Reactive Oxygen and Nitrogen Species in the Development of Pulmonary Hypertension

 ,
,  and
and
Abstract
:1. Introduction
2. Pulmonary Hypertension
3. The Lung Vasculature
4. Reactive Oxygen and Nitrogen Species
5. The Cellular Origins of Reactive Oxygen Species (ROS)
5.1. Nox Family of Enzymes
5.2. Mitochondria
5.3. Other Sources of ROS
6. The Cellular Origins of Reactive Nitrogen Species (RNS)
7. The Importance of ROS and RNS in Pulmonary Hypertension
7.1. ROS
7.1.1. Nox Enzymes
7.1.2. Mitochondrial ROS
7.1.3. Other Sources of ROS
7.1.4. Anti-Oxidant Pathways
7.1.5. TGFβ Superfamily Receptors
7.2. The Importance of RNS in Pulmonary Hypertension
7.3. Targeting ROS or RNS in the Treatment of Pulmonary Hypertension
8. Conclusions
Conflicts of Interest
References
- Lai, Y.C.; Potoka, K.C.; Champion, H.C.; Mora, A.L.; Gladwin, M.T. Pulmonary arterial hypertension: The clinical syndrome. Circ. Res. 2014, 115, 115–130. [Google Scholar] [CrossRef] [PubMed]
- Fulton, R.M.; Hutchinson, E.C.; Jones, A.M. Ventricular weight in cardiac hypertrophy. Br. Heart J. 1952, 14, 413–420. [Google Scholar] [CrossRef] [PubMed]
- Simonneau, G.; Gatzoulis, M.A.; Adatia, I.; Celermajer, D.; Denton, C.; Ghofrani, A.; Gomez Sanchez, M.A.; Krishna Kumar, R.; Landzberg, M.; Machado, R.F.; et al. Updated clinical classification of pulmonary hypertension. J. Am. Coll. Cardiol. 2013, 62, D34–D41. [Google Scholar] [CrossRef] [PubMed]
- Ryan, J.J.; Thenappan, T.; Luo, N.; Ha, T.; Patel, A.R.; Rich, S.; Archer, S.L. The who classification of pulmonary hypertension: A case-based imaging compendium. Pulm. Circ. 2012, 2, 107–121. [Google Scholar] [CrossRef] [PubMed]
- Tate, R.M.; Repine, J.E. Hydrogen peroxide causes permeability edema and hypertension in isolated salt-perfused rabbit lungs. Chest 1983, 83, 48S–50S. [Google Scholar] [CrossRef] [PubMed]
- Rhoades, R.A.; Packer, C.S.; Meiss, R.A. Pulmonary vascular smooth muscle contractility. Effect of free radicals. Chest 1988, 93, 94S–95S. [Google Scholar] [CrossRef] [PubMed]
- Rhoades, R.A.; Packer, C.S.; Roepke, D.A.; Jin, N.; Meiss, R.A. Reactive oxygen species alter contractile properties of pulmonary arterial smooth muscle. Can. J. Physiol. Pharmacol. 1990, 68, 1581–1589. [Google Scholar] [CrossRef] [PubMed]
- Moudgil, R.; Michelakis, E.D.; Archer, S.L. Hypoxic pulmonary vasoconstriction. J. Appl. Physiol. (Bethesda, Md. 1985) 2005, 98, 390–403. [Google Scholar] [CrossRef] [PubMed]
- Veit, F.; Pak, O.; Brandes, R.P.; Weissmann, N. Hypoxia-dependent reactive oxygen species signaling in the pulmonary circulation: Focus on ion channels. Antioxid. Redox Signal. 2015, 22, 537–552. [Google Scholar] [CrossRef] [PubMed]
- Dunham-Snary, K.J.; Wu, D.; Sykes, E.A.; Thakrar, A.; Parlow, L.R.; Mewburn, J.D.; Parlow, J.L.; Archer, S.L. Hypoxic pulmonary vasoconstriction: From molecular mechanisms to medicine. Chest 2017, 151, 181–192. [Google Scholar] [CrossRef] [PubMed]
- Adnot, S.; Raffestin, B.; Eddahibi, S.; Braquet, P.; Chabrier, P.E. Loss of endothelium-dependent relaxant activity in the pulmonary circulation of rats exposed to chronic hypoxia. J. Clin. Investig. 1991, 87, 155–162. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.F.; Crawley, D.E.; Barnes, P.J.; Evans, T.W. Endothelium-derived relaxing factor inhibits hypoxic pulmonary vasoconstriction in rats. Am. Rev. Respir. Dis. 1991, 143, 32–37. [Google Scholar] [CrossRef] [PubMed]
- Bedard, K.; Krause, K.H. The NOX family of ROS-generating NADPH oxidases: Physiology and pathophysiology. Physiol. Rev. 2007, 87, 245–313. [Google Scholar] [CrossRef] [PubMed]
- Beckman, J.S.; Koppenol, W.H. Nitric oxide, superoxide, and peroxynitrite: The good, the bad, and ugly. Am. J. Physiol. 1996, 271, C1424–C1437. [Google Scholar] [PubMed]
- Frazziano, G.; Champion, H.C.; Pagano, P.J. NADPH oxidase-derived ROS and the regulation of pulmonary vessel tone. Am. J. Physiol. Heart Circ. Physiol. 2012, 302, H2166–H2177. [Google Scholar] [CrossRef] [PubMed]
- Dinauer, M.C.; Pierce, E.A.; Bruns, G.A.; Curnutte, J.T.; Orkin, S.H. Human neutrophil cytochrome b light chain (p22-phox). Gene structure, chromosomal location, and mutations in cytochrome-negative autosomal recessive chronic granulomatous disease. J. Clin. Investig. 1990, 86, 1729–1737. [Google Scholar] [CrossRef] [PubMed]
- Schroder, K.; Weissmann, N.; Brandes, R.P. Organizers and activators: Cytosolic Nox proteins impacting on vascular function. Free Radic. Biol. Med. 2017, 109, 22–32. [Google Scholar] [CrossRef] [PubMed]
- Martyn, K.D.; Frederick, L.M.; von Loehneysen, K.; Dinauer, M.C.; Knaus, U.G. Functional analysis of NOX4 reveals unique characteristics compared to other NADPH oxidases. Cell Signal. 2006, 18, 69–82. [Google Scholar] [CrossRef] [PubMed]
- Nisimoto, Y.; Jackson, H.M.; Ogawa, H.; Kawahara, T.; Lambeth, J.D. Constitutive NADPH-dependent electron transferase activity of the Nox4 dehydrogenase domain. Biochemistry 2010, 49, 2433–2442. [Google Scholar] [CrossRef] [PubMed]
- Serrander, L.; Cartier, L.; Bedard, K.; Banfi, B.; Lardy, B.; Plastre, O.; Sienkiewicz, A.; Forro, L.; Schlegel, W.; Krause, K.H. Nox4 activity is determined by mRNA levels and reveals a unique pattern of ROS generation. Biochem. J. 2007, 406, 105–114. [Google Scholar] [CrossRef] [PubMed]
- Takac, I.; Schroder, K.; Zhang, L.; Lardy, B.; Anilkumar, N.; Lambeth, J.D.; Shah, A.M.; Morel, F.; Brandes, R.P. The E-loop is involved in hydrogen peroxide formation by the NADPH oxidase Nox4. J. Biol. Chem. 2011, 286, 13304–13313. [Google Scholar] [CrossRef] [PubMed]
- Chen, F.; Haigh, S.; Yu, Y.; Benson, T.; Wang, Y.; Li, X.; Dou, H.; Bagi, Z.; Verin, A.D.; Stepp, D.W.; et al. Nox5 stability and superoxide production is regulated by C-terminal binding of Hsp90 and CO-chaperones. Free Radic. Biol. Med. 2015, 89, 793–805. [Google Scholar] [CrossRef] [PubMed]
- Chen, F.; Pandey, D.; Chadli, A.; Catravas, J.D.; Chen, T.; Fulton, D.J. Hsp90 regulates NADPH oxidase activity and is necessary for superoxide but not hydrogen peroxide production. Antioxid. Redox Signal. 2011, 14, 2107–2119. [Google Scholar] [CrossRef] [PubMed]
- Chen, F.; Yu, Y.; Qian, J.; Wang, Y.; Cheng, B.; Dimitropoulou, C.; Patel, V.; Chadli, A.; Rudic, R.D.; Stepp, D.W.; et al. Opposing actions of heat shock protein 90 and 70 regulate nicotinamide adenine dinucleotide phosphate oxidase stability and reactive oxygen species production. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 2989–2999. [Google Scholar] [CrossRef] [PubMed]
- Madrigal-Matute, J.; Fernandez-Garcia, C.E.; Gomez-Guerrero, C.; Lopez-Franco, O.; Munoz-Garcia, B.; Egido, J.; Blanco-Colio, L.M.; Martin-Ventura, J.L. HSP90 inhibition by 17-DMAG attenuates oxidative stress in experimental atherosclerosis. Cardiovasc. Res. 2012, 95, 116–123. [Google Scholar] [CrossRef] [PubMed]
- Lyle, A.N.; Deshpande, N.N.; Taniyama, Y.; Seidel-Rogol, B.; Pounkova, L.; Du, P.; Papaharalambus, C.; Lassegue, B.; Griendling, K.K. Poldip2, a novel regulator of Nox4 and cytoskeletal integrity in vascular smooth muscle cells. Circ. Res. 2009, 105, 249–259. [Google Scholar] [CrossRef] [PubMed]
- Prior, K.K.; Wittig, I.; Leisegang, M.S.; Groenendyk, J.; Weissmann, N.; Michalak, M.; Jansen-Durr, P.; Shah, A.M.; Brandes, R.P. The endoplasmic reticulum chaperone calnexin is a NADPH oxidase NOX4 interacting protein. J. Biol. Chem. 2016, 291, 7045–7059. [Google Scholar] [CrossRef] [PubMed]
- Nisimoto, Y.; Diebold, B.A.; Cosentino-Gomes, D.; Lambeth, J.D. Nox4: A hydrogen peroxide-generating oxygen sensor. Biochemistry 2014, 53, 5111–5120. [Google Scholar] [CrossRef] [PubMed]
- Fulton, D.J. NOX5 and the regulation of cellular function. Antioxid. Redox Signal. 2009, 11, 2443–2452. [Google Scholar] [CrossRef] [PubMed]
- Drummond, G.R.; Sobey, C.G. Endothelial nadph oxidases: Which Nox to target in vascular disease? Trends Endocrinol. Metab. TEM 2014, 25, 452–463. [Google Scholar] [CrossRef] [PubMed]
- Murphy, M.P. How mitochondria produce reactive oxygen species. Biochem. J. 2009, 417, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Maranzana, E.; Barbero, G.; Falasca, A.I.; Lenaz, G.; Genova, M.L. Mitochondrial respiratory supercomplex association limits production of reactive oxygen species from complex I. Antioxid. Redox Signal. 2013, 19, 1469–1480. [Google Scholar] [CrossRef] [PubMed]
- Scialo, F.; Sriram, A.; Fernandez-Ayala, D.; Gubina, N.; Lohmus, M.; Nelson, G.; Logan, A.; Cooper, H.M.; Navas, P.; Enriquez, J.A.; et al. Mitochondrial ROS produced via reverse electron transport extend animal lifespan. Cell Metab. 2016, 23, 725–734. [Google Scholar] [CrossRef] [PubMed]
- Chouchani, E.T.; Pell, V.R.; James, A.M.; Work, L.M.; Saeb-Parsy, K.; Frezza, C.; Krieg, T.; Murphy, M.P. A unifying mechanism for mitochondrial superoxide production during ischemia-reperfusion injury. Cell Metab. 2016, 23, 254–263. [Google Scholar] [CrossRef] [PubMed]
- Coughlan, M.T.; Thorburn, D.R.; Penfold, S.A.; Laskowski, A.; Harcourt, B.E.; Sourris, K.C.; Tan, A.L.; Fukami, K.; Thallas-Bonke, V.; Nawroth, P.P.; et al. Rage-induced cytosolic ROS promote mitochondrial superoxide generation in diabetes. J. Am. Soc. Nephrol. 2009, 20, 742–752. [Google Scholar] [CrossRef] [PubMed]
- Frazziano, G.; Al Ghouleh, I.; Baust, J.; Shiva, S.; Champion, H.C.; Pagano, P.J. Nox-derived ROS are acutely activated in pressure overload pulmonary hypertension: Indications for a seminal role for mitochondrial NOX4. Am. J. Physiol. Heart Circ. Physiol. 2014, 306, H197–H205. [Google Scholar] [CrossRef] [PubMed]
- Kontos, H.A.; Wei, E.P.; Ellis, E.F.; Jenkins, L.W.; Povlishock, J.T.; Rowe, G.T.; Hess, M.L. Appearance of superoxide anion radical in cerebral extracellular space during increased prostaglandin synthesis in cats. Circ. Res. 1985, 57, 142–151. [Google Scholar] [CrossRef] [PubMed]
- Fulton, D.; McGiff, J.C.; Wolin, M.S.; Kaminski, P.; Quilley, J. Evidence against a cytochrome p450-derived reactive oxygen species as the mediator of the nitric oxide-independent vasodilator effect of bradykinin in the perfused heart of the rat. J. Pharmacol. Exp. Ther. 1997, 280, 702–709. [Google Scholar] [PubMed]
- Amaya, Y.; Yamazaki, K.; Sato, M.; Noda, K.; Nishino, T.; Nishino, T. Proteolytic conversion of xanthine dehydrogenase from the NAD-dependent type to the O2-dependent type. Amino acid sequence of rat liver xanthine dehydrogenase and identification of the cleavage sites of the enzyme protein during irreversible conversion by trypsin. J. Biol. Chem. 1990, 265, 14170–14175. [Google Scholar] [PubMed]
- Vasquez-Vivar, J.; Kalyanaraman, B.; Martasek, P.; Hogg, N.; Masters, B.S.; Karoui, H.; Tordo, P.; Pritchard, K.A., Jr. Superoxide generation by endothelial nitric oxide synthase: The influence of cofactors. Proc. Natl. Acad. Sci. USA 1998, 95, 9220–9225. [Google Scholar] [CrossRef] [PubMed]
- Wever, R.M.; van Dam, T.; van Rijn, H.J.; de Groot, F.; Rabelink, T.J. Tetrahydrobiopterin regulates superoxide and nitric oxide generation by recombinant endothelial nitric oxide synthase. Biochem. Biophys. Res. Commun. 1997, 237, 340–344. [Google Scholar] [CrossRef] [PubMed]
- Ma, Q.L.; Zhang, G.G.; Peng, J. Vascular peroxidase 1: A novel enzyme in promoting oxidative stress in cardiovascular system. Trends Cardiovasc. Med. 2013, 23, 179–183. [Google Scholar] [CrossRef] [PubMed]
- Kim-Shapiro, D.B.; Gladwin, M.T. Mechanisms of nitrite bioactivation. Nitric Oxide 2014, 38, 58–68. [Google Scholar] [CrossRef] [PubMed]
- Gryglewski, R.J.; Palmer, R.M.; Moncada, S. Superoxide anion is involved in the breakdown of endothelium-derived vascular relaxing factor. Nature 1986, 320, 454–456. [Google Scholar] [CrossRef] [PubMed]
- Stamler, J.S.; Simon, D.I.; Osborne, J.A.; Mullins, M.E.; Jaraki, O.; Michel, T.; Singel, D.J.; Loscalzo, J. S-nitrosylation of proteins with nitric oxide: Synthesis and characterization of biologically active compounds. Proc. Natl. Acad. Sci. USA 1992, 89, 444–448. [Google Scholar] [CrossRef] [PubMed]
- Lancaster, J.R., Jr.; Hibbs, J.B., Jr. EPR demonstration of iron-nitrosyl complex formation by cytotoxic activated macrophages. Proc. Natl. Acad. Sci. USA 1990, 87, 1223–1227. [Google Scholar] [CrossRef] [PubMed]
- Lim, D.G.; Sweeney, S.; Bloodsworth, A.; White, C.R.; Chumley, P.H.; Krishna, N.R.; Schopfer, F.; O’Donnell, V.B.; Eiserich, J.P.; Freeman, B.A. Nitrolinoleate, a nitric oxide-derived mediator of cell function: Synthesis, characterization, and vasomotor activity. Proc. Natl. Acad. Sci. USA 2002, 99, 15941–15946. [Google Scholar] [CrossRef] [PubMed]
- Hughes, M.N. Relationships between nitric oxide, nitroxyl ion, nitrosonium cation and peroxynitrite. Biochim. Biophys. Acta 1999, 1411, 263–272. [Google Scholar] [CrossRef]
- Hoshikawa, Y.; Ono, S.; Suzuki, S.; Tanita, T.; Chida, M.; Song, C.; Noda, M.; Tabata, T.; Voelkel, N.F.; Fujimura, S. Generation of oxidative stress contributes to the development of pulmonary hypertension induced by hypoxia. J. Appl. Phys. (Bethesda, Md. 1985) 2001, 90, 1299–1306. [Google Scholar]
- Reis, G.S.; Augusto, V.S.; Silveira, A.P.; Jordao, A.A., Jr.; Baddini-Martinez, J.; Poli Neto, O.; Rodrigues, A.J.; Evora, P.R. Oxidative-stress biomarkers in patients with pulmonary hypertension. Pulm. Circ. 2013, 3, 856–861. [Google Scholar] [CrossRef] [PubMed]
- Cracowski, J.L.; Cracowski, C.; Bessard, G.; Pepin, J.L.; Bessard, J.; Schwebel, C.; Stanke-Labesque, F.; Pison, C. Increased lipid peroxidation in patients with pulmonary hypertension. Am. J. Respir. Crit. Care Med. 2001, 164, 1038–1042. [Google Scholar] [CrossRef] [PubMed]
- Irodova, N.L.; Lankin, V.Z.; Konovalova, G.K.; Kochetov, A.G.; Chazova, I.E. Oxidative stress in patients with primary pulmonary hypertension. Bull. Exp. Biol. Med. 2002, 133, 580–582. [Google Scholar] [CrossRef] [PubMed]
- Bowers, R.; Cool, C.; Murphy, R.C.; Tuder, R.M.; Hopken, M.W.; Flores, S.C.; Voelkel, N.F. Oxidative stress in severe pulmonary hypertension. Am. J. Respir. Crit. Care Med. 2004, 169, 764–769. [Google Scholar] [CrossRef] [PubMed]
- Csiszar, A.; Labinskyy, N.; Olson, S.; Pinto, J.T.; Gupte, S.; Wu, J.M.; Hu, F.; Ballabh, P.; Podlutsky, A.; Losonczy, G.; et al. Resveratrol prevents monocrotaline-induced pulmonary hypertension in rats. Hypertension (Dallas, Tex. 1979) 2009, 54, 668–675. [Google Scholar] [CrossRef] [PubMed]
- Barman, S.A.; Chen, F.; Su, Y.; Dimitropoulou, C.; Wang, Y.; Catravas, J.D.; Han, W.; Orfi, L.; Szantai-Kis, C.; Keri, G.; et al. NADPH oxidase 4 is expressed in pulmonary artery adventitia and contributes to hypertensive vascular remodeling. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 1704–1715. [Google Scholar] [CrossRef] [PubMed]
- Muzaffar, S.; Shukla, N.; Angelini, G.D.; Jeremy, J.Y. Acute hypoxia simultaneously induces the expression of gp91phox and endothelial nitric oxide synthase in the porcine pulmonary artery. Thorax 2005, 60, 305–313. [Google Scholar] [CrossRef] [PubMed]
- Nisbet, R.E.; Graves, A.S.; Kleinhenz, D.J.; Rupnow, H.L.; Reed, A.L.; Fan, T.H.; Mitchell, P.O.; Sutliff, R.L.; Hart, C.M. The role of NADPH oxidase in chronic intermittent hypoxia-induced pulmonary hypertension in mice. Am. J. Respir. Cell Mol. Biol. 2009, 40, 601–609. [Google Scholar] [CrossRef] [PubMed]
- Seta, F.; Rahmani, M.; Turner, P.V.; Funk, C.D. Pulmonary oxidative stress is increased in cyclooxygenase-2 knockdown mice with mild pulmonary hypertension induced by monocrotaline. PLoS ONE 2011, 6, e23439. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.Q.; Erbynn, E.M.; Folz, R.J. Chronic hypoxia-enhanced murine pulmonary vasoconstriction: Role of superoxide and gp91phox. Chest 2005, 128, 594s–596s. [Google Scholar] [CrossRef]
- Fresquet, F.; Pourageaud, F.; Leblais, V.; Brandes, R.P.; Savineau, J.P.; Marthan, R.; Muller, B. Role of reactive oxygen species and gp91phox in endothelial dysfunction of pulmonary arteries induced by chronic hypoxia. Br. J. Pharmacol. 2006, 148, 714–723. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.Q.; Zelko, I.N.; Erbynn, E.M.; Sham, J.S.; Folz, R.J. Hypoxic pulmonary hypertension: Role of superoxide and NADPH oxidase (gp91phox). Am. J. Physiol. Lung Cell. Mol. Physiol. 2006, 290, L2–L10. [Google Scholar] [CrossRef] [PubMed]
- Brennan, L.A.; Steinhorn, R.H.; Wedgwood, S.; Mata-Greenwood, E.; Roark, E.A.; Russell, J.A.; Black, S.M. Increased superoxide generation is associated with pulmonary hypertension in fetal lambs: A role for NADPH oxidase. Circ. Res. 2003, 92, 683–691. [Google Scholar] [CrossRef] [PubMed]
- Guo, D.; Gu, J.; Jiang, H.; Ahmed, A.; Zhang, Z.; Gu, Y. Inhibition of pyruvate kinase M2 by reactive oxygen species contributes to the development of pulmonary arterial hypertension. J. Mol. Cell. Cardiol. 2016, 91, 179–187. [Google Scholar] [CrossRef] [PubMed]
- Rathore, R.; Zheng, Y.M.; Niu, C.F.; Liu, Q.H.; Korde, A.; Ho, Y.S.; Wang, Y.X. Hypoxia activates NADPH oxidase to increase [ROS]i and [Ca2+]i through the mitochondrial ROS-PKCε signaling axis in pulmonary artery smooth muscle cells. Free Radic. Biol. Med. 2008, 45, 1223–1231. [Google Scholar] [CrossRef] [PubMed]
- Dennis, K.E.; Aschner, J.L.; Milatovic, D.; Schmidt, J.W.; Aschner, M.; Kaplowitz, M.R.; Zhang, Y.; Fike, C.D. NADPH oxidases and reactive oxygen species at different stages of chronic hypoxia-induced pulmonary hypertension in newborn piglets. Am. J. Physiol. Lung Cell. Mol. Physiol. 2009, 297, L596–L607. [Google Scholar] [CrossRef] [PubMed]
- Mittal, M.; Gu, X.Q.; Pak, O.; Pamenter, M.E.; Haag, D.; Fuchs, D.B.; Schermuly, R.T.; Ghofrani, H.A.; Brandes, R.P.; Seeger, W.; et al. Hypoxia induces Kv channel current inhibition by increased NADPH oxidase-derived reactive oxygen species. Free Radic. Biol. Med. 2012, 52, 1033–1042. [Google Scholar] [CrossRef] [PubMed]
- Ogura, S.; Shimosawa, T.; Mu, S.; Sonobe, T.; Kawakami-Mori, F.; Wang, H.; Uetake, Y.; Yoshida, K.; Yatomi, Y.; Shirai, M.; et al. Oxidative stress augments pulmonary hypertension in chronically hypoxic mice overexpressing the oxidized ldl receptor. Am. J. Physiol. Heart Circ. Physiol. 2013, 305, H155–H162. [Google Scholar] [CrossRef] [PubMed]
- Miriyala, S.; Gongora Nieto, M.C.; Mingone, C.; Smith, D.; Dikalov, S.; Harrison, D.G.; Jo, H. Bone morphogenic protein-4 induces hypertension in mice: Role of noggin, vascular NADPH oxidases, and impaired vasorelaxation. Circulation 2006, 113, 2818–2825. [Google Scholar] [CrossRef] [PubMed]
- Hood, K.Y.; Montezano, A.C.; Harvey, A.P.; Nilsen, M.; MacLean, M.R.; Touyz, R.M. Nicotinamide adenine dinucleotide phosphate oxidase-mediated redox signaling and vascular remodeling by 16α-hydroxyestrone in human pulmonary artery cells: Implications in pulmonary arterial hypertension. Hypertension (Dallas, Tex. 1979) 2016, 68, 796–808. [Google Scholar] [CrossRef] [PubMed]
- Veit, F.; Pak, O.; Egemnazarov, B.; Roth, M.; Kosanovic, D.; Seimetz, M.; Sommer, N.; Ghofrani, H.A.; Seeger, W.; Grimminger, F.; et al. Function of NADPH oxidase 1 in pulmonary arterial smooth muscle cells after monocrotaline-induced pulmonary vascular remodeling. Antioxid. Redox Signal. 2013, 19, 2213–2231. [Google Scholar] [CrossRef] [PubMed]
- Iwata, K.; Ikami, K.; Matsuno, K.; Yamashita, T.; Shiba, D.; Ibi, M.; Matsumoto, M.; Katsuyama, M.; Cui, W.; Zhang, J.; et al. Deficiency of Nox1/nicotinamide adenine dinucleotide phosphate, reduced form oxidase leads to pulmonary vascular remodeling. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 110–119. [Google Scholar] [CrossRef] [PubMed]
- Dorfmuller, P.; Chaumais, M.C.; Giannakouli, M.; Durand-Gasselin, I.; Raymond, N.; Fadel, E.; Mercier, O.; Charlotte, F.; Montani, D.; Simonneau, G.; et al. Increased oxidative stress and severe arterial remodeling induced by permanent high-flow challenge in experimental pulmonary hypertension. Respir. Res. 2011, 12, 119. [Google Scholar] [CrossRef] [PubMed]
- Mittal, M.; Roth, M.; Konig, P.; Hofmann, S.; Dony, E.; Goyal, P.; Selbitz, A.C.; Schermuly, R.T.; Ghofrani, H.A.; Kwapiszewska, G.; et al. Hypoxia-dependent regulation of nonphagocytic nadph oxidase subunit Nox4 in the pulmonary vasculature. Circ. Res. 2007, 101, 258–267. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Tabar, S.S.; Malec, V.; Eul, B.G.; Klepetko, W.; Weissmann, N.; Grimminger, F.; Seeger, W.; Rose, F.; Hanze, J. Nox4 regulates ROS levels under normoxic and hypoxic conditions, triggers proliferation, and inhibits apoptosis in pulmonary artery adventitial fibroblasts. Antioxid. Redox Signal. 2008, 10, 1687–1698. [Google Scholar] [CrossRef] [PubMed]
- Ismail, S.; Sturrock, A.; Wu, P.; Cahill, B.; Norman, K.; Huecksteadt, T.; Sanders, K.; Kennedy, T.; Hoidal, J. Nox4 mediates hypoxia-induced proliferation of human pulmonary artery smooth muscle cells: The role of autocrine production of transforming growth factor-β1 and insulin-like growth factor binding protein-3. Am. J. Physiol. Lung Cell. Mol. Physiol. 2009, 296, L489–L499. [Google Scholar] [CrossRef] [PubMed]
- Teng, R.J.; Eis, A.; Bakhutashvili, I.; Arul, N.; Konduri, G.G. Increased superoxide production contributes to the impaired angiogenesis of fetal pulmonary arteries with in utero pulmonary hypertension. Am. J. Physiol. Lung Cell. Mol. Physiol. 2009, 297, L184–l195. [Google Scholar] [CrossRef] [PubMed]
- Green, D.E.; Murphy, T.C.; Kang, B.Y.; Kleinhenz, J.M.; Szyndralewiez, C.; Page, P.; Sutliff, R.L.; Hart, C.M. The Nox4 inhibitor GKT137831 attenuates hypoxia-induced pulmonary vascular cell proliferation. Am. J. Respir. Cell Mol. Biol. 2012, 47, 718–726. [Google Scholar] [CrossRef] [PubMed]
- Veith, C.; Kraut, S.; Wilhelm, J.; Sommer, N.; Quanz, K.; Seeger, W.; Brandes, R.P.; Weissmann, N.; Schroder, K. NADPH oxidase 4 is not involved in hypoxia-induced pulmonary hypertension. Pulm. Circ. 2016, 6, 397–400. [Google Scholar] [CrossRef] [PubMed]
- Goncharov, D.A.; Kudryashova, T.V.; Ziai, H.; Ihida-Stansbury, K.; DeLisser, H.; Krymskaya, V.P.; Tuder, R.M.; Kawut, S.M.; Goncharova, E.A. Mammalian target of rapamycin complex 2 (mTORC2) coordinates pulmonary artery smooth muscle cell metabolism, proliferation, and survival in pulmonary arterial hypertension. Circulation 2014, 129, 864–874. [Google Scholar] [CrossRef] [PubMed]
- Adesina, S.E.; Kang, B.Y.; Bijli, K.M.; Ma, J.; Cheng, J.; Murphy, T.C.; Michael Hart, C.; Sutliff, R.L. Targeting mitochondrial reactive oxygen species to modulate hypoxia-induced pulmonary hypertension. Free Radic. Biol. Med. 2015, 87, 36–47. [Google Scholar] [CrossRef] [PubMed]
- Chen, F.; Haigh, S.; Barman, S.; Fulton, D.J. From form to function: The role of Nox4 in the cardiovascular system. Front. Physiol. 2012, 3, 412. [Google Scholar] [CrossRef] [PubMed]
- Hecker, L.; Vittal, R.; Jones, T.; Jagirdar, R.; Luckhardt, T.R.; Horowitz, J.C.; Pennathur, S.; Martinez, F.J.; Thannickal, V.J. NADPH oxidase-4 mediates myofibroblast activation and fibrogenic responses to lung injury. Nat. Med. 2009, 15, 1077–1081. [Google Scholar] [CrossRef] [PubMed]
- Chen, F.; Barman, S.; Yu, Y.; Haigh, S.; Wang, Y.; Black, S.M.; Rafikov, R.; Dou, H.; Bagi, Z.; Han, W.; et al. Caveolin-1 is a negative regulator of NADPH oxidase-derived reactive oxygen species. Free Radic. Biol. Med. 2014, 73, 201–213. [Google Scholar] [CrossRef] [PubMed]
- Bernard, K.; Hecker, L.; Luckhardt, T.R.; Cheng, G.; Thannickal, V.J. NADPH oxidases in lung health and disease. Antioxid. Redox Signal. 2014, 20, 2838–2853. [Google Scholar] [CrossRef] [PubMed]
- Bonnet, S.; Michelakis, E.D.; Porter, C.J.; Andrade-Navarro, M.A.; Thebaud, B.; Bonnet, S.; Haromy, A.; Harry, G.; Moudgil, R.; McMurtry, M.S.; et al. An abnormal mitochondrial-hypoxia inducible factor-1α-kv channel pathway disrupts oxygen sensing and triggers pulmonary arterial hypertension in fawn hooded rats: Similarities to human pulmonary arterial hypertension. Circulation 2006, 113, 2630–2641. [Google Scholar] [CrossRef] [PubMed]
- Pak, O.; Sommer, N.; Hoeres, T.; Bakr, A.; Waisbrod, S.; Sydykov, A.; Haag, D.; Esfandiary, A.; Kojonazarov, B.; Veit, F.; et al. Mitochondrial hyperpolarization in pulmonary vascular remodeling. Mitochondrial uncoupling protein deficiency as disease model. Am. J. Respir. Cell Mol. Biol. 2013, 49, 358–367. [Google Scholar] [CrossRef] [PubMed]
- Ryan, J.; Dasgupta, A.; Huston, J.; Chen, K.H.; Archer, S.L. Mitochondrial dynamics in pulmonary arterial hypertension. J. Mol. Med. (Berl. Ger.) 2015, 93, 229–242. [Google Scholar] [CrossRef] [PubMed]
- Waypa, G.B.; Smith, K.A.; Schumacker, P.T. O2 sensing, mitochondria and ROS signaling: The fog is lifting. Mol. Asp. Med. 2016, 47–48, 76–89. [Google Scholar] [CrossRef] [PubMed]
- Archer, S.L.; Marsboom, G.; Kim, G.H.; Zhang, H.J.; Toth, P.T.; Svensson, E.C.; Dyck, J.R.; Gomberg-Maitland, M.; Thebaud, B.; Husain, A.N.; et al. Epigenetic attenuation of mitochondrial superoxide dismutase 2 in pulmonary arterial hypertension: A basis for excessive cell proliferation and a new therapeutic target. Circulation 2010, 121, 2661–2671. [Google Scholar] [CrossRef] [PubMed]
- Xu, M.; Xu, M.; Han, L.; Yuan, C.; Mei, Y.; Zhang, H.; Chen, S.; Sun, K.; Zhu, B. Role for functional SOD2 polymorphism in pulmonary arterial hypertension in a chinese population. Int. J. Environ. Res. Public Health 2017, 14, 266. [Google Scholar] [CrossRef] [PubMed]
- Ohman, T.; Parish, G.; Jackson, R.M. Hypoxic modulation of manganese superoxide dismutase promoter activity and gene expression in lung epithelial cells. Am. J. Respir. Cell Mol. Biol. 1999, 21, 119–127. [Google Scholar] [CrossRef] [PubMed]
- Terada, L.S.; Guidot, D.M.; Leff, J.A.; Willingham, I.R.; Hanley, M.E.; Piermattei, D.; Repine, J.E. Hypoxia injures endothelial cells by increasing endogenous xanthine oxidase activity. Proc. Natl. Acad. Sci. USA 1992, 89, 3362–3366. [Google Scholar] [CrossRef] [PubMed]
- Jankov, R.P.; Kantores, C.; Pan, J.; Belik, J. Contribution of xanthine oxidase-derived superoxide to chronic hypoxic pulmonary hypertension in neonatal rats. Am. J. Physiol. Lung Cell. Mol. Physiol. 2008, 294, L233–L245. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Luo, X.J.; Yang, Z.B.; Zhang, J.J.; Li, T.B.; Zhang, X.J.; Ma, Q.L.; Zhang, G.G.; Hu, C.P.; Peng, J. Inhibition of Nox/VPO1 pathway and inflammatory reaction by trimethoxystilbene in prevention of cardiovascular remodeling in hypoxia-induced pulmonary hypertensive rats. J. Cardiovasc. Pharmacol. 2014, 63, 567–576. [Google Scholar] [CrossRef] [PubMed]
- Ramiro-Diaz, J.M.; Nitta, C.H.; Maston, L.D.; Codianni, S.; Giermakowska, W.; Resta, T.C.; Gonzalez Bosc, L.V. NFAT is required for spontaneous pulmonary hypertension in superoxide dismutase 1 knockout mice. Am. J. Physiol. Lung Cell. Mol. Physiol. 2013, 304, L613–L625. [Google Scholar] [CrossRef] [PubMed]
- Xu, D.; Guo, H.; Xu, X.; Lu, Z.; Fassett, J.; Hu, X.; Xu, Y.; Tang, Q.; Hu, D.; Somani, A.; et al. Exacerbated pulmonary arterial hypertension and right ventricular hypertrophy in animals with loss of function of extracellular superoxide dismutase. Hypertension (Dallas, Tex. 1979) 2011, 58, 303–309. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.Y.; Zhao, Y.D.; Mirza, M.K.; Huang, J.H.; Potula, H.H.; Vogel, S.M.; Brovkovych, V.; Yuan, J.X.; Wharton, J.; Malik, A.B. Persistent eNOS activation secondary to caveolin-1 deficiency induces pulmonary hypertension in mice and humans through pkg nitration. J. Clin. Investig. 2009, 119, 2009–2018. [Google Scholar] [CrossRef] [PubMed]
- Christou, H.; Morita, T.; Hsieh, C.M.; Koike, H.; Arkonac, B.; Perrella, M.A.; Kourembanas, S. Prevention of hypoxia-induced pulmonary hypertension by enhancement of endogenous heme oxygenase-1 in the rat. Circ. Res. 2000, 86, 1224–1229. [Google Scholar] [CrossRef] [PubMed]
- Yet, S.F.; Perrella, M.A.; Layne, M.D.; Hsieh, C.M.; Maemura, K.; Kobzik, L.; Wiesel, P.; Christou, H.; Kourembanas, S.; Lee, M.E. Hypoxia induces severe right ventricular dilatation and infarction in heme oxygenase-1 null mice. J. Clin. Investig. 1999, 103, R23–R29. [Google Scholar] [CrossRef] [PubMed]
- Masri, F.A.; Comhair, S.A.; Dostanic-Larson, I.; Kaneko, F.T.; Dweik, R.A.; Arroliga, A.C.; Erzurum, S.C. Deficiency of lung antioxidants in idiopathic pulmonary arterial hypertension. Clin. Trans. Sci. 2008, 1, 99–106. [Google Scholar] [CrossRef] [PubMed]
- Adesina, S.E.; Wade, B.E.; Bijli, K.M.; Kang, B.Y.; Williams, C.R.; Ma, J.; Go, Y.M.; Hart, C.M.; Sutliff, R.L. Hypoxia inhibits expression and function of mitochondrial thioredoxin 2 to promote pulmonary hypertension. Am. J. Physiol. Lung Cell. Mol. Physiol. 2017, 312, L599–L608. [Google Scholar] [CrossRef] [PubMed]
- Sturrock, A.; Cahill, B.; Norman, K.; Huecksteadt, T.P.; Hill, K.; Sanders, K.; Karwande, S.V.; Stringham, J.C.; Bull, D.A.; Gleich, M.; et al. Transforming growth factor-beta1 induces Nox4 NAD(P)H oxidase and reactive oxygen species-dependent proliferation in human pulmonary artery smooth muscle cells. Am. J. Physiol. Lung Cell. Mol. Physiol. 2006, 290, L661–L673. [Google Scholar] [CrossRef] [PubMed]
- Atkinson, C.; Stewart, S.; Upton, P.D.; Machado, R.; Thomson, J.R.; Trembath, R.C.; Morrell, N.W. Primary pulmonary hypertension is associated with reduced pulmonary vascular expression of type II bone morphogenetic protein receptor. Circulation 2002, 105, 1672–1678. [Google Scholar] [CrossRef] [PubMed]
- Lane, K.L.; Talati, M.; Austin, E.; Hemnes, A.R.; Johnson, J.A.; Fessel, J.P.; Blackwell, T.; Mernaugh, R.L.; Robinson, L.; Fike, C.; et al. Oxidative injury is a common consequence of BMPR2 mutations. Pulm. Circ. 2011, 1, 72–83. [Google Scholar] [CrossRef] [PubMed]
- Soon, E.; Crosby, A.; Southwood, M.; Yang, P.; Tajsic, T.; Toshner, M.; Appleby, S.; Shanahan, C.M.; Bloch, K.D.; Pepke-Zaba, J.; et al. Bone morphogenetic protein receptor type II deficiency and increased inflammatory cytokine production. A gateway to pulmonary arterial hypertension. Am. J. Respir. Crit. Care Med. 2015, 192, 859–872. [Google Scholar] [CrossRef] [PubMed]
- Yao, C.; Yu, J.; Taylor, L.; Polgar, P.; McComb, M.E.; Costello, C.E. Protein expression by human pulmonary artery smooth muscle cells containing a BMPR2 mutation and the action of ET-1 as determined by proteomic mass spectrometry. Int. J. Mass Spectrom. 2015, 378, 347–359. [Google Scholar] [CrossRef] [PubMed]
- Trembath, R.C.; Thomson, J.R.; Machado, R.D.; Morgan, N.V.; Atkinson, C.; Winship, I.; Simonneau, G.; Galie, N.; Loyd, J.E.; Humbert, M.; et al. Clinical and molecular genetic features of pulmonary hypertension in patients with hereditary hemorrhagic telangiectasia. N. Engl. J. Med. 2001, 345, 325–334. [Google Scholar] [CrossRef] [PubMed]
- Jerkic, M.; Kabir, M.G.; Davies, A.; Yu, L.X.; McIntyre, B.A.; Husain, N.W.; Enomoto, M.; Sotov, V.; Husain, M.; Henkelman, M.; et al. Pulmonary hypertension in adult Alk1 heterozygous mice due to oxidative stress. Cardiovasc. Res. 2011, 92, 375–384. [Google Scholar] [CrossRef] [PubMed]
- Cherry, P.D.; Gillis, C.N. Evidence for the role of endothelium-derived relaxing factor in acetylcholine-induced vasodilatation in the intact lung. J. Pharmacol. Exp. Ther. 1987, 241, 516–520. [Google Scholar] [PubMed]
- Brashers, V.L.; Peach, M.J.; Rose, C.E., Jr. Augmentation of hypoxic pulmonary vasoconstriction in the isolated perfused rat lung by in vitro antagonists of endothelium-dependent relaxation. J. Clin. Investig. 1988, 82, 1495–1502. [Google Scholar] [CrossRef] [PubMed]
- Fineman, J.R.; Heymann, M.A.; Soifer, S.J. N omega-nitro-L-arginine attenuates endothelium-dependent pulmonary vasodilation in lambs. Am. J. Physiol. 1991, 260, H1299–H1306. [Google Scholar] [PubMed]
- Cremona, G.; Wood, A.M.; Hall, L.W.; Bower, E.A.; Higenbottam, T. Effect of inhibitors of nitric oxide release and action on vascular tone in isolated lungs of pig, sheep, dog and man. J. Physiol. 1994, 481, 185–195. [Google Scholar] [CrossRef] [PubMed]
- Ignarro, L.J.; Buga, G.M.; Wood, K.S.; Byrns, R.E.; Chaudhuri, G. Endothelium-derived relaxing factor produced and released from artery and vein is nitric oxide. Proc. Natl. Acad. Sci. USA 1987, 84, 9265–9269. [Google Scholar] [CrossRef] [PubMed]
- Tabima, D.M.; Frizzell, S.; Gladwin, M.T. Reactive oxygen and nitrogen species in pulmonary hypertension. Free Radic. Biol. Med. 2012, 52, 1970–1986. [Google Scholar] [CrossRef] [PubMed]
- Denninger, J.W.; Marletta, M.A. Guanylate cyclase and the ⋅NO/cGMP signaling pathway. Biochim. Biophys. Acta 1999, 1411, 334–350. [Google Scholar] [CrossRef]
- Steudel, W.; Ichinose, F.; Huang, P.L.; Hurford, W.E.; Jones, R.C.; Bevan, J.A.; Fishman, M.C.; Zapol, W.M. Pulmonary vasoconstriction and hypertension in mice with targeted disruption of the endothelial nitric oxide synthase (NOS 3) gene. Circ. Res. 1997, 81, 34–41. [Google Scholar] [CrossRef] [PubMed]
- Fagan, K.A.; Tyler, R.C.; Sato, K.; Fouty, B.W.; Morris, K.G., Jr.; Huang, P.L.; McMurtry, I.F.; Rodman, D.M. Relative contributions of endothelial, inducible, and neuronal NOS to tone in the murine pulmonary circulation. Am. J. Physiol. 1999, 277, L472–L478. [Google Scholar] [PubMed]
- Altiere, R.J.; Olson, J.W.; Gillespie, M.N. Altered pulmonary vascular smooth muscle responsiveness in monocrotaline-induced pulmonary hypertension. J. Pharmacol. Exp. Ther. 1986, 236, 390–395. [Google Scholar] [PubMed]
- Mathew, R.; Zeballos, G.A.; Tun, H.; Gewitz, M.H. Role of nitric oxide and endothelin-1 in monocrotaline-induced pulmonary hypertension in rats. Cardiovasc. Res. 1995, 30, 739–746. [Google Scholar] [CrossRef]
- Mathew, R. Pathogenesis of pulmonary hypertension: A case for caveolin-1 and cell membrane integrity. Am. J. Physiol. Heart Circ. Physiol. 2014, 306, H15–H25. [Google Scholar] [CrossRef] [PubMed]
- Giaid, A.; Saleh, D. Reduced expression of endothelial nitric oxide synthase in the lungs of patients with pulmonary hypertension. N. Engl. J. Med. 1995, 333, 214–221. [Google Scholar] [CrossRef] [PubMed]
- Kaneko, F.T.; Arroliga, A.C.; Dweik, R.A.; Comhair, S.A.; Laskowski, D.; Oppedisano, R.; Thomassen, M.J.; Erzurum, S.C. Biochemical reaction products of nitric oxide as quantitative markers of primary pulmonary hypertension. Am. J. Respir. Crit. Care Med. 1998, 158, 917–923. [Google Scholar] [CrossRef] [PubMed]
- Ozkan, M.; Dweik, R.A.; Laskowski, D.; Arroliga, A.C.; Erzurum, S.C. High levels of nitric oxide in individuals with pulmonary hypertension receiving epoprostenol therapy. Lung 2001, 179, 233–243. [Google Scholar] [CrossRef] [PubMed]
- Tuder, R.M.; Cool, C.D.; Geraci, M.W.; Wang, J.; Abman, S.H.; Wright, L.; Badesch, D.; Voelkel, N.F. Prostacyclin synthase expression is decreased in lungs from patients with severe pulmonary hypertension. Am. J. Respir. Crit. Care Med. 1999, 159, 1925–1932. [Google Scholar] [CrossRef] [PubMed]
- Xue, C.; Johns, R.A. Endothelial nitric oxide synthase in the lungs of patients with pulmonary hypertension. N. Engl. J. Med. 1995, 333, 1642–1644. [Google Scholar] [PubMed]
- Mason, N.A.; Springall, D.R.; Burke, M.; Pollock, J.; Mikhail, G.; Yacoub, M.H.; Polak, J.M. High expression of endothelial nitric oxide synthase in plexiform lesions of pulmonary hypertension. J. Pathol. 1998, 185, 313–318. [Google Scholar] [CrossRef]
- Fagan, K.A.; Morrissey, B.; Fouty, B.W.; Sato, K.; Harral, J.W.; Morris, K.G., Jr.; Hoedt-Miller, M.; Vidmar, S.; McMurtry, I.F.; Rodman, D.M. Upregulation of nitric oxide synthase in mice with severe hypoxia-induced pulmonary hypertension. Respir. Res. 2001, 2, 306–313. [Google Scholar] [CrossRef] [PubMed]
- Ischiropoulos, H.; Zhu, L.; Chen, J.; Tsai, M.; Martin, J.C.; Smith, C.D.; Beckman, J.S. Peroxynitrite-mediated tyrosine nitration catalyzed by superoxide dismutase. Arch. Biochem. Biophys. 1992, 298, 431–437. [Google Scholar] [CrossRef]
- Karuppiah, K.; Druhan, L.J.; Chen, C.A.; Smith, T.; Zweier, J.L.; Sessa, W.C.; Cardounel, A.J. Suppression of eNOS-derived superoxide by caveolin-1: A biopterin-dependent mechanism. Am. J. Physiol. Heart Circ. Physiol. 2011, 301, H903–H911. [Google Scholar] [CrossRef] [PubMed]
- Millatt, L.J.; Whitley, G.S.; Li, D.; Leiper, J.M.; Siragy, H.M.; Carey, R.M.; Johns, R.A. Evidence for dysregulation of dimethylarginine dimethylaminohydrolase I in chronic hypoxia-induced pulmonary hypertension. Circulation 2003, 108, 1493–1498. [Google Scholar] [CrossRef] [PubMed]
- Kielstein, J.T.; Bode-Boger, S.M.; Hesse, G.; Martens-Lobenhoffer, J.; Takacs, A.; Fliser, D.; Hoeper, M.M. Asymmetrical dimethylarginine in idiopathic pulmonary arterial hypertension. Arterioscler. Thromb. Vasc. Biol. 2005, 25, 1414–1418. [Google Scholar] [CrossRef] [PubMed]
- Druhan, L.J.; Forbes, S.P.; Pope, A.J.; Chen, C.A.; Zweier, J.L.; Cardounel, A.J. Regulation of eNOS-derived superoxide by endogenous methylarginines. Biochemistry 2008, 47, 7256–7263. [Google Scholar] [CrossRef] [PubMed]
- Khoo, J.P.; Zhao, L.; Alp, N.J.; Bendall, J.K.; Nicoli, T.; Rockett, K.; Wilkins, M.R.; Channon, K.M. Pivotal role for endothelial tetrahydrobiopterin in pulmonary hypertension. Circulation 2005, 111, 2126–2133. [Google Scholar] [CrossRef] [PubMed]
- Francis, B.N.; Hale, A.; Channon, K.M.; Wilkins, M.R.; Zhao, L. Effects of tetrahydrobiopterin oral treatment in hypoxia-induced pulmonary hypertension in rat. Pulm. Circ. 2014, 4, 462–470. [Google Scholar] [CrossRef] [PubMed]
- Hampl, V.; Bibova, J.; Banasova, A.; Uhlik, J.; Mikova, D.; Hnilickova, O.; Lachmanova, V.; Herget, J. Pulmonary vascular iNOS induction participates in the onset of chronic hypoxic pulmonary hypertension. Am. J. Physiol. Lung Cell. Mol. Physiol. 2006, 290, L11–L20. [Google Scholar] [CrossRef] [PubMed]
- Murata, T.; Lin, M.I.; Huang, Y.; Yu, J.; Bauer, P.M.; Giordano, F.J.; Sessa, W.C. Reexpression of caveolin-1 in endothelium rescues the vascular, cardiac, and pulmonary defects in global caveolin-1 knockout mice. J. Exp. Med. 2007, 204, 2373–2382. [Google Scholar] [CrossRef] [PubMed]
- Shiroto, T.; Romero, N.; Sugiyama, T.; Sartoretto, J.L.; Kalwa, H.; Yan, Z.; Shimokawa, H.; Michel, T. Caveolin-1 is a critical determinant of autophagy, metabolic switching, and oxidative stress in vascular endothelium. PLoS ONE 2014, 9, e87871. [Google Scholar] [CrossRef] [PubMed]
- Pritchard, K.A., Jr.; Ackerman, A.W.; Gross, E.R.; Stepp, D.W.; Shi, Y.; Fontana, J.T.; Baker, J.E.; Sessa, W.C. Heat shock protein 90 mediates the balance of nitric oxide and superoxide anion from endothelial nitric-oxide synthase. J. Biol. Chem. 2001, 276, 17621–17624. [Google Scholar] [CrossRef] [PubMed]
- Kondrikov, D.; Elms, S.; Fulton, D.; Su, Y. eNOS-β-actin interaction contributes to increased peroxynitrite formation during hyperoxia in pulmonary artery endothelial cells and mouse lungs. J. Biol. Chem. 2010, 285, 35479–35487. [Google Scholar] [CrossRef] [PubMed]
- Konduri, G.G.; Ou, J.; Shi, Y.; Pritchard, K.A., Jr. Decreased association of HSP90 impairs endothelial nitric oxide synthase in fetal lambs with persistent pulmonary hypertension. Am. J. Physiol. Heart Circ. Physiol. 2003, 285, H204–H211. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.K.; Li, S.H.; Zhao, Z.M.; Liu, S.X.; Zhang, G.X.; Yang, F.; Wang, Y.; Wu, F.; Zhao, X.X.; Xu, Z.Y. Inhibition of heat shock protein 90 improves pulmonary arteriole remodeling in pulmonary arterial hypertension. Oncotarget 2016, 7, 54263–54273. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Malik, P.; Pandey, D.; Gupta, S.; Jagnandan, D.; Belin de Chantemele, E.; Banfi, B.; Marrero, M.B.; Rudic, R.D.; Stepp, D.W.; et al. Paradoxical activation of endothelial nitric oxide synthase by NADPH oxidase. Arterioscler. Thromb. Vasc. Biol. 2008, 28, 1627–1633. [Google Scholar] [CrossRef] [PubMed]
- Wedgwood, S.; Lakshminrusimha, S.; Farrow, K.N.; Czech, L.; Gugino, S.F.; Soares, F.; Russell, J.A.; Steinhorn, R.H. Apocynin improves oxygenation and increases eNOS in persistent pulmonary hypertension of the newborn. Am. J. Physiol. Lung Cell. Mol. Physiol. 2012, 302, L616–L626. [Google Scholar] [CrossRef] [PubMed]
- Schroder, K.; Zhang, M.; Benkhoff, S.; Mieth, A.; Pliquett, R.; Kosowski, J.; Kruse, C.; Luedike, P.; Michaelis, U.R.; Weissmann, N.; et al. Nox4 is a protective reactive oxygen species generating vascular NADPH oxidase. Circ. Res. 2012, 110, 1217–1225. [Google Scholar] [CrossRef] [PubMed]
- Craige, S.M.; Chen, K.; Pei, Y.; Li, C.; Huang, X.; Chen, C.; Shibata, R.; Sato, K.; Walsh, K.; Keaney, J.F., Jr. NADPH oxidase 4 promotes endothelial angiogenesis through endothelial nitric oxide synthase activation. Circulation 2011, 124, 731–740. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.D.; Courtman, D.W.; Deng, Y.; Kugathasan, L.; Zhang, Q.; Stewart, D.J. Rescue of monocrotaline-induced pulmonary arterial hypertension using bone marrow-derived endothelial-like progenitor cells: Efficacy of combined cell and eNOS gene therapy in established disease. Circ. Res. 2005, 96, 442–450. [Google Scholar] [CrossRef] [PubMed]
- Fullerton, D.A.; Hahn, A.R.; McIntyre, R.C., Jr. Mechanistic imbalance of pulmonary vasomotor control in progressive lung injury. Surgery 1996, 119, 98–103. [Google Scholar] [CrossRef]
- Mam, V.; Tanbe, A.F.; Vitali, S.H.; Arons, E.; Christou, H.A.; Khalil, R.A. Impaired vasoconstriction and nitric oxide-mediated relaxation in pulmonary arteries of hypoxia- and monocrotaline-induced pulmonary hypertensive rats. J. Pharmacol. Exp. Ther. 2010, 332, 455–462. [Google Scholar] [CrossRef] [PubMed]
- Schermuly, R.T.; Stasch, J.P.; Pullamsetti, S.S.; Middendorff, R.; Muller, D.; Schluter, K.D.; Dingendorf, A.; Hackemack, S.; Kolosionek, E.; Kaulen, C.; et al. Expression and function of soluble guanylate cyclase in pulmonary arterial hypertension. Eur. Respir. J. 2008, 32, 881–891. [Google Scholar] [CrossRef] [PubMed]
- Ghofrani, H.A.; Wiedemann, R.; Rose, F.; Weissmann, N.; Schermuly, R.T.; Quanz, K.; Grimminger, F.; Seeger, W.; Olschewski, H. Lung cGMP release subsequent to NO inhalation in pulmonary hypertension: Responders versus nonresponders. Eur. Respir. J. 2002, 19, 664–671. [Google Scholar] [CrossRef] [PubMed]
- Kodama, K.; Adachi, H. Improvement of mortality by long-term E4010 treatment in monocrotaline-induced pulmonary hypertensive rats. J. Pharmacol. Exper. Ther. 1999, 290, 748–752. [Google Scholar]
- Pezzuto, B.; Badagliacca, R.; Poscia, R.; Ghio, S.; D’Alto, M.; Vitulo, P.; Mule, M.; Albera, C.; Volterrani, M.; Fedele, F.; et al. Circulating biomarkers in pulmonary arterial hypertension: Update and future direction. J. Heart Lung Trans. Off. Publ. Int. Soc. Heart Trans. 2015, 34, 282–305. [Google Scholar] [CrossRef] [PubMed]
- Stasch, J.P.; Schmidt, P.M.; Nedvetsky, P.I.; Nedvetskaya, T.Y.; H, S.A.; Meurer, S.; Deile, M.; Taye, A.; Knorr, A.; Lapp, H.; et al. Targeting the heme-oxidized nitric oxide receptor for selective vasodilatation of diseased blood vessels. J. Clin. Investig. 2006, 116, 2552–2561. [Google Scholar] [CrossRef] [PubMed]
- Stasch, J.P.; Evgenov, O.V. Soluble guanylate cyclase stimulators in pulmonary hypertension. Handb. Exp. Pharmacol. 2013, 218, 279–313. [Google Scholar] [PubMed]
- Chester, M.; Seedorf, G.; Tourneux, P.; Gien, J.; Tseng, N.; Grover, T.; Wright, J.; Stasch, J.P.; Abman, S.H. Cinaciguat, a soluble guanylate cyclase activator, augments cGMP after oxidative stress and causes pulmonary vasodilation in neonatal pulmonary hypertension. Am. J. Physiol. Lung Cell. Mol. Physiol. 2011, 301, L755–L764. [Google Scholar] [CrossRef] [PubMed]
- Bender, A.T.; Beavo, J.A. Cyclic nucleotide phosphodiesterases: Molecular regulation to clinical use. Pharmacol. Rev. 2006, 58, 488–520. [Google Scholar] [CrossRef] [PubMed]
- Murray, F.; MacLean, M.R.; Pyne, N.J. Increased expression of the cGMP-inhibited cAMP-specific (PDE3) and cGMP binding cGMP-specific (PDE5) phosphodiesterases in models of pulmonary hypertension. Br. J. Pharmacol. 2002, 137, 1187–1194. [Google Scholar] [CrossRef] [PubMed]
- Sebkhi, A.; Strange, J.W.; Phillips, S.C.; Wharton, J.; Wilkins, M.R. Phosphodiesterase type 5 as a target for the treatment of hypoxia-induced pulmonary hypertension. Circulation 2003, 107, 3230–3235. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.D.; Cai, L.; Mirza, M.K.; Huang, X.; Geenen, D.L.; Hofmann, F.; Yuan, J.X.; Zhao, Y.Y. Protein kinase G-I deficiency induces pulmonary hypertension through Rho A/Rho kinase activation. Am. J. Pathol. 2012, 180, 2268–2275. [Google Scholar] [CrossRef] [PubMed]
- Palacios-Callender, M.; Quintero, M.; Hollis, V.S.; Springett, R.J.; Moncada, S. Endogenous NO regulates superoxide production at low oxygen concentrations by modifying the redox state of cytochrome c oxidase. Proc. Natl. Acad. Sci. USA 2004, 101, 7630–7635. [Google Scholar] [CrossRef] [PubMed]
- Qian, J.; Chen, F.; Kovalenkov, Y.; Pandey, D.; Moseley, M.A.; Foster, M.W.; Black, S.M.; Venema, R.C.; Stepp, D.W.; Fulton, D.J. Nitric oxide reduces NADPH oxidase 5 (Nox5) activity by reversible S-nitrosylation. Free Radic. Biol. Med. 2012, 52, 1806–1819. [Google Scholar] [CrossRef] [PubMed]
- Burwell, L.S.; Nadtochiy, S.M.; Tompkins, A.J.; Young, S.; Brookes, P.S. Direct evidence for S-nitrosation of mitochondrial complex I. Biochem. J. 2006, 394, 627–634. [Google Scholar] [CrossRef] [PubMed]
- Tuder, R.M.; Groves, B.; Badesch, D.B.; Voelkel, N.F. Exuberant endothelial cell growth and elements of inflammation are present in plexiform lesions of pulmonary hypertension. Am. J. Pathol. 1994, 144, 275–285. [Google Scholar] [PubMed]
- Jonigk, D.; Golpon, H.; Bockmeyer, C.L.; Maegel, L.; Hoeper, M.M.; Gottlieb, J.; Nickel, N.; Hussein, K.; Maus, U.; Lehmann, U.; et al. Plexiform lesions in pulmonary arterial hypertension composition, architecture, and microenvironment. Am. J. Pathol. 2011, 179, 167–179. [Google Scholar] [CrossRef] [PubMed]
- Tuder, R.M.; Chacon, M.; Alger, L.; Wang, J.; Taraseviciene-Stewart, L.; Kasahara, Y.; Cool, C.D.; Bishop, A.E.; Geraci, M.; Semenza, G.L.; et al. Expression of angiogenesis-related molecules in plexiform lesions in severe pulmonary hypertension: Evidence for a process of disordered angiogenesis. J. Pathol. 2001, 195, 367–374. [Google Scholar] [CrossRef] [PubMed]
- Nair, J.; Lakshminrusimha, S. Update on PPHN: Mechanisms and treatment. Semin. Perinatol. 2014, 38, 78–91. [Google Scholar] [CrossRef] [PubMed]
- Zuckerbraun, B.S.; Shiva, S.; Ifedigbo, E.; Mathier, M.A.; Mollen, K.P.; Rao, J.; Bauer, P.M.; Choi, J.J.; Curtis, E.; Choi, A.M.; et al. Nitrite potently inhibits hypoxic and inflammatory pulmonary arterial hypertension and smooth muscle proliferation via xanthine oxidoreductase-dependent nitric oxide generation. Circulation 2010, 121, 98–109. [Google Scholar] [CrossRef] [PubMed]
- Hunter, C.J.; Dejam, A.; Blood, A.B.; Shields, H.; Kim-Shapiro, D.B.; Machado, R.F.; Tarekegn, S.; Mulla, N.; Hopper, A.O.; Schechter, A.N.; et al. Inhaled nebulized nitrite is a hypoxia-sensitive NO-dependent selective pulmonary vasodilator. Nat. Med. 2004, 10, 1122–1127. [Google Scholar] [CrossRef] [PubMed]
- Nishimura, T.; Faul, J.L.; Berry, G.J.; Vaszar, L.T.; Qiu, D.; Pearl, R.G.; Kao, P.N. Simvastatin attenuates smooth muscle neointimal proliferation and pulmonary hypertension in rats. Am. J. Respir. Crit. Care Med. 2002, 166, 1403–1408. [Google Scholar] [CrossRef] [PubMed]
- Oesterle, A.; Laufs, U.; Liao, J.K. Pleiotropic effects of statins on the cardiovascular system. Circ. Res. 2017, 120, 229–243. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Qu, M.; Chen, Y.; Zhou, Y.; Wan, Z. Statins have no additional benefit for pulmonary hypertension: A meta-analysis of randomized controlled trials. PLoS ONE 2016, 11, e0168101. [Google Scholar] [CrossRef] [PubMed]
- Dubois, M.; Delannoy, E.; Duluc, L.; Closs, E.; Li, H.; Toussaint, C.; Gadeau, A.P.; Godecke, A.; Freund-Michel, V.; Courtois, A.; et al. Biopterin metabolism and eNOS expression during hypoxic pulmonary hypertension in mice. PLoS ONE 2013, 8, e82594. [Google Scholar] [CrossRef] [PubMed]
- Jasmin, J.F.; Mercier, I.; Dupuis, J.; Tanowitz, H.B.; Lisanti, M.P. Short-term administration of a cell-permeable caveolin-1 peptide prevents the development of monocrotaline-induced pulmonary hypertension and right ventricular hypertrophy. Circulation 2006, 114, 912–920. [Google Scholar] [CrossRef] [PubMed]
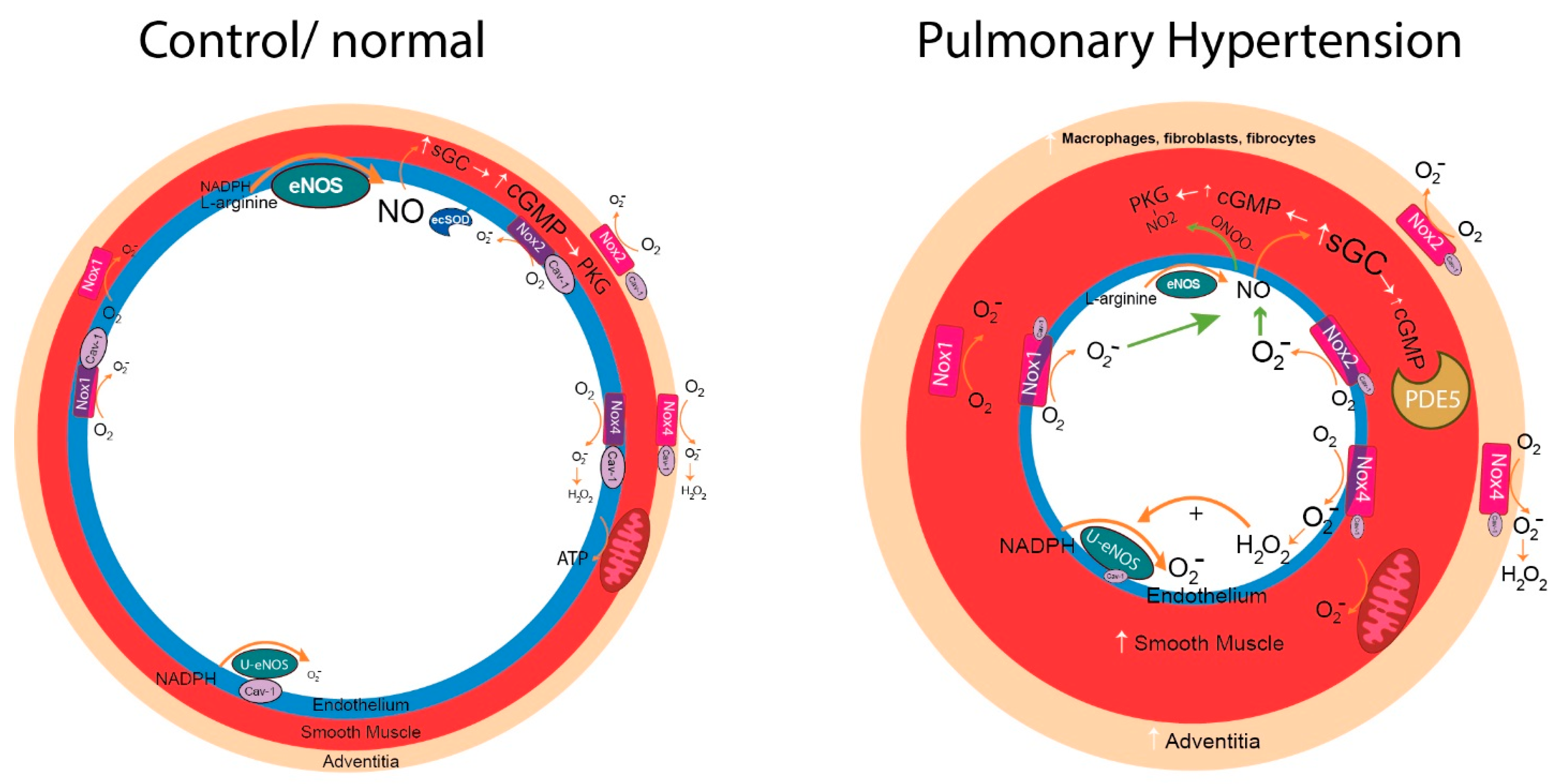

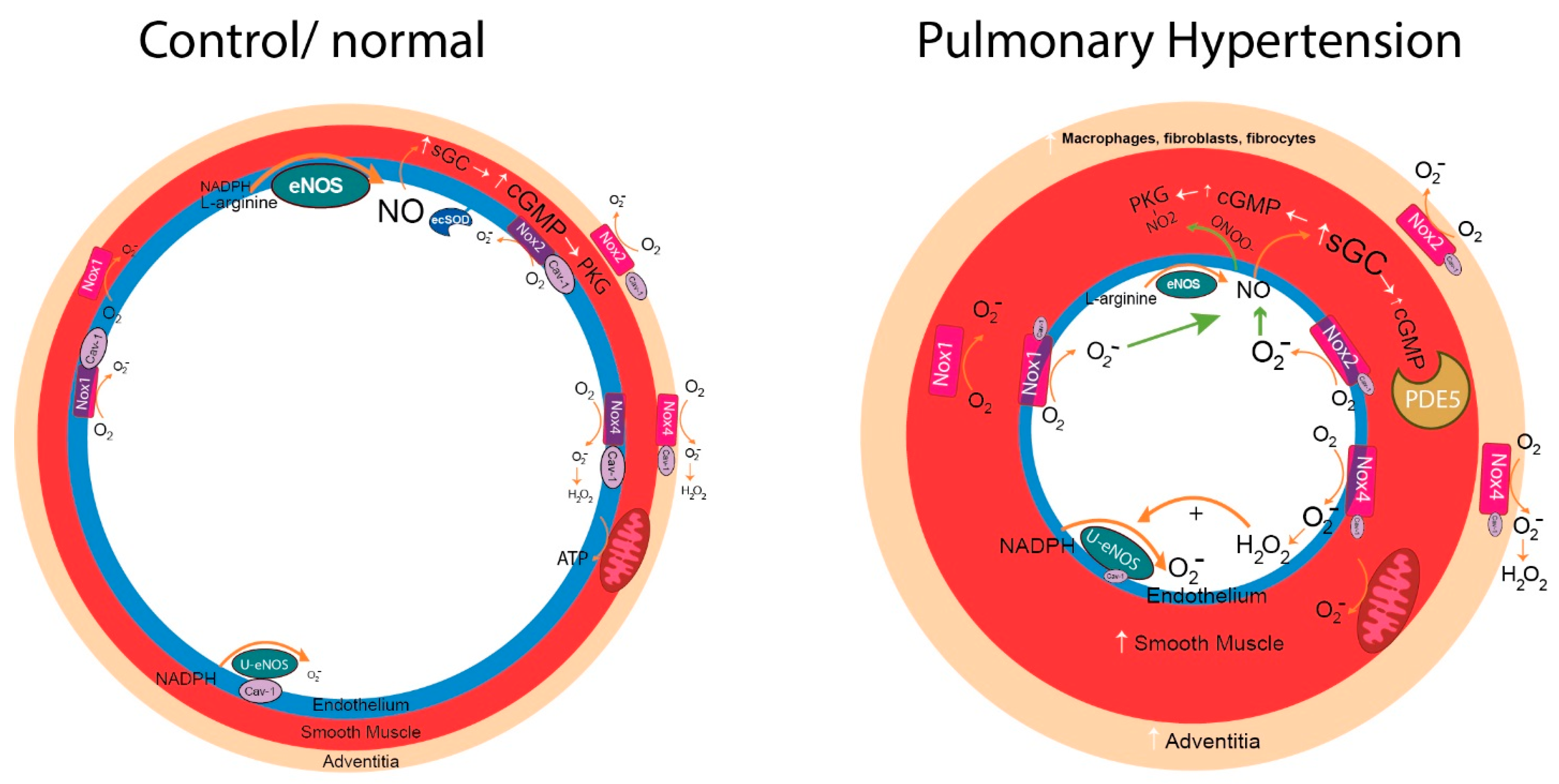{kind=link}
| Type of PH | Organism | Nox mRNA | Nox Protein | Intervention | Effect on Disease | Reference |
|---|---|---|---|---|---|---|
| Hypoxia, IPAH | Mouse, human | ↑ Nox4 No change Nox1, Nox2 | ↑ Nox4 | Nox4 siRNA PASMC | ↓ SMC proliferation | [73] |
| Hypoxia (in vitro) | Human | ↑ Nox4, Nox1 | ↑ Nox4 | Nox4 siRNA in PAFB | ↓ PAFB proliferation, ↑ apoptosis | [74] |
| Hypoxia | Pig | - | ↑ Nox1 No change Nox4 | Apocynin, ROS scavengers | ↑ PA function | [65] |
| Hypoxia (in vitro) | Human | ↑ Nox4 | - | Nox4 siRNA PASMC | ↓ SMC proliferation | [75] |
| CIH | Mouse | ↑ Nox4, p22 | Nox2 KO | ↓ pulmonary hypertension | [57] | |
| PPHN | Sheep | ↑ Nox2, Nox4 | - | Apocynin, ROS scavengers | ↑ PAEC angiogenesis, decreased apoptosis | [76] |
| MCT, MCT/PN | Rat | ↑ Nox2, Nox4, no change Nox1 | - | - | - | [72] |
| Hypoxia | Mouse, Human EC | ↑ Nox4 | - | GKT137831 (Nox4/Nox1) | ↓ pulmonary hypertension, ↓ PASMC proliferation | [77] |
| MCT | RAT PASMC | ↑ Nox1, no change Nox2, Nox4 | ↑ Nox1 | Nox1 siRNA PASMC | ↓ SMC proliferation, migration | [70] |
| MCT, FHR, Sugen Hypoxia, IPAH | Rat PA, Human lung | ↑ Nox4, Nox2, no change Nox1 | ↑ Nox4 | GKT136901 (Nox4/Nox1), VCC588646, VCC202273 | ↓ pulmonary hypertension, ↓ remodeling/PAFB proliferation | [55] |
| Hypoxia | Mouse | - | - | Nox1 KO | ↑ pulmonary hypertension | [71] |
| Hypoxia | Mouse | no change Nox2 | - | Nox2 KO | ↓ pulmonary hypertension | [61] |
| MCT | Mouse | - | - | Nox4 transgenic, inducible KO | No change in pulmonary hypertension | [78] |
| MCT | Rat | - | ↑ Nox1, Nox2 | Resveratrol | ↓ pulmonary hypertension, ↓ PASMC proliferation | [54] |
| iPAH, hypoxia | Human, rat | - | ↑ Nox4 | PP242 (mTOR) | ↓PAremodeling, ↑ SMC apoptosis | [79] |
| Hypoxia | Mouse | - | - | Nox2 KO | ↑ endothelial function | [60] |
| Hypoxia | Mouse, human PAEC | ↑ Nox4, Nox2 (HPAEC) | ↑ Nox4, Nox2 (HPAEC) | Mitochondrial-targeted catalase | ↓ pulmonary hypertension | [80] |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fulton, D.J.R.; Li, X.; Bordan, Z.; Haigh, S.; Bentley, A.; Chen, F.; Barman, S.A. Reactive Oxygen and Nitrogen Species in the Development of Pulmonary Hypertension. Antioxidants 2017, 6, 54. https://doi.org/10.3390/antiox6030054
Fulton DJR, Li X, Bordan Z, Haigh S, Bentley A, Chen F, Barman SA. Reactive Oxygen and Nitrogen Species in the Development of Pulmonary Hypertension. Antioxidants. 2017; 6(3):54. https://doi.org/10.3390/antiox6030054
Chicago/Turabian StyleFulton, David J.R., Xueyi Li, Zsuzsanna Bordan, Stephen Haigh, Austin Bentley, Feng Chen, and Scott A. Barman. 2017. "Reactive Oxygen and Nitrogen Species in the Development of Pulmonary Hypertension" Antioxidants 6, no. 3: 54. https://doi.org/10.3390/antiox6030054
APA StyleFulton, D. J. R., Li, X., Bordan, Z., Haigh, S., Bentley, A., Chen, F., & Barman, S. A. (2017). Reactive Oxygen and Nitrogen Species in the Development of Pulmonary Hypertension. Antioxidants, 6(3), 54. https://doi.org/10.3390/antiox6030054