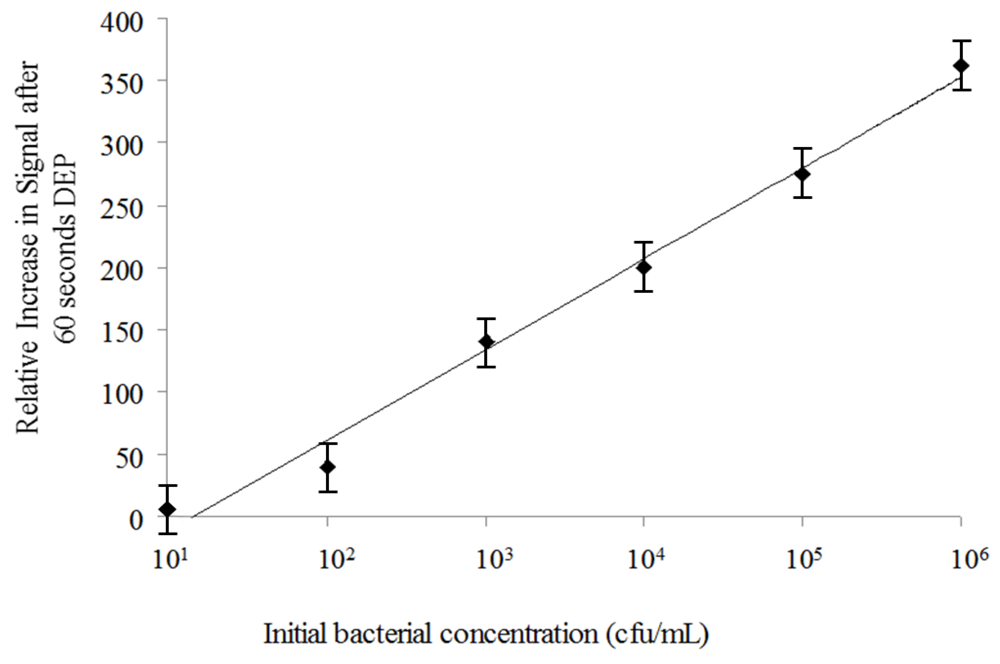
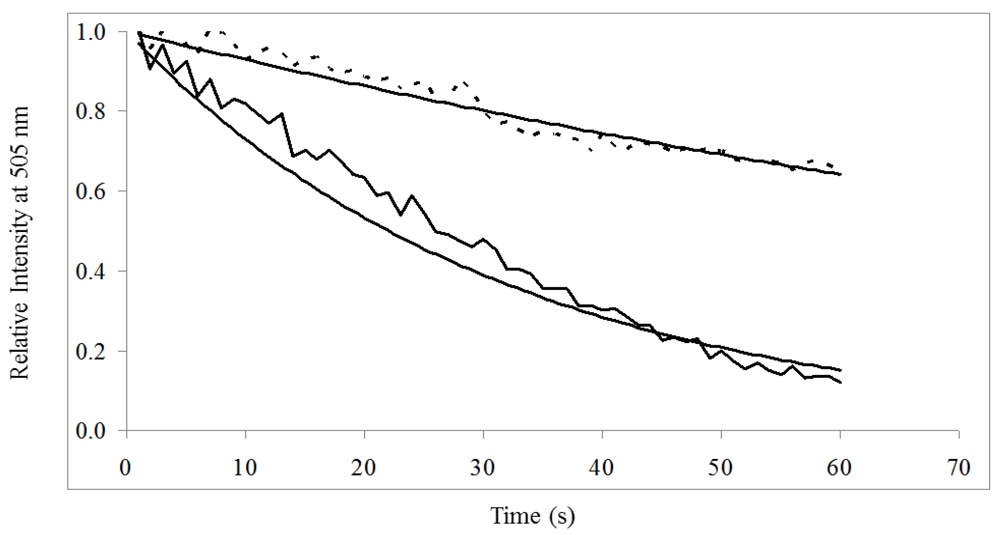
3.3. On-Chip Probe-Based Identification: FRET-ISH
 ) was significantly greater than donor in the presence of acceptor (- - -) when excited at 485/20 nm for 60 s.
) was significantly greater than donor in the presence of acceptor (- - -) when excited at 485/20 nm for 60 s.
) was significantly greater than donor in the presence of acceptor (- - -) when excited at 485/20 nm for 60 s.
) was significantly greater than donor in the presence of acceptor (- - -) when excited at 485/20 nm for 60 s.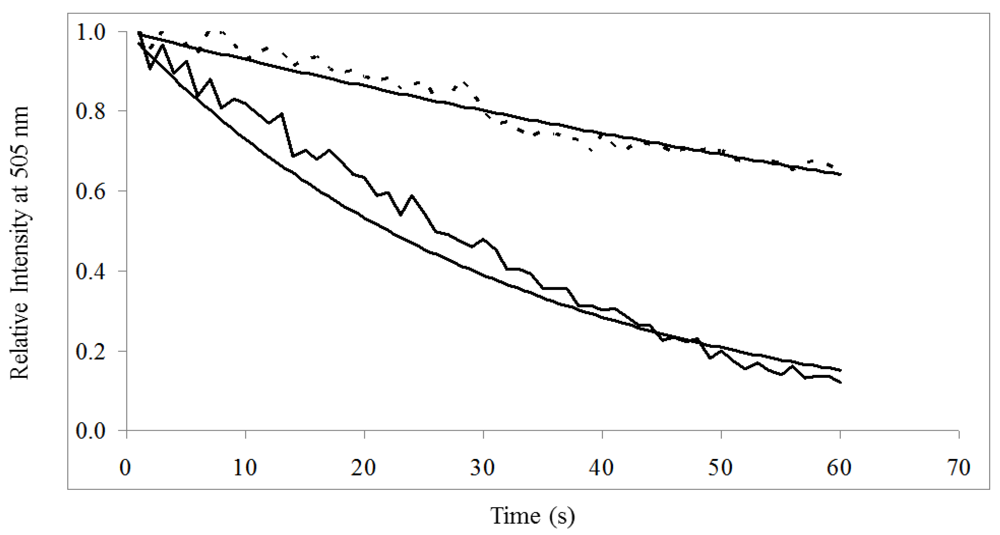
| SYTO®-9 alone photobleaching decay time constant (τpb) | 31.8 s |
| SYTO®-9 with bound probe photobleaching decay time constant (τ'pb) | 135.1 s |
| FRET Efficiency (E) | 76.4% |
4. Conclusions
Acknowledgments
References
- Torres, C.E.; Gibello, A.; Nande, M.; Martin, M.; Blanco, A. Fluorescent in situ hybridization and flow cytometry as tools to evaluate the treatments for the control of slime-forming enterobacteria in paper mills. Appl. Microbiol. Biotechnol. 2008, 78, 889–897. [Google Scholar] [CrossRef]
- Amann, R.; Fuchs, B.M. Single-cell identification in microbial communities by improved fluorescence in situ hybridization techniques. Nat. Rev. Micro. 2008, 6, 339–348. [Google Scholar] [CrossRef]
- Chen, X.; Zhang, X.E.; Chai, Y.Q.; Hu, W.P.; Zhang, Z.P.; Zhang, X.M.; Cass, A.E. DNA optical sensor: A rapid method for the detection of DNA hybridization. Biosens. Bioelectron. 1998, 13, 451–458. [Google Scholar] [CrossRef]
- Liu, W.; Kim, H.J.; Lucchetta, E.M.; Du, W.; Ismagilov, R.F. Isolation, incubation, and parallel functional testing and identification by FISH of rare microbial single-copy cells from multi-species mixtures using the combination of chemistrode and stochastic confinement. Lab Chip 2009, 9, 2153–2162. [Google Scholar] [CrossRef]
- Matsunaga, T.; Hosokawa, M.; Arakaki, A.; Taguchi, T.; Mori, T.; Tanaka, T.; Takeyama, H. High-efficiency single-cell entrapment and fluorescence in situ hybridization analysis using a poly(dimethylsiloxane) microfluidic device integrated with a black poly(ethylene terephthalate) micromesh. Anal. Chem. 2008, 80, 5139–5145. [Google Scholar]
- Chen, L.; Lee, S.; Lee, M.; Lim, C.; Choo, J.; Park, J.Y.; Lee, S.; Joo, S.W.; Lee, K.H.; Choi, Y.W. DNA hybridization detection in a microfluidic channel using two fluorescently labelled nucleic acid probes. Biosens. Bioelectron. 2008, 23, 1878–1882. [Google Scholar] [CrossRef]
- Lantz, A.W.; Brehm-Stecher, B.F.; Armstrong, D.W. Combined capillary electrophoresis and DNA-fluorescence in situ hybridization for rapid molecular identification of Salmonella Typhimurium in mixed culture. Electrophoresis 2008, 29, 2477–2484. [Google Scholar] [CrossRef]
- Sieben, V.J.; Debes Marun, C.S.; Pilarski, P.M.; Kaigala, G.V.; Pilarski, L.M.; Backhouse, C.J. FISH and chips: Chromosomal analysis on microfluidic platforms. IET Nanobiotechnol. 2007, 1, 27–35. [Google Scholar] [CrossRef]
- Sieben, V.J.; Debes-Marun, C.S.; Pilarski, L.M.; Backhouse, C.J. An integrated microfluidic chip for chromosome enumeration using fluorescence in situ hybridization. Lab Chip 2008, 8, 2151–2156. [Google Scholar] [CrossRef]
- Voldman, J. Electrical forces for microscale cell manipulation. Annu. Rev. Biomed. Eng. 2006, 8, 425–454. [Google Scholar] [CrossRef]
- Li, H.; Bashir, R. Dielectrophoretic separation and manipulation of live and heat-treated cells of Listeria on microfabricated devices with interdigitated electrodes. Sens. Actuator. B 2002, 86, 215–221. [Google Scholar] [CrossRef]
- Kim, J.; Johnson, M.; Hill, P.; Gale, B.K. Microfluidic sample preparation: Cell lysis and nucleic acid purification. Integr. Biol. (Camb) 2009, 1, 574–586. [Google Scholar]
- Bao, N.; Lu, C. A microfluidic device for physical trapping and electrical lysis of bacterial cells. Appl. Phys. Lett. 2008, 92, 1–3. [Google Scholar]
- Brown, R.B.; Audet, J. Current techniques for single-cell lysis. J. R. Soc. Interface 2008, 5, S131–S138. [Google Scholar] [CrossRef]
- Chen, D.; Mauk, M.; Qiu, X.; Liu, C.; Kim, J.; Ramprasad, S.; Ongagna, S.; Abrams, W.R.; Malamud, D.; Corstjens, P.L.; Bau, H.H. An integrated, self-contained microfluidic cassette for isolation, amplification, and detection of nucleic acids. Biomed. Microdevices 2010, 12, 705–719. [Google Scholar] [CrossRef]
- Kremers, G.J.; van Munster, E.B.; Goedhart, J.; Gadella, T.W., Jr. Quantitative lifetime unmixing of multiexponentially decaying fluorophores using single-frequency fluorescence lifetime imaging microscopy. Biophys. J. 2008, 95, 378–389. [Google Scholar]
- Young, R.M.; Arnette, J.K.; Roess, D.A.; Barisas, B.G. Quantitation of fluorescence energy transfer between cell surface proteins via fluorescence donor photobleaching kinetics. Biophys. J. 1994, 67, 881–888. [Google Scholar] [CrossRef]
- Berney, M.; Weilenmann, H.U.; Egli, T. Flow-cytometric study of vital cellular functions in Escherichia coli during solar disinfection (SODIS). Microbiology 2006, 152, 1719–1729. [Google Scholar] [CrossRef]
- Abramoff, M.D.; Magalhaes, P.J.; Ram, S.J. Image processing with ImageJ. Biophotonics Int. 2004, 11, 36–42. [Google Scholar]
- Landini, G. Threshold_Colour 1.12. Available online: http://www.dentistry.bham.ac.uk/landinig/software/software.html (accessed on 20 August 2012).
- Gordon, G.W.; Berry, G.; Liang, X.H.; Levine, B.; Herman, B. Quantitative fluorescence resonance energy transfer measurements using fluorescence microscopy. Biophys. J. 1998, 74, 2702–2713. [Google Scholar]
- Kubitscheck, U.; Kircheis, M.; Schweitzerstenner, R.; Dreybrodt, W.; Jovin, T.M.; Pecht, I. Fluorescence resonance energy-transfer on single living cells. Biophys. J. 1991, 60, 307–318. [Google Scholar] [CrossRef]
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Packard, M.M.; Shusteff, M.; Alocilja, E.C. Microfluidic-Based Amplification-Free Bacterial DNA Detection by Dielectrophoretic Concentration and Fluorescent Resonance Energy Transfer Assisted in Situ Hybridization (FRET-ISH). Biosensors 2012, 2, 405-416. https://doi.org/10.3390/bios2040405
Packard MM, Shusteff M, Alocilja EC. Microfluidic-Based Amplification-Free Bacterial DNA Detection by Dielectrophoretic Concentration and Fluorescent Resonance Energy Transfer Assisted in Situ Hybridization (FRET-ISH). Biosensors. 2012; 2(4):405-416. https://doi.org/10.3390/bios2040405
Chicago/Turabian StylePackard, Michelle M., Maxim Shusteff, and Evangelyn C. Alocilja. 2012. "Microfluidic-Based Amplification-Free Bacterial DNA Detection by Dielectrophoretic Concentration and Fluorescent Resonance Energy Transfer Assisted in Situ Hybridization (FRET-ISH)" Biosensors 2, no. 4: 405-416. https://doi.org/10.3390/bios2040405