Cross-talk between Bcr-abl and the Thioredoxin System in Chronic Myeloid Leukaemia: Implications for CML Treatment

 and
and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Lines and Reagents
2.2. Cell Viability Assay
2.3. Development of Imatinib Resistant CML Cell Lines
2.4. Transient Transfections
2.5. MTT Cell Proliferation Assay
2.6. Caspase-3 Activity Assay
2.7. Thioredoxin Reductase Activity Assay
2.8. ROS Levels
2.9. Reverse Transcriptase Quantitative PCR (RT-qPCR)
2.10. Western Blotting
2.11. Bioinformatics
2.12. Statistical Analysis
3. Results
3.1. Auranofin and [Au(d2pype)2]Cl Induce Apoptosis in CML Cells
3.2. Lowered TrxR Activity Via Auranofin and [Au(d2pype)2]Cl Results in Increased ROS
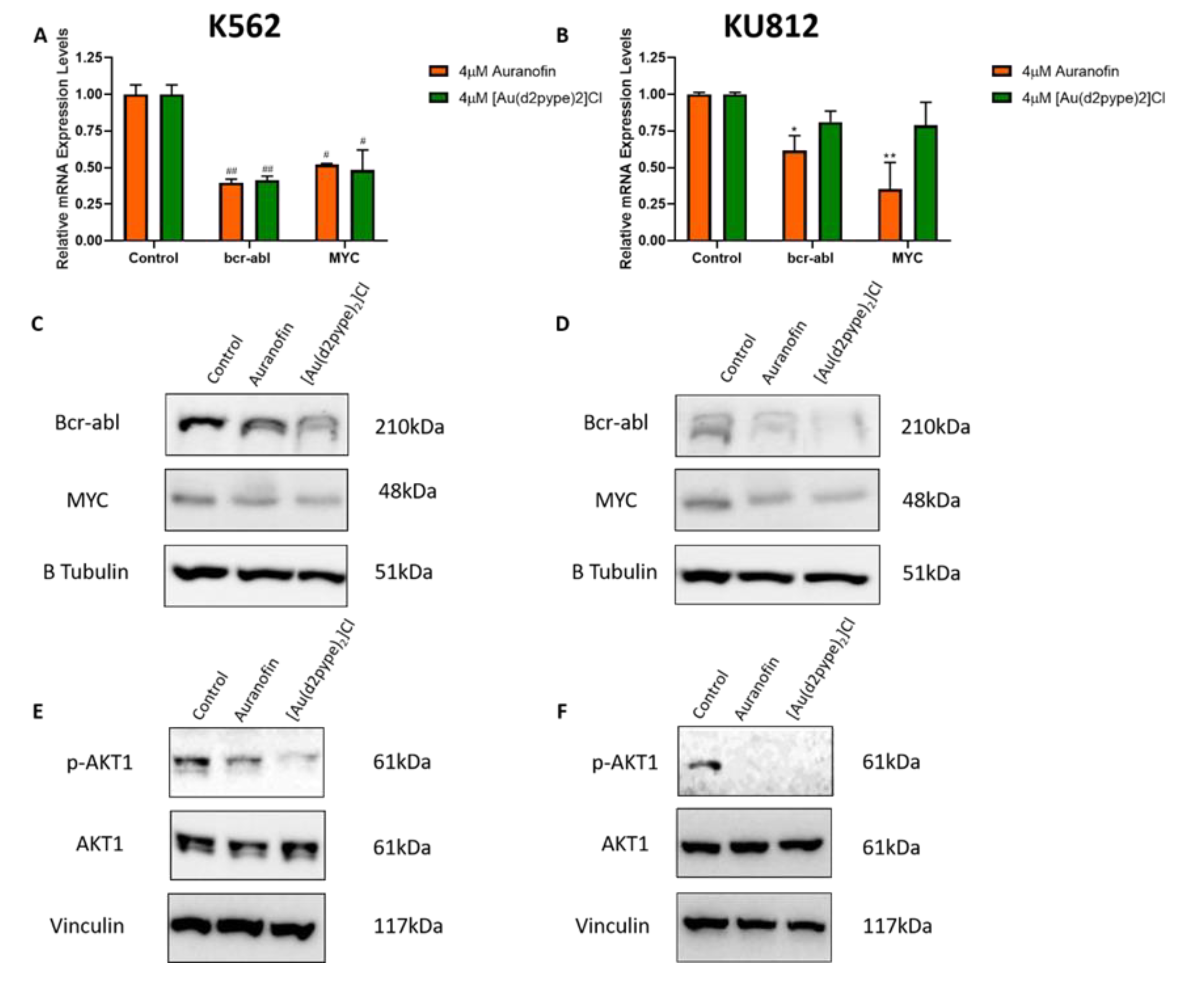
3.3. Bcr-abl Expression is Linked to Activity of the Trx System
3.4. Knockdown of TrxR1 in CML cells Results in the Decreased Expression of Bcr-abl
3.5. Imatinib Resistant CML Cells have Upregulated Levels of the Trx System
3.6. Imatinib Resistant CML Cells are Sensitive to TrxR Inhibitors
4. Discussion
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Apperley, J.F. Part I: Mechanisms of resistance to imatinib in chronic myeloid leukaemia. Lancet Oncol. 2007, 8, 1018–1029. [Google Scholar] [CrossRef]
- Mahon, G.M.; Wang, Y.; Korus, M.; Kostenko, E.; Cheng, L.; Sun, T.; Arlinghaus, R.B.; Whitehead, I.P. The c-Myc Oncoprotein Interacts with Bcr. Curr. Biol. 2003, 13, 437–441. [Google Scholar] [CrossRef] [Green Version]
- Sharma, N.; Magistroni, V.; Piazza, R.; Citterio, S.; Mezzatesta, C.; Khandelwal, P.; Pirola, A.; Gambacorti-Passerini, C. BCR/ABL1 and BCR are under the transcriptional control of the MYC oncogene. Mol. Cancer 2015, 14, 132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gomez-Casares, M.T.; Garcia-Alegria, E.; Lopez-Jorge, C.E.; Ferrandiz, N.; Blanco, R.; Alvarez, S.; Vaque, J.P.; Bretones, G.; Caraballo, J.M.; Sanchez-Bailon, P.; et al. MYC antagonizes the differentiation induced by imatinib in chronic myeloid leukemia cells through downregulation of p27(KIP1.). Oncogene 2013, 32, 2239–2246. [Google Scholar] [CrossRef]
- Albajar, M.; Gomez-Casares, M.T.; Llorca, J.; Mauleon, I.; Vaque, J.P.; Acosta, J.C.; Bermudez, A.; Donato, N.; Delgado, M.D.; Leon, J. MYC in chronic myeloid leukemia: Induction of aberrant DNA synthesis and association with poor response to imatinib. Mol. Cancer Res. 2011, 9, 564–576. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Radich, J.P. The Biology of CML blast crisis. Hematol. Am. Soc. Hematol. Educ. Program 2007. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saussele, S.; Silver, R.T. Management of chronic myeloid leukemia in blast crisis. Ann. Hematol. 2015, 94, S159–S165. [Google Scholar] [CrossRef]
- Apperley, J.F. Chronic myeloid leukaemia. Lancet 2015, 385, 1447–1459. [Google Scholar] [CrossRef]
- Geelen, I.G.P.; Thielen, N.; Janssen, J.; Hoogendoorn, M.; Roosma, T.J.A.; Willemsen, S.P.; Visser, O.; Cornelissen, J.J.; Westerweel, P.E. Treatment outcome in a population-based, ‘real-world’ cohort of patients with chronic myeloid leukemia. Haematologica 2017, 102, 1842–1849. [Google Scholar] [CrossRef]
- Gorre, M.E.; Mohammed, M.; Ellwood, K.; Hsu, N.; Paquette, R.; Rao, P.N.; Sawyers, C.L. Clinical resistance to STI-571 cancer therapy caused by BCR-ABL gene mutation or amplification. Science 2001, 293, 876–880. [Google Scholar] [CrossRef] [Green Version]
- Chahardouli, B.; Zaker, F.; Mousavi, S.A.; Kazemi, A.; Ostadali, M.; Nadali, F.; Rostami, S.; Alimoghaddam, K.; Ghavamzade, A. Evaluation of T315I mutation frequency in chronic myeloid leukemia patients after imatinib resistance. Hematology 2013, 18, 158–162. [Google Scholar] [CrossRef] [PubMed]
- Nicolini, F.E.; Corm, S.; Le, Q.H.; Sorel, N.; Hayette, S.; Bories, D.; Leguay, T.; Roy, L.; Giraudier, S.; Tulliez, M.; et al. Mutation status and clinical outcome of 89 imatinib mesylate-resistant chronic myelogenous leukemia patients: A retrospective analysis from the French intergroup of CML (Fi(phi)-LMC GROUP). Leukemia 2006, 20, 1061–1066. [Google Scholar] [CrossRef] [PubMed]
- Sze, J.H.; Raninga, P.V.; Nakamura, K.; Casey, M.; Khanna, K.K.; Berners-Price, S.J.; Di Trapani, G.; Tonissen, K.F. Anticancer activity of a Gold(I) phosphine thioredoxin reductase inhibitor in multiple myeloma. Redox Biol. 2020, 28. [Google Scholar] [CrossRef] [PubMed]
- Marzano, C.; Gandin, V.; Folda, A.; Scutari, G.; Bindoli, A.; Rigobello, M.P. Inhibition of thioredoxin reductase by auranofin induces apoptosis in cisplatin-resistant human ovarian cancer cells. Free Radic. Biol. Med. 2007, 42, 872–881. [Google Scholar] [CrossRef]
- Pessetto, Z.Y.; Weir, S.J.; Sethi, G.; Broward, M.A.; Godwin, A.K. Drug repurposing for gastrointestinal stromal tumor. Mol. Cancer Ther. 2013, 12, 1299–1309. [Google Scholar] [CrossRef] [Green Version]
- Hwang-Bo, H.; Jeong, J.W.; Han, M.H.; Park, C.; Hong, S.H.; Kim, G.Y.; Moon, S.K.; Cheong, J.; Kim, W.J.; Yoo, Y.H.; et al. Auranofin, an inhibitor of thioredoxin reductase, induces apoptosis in hepatocellular carcinoma Hep3B cells by generation of reactive oxygen species. Gen. Physiol. Biophys. 2017, 36, 117–128. [Google Scholar] [CrossRef] [Green Version]
- Rackham, O.; Shearwood, A.M.; Thyer, R.; McNamara, E.; Davies, S.M.; Callus, B.A.; Miranda-Vizuete, A.; Berners-Price, S.J.; Cheng, Q.; Arner, E.S.; et al. Substrate and inhibitor specificities differ between human cytosolic and mitochondrial thioredoxin reductases: Implications for development of specific inhibitors. Free Radic. Biol. Med. 2011, 50, 689–699. [Google Scholar] [CrossRef]
- Stafford, W.C.; Peng, X.; Olofsson, M.H.; Zhang, X.; Luci, D.K.; Lu, L.; Cheng, Q.; Tresaugues, L.; Dexheimer, T.S.; Coussens, N.P.; et al. Irreversible inhibition of cytosolic thioredoxin reductase 1 as a mechanistic basis for anticancer therapy. Sci. Transl. Med. 2018, 10. [Google Scholar] [CrossRef] [Green Version]
- Raninga, P.V.; Di Trapani, G.; Vuckovic, S.; Tonissen, K.F. TrxR1 inhibition overcomes both hypoxia-induced and acquired bortezomib resistance in multiple myeloma through NF-kB Inhibition. Cell Cycle 2016, 15, 559–572. [Google Scholar] [CrossRef] [Green Version]
- Jost, P.J.; Ruland, J. Aberrant NF-kappaB signaling in lymphoma: Mechanisms, consequences, and therapeutic implications. Blood 2007, 109, 2700–2707. [Google Scholar] [CrossRef] [Green Version]
- Soni, S.; Padwad, Y.S. HIF-1 in cancer therapy: Two decade long story of a transcription factor. Acta Oncol. 2017, 56, 503–515. [Google Scholar] [CrossRef] [PubMed]
- Evens, A.M.; Schumacker, P.T.; Helenowski, I.B.; Singh, A.T.; Dokic, D.; Keswani, A.; Kordeluk, E.; Raji, A.; Winter, J.N.; Jovanovic, B.D.; et al. Hypoxia inducible factor-alpha activation in lymphoma and relationship to the thioredoxin family. Br. J. Haematol. 2008, 141, 676–680. [Google Scholar] [CrossRef] [Green Version]
- Patwari, P.; Higgins, L.J.; Chutkow, W.A.; Yoshioka, J.; Lee, R.T. The interaction of thioredoxin with Txnip. Evidence for formation of a mixed disulfide by disulfide exchange. J. Biol. Chem. 2006, 281, 21884–21891. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morrison, J.A.; Pike, L.A.; Sams, S.B.; Sharma, V.; Zhou, Q.; Severson, J.J.; Tan, A.C.; Wood, W.M.; Haugen, B.R. Thioredoxin interacting protein (TXNIP) is a novel tumor suppressor in thyroid cancer. Mol. Cancer 2014, 13, 62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, P.; Gao, J.; Wang, X.; Wen, W.; Yang, H.; Tian, Y.; Liu, N.; Wang, Z.; Liu, H.; Zhang, Y.; et al. A novel indication of thioredoxin-interacting protein as a tumor suppressor gene in malignant glioma. Oncol. Lett. 2017, 14, 2053–2058. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bian, M.; Fan, R.; Zhao, S.; Liu, W. Targeting the Thioredoxin System as a Strategy for Cancer Therapy. J. Med. Chem. 2019, 62, 7309–7321. [Google Scholar] [CrossRef]
- Zhang, J.; Li, X.; Han, X.; Liu, R.; Fang, J. Targeting the Thioredoxin System for Cancer Therapy. Trends Pharmacol. Sci. 2017, 38, 794–808. [Google Scholar] [CrossRef]
- Casini, A.; Gabbiani, C.; Sorrentino, F.; Rigobello, M.P.; Bindoli, A.; Geldbach, T.J.; Marrone, A.; Re, N.; Hartinger, C.G.; Dyson, P.J.; et al. Emerging protein targets for anticancer metallodrugs: Inhibition of thioredoxin reductase and cathepsin B by antitumor ruthenium(II)-arene compounds. J. Med. Chem. 2008, 51, 6773–6781. [Google Scholar] [CrossRef]
- Prast-Nielsen, S.; Cebula, M.; Pader, I.; Arner, E.S. Noble metal targeting of thioredoxin reductase—Covalent complexes with thioredoxin and thioredoxin-related protein of 14 kDa triggered by cisplatin. Free Radic. Biol. Med. 2010, 49, 1765–1778. [Google Scholar] [CrossRef]
- Rackham, O.; Nichols, S.J.; Leedman, P.J.; Berners-Price, S.J.; Filipovska, A. A gold(I) phosphine complex selectively induces apoptosis in breast cancer cells: Implications for anticancer therapeutics targeted to mitochondria. Biochem. Pharmacol. 2007, 74, 992–1002. [Google Scholar] [CrossRef]
- Rigobello, M.P.; Folda, A.; Baldoin, M.C.; Scutari, G.; Bindoli, A. Effect of auranofin on the mitochondrial generation of hydrogen peroxide. Role of thioredoxin reductase. Free Radic. Res. 2005, 39, 687–695. [Google Scholar] [CrossRef] [PubMed]
- Rigobello, M.P.; Scutari, G.; Folda, A.; Bindoli, A. Mitochondrial thioredoxin reductase inhibition by gold(I) compounds and concurrent stimulation of permeability transition and release of cytochrome c. Biochem. Pharmacol. 2004, 67, 689–696. [Google Scholar] [CrossRef] [PubMed]
- Parsonage, D.; Sheng, F.; Hirata, K.; Debnath, A.; McKerrow, J.H.; Reed, S.L.; Abagyan, R.; Poole, L.B.; Podust, L.M. X-ray structures of thioredoxin and thioredoxin reductase from Entamoeba histolytica and prevailing hypothesis of the mechanism of Auranofin action. J. Struct. Biol. 2016, 194, 180–190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pickering, I.J.; Cheng, Q.; Rengifo, E.M.; Nehzati, S.; Dolgova, N.V.; Kroll, T.; Sokaras, D.; George, G.N.; Arnér, E.S.J. Direct Observation of Methylmercury and Auranofin Binding to Selenocysteine in Thioredoxin Reductase. Inorg. Chem. 2020. [Google Scholar] [CrossRef]
- Park, S.H.; Lee, J.H.; Berek, J.S.; Hu, M.C. Auranofin displays anticancer activity against ovarian cancer cells through FOXO3 activation independent of p53. Int. J. Oncol. 2014, 45, 1691–1698. [Google Scholar] [CrossRef] [Green Version]
- Hatem, E.; Azzi, S.; El Banna, N.; He, T.; Heneman-Masurel, A.; Vernis, L.; Baille, D.; Masson, V.; Dingli, F.; Loew, D.; et al. Auranofin/Vitamin C: A Novel Drug Combination Targeting Triple-Negative Breast Cancer. J. Natl. Cancer Inst. 2018. [Google Scholar] [CrossRef]
- Lozzio, C.B.; Lozzio, B.B. Human chronic myelogenous leukemia cell-line with positive Philadelphia chromosome. Blood 1975, 45, 321–334. [Google Scholar] [CrossRef] [Green Version]
- Kishi, K. A new leukemia cell line with Philadelphia chromosome characterized as basophil precursors. Leuk. Res. 1985, 9, 381–390. [Google Scholar] [CrossRef]
- Berners-Price, S.J.; Bowen, R.J.; Hambley, T.W.; Healy, P.C. NMR and structural studies of gold(I) chloride adducts with bidentate 2-, 3- and 4-pyridyl phosphines. J. Chem. Soc. Dalton Trans. 1999. [Google Scholar] [CrossRef]
- Raninga, P.V.; Di Trapani, G.; Vuckovic, S.; Bhatia, M.; Tonissen, K.F. Inhibition of thioredoxin 1 leads to apoptosis in drug-resistant multiple myeloma. Oncotarget 2015, 6, 15410–15424. [Google Scholar] [CrossRef] [Green Version]
- Cox, A.G.; Brown, K.K.; Arner, E.S.; Hampton, M.B. The thioredoxin reductase inhibitor auranofin triggers apoptosis through a Bax/Bak-dependent process that involves peroxiredoxin 3 oxidation. Biochem. Pharmacol. 2008, 76, 1097–1109. [Google Scholar] [CrossRef]
- Smith, A.D.; Levander, O.A. High-throughput 96-well microplate assays for determining specific activities of glutathione peroxidase and thioredoxin reductase. Methods Enzymol. 2002, 347, 113–121. [Google Scholar]
- Rushworth, S.A.; MacEwan, D.J.; O’Connell, M.A. Lipopolysaccharide-induced expression of NAD(P)H:quinone oxidoreductase 1 and heme oxygenase-1 protects against excessive inflammatory responses in human monocytes. J. Immunol. 2008, 181, 6730–6737. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karlenius, T.C.; Shah, F.; Di Trapani, G.; Clarke, F.M.; Tonissen, K.F. Cycling hypoxia up-regulates thioredoxin levels in human MDA-MB-231 breast cancer cells. Biochem. Biophys. Res. Commun. 2012, 419, 350–355. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crossman, L.C.; Mori, M.; Hsieh, Y.C.; Lange, T.; Paschka, P.; Harrington, C.A.; Krohn, K.; Niederwieser, D.W.; Hehlmann, R.; Hochhaus, A.; et al. In chronic myeloid leukemia white cells from cytogenetic responders and non-responders to imatinib have very similar gene expression signatures. Haematologica 2005, 90, 459–464. [Google Scholar]
- Liu, J.J.; Liu, Q.; Wei, H.L.; Yi, J.; Zhao, H.S.; Gao, L.P. Inhibition of thioredoxin reductase by auranofin induces apoptosis in adriamycin-resistant human K562 chronic myeloid leukemia cells. Pharmazie 2011, 66, 440–444. [Google Scholar]
- Tonissen, K.F.; Di Trapani, G. Thioredoxin system inhibitors as mediators of apoptosis for cancer therapy. Mol. Nutr. Food Res. 2009, 53, 87–103. [Google Scholar] [CrossRef]
- Chen, X.; Shi, X.; Zhao, C.; Li, X.; Lan, X.; Liu, S.; Huang, H.; Liu, N.; Liao, S.; Zang, D.; et al. Anti-rheumatic agent auranofin induced apoptosis in chronic myeloid leukemia cells resistant to imatinib through both Bcr/Abl-dependent and -independent mechanisms. Oncotarget 2014, 5, 9118–9132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Butler, A.M.; Scotti Buzhardt, M.L.; Erdogan, E.; Li, S.; Inman, K.S.; Fields, A.P.; Murray, N.R. A small molecule inhibitor of atypical protein kinase C signaling inhibits pancreatic cancer cell transformed growth and invasion. Oncotarget 2015, 6, 15297–15310. [Google Scholar] [CrossRef]
- Abraham, J.; Salama, N.N.; Azab, A.K. The role of P-glycoprotein in drug resistance in multiple myeloma. Leuk. Lymphoma 2015, 56, 26–33. [Google Scholar] [CrossRef]
- Circu, M.L.; Aw, T.Y. Reactive oxygen species, cellular redox systems, and apoptosis. Free Radic. Biol. Med. 2010, 48, 749–762. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Redza-Dutordoir, M.; Averill-Bates, D.A. Activation of apoptosis signalling pathways by reactive oxygen species. Biochim. Biophys. Acta 2016, 1863, 2977–2992. [Google Scholar] [CrossRef] [PubMed]
- Gorrini, C.; Harris, I.S.; Mak, T.W. Modulation of oxidative stress as an anticancer strategy. Nat. Rev. Drug Discov. 2013, 12, 931–947. [Google Scholar] [CrossRef] [PubMed]
- Locy, M.L.; Rogers, L.K.; Prigge, J.R.; Schmidt, E.E.; Arner, E.S.; Tipple, T.E. Thioredoxin reductase inhibition elicits Nrf2-mediated responses in Clara cells: Implications for oxidant-induced lung injury. Antioxid Redox Signal 2012, 17, 1407–1416. [Google Scholar] [CrossRef] [Green Version]
- Lu, J.; Chew, E.H.; Holmgren, A. Targeting thioredoxin reductase is a basis for cancer therapy by arsenic trioxide. Proc. Natl. Acad. Sci. USA 2007, 104, 12288–12293. [Google Scholar] [CrossRef] [Green Version]
- Meyer, N.; Penn, L.Z. Reflecting on 25 years with MYC. Nat. Rev. Cancer 2008, 8, 976–990. [Google Scholar] [CrossRef]
- Schaub, F.X.; Dhankani, V.; Berger, A.C.; Trivedi, M.; Richardson, A.B.; Shaw, R.; Zhao, W.; Zhang, X.; Ventura, A.; Liu, Y.; et al. Pan-cancer Alterations of the MYC Oncogene and Its Proximal Network across the Cancer Genome Atlas. Cell Syst. 2018, 6, 282–300. [Google Scholar] [CrossRef] [Green Version]
- Cancer Genome Atlas, N. Comprehensive molecular characterization of human colon and rectal cancer. Nature 2012, 487, 330–337. [Google Scholar] [CrossRef] [Green Version]
- Tsiatis, A.C.; Herceg, M.E.; Keedy, V.L.; Halpern, J.L.; Holt, G.E.; Schwartz, H.S.; Cates, J.M. Prognostic significance of c-Myc expression in soft tissue leiomyosarcoma. Mod. Pathol. 2009, 22, 1432–1438. [Google Scholar] [CrossRef] [Green Version]
- Manning, B.D.; Toker, A. AKT/PKB Signaling: Navigating the Network. Cell 2017, 169, 381–405. [Google Scholar] [CrossRef] [Green Version]
- Angeles-Velazquez, J.L.; Hurtado-Monroy, R.; Vargas-Viveros, P.; Carrillo-Munoz, S.; Candelaria-Hernandez, M. Imatinib Intolerance Is Associated With Blastic Phase Development in Philadelphia Chromosome-Positive Chronic Myeloid Leukemia. Clin. Lymphoma Myeloma Leuk. 2016, 16, S82–S85. [Google Scholar] [CrossRef]
- Kim, S.J.; Miyoshi, Y.; Taguchi, T.; Tamaki, Y.; Nakamura, H.; Yodoi, J.; Kato, K.; Noguchi, S. High thioredoxin expression is associated with resistance to docetaxel in primary breast cancer. Clin. Cancer Res. 2005, 11, 8425–8430. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iwasawa, S.; Yamano, Y.; Takiguchi, Y.; Tanzawa, H.; Tatsumi, K.; Uzawa, K. Upregulation of thioredoxin reductase 1 in human oral squamous cell carcinoma. Oncol. Rep. 2011, 25, 637–644. [Google Scholar] [CrossRef]
- Welsh, S.J.; Bellamy, W.T.; Briehl, M.M.; Powis, G. The redox protein thioredoxin-1 (Trx-1) increases hypoxia-inducible factor 1alpha protein expression: Trx-1 overexpression results in increased vascular endothelial growth factor production and enhanced tumor angiogenesis. Cancer Res. 2002, 62, 5089–5095. [Google Scholar] [PubMed]
- Naranjo-Suarez, S.; Carlson, B.A.; Tobe, R.; Yoo, M.H.; Tsuji, P.A.; Gladyshev, V.N.; Hatfield, D.L. Regulation of HIF-1alpha activity by overexpression of thioredoxin is independent of thioredoxin reductase status. Mol. Cells 2013, 36, 151–157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hayashi, T.; Ueno, Y.; Okamoto, T. Oxidoreductive regulation of nuclear factor kappa B. Involvement of a cellular reducing catalyst thioredoxin. J. Biol. Chem. 1993, 268, 11380–11388. [Google Scholar]
- Morgan, M.J.; Liu, Z.G. Crosstalk of reactive oxygen species and NF-kappaB signaling. Cell Res. 2011, 21, 103–115. [Google Scholar] [CrossRef] [Green Version]
- Nagaraj, K.; Lapkina-Gendler, L.; Sarfstein, R.; Gurwitz, D.; Pasmanik-Chor, M.; Laron, Z.; Yakar, S.; Werner, H. Identification of thioredoxin-interacting protein (TXNIP) as a downstream target for IGF1 action. Proc. Natl. Acad. Sci. USA 2018, 115, 1045–1050. [Google Scholar] [CrossRef] [Green Version]






© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Clapper, E.; Wang, S.; Raninga, P.V.; Di Trapani, G.; Tonissen, K.F. Cross-talk between Bcr-abl and the Thioredoxin System in Chronic Myeloid Leukaemia: Implications for CML Treatment. Antioxidants 2020, 9, 207. https://doi.org/10.3390/antiox9030207
Clapper E, Wang S, Raninga PV, Di Trapani G, Tonissen KF. Cross-talk between Bcr-abl and the Thioredoxin System in Chronic Myeloid Leukaemia: Implications for CML Treatment. Antioxidants. 2020; 9(3):207. https://doi.org/10.3390/antiox9030207
Chicago/Turabian StyleClapper, Erin, Sicong Wang, Prahlad V. Raninga, Giovanna Di Trapani, and Kathryn F. Tonissen. 2020. "Cross-talk between Bcr-abl and the Thioredoxin System in Chronic Myeloid Leukaemia: Implications for CML Treatment" Antioxidants 9, no. 3: 207. https://doi.org/10.3390/antiox9030207