Dried Yeast Extracts Curtails Pulmonary Oxidative Stress, Inflammation and Tissue Destruction in a Model of Experimental Emphysema

Abstract
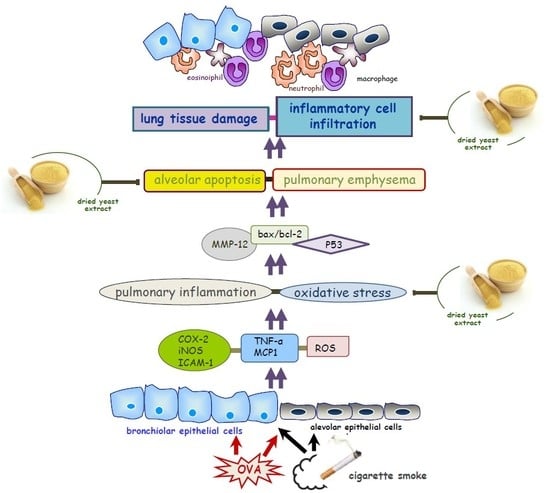
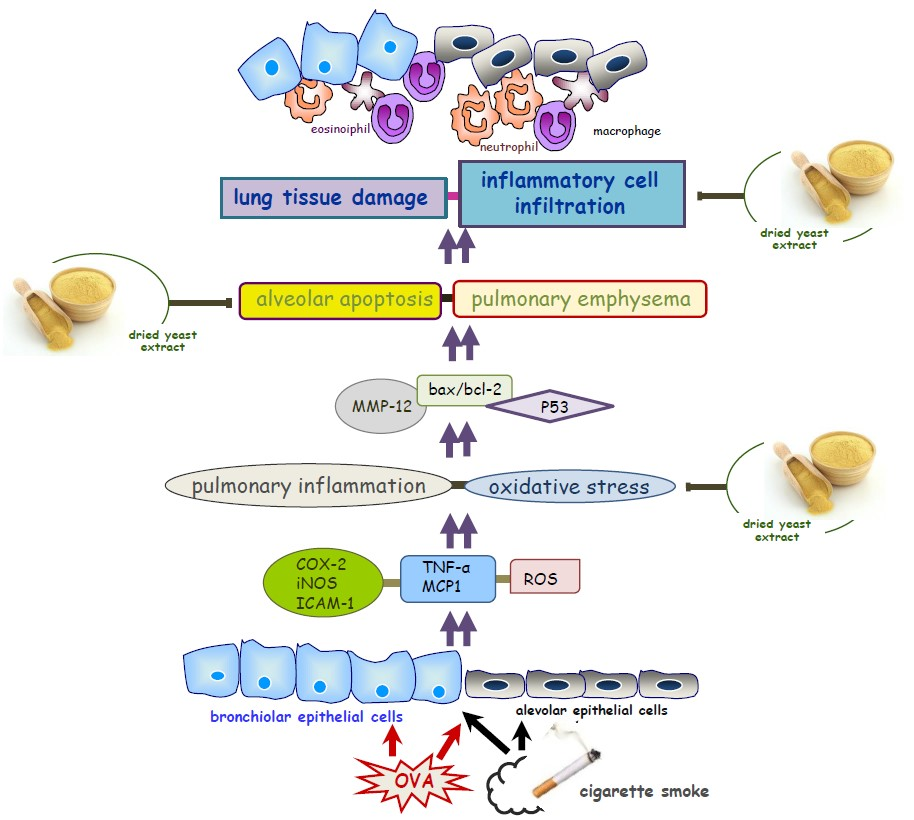{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Chemicals
2.2. Preparation of YE
2.3. Animal Experiments
2.4. Preparation of CSE for Cell Culture
2.5. A549 Cell Culture and Viability
2.6. Staining with Hematoxylin and Eosin (H&E)
2.7. Western Blot Analysis
2.8. Immunohistochemical Staining
2.9. Dihydroethidium (DHE) Staining for Reactive Oxygen Species (ROS) Production
2.10. Hoechst 33258 Staining
2.11. Terminal Deoxynucleotidyl Transferase dUTP Nick End Labeling (TUNEL) Assay
2.12. Enzyme-Linked Immunosorbent Assay (ELISA)
2.13. Immunocytochemical Staining
2.14. Statistical Analysis
3. Results
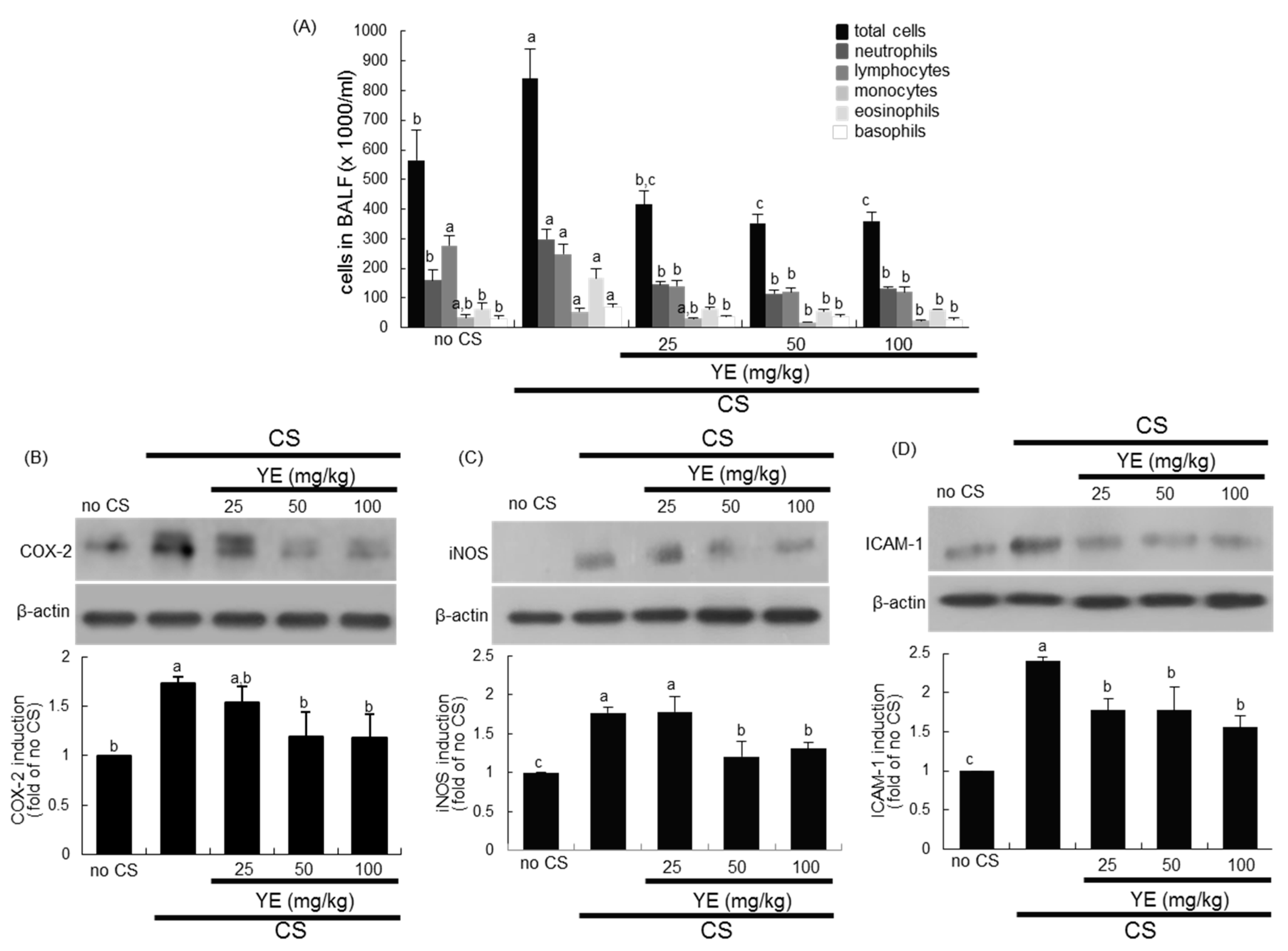
3.1. Inhibition of Airway Inflammation of CS-Exposed Airways by YE
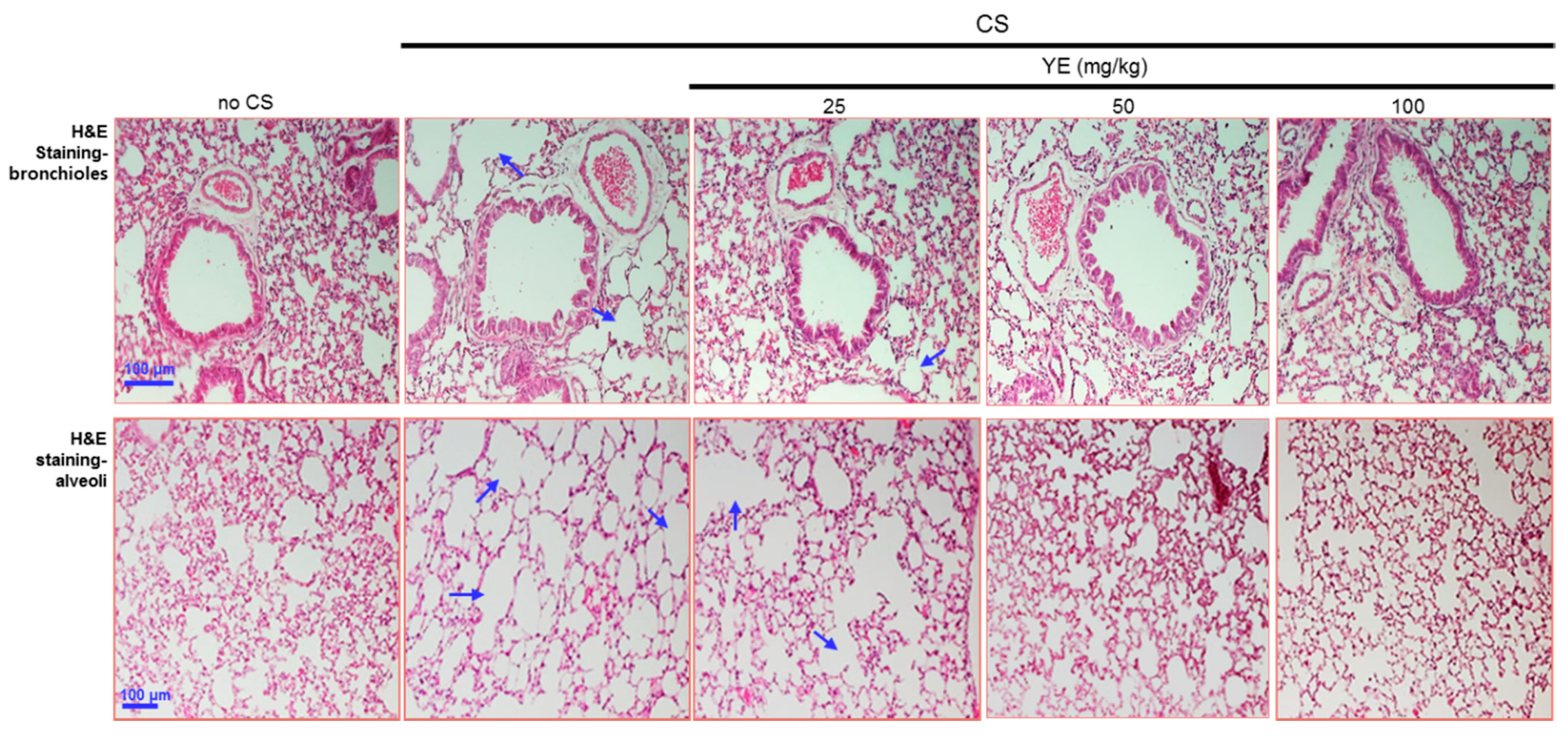
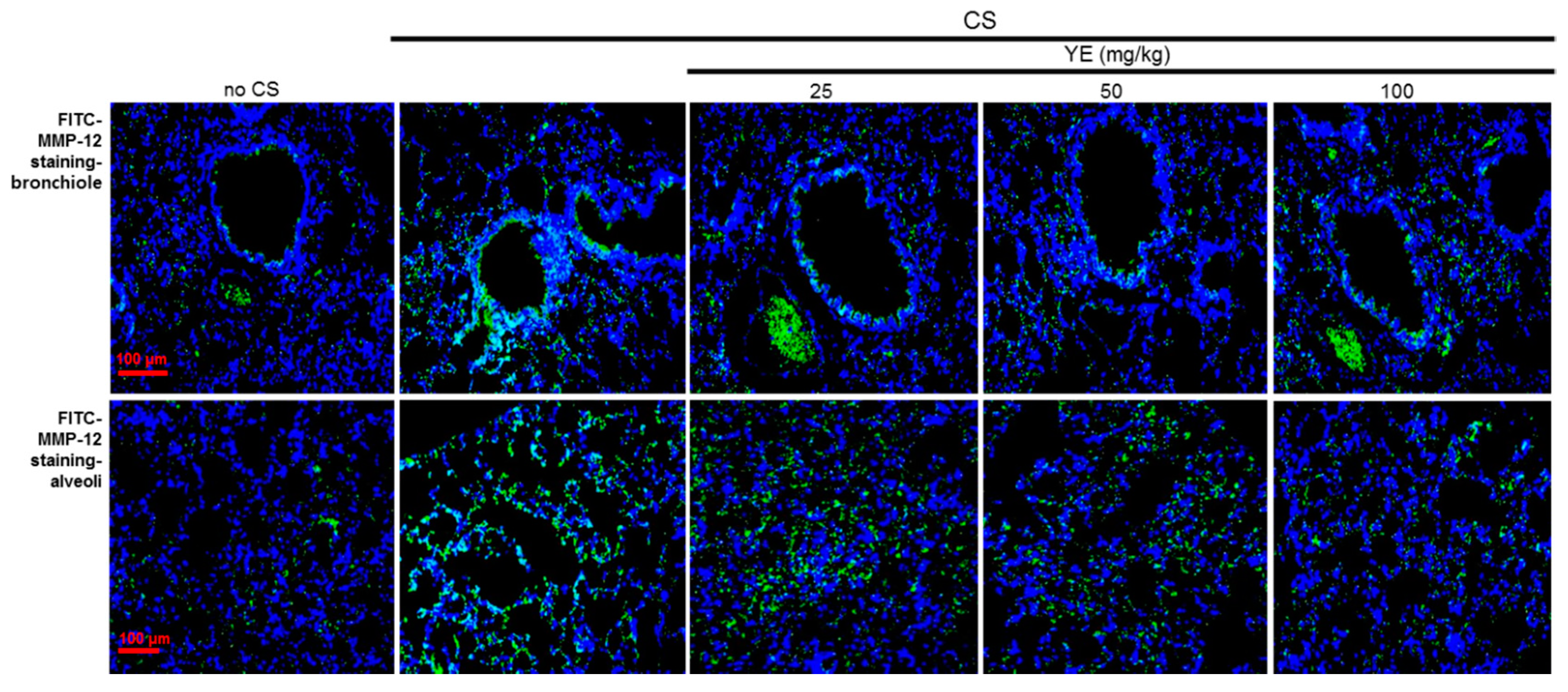
3.2. Blockade of Emphysematous Injury of CS-Challenged Airways by YE
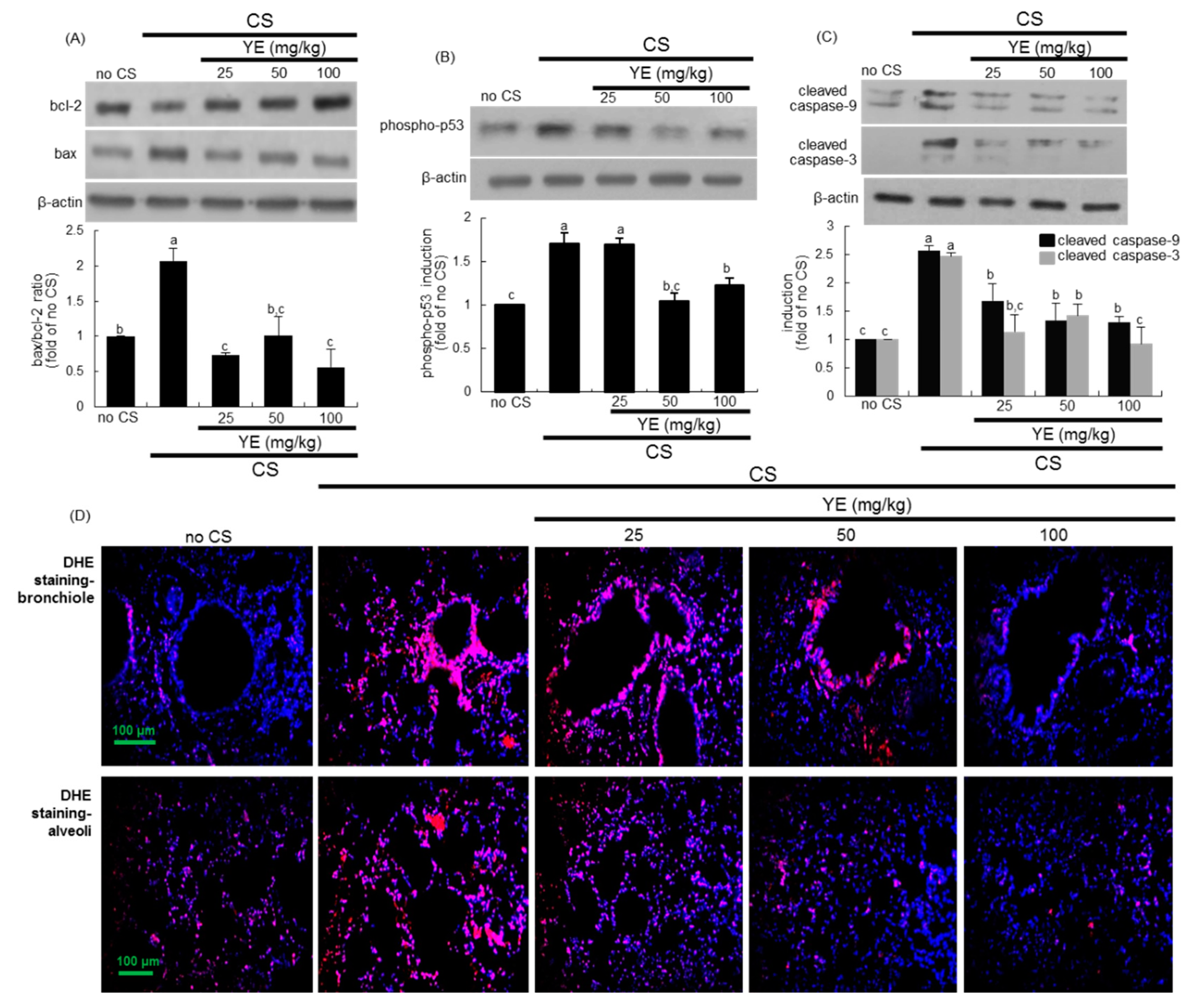
3.3. Inhibitory Effects of YE on CS-Induced Pulmonary Apoptosis and Oxidative Stress
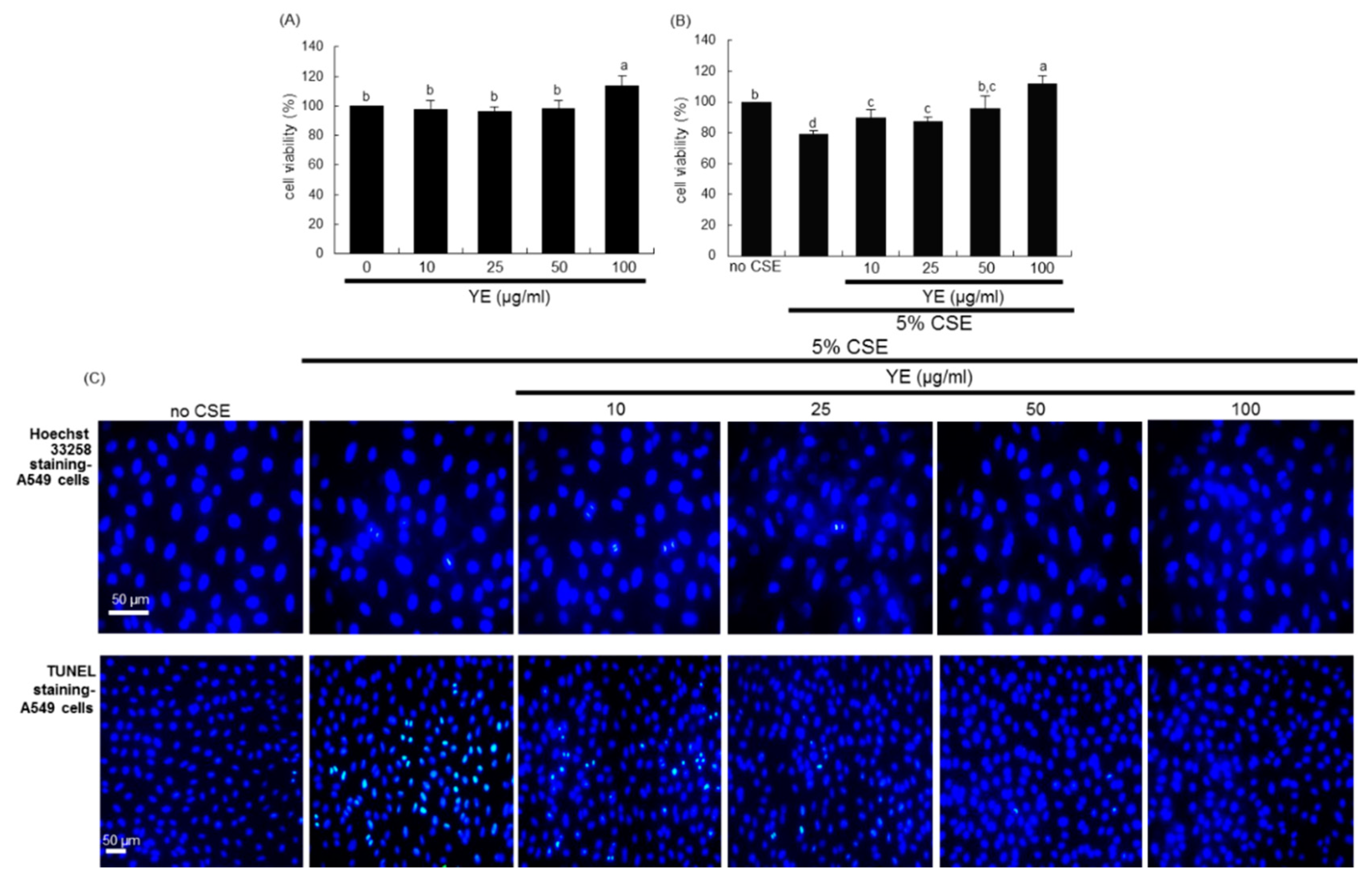
3.4. Suppressive Effects of YE on CSE-Loaded Alveolar Apoptotic Injury
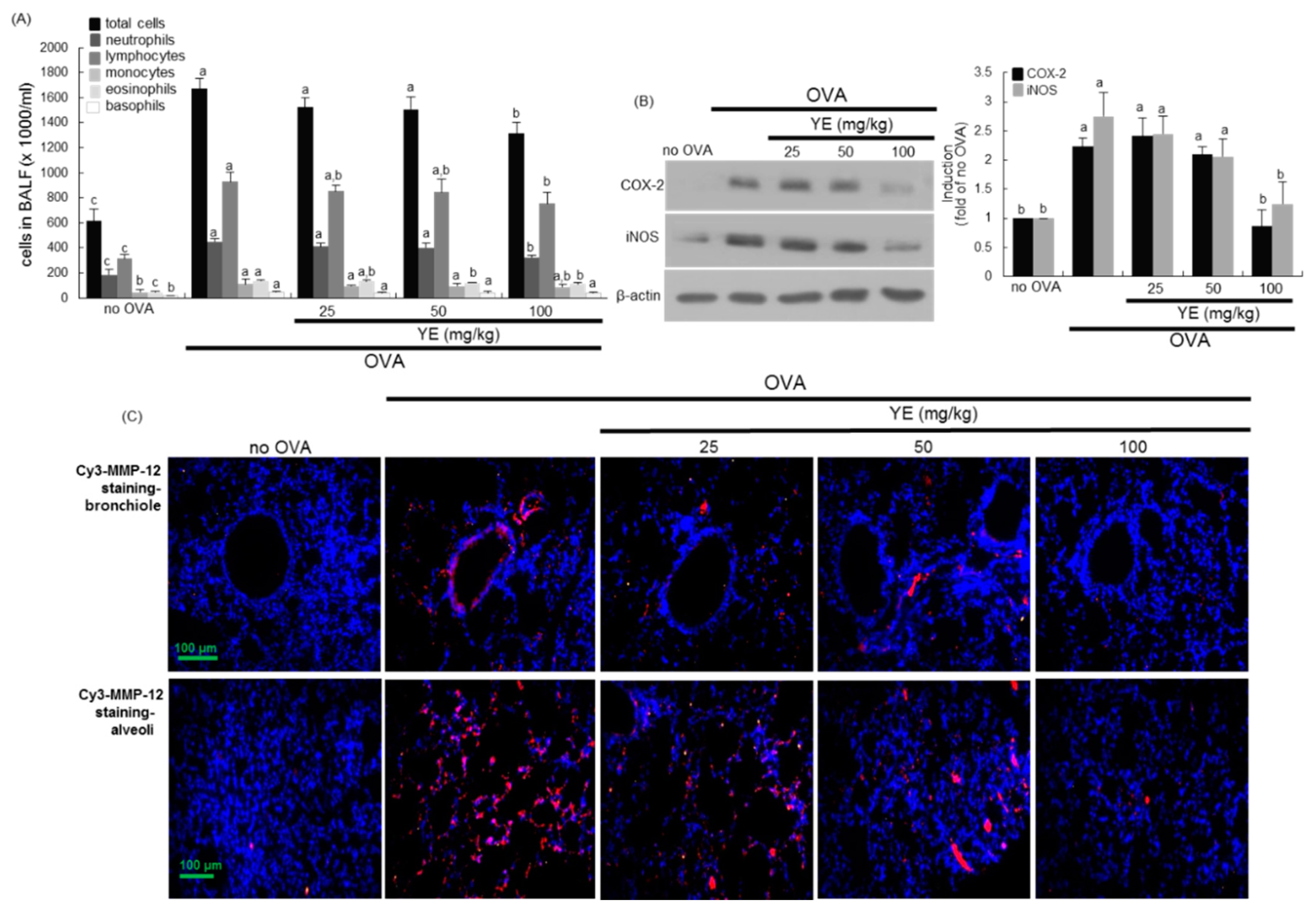
3.5. Inhibition of Airway Inflammation of OVA-Exposed Airways by YE
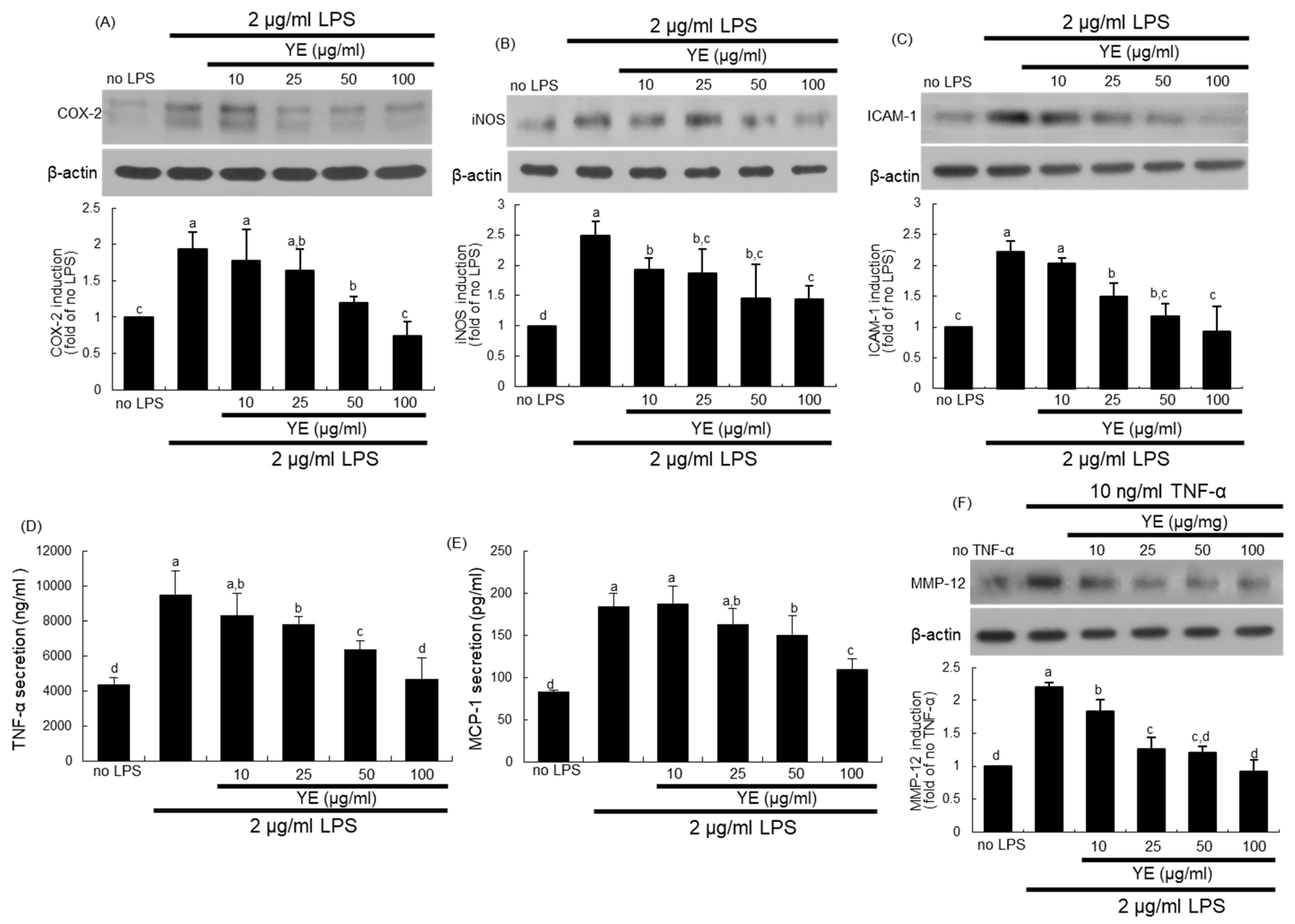
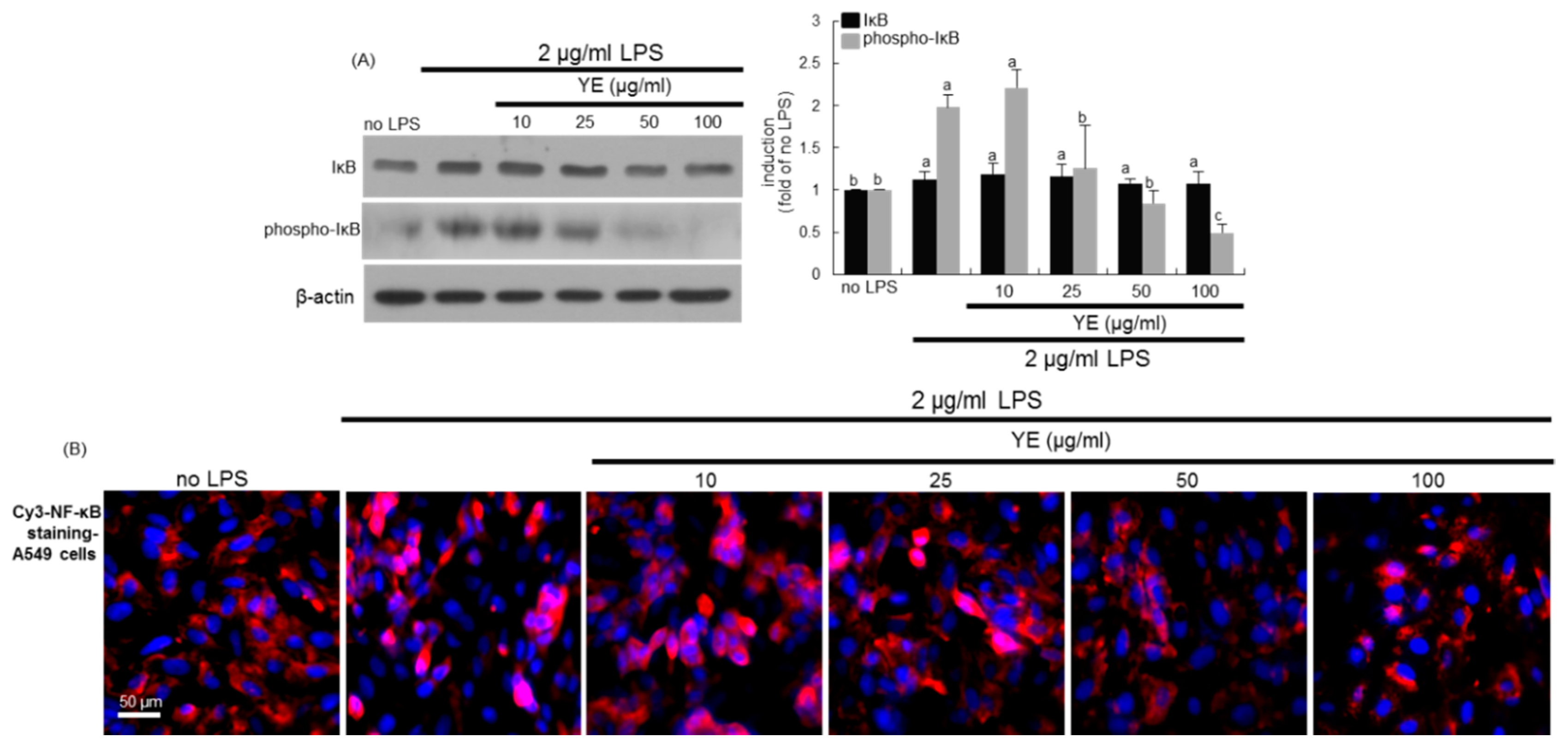
3.6. Blockade of LPS-Triggered Airway Inflammation by YE
4. Discussion
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| COPD | chronic obstructive pulmonary disease |
| COX-2 | cyclooxygenase-2 |
| CS | cigarette smoke |
| CSE | cigarette smoke extracts |
| YE | yeast extracts |
| IL | interleukin |
| iNOS | inducible nitric oxide synthase |
| LPS | lipopolysaccharide |
| MCP-1 | monocyte chemoattractant protein-1 |
| MMP-12 | matrix metalloproteinase-12 |
| NF-κB | nuclear factor-κB |
| OVA | ovalbumin |
| ROS | reactive oxygen species |
| TNF-α | tumor necrosis factor-α |
References
- O’Donnell, R.; Breen, D.; Wilson, S.; Djukanovic, R. Inflammatory cells in the airways in COPD. Thorax 2006, 61, 448–454. [Google Scholar] [CrossRef] [PubMed]
- Rovina, N.; Koutsoukou, A.; Koulouri, N.G. Inflammation and immune response in COPD: Where do we stand? Mediat. Inflamm. 2013, 2013, 413735. [Google Scholar] [CrossRef] [PubMed]
- Angelis, N.; Porpodis, K.; Zarogoulidis, P.; Spyratos, D.; Kioumis, I.; Papaiwannou, A.; Pitsiou, G.; Tsakiridis, K.; Mpakas, A.; Arikas, S.; et al. Airway inflammation in chronic obstructive pulmonary disease. J. Thorac. Dis. 2014, 6, S167–S172. [Google Scholar] [PubMed]
- Yao, H.; Rahman, I. Current concepts on oxidative/carbonyl stress, inflammation and epigenetics in pathogenesis of chronic obstructive pulmonary disease. Toxicol. Appl. Pharmacol. 2011, 254, 72–85. [Google Scholar] [CrossRef] [PubMed]
- Laniado-Laborín, R. Smoking and chronic obstructive pulmonary disease (COPD). Parallel epidemics of the 21st century. Int. J. Environ. Res. Public Health 2009, 6, 209–224. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Xu, J.; Meng, Y.; Adcock, I.M.; Yao, X. Role of inflammatory cells in airway remodeling in COPD. Int. J. Chron. Obstr. Pulm. Dis. 2018, 13, 3341–3348. [Google Scholar] [CrossRef] [PubMed]
- Huang, G.; Xu, X.C.; Zhou, J.S.; Li, Z.Y.; Chen, H.P.; Wang, Y.; Li, W.; Shen, H.H.; Chen, Z.H. Neutrophilic inflammation in the immune responses of chronic obstructive pulmonary disease: Lessons from animal models. J. Immunol. Res. 2017, 2017, 7915975. [Google Scholar] [CrossRef]
- Alkhouri, H.; Poppinga, W.J.; Tania, N.P.; Ammit, A.; Schuliga, M. Regulation of pulmonary inflammation by mesenchymal cells. Pulm. Pharmacol. Ther. 2014, 29, 156–165. [Google Scholar] [CrossRef]
- Meijer, M.; Rijkers, G.T.; van Overveld, F.J. Neutrophils and emerging targets for treatment in chronic obstructive pulmonary disease. Expert Rev. Clin. Immunol. 2013, 9, 1055–1068. [Google Scholar] [CrossRef]
- Fu, P.; Natarajan, V.; Harijith, A. The role of nicotinamide adenine dinucleotide phosphate oxidases in lung architecture remodeling. Antioxidants 2017, 6, 104. [Google Scholar]
- Barnes, P.J. Cellular and molecular mechanisms of chronic obstructive pulmonary disease. Clin. Chest Med. 2014, 35, 71–86. [Google Scholar] [CrossRef]
- Tuder, R.M.; Yoshida, T.; Arap, W.; Pasqualini, R.; Petrache, I. Cellular and molecular mechanisms of alveolar destruction in emphysema. An evolutionary perspective. Proc. Am. Thorac. Soc. 2006, 3, 503–510. [Google Scholar] [CrossRef]
- Kim, V.; Criner, G.J. Chronic bronchitis and chronic obstructive pulmonary disease. Am. J. Respir. Crit. Care Med. 2013, 187, 228–237. [Google Scholar] [CrossRef]
- Roche, N.; Marthan, R.; Berger, P.; Chambellan, A.; Chanez, P.; Aguilaniu, B.; Brillet, P.Y.; Burgel, P.R.; Chaouat, A.; Devillier, P.; et al. Beyond corticosteroids: Future prospects in the management of inflammation in COPD. Eur. Respir. Rev. 2011, 20, 175–182. [Google Scholar] [CrossRef]
- Brusselle, G.; Bracke, K. Targeting immune pathways for therapy in asthma and chronic obstructive pulmonary disease. Ann. Am. Thorac. Soc. 2014, 11 (Suppl. 5), S322–S328. [Google Scholar] [CrossRef]
- Fischer, B.M.; Pavlisko, E.; Voynow, J.A. Pathogenic triad in COPD: Oxidative stress, protease-antiprotease imbalance, and inflammation. Int. J. Chron. Obstr. Pulm. Dis. 2011, 6, 413–421. [Google Scholar] [CrossRef]
- Kirkham, P.A.; Barnes, P.J. Oxidative stress in COPD. Chest 2013, 144, 266–273. [Google Scholar] [CrossRef]
- Lomas, D.A. Does protease-antiprotease imbalance explain chronic obstructive pulmonary disease? Ann. Am. Thorac. Soc. 2016, 13 (Suppl. 2), S130–S137. [Google Scholar]
- Loukides, S.; Bartziokas, K.; Vestbo, J.; Singh, D. Novel anti-inflammatory agents in COPD: Targeting lung and systemic inflammation. Curr. Drug Targets 2013, 14, 235–245. [Google Scholar] [CrossRef]
- Mapel, D.W.; Roberts, M.H. Management of asthma and chronic obstructive pulmonary disease with combination inhaled corticosteroids and long-acting β-agonists: A review of comparative effectiveness research. Drugs 2014, 74, 737–755. [Google Scholar] [CrossRef][Green Version]
- Sharafkhaneh, A.; Velamuri, S.; Badmaev, V.; Lan, C.; Hanania, N. The potential role of natural agents in treatment of airway inflammation. Ther. Adv. Respir. Dis. 2007, 1, 105–120. [Google Scholar] [CrossRef]
- Liu, X.J.; Bao, H.R.; Zeng, X.L.; Wei, J.M. Effects of resveratrol and genistein on nuclear factor-κB, tumor necrosis factor-α and matrix metalloproteinase-9 in patients with chronic obstructive pulmonary disease. Mol. Med. Reports. 2016, 13, 4266–4272. [Google Scholar] [CrossRef]
- Yang, Y.L.; Hsu, H.T.; Wang, K.H.; Wang, C.S.; Chen, C.M.; Ko, W.C. Hesperidin-3′-o-methylether is more potent than hesperidin in phosphodiesterase inhibition and suppression of ovalbumin-induced airway hyperresponsiveness. Evid. Based Complement. Alternat. Med. 2012, 2012, 908562. [Google Scholar] [CrossRef]
- Impellizzeri, D.; Talero, E.; Siracusa, R.; Alcaide, A.; Cordaro, M.; Zubelia, J.; Bruschetta, G.; Crupi, R.; Esposito, E.; Cuzzocrea, S.; et al. Protective effect of polyphenols in an inflammatory process associated with experimental pulmonary fibrosis in mice. Br. J. Nutr. 2015, 114, 853–865. [Google Scholar] [CrossRef]
- Biswas, S.; Hwang, J.W.; Kirkham, P.A.; Rahman, I. Pharmacological and dietary antioxidant therapies for chronic obstructive pulmonary disease. Curr. Med. Chem. 2013, 20, 1496–1530. [Google Scholar] [CrossRef]
- Tabak, C.; Arts, I.C.; Smit, H.A.; Heederik, D.; Kromhout, D. Chronic obstructive pulmonary disease and intake of catechins, flavonols, and flavones: The MORGEN Study. Am. J. Respir. Crit. Care Med. 2001, 164, 61–64. [Google Scholar] [CrossRef]
- Kim, Y.H.; Choi, Y.J.; Kang, M.K.; Park, S.H.; Antika, L.D.; Lee, E.J.; Kim, D.Y.; Kang, Y.H. Astragalin inhibits allergic inflammation and airway thickening in ovalbumin-challenged mice. J. Agric. Food Chem. 2017, 65, 836–845. [Google Scholar] [CrossRef]
- Gong, J.H.; Cho, I.H.; Shin, D.; Han, S.Y.; Park, S.H.; Kang, Y.H. Inhibition of airway epithelial-to-mesenchymal transition and fibrosis by kaempferol in endotoxin-induced epithelial cells and ovalbumin-sensitized mice. Lab. Investig. 2014, 94, 297–308. [Google Scholar] [CrossRef]
- Kim, Y.H.; Choi, Y.J.; Kang, M.K.; Lee, E.J.; Kim, D.Y.; Oh, H.; Kang, Y.H. Oleuropein curtails pulmonary inflammation and tissue destruction in models of experimental asthma and emphysema. J. Agric. Food Chem. 2018, 66, 7643–7654. [Google Scholar] [CrossRef]
- Kim, Y.H.; Choi, Y.J.; Lee, E.J.; Kang, M.K.; Park, S.H.; Kim, D.Y.; Oh, H.; Park, S.J.; Kang, Y.H. Novel glutathione-containing yeast extracts inhibit eosinophilia and mucus overproduction in a murine model of asthma. Nutr. Res. Pract. 2017, 11, 461–469. [Google Scholar] [CrossRef]
- Chipuk, J.E.; Kuwana, T.; Bouchier-Hayes, L.; Droin, N.M.; Newmeyer, D.D.; Schuler, M.; Green, D.R. Direct activation of Bax by p53 mediates mitochondrial membrane permeabilization and apoptosis. Science 2004, 303, 1010–1014. [Google Scholar] [CrossRef]
- Han, W.; Dong, Z.; Dimitropoulou, C.; Su, Y. Hydrogen Sulfide Ameliorates Tobacco Smoke-Induced Oxidative Stress and Emphysema in Mice. Antioxid. Redox Signal. 2011, 15, 2121–2134. [Google Scholar] [CrossRef]
- Schuliga, M. NF-κB signaling in chronic inflammatory airway disease. Biomolecules 2015, 5, 1266–1283. [Google Scholar] [CrossRef]
- Edwards, M.R.; Bartlett, N.W.; Clarke, D.; Birrell, M.; Belvisi, M.; Johnston, S.L. Targeting the NF-κB pathway in asthma and chronic obstructive pulmonary disease. Pharmacol. Ther. 2009, 121, 1–13. [Google Scholar] [CrossRef]
- Thangapandiyan, S.; Miltonprabu, S. Epigallocatechin gallate supplementation protects against renal injury induced by fluoride intoxication in rats: Role of Nrf2/HO-1 signaling. Toxicol. Rep. 2014, 1, 12–30. [Google Scholar] [CrossRef]
- Yildirim, A.O.; Ince, M.; Eyi, Y.E.; Tuncer, S.K.; Kaldirim, U.; Eroglu, M.; Oztas, E.; Cayci, T.; Kilic, A.; Inal, V.; et al. The effects of glycyrrhizin on experimental acute pancreatitis in rats. Eur. Rev. Med. Pharmacol. Sci. 2013, 17, 2981–2987. [Google Scholar]
- Zuo, L.; He, F.; Sergakis, G.G.; Koozehchian, M.S.; Stimpfl, J.N.; Rong, Y.; Diaz, P.T.; Best, T.M. Interrelated role of cigarette smoking, oxidative stress, and immune response in COPD and corresponding treatments. Am. J. Physiol. Cell. Mol. Physiol. 2014, 307, L205–L218. [Google Scholar] [CrossRef]
- Barreiro, E.; Peinado, V.I.; Galdiz, J.B.; Ferrer, E.; Marin-Corral, J.; Sánchez, F.; Gea, J.; Barberà, J.A. Enigma in COPD project. Cigarette smoke-induced oxidative stress: A role in chronic obstructive pulmonary disease skeletal muscle dysfunction. Am. J. Respir. Crit. Care Med. 2010, 182, 477–488. [Google Scholar] [CrossRef]
- Demedts, I.K.; Demoor, T.; Bracke, K.R.; Joos, G.F.; Brusselle, G.G. Role of apoptosis in the pathogenesis of COPD and pulmonary emphysema. Respir. Res. 2006, 7, 53. [Google Scholar] [CrossRef]
- Tuder, R.M.; Petrache, I.; Elias, J.A.; Voelkel, N.F.; Henson, P.M. Apoptosis and emphysema: The missing link. Am. J. Respir. Cell. Mol. Biol. 2003, 28, 551–554. [Google Scholar] [CrossRef]
- Durham, A.L.; Caramori, G.; Chung, K.F.; Adcock, I.M. Targeted anti-inflammatory therapeutics in asthma and chronic obstructive lung disease. Transl. Res. 2016, 167, 192–203. [Google Scholar] [CrossRef]
- Bernardo, I.; Bozinovski, S.; Vlahos, R. Targeting oxidant-dependent mechanisms for the treatment of COPD and its comorbidities. Pharmacol. Ther. 2015, 155, 60–79. [Google Scholar] [CrossRef]
- Gonçalves, P.B.; Romeiro, N.C. Multi-target natural products as alternatives against oxidative stress in chronic obstructive pulmonary disease (COPD). Eur. J. Med. Chem. 2019, 163, 911–931. [Google Scholar] [CrossRef]
- Tanaka, T.; Takahashi, R. Flavonoids and asthma. Nutrients 2013, 5, 2128–2143. [Google Scholar] [CrossRef]








© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, Y.-H.; Kang, M.-K.; Lee, E.-J.; Kim, D.Y.; Oh, H.; Kim, S.-I.; Oh, S.Y.; Kim, K.-H.; Park, S.-J.; Choi, Y.-J.; et al. Dried Yeast Extracts Curtails Pulmonary Oxidative Stress, Inflammation and Tissue Destruction in a Model of Experimental Emphysema. Antioxidants 2019, 8, 349. https://doi.org/10.3390/antiox8090349
Kim Y-H, Kang M-K, Lee E-J, Kim DY, Oh H, Kim S-I, Oh SY, Kim K-H, Park S-J, Choi Y-J, et al. Dried Yeast Extracts Curtails Pulmonary Oxidative Stress, Inflammation and Tissue Destruction in a Model of Experimental Emphysema. Antioxidants. 2019; 8(9):349. https://doi.org/10.3390/antiox8090349
Chicago/Turabian StyleKim, Yun-Ho, Min-Kyung Kang, Eun-Jung Lee, Dong Yeon Kim, Hyeongjoo Oh, Soo-Il Kim, Su Yeon Oh, Kyung-Hee Kim, Sang-Jae Park, Yean-Jung Choi, and et al. 2019. "Dried Yeast Extracts Curtails Pulmonary Oxidative Stress, Inflammation and Tissue Destruction in a Model of Experimental Emphysema" Antioxidants 8, no. 9: 349. https://doi.org/10.3390/antiox8090349
APA StyleKim, Y.-H., Kang, M.-K., Lee, E.-J., Kim, D. Y., Oh, H., Kim, S.-I., Oh, S. Y., Kim, K.-H., Park, S.-J., Choi, Y.-J., & Kang, Y.-H. (2019). Dried Yeast Extracts Curtails Pulmonary Oxidative Stress, Inflammation and Tissue Destruction in a Model of Experimental Emphysema. Antioxidants, 8(9), 349. https://doi.org/10.3390/antiox8090349