
Mitochondrial Dysfunction in Cancer and Neurodegenerative Diseases: Spotlight on Fatty Acid Oxidation and Lipoperoxidation Products


 ,
,  , and
, and 
Abstract
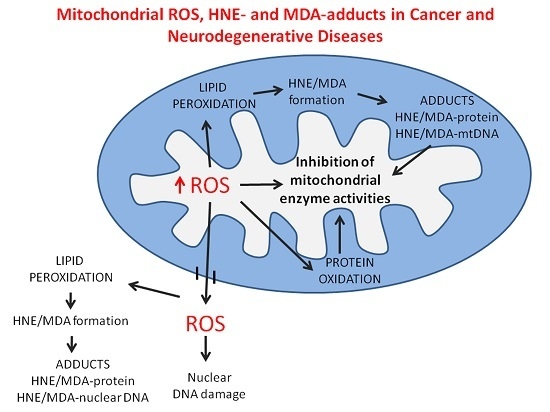
:
1. Introduction
2. Mitochondrial Dysfunction in Cancer
2.1. Aerobic Glycolysis and Oxidative Phosphorylation
2.2. MtDNA Mutations in Cancer Cells
3. Lipid Composition and Lipoperoxidation in Mitochondria of Cancer Cells
3.1. Lipid Composition and Mitochondrial Functions in Normal and Cancer Cells
3.2. Lipid Catabolism and Reactive Oxygen Species (ROS) Production in Cancer Cells
3.3. Lipid Peroxidation and Its Products
3.4. 4-Hydroxynonenal (HNE)-Protein Adducts in Cancer Cell Mitochondria
4. Oxidative Stress and Mitochondrial Dysfunction in Neurodegenerative Disease
5. Role of the Adducts of Aldehydes Derived from Lipid Peroxidation with Mitochondrial Proteins in Neurodegeneration
5.1. HNE-Modified Mitochondrial Proteins
5.1.1. Mitochondrial Aconitate Hydratase (Aconitase 2, ACO2)
5.1.2. Mitochondrial Adenosine Triphosphate (ATP) Synthase (Respiratory Complex V), Alpha Subunit 1 (ATP5A1)
- (a)
- Leigh syndrome (LS, Necrotizing encephalopathy, infantile subacute, SNE, MIM 256000). This is an early-onset, progressive neurodegenerative disorder, with bilateral foci of demyelination, necrosis, gliosis, spongiosis or capillary proliferation in one or more areas of the central nervous system (brainstem, thalamus, basal ganglia, cerebellum and spinal cord). Patients exhibit psychomotor retardation, epilepsy, cerebellar and motor disturbances, extraocular muscle incoordination, neurogenic breathing disorders, neural deafness, retinitis pigmentosa, polyneuropathy, lactic acidosis, and cardiomyopathy. LS is genetically heterogeneous, being associated with the defects of several mitochondrial- and nuclear-encoded subunits and assembly factors of respiratory complexes IV, I (most often), but also V, III, mitochondrial tRNAs and other nuclear-encoded mitochondrial proteins, such as pyruvate dehydrogenase E-1α subunit.
- (b)
- LHON (Leber Hereditary Optic Neuropathy, MIM 535000). LHON presents in midlife as acute or subacute central vision loss, with peripapillary microangiopathy and telangiectasia, to central scotoma and blindness, inherited with a maternal pattern of transmission. Disturbances of cardiac conduction and dystonia may coexist. The disease has been associated with many allelic missense mutations in the mtDNA that can act autonomously or in association with each other to cause the disease. LHON is also genetically heterogeneous, possibly reflecting mutations of most mitochondrially encoded subunits of respiratory complexes I, III, IV and V.
- (c)
- NARP (Neurogenic muscle weakness, Ataxia, and Retinitis Pigmentosa, MIM 551500). This syndrome entails a combination of developmental and psychomotor retardation, retinitis pigmentosa, dementia, seizures, clonic spasms, ataxia, proximal neurogenic muscle weakness, sensory neuropathy, hearing loss and optic atrophy, with a maternal pattern of transmission, with no histochemical evidence of myopathy. Mattiazzi et al. [139] showed that the T8993G mutation of the ATP6 gene inhibited oxidative phosphorylation and enhanced free radical production. Antioxidants restored respiration and partially rescued ATP synthesis in cells harboring the mutation, suggesting that free radicals may play an important pathogenetic role.
5.1.3. Mitochondrial Translation Elongation Factor Tu (EF-Tu, TUFM), Malate Dehydrogenase 2 (MDH2), and Mn Superoxide Dismutase (SOD2)
5.2. Malondialdehyde-Modified Mitochondrial Proteins
Ubiquinol-Cytochrome c Reductase (Respiratory Complex III, Cytochrome B-C1) Core Protein 1 (UQCRC1), ATP Synthase (Respiratory Complex V) Beta Subunit (ATP5B), and 60-kDa Heat Shock Protein (HSPD1, HSP60, GroEL), and Mitochondrial Glutamate Dehydrogenase 1 (GDH1)
5.3. Mitochondrial Proteins with Increased Content of DPNH-Reactive Groups
5.3.1. Mitochondrial Aconitate Hydratase (Aconitase 2, ACO2)
5.3.2. Mitochondrial Citrate Synthase (CS), Ubiquitous Creatine Kinase (CKMT1A), Ubiquinol-Cytochrome c Reductase (Respiratory Complex III, Cytochrome B-C1) Core Protein 2 (UQCRC2), and ATP Synthase (Respiratory Complex V) Alpha Subunit 1 (ATP5A1)
5.3.3. Antioxidant Defense Protein DJ-1 (Parkinson Protein 7, PARK7)
5.3.4. Mitochondrial NADH-Ubiquinone Oxidoreductase (Respiratory Complex I)
6. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Pandey, P.R.; Liu, W.; Xing, F.; Fukuda, K.; Watabe, K. Anti-cancer drugs targeting fatty acid synthase (FAS). Recent Pat. Anticancer Drug Discov. 2012, 7, 185–197. [Google Scholar] [CrossRef] [PubMed]
- Wise, D.R.; Thompson, C.B. Glutamine addiction: A new therapeutic target in cancer. Trends Biochem. Sci. 2010, 35, 427–433. [Google Scholar] [CrossRef] [PubMed]
- Warburg, O. On the origin of cancer cells. Science 1956, 123, 309–314. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Yang, C.; Yang, Z.; Zhang, D.; Ma, X.; Mills, G.; Liu, Z. Homeostasis of redox status derived from glucose metabolic pathway could be the key to understanding the Warburg effect. Am. J. Cancer Res. 2015, 5, 1265–1280. [Google Scholar] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Samudio, I.; Harmancey, R.; Fiegl, M.; Kantarjian, H.; Konopleva, M.; Korchin, B.; Kaluarachchi, K.; Bornmann, W.; Duvvuri, S.; Taegtmeyer, H.; et al. Pharmacologic inhibition of fatty acid oxidation sensitizes human leukemia cells to apoptosis induction. J. Clin. Investig. 2010, 120, 142–156. [Google Scholar] [CrossRef] [PubMed]
- Tirado-Vélez, J.M.; Joumady, I.; Sáez-Benito, A.; Cózar-Castellano, I.; Perdomo, G. Inhibition of fatty acid metabolism reduces human myeloma cells proliferation. PLoS ONE 2012, 7. [Google Scholar] [CrossRef] [PubMed]
- West, D.; Marnett, L.J. Endogenous reactive intermediates as modulators of cell signaling and cell death. Chem. Res. Toxicol. 2006, 19, 173–194. [Google Scholar] [CrossRef] [PubMed]
- Barrera, G.; Pizzimenti, S.; Ciamporcero, E.S.; Daga, M.; Ullio, C.; Arcaro, A.; Cetrangolo, G.P.; Ferretti, C.; Dianzani, C.; Lepore, A.; Gentile, F. Role of 4-hydroxynonenal-protein adducts in human diseases. Antioxid. Redox Signal. 2015, 22, 1681–1702. [Google Scholar] [CrossRef] [PubMed]
- Chandra, D.; Singh, K.K. Genetic insights into OXPHOS defect and its role in cancer. Biochim. Biophys. Acta 2011, 1807, 620–625. [Google Scholar] [CrossRef] [PubMed]
- Yadav, N.; Chandra, D. Mitochondrial DNA mutations and breast tumorigenesis. Biochim. Biophys. Acta 2013, 1836, 336–344. [Google Scholar] [CrossRef] [PubMed]
- Wallace, D.C. Mitochondria and cancer. Nat. Rev. Cancer 2012, 12, 685–698. [Google Scholar] [CrossRef] [PubMed]
- Jang, M.; Kim, S.S.; Lee, J. Cancer cell metabolism: Implications for therapeutic targets. Exp. Mol. Med. 2013, 45. [Google Scholar] [CrossRef] [PubMed]
- Yadava, N.; Schneider, S.S.; Jerry, D.J.; Kim, C. Impaired mitochondrial metabolism and mammary carcinogenesis. J. Mammary Gland Biol. Neoplasia 2013, 18, 75–87. [Google Scholar] [CrossRef] [PubMed]
- Brand, K.A.; Hermfisse, U. Aerobic glycolysis by proliferating cells: A protective strategy against reactive oxygen species. FASEB J. 1997, 11, 388–395. [Google Scholar] [PubMed]
- Sabharwal, S.S.; Schumacker, P.T. Mitochondrial ROS in cancer: Initiators, amplifiers or an Achilles’ heel? Nat. Rev. Cancer. 2014, 14, 709–721. [Google Scholar] [CrossRef] [PubMed]
- Viale, A.; Corti, D.; Draetta, G.F. Tumors and Mitochondrial Respiration: A Neglected Connection. Cancer Res. 2015, 75, 3685–3686. [Google Scholar] [CrossRef] [PubMed]
- Xekouki, P.; Stratakis, C.A. Succinate dehydrogenase (SDHx) mutations in pituitary tumors: Could this be a new role for mitochondrial complex II and/or Krebs cycle defects? Endocrinol. Relat. Cancer 2012, 19. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.; Soga, T.; Pollard, P.J.; Adam, J. The emerging role of fumarate as an oncometabolite. Front. Oncol. 2012, 2. [Google Scholar] [CrossRef] [PubMed]
- Mayr, J.A.; Meierhofer, D.; Zimmerman, F.; Feichtinger, R.; Kögler, C.; Ratschek, M.; Schmeller, N.; Sperl, W.; Kofler, B. Loss of complex I due to mitochondrial DNA mutations in renal oncocytoma. Clin. Cancer Res. 2008, 14, 2270–2275. [Google Scholar] [CrossRef] [PubMed]
- Chiaradonna, F.; Gaglio, D.; Vanoni, M.; Alberghina, L. Expression oftransforming K-Ras oncogene affects mitochondrial function and morphologyin mouse fibroblasts. Biochim. Biophys. Acta 2006, 1757, 1338–1356. [Google Scholar] [CrossRef] [PubMed]
- Fridovich, I. Superoxide radical and superoxide dismutases. Annu. Rev. Biochem. 1995, 64, 97–112. [Google Scholar] [CrossRef] [PubMed]
- Okado-Matsumoto, A.; Fridovich, I. Subcellular distribution of superoxide dismutase (SOD) in rat liver: Cu, Zn-SOD in mitochondria. J. Biol. Chem. 2001, 276, 38388–38393. [Google Scholar] [CrossRef] [PubMed]
- Weinberg, F.; Hamanaka, R.; Wheaton, W.W.; Weinberg, S.; Joseph, J.; Lopez, M.; Kalyanaraman, B.; Mutlu, G.M.; Budinger, G.R.; Chandel, N.S. Mitochondrial metabolism and ROS generation are essential for Kras-mediated tumorigenicity. Proc. Natl Acad. Sci. USA 2010, 107, 8788–8793. [Google Scholar] [CrossRef] [PubMed]
- Patel, H.; Pietro, E.D.; MacKenzie, R.E. Mammalian fibroblasts lacking mitochondrialNAD+-dependent methylenetetrahydrofolate dehydrogenase cyclohydrolase are glycine auxotrophs. J. Biol. Chem. 2003, 278, 19436–19441. [Google Scholar] [CrossRef] [PubMed]
- Christensen, K.E.; Mackenzie, R.E. Mitochondrial methylenetetrahydrofolate dehydrogenase, methenyltetrahydrofolate cyclohydrolase, and formyltetrahydrofolate synthetases. Vitam. Horm. 2008, 79, 393–410. [Google Scholar] [PubMed]
- Fan, J.; Ye, J.; Kamphorst, J.J.; Shlomi, T.; Thompson, C.B.; Rabinowitz, J.D. Quantitative flux analysis reveals folate-dependent NADPH production. Nature 2014, 510, 298–302. [Google Scholar] [CrossRef] [PubMed]
- Nilsson, R.; Jain, M.; Madhusudhan, N.; Sheppard, N.G.; Strittmatter, L.; Kampf, C.; Huang, J.; Asplund, A.; Mootha, V.K. Metabolic enzyme expression highlights a key role for MTHFD2 and the mitochondrial folate pathway in cancer. Nat. Commun. 2014, 5. [Google Scholar] [CrossRef] [PubMed]
- Craigen, W.J. Mitochondrial DNA mutations: An overview of clinical and molecular aspects. Methods Mol. Biol. 2012, 837, 3–15. [Google Scholar] [PubMed]
- He, Y.; Wu, J.; Dressman, D.C.; Iacobuzio-Donahue, C.; Markowitz, S.D.; Velculescu, V.E.; Diaz, L.A., Jr.; Kinzler, K.W.; Vogelstein, B.; Papadopoulos, N. Heteroplasmic mitochondrial DNA mutations in normal and tumour cells. Nature 2010, 464, 610–614. [Google Scholar] [CrossRef] [PubMed]
- Lynch, M.; Koskella, B.; Schaack, S. Mutation pressure and the evolution of organelle genomic architecture. Science 2006, 311, 1727–1730. [Google Scholar] [CrossRef] [PubMed]
- Johnson, A.A.; Johnson, K.A. Exonuclease proofreading by human mitochondrial DNA polymerase. J. Biol. Chem. 2001, 276, 38097–38107. [Google Scholar] [CrossRef] [PubMed]
- Kornberg, R.D. Chromatin structure: A repeating unit of histones and DNA. Science 1974, 184, 868–871. [Google Scholar] [CrossRef] [PubMed]
- Yakes, F.M.; van Houten, B. Mitochondrial DNA damage is more extensive and persists longer than nuclear DNA damage in human cells following oxidative stress. Proc. Natl. Acad. Sci. USA 1997, 94, 514–519. [Google Scholar] [CrossRef] [PubMed]
- Polyak, K.; Li, Y.; Zhu, H.; Lengauer, C.; Willson, J.K.; Markowitz, S.D.; Trush, M.A.; Kinzler, K.W.; Vogelstein, B. Somatic mutations of the mitochondrial genome in human colorectal tumours. Nat. Genet. 1998, 20, 291–293. [Google Scholar] [PubMed]
- Kulawiec, M.; Salk, J.J.; Ericson, N.G.; Wanagat, J.; Bielas, J.H. Generation, function, and prognostic utility of somatic mitochondrial DNA mutations in cancer. Environ. Mol. Mutagen. 2010, 51, 427–439. [Google Scholar] [CrossRef] [PubMed]
- Liu, V.W.; Shi, H.H.; Cheung, A.N.; Chiu, P.M.; Leung, T.W.; Nagley, P.; Wong, L.C.; Ngan, H.Y. High incidence of somatic mitochondrial DNA mutations in human ovarian carcinomas. Cancer Res. 2001, 61, 5998–6001. [Google Scholar] [PubMed]
- Van Gisbergen, M.W.; Voets, A.M.; Starmans, M.H.; de Coo, I.F.; Yadak, R.; Hoffmann, R.F.; Boutros, P.C.; Smeets, H.J.; Dubois, L.; Lambin, P. How do changes in the mtDNA and mitochondrial dysfunction influence cancer and cancer therapy? Challenges, opportunities and models. Mutat. Res. Rev. Mutat. Res. 2015, 764, 16–30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schon, E.A.; DiMauro, S.; Hirano, M. Human mitochondrial DNA: Roles of inherited and somatic mutations. Nat. Rev. Genet. 2012, 13, 878–890. [Google Scholar] [CrossRef] [PubMed]
- Petros, J.A.; Baumann, A.K.; Ruiz-Pesini, E.; Amin, M.B.; Sun, C.Q.; Hall, J.; Lim, S.; Issa, M.M.; Flanders, W.D.; Hosseini, S.H.; et al. mtDNA mutations increase tumorigenicity in prostate cancer. Proc. Natl. Acad. Sci. USA 2005, 102, 719–724. [Google Scholar] [CrossRef] [PubMed]
- Ishikawa, K.; Takenaga, K.; Akimoto, M.; Koshikawa, N.; Yamaguchi, A.; Imanishi, H.; Nakada, K.; Honma, Y.; Hayashi, J. ROS-generating mitochondrial DNA mutations can regulate tumor cell metastasis. Science 2008, 320, 661–664. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sullivan, L.B.; Chandel, N.S. Mitochondrial reactive oxygen species and cancer. Cancer Metab. 2014, 2. [Google Scholar] [CrossRef] [PubMed]
- Schug, Z.T.; Frezza, C.; Galbraith, L.C.; Gottlieb, E. The music of lipids: How lipid composition orchestrates cellular behaviour. Acta Oncol. 2012, 51, 301–310. [Google Scholar] [CrossRef] [PubMed]
- Schenkel, L.C.; Bakovic, M. Formation and regulation of mitochondrial membranes. Int. J. Cell Biol. 2014, 2014. [Google Scholar] [CrossRef] [PubMed]
- Horvath, S.E.; Daum, G. Lipids of mitochondria. Prog. Lipid Res. 2013, 52, 590–614. [Google Scholar] [CrossRef] [PubMed]
- Daum, G.; Vance, J.E. Import of lipids into mitochondria. Prog. Lipid Res. 1997, 36, 103–130. [Google Scholar] [CrossRef]
- Ren, M.; Phoon, C.K.; Schlame, M. Metabolism and function of mitochondrial cardiolipin. Prog. Lipid Res. 2014, 55, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Chu, C.T.; Ji, J.; Dagda, R.K.; Jiang, J.F.; Tyurina, Y.Y.; Kapralov, A.A.; Tyurin, V.A.; Yanamala, N.; Shrivastava, I.H.; Mohammadyani, D.; et al. Cardiolipin externalization to the outer mitochondrial membrane acts as an elimination signal for mitophagy in neuronal cells. Nat. Cell Biol. 2013, 15, 1197–1205. [Google Scholar] [CrossRef] [PubMed]
- Osman, C.; Voelker, D.R.; Langer, T. Making heads or tails of phospholipids in mitochondria. J. Cell Biol. 2011, 192, 7–16. [Google Scholar] [CrossRef] [PubMed]
- Shiao, Y.-J.; Vance, J.E. Evidence for an ethanolamine cycle: Differential recycling of the ethanolamine moiety of phosphatidylethanolamine derived from phosphatidylserine and ethanolamine. Biochem. J. 1995, 310, 673–679. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Dai, Q.; Chen, J.; Durrant, D.; Freeman, A.; Liu, T.; Grossman, D.; Lee, R.M. Phospholipid scramblase 3 controls mitochondrial structure, function, and apoptotic response. Mol. Cancer Res. 2003, 1, 892–902. [Google Scholar] [PubMed]
- Mayr, J.A. Lipid metabolism in mitochondrial membranes. J. Inherit. Metab. Dis. 2015, 38, 137–144. [Google Scholar] [CrossRef] [PubMed]
- Hovius, R.; Thijssen, J.; van der Linden, P.; Nicolay, K.; de Kruijff, B. Phospholipid asymmetry of the outer membrane of rat liver mitochondria. Evidence for the presence of cardiolipin on the outside of the outer membrane. FEBS Lett. 1993, 330, 71–76. [Google Scholar] [CrossRef]
- Holloway, G.P.; Fajardo, V.A.; McMeekin, L.; LeBlanc, P.J. Unsaturation of mitochondrial membrane lipids is related to palmitate oxidation in subsarcolemmal and intermyofibrillar mitochondria. J. Membr. Biol. 2012, 245, 165–176. [Google Scholar] [CrossRef] [PubMed]
- Yamaoka, S.; Urade, R.; Kito, M. Cardiolipin molecular species in rat heart mitochondria are sensitive to essential fatty acid-deficient dietary lipids. J. Nutr. 1990, 120, 415–421. [Google Scholar] [PubMed]
- Fajardo, V.A.; McMeekin, L.; Saint, C.; LeBlanc, P.J. Cardiolipin linoleic acid content and mitochondrial cytochrome c oxidase activity are associated in rat skeletal muscle. Chem. Phys. Lipids 2015, 187, 50–55. [Google Scholar] [CrossRef] [PubMed]
- Soni, S.P.; LoCascio, D.S.; Liu, Y.; Williams, J.A.; Bittman, R.; Stillwell, W.; Wassall, S.R. Docosahexaenoic acid enhances segregation of lipids between: 2H-NMR study. Biophys. J. 2008, 95, 203–214. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.Y.; Jackson, S.N.; Woods, A.S. Direct MALDI-MS analysis of cardiolipin from rat organs sections. J. Am. Soc. Mass. Spectrom. 2007, 18, 567–577. [Google Scholar] [CrossRef] [PubMed]
- Feillet-Coudray, C.; Fouret, G.; Casas, F.; Coudray, C. Impact of high dietary lipid intake and related metabolic disorders on the abundance and acyl composition of the unique mitochondrial phospholipid, cardiolipin. J. Bioenerg. Biomembr. 2014, 46, 447–457. [Google Scholar] [CrossRef] [PubMed]
- Guderley, H.; Kraffe, E.; Bureau, W.; Bureau, D.P. Dietary fatty acid composition changes mitochondrial phospholipids and oxidative capacities in rainbow trout red muscle. J. Comp. Physiol. 2008, 178, 385–399. [Google Scholar] [CrossRef] [PubMed]
- Hiltunen, J.K.; Chen, Z.; Haapalainen, A.M.; Wierenga, R.K.; Kastaniotis, A.J. Mitochondrial fatty acid synthesis—An adopted set of enzymes making a pathway of major importance for the cellular metabolism. Prog. Lipid Res. 2010, 49, 27–45. [Google Scholar] [CrossRef] [PubMed]
- Hiltunen, J.K.; Schonauer, M.S.; Autio, K.J.; Mittelmeier, T.M.; Kastaniotis, A.J.; Dieckmann, C.L. Mitochondrial fatty acid synthesis type II: More than just fatty acids. J. Biol. Chem. 2009, 284, 9011–9015. [Google Scholar] [CrossRef] [PubMed]
- Stavrovskaya, I.G.; Bird, S.S.; Marur, V.R.; Sniatynski, M.J.; Baranov, S.V.; Greenberg, H.K.; Porter, C.L.; Kristal, B.S. Dietary macronutrients modulate the fatty acyl composition of rat liver mitochondrial cardiolipins. J. Lipid Res. 2013, 54, 2623–2635. [Google Scholar] [CrossRef] [PubMed]
- Stanley, W.C.; Khairallah, R.J.; Dabkowski, E.R. Update on lipids and mitochondrial function: Impact of dietary n-3 polyunsaturated fatty acids. Curr. Opin. Clin. Nutr. Metab. Care. 2012, 15, 122–126. [Google Scholar] [CrossRef] [PubMed]
- Hulbert, A.; Turner, J.N.L.; Storlien, H.; Else, P.L. Dietary fats and membrane function: Implications for metabolism and disease. Biol. Rev. Camb. Philos. Soc. 2005, 1, 155–169. [Google Scholar] [CrossRef]
- Herbst, E.A.; Paglialunga, S.; Gerling, C.; Whitfield, J.; Mukai, K.; Chabowski, A.; Heigenhauser, G.J.; Spriet, L.L.; Holloway, G.P. Omega-3 supplementation alters mitochondrial membrane composition and respiration kinetics in human skeletal muscle. J. Physiol. 2014, 592, 1341–1352. [Google Scholar] [CrossRef] [PubMed]
- Pepe, S.; Tsuchiya, N.; Lakatta, E.G.; Hansford, R.G. PUFA and aging modulate cardiac mitochondrial membrane lipid composition and Ca2+ activation of PDH. Am. J. Physiol. 1999, 276, H149–H158. [Google Scholar] [PubMed]
- Canuto, R.A.; Biocca, M.E.; Muzio, G.; Dianzani, M.U. Fatty acid composition of phospholipids in mitochondria and microsomes during diethylnitrosamine carcinogenesis in rat liver. Cell Biochem. Funct. 1989, 7, 11–19. [Google Scholar] [CrossRef] [PubMed]
- Olsson, J.M.; Eriksson, L.C.; Daliner, G. Lipid Compositions of Intracellular Membranes Isolated from Rat Liver Nodules in Wistar Rats. Cancer Res. 1991, 51, 3774–3780. [Google Scholar] [PubMed]
- Hartz, J.W.; Morton, R.E.; Waite, M.M.; Morris, H.P. Correlation of fatty acyl composition of mitochondrial and microsomal phospholipid with growth rate of rat hepatomas. Lab. Investig. 1982, 46, 73–78. [Google Scholar] [PubMed]
- Canuto, R.A.; Ferro, M.; Salvo, R.A.; Bassi, A.M.; Trombetta, A.; Maggiora, M.; Martinasso, G.; Lindahl, R.; Muzio, G. Increase in class 2 aldehyde dehydrogenase expression by arachidonic acid in rat hepatoma cells. Biochem. J. 2001, 357, 811–818. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, K.; Blaudszun, J.; Brunken, C.; Höpker, W.W.; Tauber, R.; Steinhart, H. Distribution of polyunsaturated fatty acids including conjugated linoleic acids in total and subcellular fractions from healthy and cancerous parts of human kidneys. Lipids 2005, 40, 309–315. [Google Scholar] [CrossRef] [PubMed]
- Wong, D.A.; Bassilian, S.; Lim, S.; Paul Lee, W.N. Coordination of peroxisomal beta-oxidation and fatty acid elongation in HepG2 cells. J. Biol. Chem. 2004, 279, 41302–41309. [Google Scholar] [CrossRef] [PubMed]
- Alfonso, P.; Núnez, A.; Madoz-Gurpide, J.; Lombardia, L.; Sánchez, L.; Casal, J.I. Proteomic expression analysis of colorectal cancer by two-dimensional differential gel electrophoresis. Proteomics 2005, 5, 2602–2611. [Google Scholar] [CrossRef] [PubMed]
- Capuano, F.; Guerrieri, F.; Papa, S. Oxidative phosphorylation enzymes innormal and neoplastic cell growth. J. Bioenerg. Biomembr. 1997, 29, 379–384. [Google Scholar] [CrossRef] [PubMed]
- Herrmann, P.C.; Gillespie, J.W.; Charboneau, L.; Bichsel, V.E.; Paweletz, C.P.; Calvert, V.S.; Kohn, E.C.; Emmert-Buck, M.R.; Liotta, L.A.; Petricoin, E.F., 3rd. Mitochondrial proteome: Altered cytochrome c oxidase subunit levels in prostate cancer. Proteomics 2003, 3, 1801–1810. [Google Scholar] [CrossRef] [PubMed]
- Buzzai, M.; Bauer, D.E.; Jones, R.G.; DeBerardinis, R.J.; Hatzivassiliou, G.; Elstrom, R.L.; Thompson, C.B. The glucose dependence of Akt-transformed cells can be reversed by pharmacologic activation of fatty acid beta-oxidation. Oncogene 2005, 24, 4165–4173. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-Enríquez, S.; Hernández-Esquivel, L.; Marín-Hernández, A.; El Hafidi, M.; Gallardo-Pérez, J.C.; Hernández-Reséndiz, I.; Rodríguez-Zavala, J.S.; Pacheco-Velázquez, S.C.; Moreno-Sánchez, R. Mitochondrial free fatty acid β-oxidation supports oxidative phosphorylation and proliferation in cancer cells. Int. J. Biochem. Cell Biol. 2015, 65, 209–221. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.W.; Tchernyshyov, I.; Semenza, G.L.; Dang, C.V. HIF-1-mediated expression of pyruvate dehydrogenase kinase: A metabolic switch required for cellular adaptation to hypoxia. Cell Metab. 2006, 3, 177–185. [Google Scholar] [CrossRef] [PubMed]
- Gogvadze, V.; Zhivotovsky, B.; Orrenius, S. The Warburg effect and mitochondrial stability in cancer cells. Mol. Aspects Med. 2010, 31, 60–74. [Google Scholar] [CrossRef] [PubMed]
- Raza, H.; John, A.; Brown, E.M.; Benedict, S.; Kambal, A. Alterations in mitochondrial respiratory functions, redox metabolism and apoptosis by oxidant 4-hydroxynonenal and antioxidants curcumin and melatonin in PC12 cells. Toxicol. Appl. Pharmacol. 2008, 226, 161–168. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Kozekov, I.D.; Kozekova, A.; Wang, H.; Lloyd, R.S.; Rizzo, C.J.; Stone, M.P. DNA cross-link induced by trans-4-hydroxynonenal. Environ. Mol. Mutagen. 2010, 51, 625–634. [Google Scholar] [CrossRef] [PubMed]
- Simonnet, H.; Alazard, N.; Pfeiffer, K.; Gallou, C.; Béroud, C.; Demont, J.; Bouvier, R.; Schägger, H.; Godinot, C. Low mitochondrial respiratory chain content correlates with tumor aggressiveness in renal cell carcinoma. Carcinogenesis 2002, 23, 759–768. [Google Scholar] [CrossRef] [PubMed]
- Barrera, G.; Pizzimenti, S.; Dianzani, M.U. Lipid peroxidation: Control of cell proliferation, cell differentiation and cell death. Mol. Aspects Med. 2008, 29, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Esterbauer, H.; Schaur, R.J.; Zollner, H. Chemistry and biochemistry of 4-hydroxynonenal, malonaldehyde and related aldehydes. Free Radic. Biol. Med. 1991, 11, 81–128. [Google Scholar] [CrossRef]
- Houtkooper, R.H.; Vaz, F.M. Cardiolipin, the heart of mitochondrial metabolism. Cell. Mol. Life Sci. 2008, 65, 2493–2506. [Google Scholar] [CrossRef] [PubMed]
- Yin, H.; Zhu, M. Free radical oxidation of cardiolipin: Chemical mechanisms, detection and implication in apoptosis, mitochondrial dysfunction and human diseases. Free Radic. Res. 2012, 46, 959–974. [Google Scholar] [CrossRef] [PubMed]
- Zhong, H.; Lu, J.; Xia, L.; Zhu, M.; Yin, H. Formation of electrophilic oxidation products from mitochondria lcardiolipin in vitro and in vivo in the context of apoptosis and atherosclerosis. Redox Biol. 2014, 2, 878–883. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.J.; Yu, B.P. Alterations in mitochondrial membrane fluidity by lipid peroxidation products. Free Radic. Biol. Med. 1994, 17, 411–418. [Google Scholar] [CrossRef]
- Chatterjee, S.N.; Agarwal, S.; Kumarjana, A.; Bose, B. Membrane lipid peroxidation and its pathological consequences. Ind. J. Biochem. Biophys. 1988, 25, 25–31. [Google Scholar]
- Borchman, D.; Lamba, O.P.; Salmassi, S.; Lou, M.; Yappert, M.C. The dual effect of oxidation on lipid bilayer structure. Lipids 1992, 27, 261–265. [Google Scholar] [CrossRef] [PubMed]
- Goldstein, R.M.; Weissmann, G. Effects of the generation of superoxide anion on permeability of liposomes. Biochem. Biophys. Res. Commun. 1977, 75, 604–609. [Google Scholar] [CrossRef]
- Anderson, E.J.; Katunga, L.A.; Willis, M.S. Mitochondria as a source and target of lipid peroxidation products in healthy and diseased heart. Clin. Exp. Pharmacol. Physiol. 2012, 39, 179–193. [Google Scholar] [CrossRef] [PubMed]
- Prasad, S.B.; Rosangkima, G.; Kharbangar, A. Structural and biochemical changes in mitochondria after cisplatin treatment of Dalton’s lymphoma-bearing mice. Mitochondrion 2010, 10, 38–45. [Google Scholar] [CrossRef] [PubMed]
- Gentile, F.; Pizzimenti, S.; Arcaro, A.; Pettazzoni, P.; Minelli, R.; D’Angelo, D.; Mamone, G.; Ferranti, P.; Toaldo, C.; Cetrangolo, G.; et al. Exposure of HL-60 human leukaemic cells to 4-hydroxynonenal promotes the formation of adduct(s) with alpha-enolase devoid of plasminogen binding activity. Biochem. J. 2009, 422, 285–294. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Miriyala, S.; Miao, L.; Mitov, M.; Schnell, D.; Dhar, S.K.; Cai, J.; Klein, J.B.; Sultana, R.; Butterfield, D.A.; et al. Redox proteomic identification of HNE-bound mitochondrial proteins in cardiac tissues reveals a systemic effect on energy metabolism after doxorubicin treatment. Free Radic. Biol. Med. 2014, 72, 55–65. [Google Scholar] [CrossRef] [PubMed]
- Doorn, J.A.; Hurley, T.D.; Petersen, D.R. Inhibition of human mitochondrial aldehyde dehydrogenase by 4-Hydroxynon-2-enal and 4-Oxonon-2-enal. Chem. Res. Toxicol. 2006, 19, 102–110. [Google Scholar] [CrossRef] [PubMed]
- Ding, J.H.; Li, S.P.; Cao, H.X.; Wu, J.Z.; Gao, C.M.; Su, P.; Liu, Y.T.; Zhou, J.N.; Chang, J.; Yao, G.H. Polymorphisms of alcohol dehydrogenase-2 andaldehyde dehydrogenase-2 and esophageal cancer risk in Southeast Chinese males. World J. Gastroenterol. 2009, 15, 2395–2400. [Google Scholar] [CrossRef] [PubMed]
- Barrera, G. Oxidative stress and lipid peroxidation products in cancer progression and therapy. ISRN Oncol. 2012, 2012. [Google Scholar] [CrossRef] [PubMed]
- Awasthi, Y.C.; Ansari, G.A.; Awasthi, S. Regulation of 4-hydroxynonenal mediated signaling by glutathione S-transferases. Methods Enzymol. 2005, 401, 379–407. [Google Scholar] [CrossRef] [PubMed]
- Oberley, T.D.; Toyokuni, S.; Szweda, L.I. Localization of hydroxynonenal protein adducts in normal human kidney and selected human kidney cancers. Free Radic. Biol. Med. 1999, 27, 695–703. [Google Scholar] [CrossRef]
- Zhao, Y.; Xue, Y.; Oberley, T.D.; Kiningham, K.K.; Lin, S.M.; Yen, H.C.; Majima, H.; Hines, J.; St Clair, D. Overexpression of manganese superoxide dismutase suppresses tumor formation by modulation of activator protein-1 signaling in a multistage skin carcinogenesis model. Cancer Res. 2001, 61, 6082–6088. [Google Scholar] [PubMed]
- Li, Y.P.; Tian, F.G.; Shi, P.C.; Guo, L.Y.; Wu, H.M.; Chen, R.Q.; Xue, J.M. 4-hydroxynonenal promotes growth and angiogenesis of breast cancer cells through HIF-1α stabilization. Asian Pac. J. Cancer Prev. 2014, 15, 10151–10156. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Du, Y.; Zhang, X.; Lu, J.; Holmgren, A. Glutaredoxin 2 reduces both thioredoxin 2 and thioredoxin 1 and protects cells from apoptosis induced by auranofin and 4-hydroxynonenal. Antioxid. Redox Signal. 2014, 21, 669–681. [Google Scholar] [CrossRef] [PubMed]
- Ji, C.; Amarnath, V.; Pietenpol, J.A.; Marnett, L.J. 4-hydroxynonenal induces apoptosis via caspase-3 activation and cytochrome c release. Chem. Res. Toxicol. 2001, 14, 1090–1096. [Google Scholar] [CrossRef] [PubMed]
- Haynes, R.L.; Brune, B.; Townsend, A.J. Apoptosis in RAW 264.7 cells exposed to 4-hydroxy-2-nonenal: Dependence on cytochrome C release but not p53 accumulation. Free Radic. Biol. Med. 2001, 30, 884–894. [Google Scholar] [CrossRef]
- Butterfield, D.A.; Reed, T.; Perluigi, M.; De Marco, C.; Coccia, R.; Cini, C.; Sultana, R. Elevated protein-bound levels of the lipid peroxidation product, 4-hydroxy-2-nonenal, in brain from persons with mild cognitive impairment. Neurosci. Lett. 2006, 397, 170–173. [Google Scholar] [CrossRef] [PubMed]
- Mangialasche, F.; Polidori, M.C.; Monastero, R.; Ercolani, S.; Camarda, C.; Cecchetti, R.; Mecucci, P. Biomarkers of oxidative and nitrosative damage in Alzheimer’s disease and mild cognitive impairment. Ageing Res. Rev. 2009, 8, 285–305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reed, T.; Perluigi, M.; Sultana, R.; Pierce, W.M.; Klein, J.B.; Turner, D.M.; Coccia, R.; Markesbery, W.R.; Butterfield, D.A. Redox proteomic identification of 4-hydroxy-2-nonenal modified brain proteins in amnestic mild cognitive impairment: Insight into the role of lipid peroxidation in the progression and pathogenesis of Alzheimer’s disease. Neurobiol. Dis. 2008, 30, 107–120. [Google Scholar] [CrossRef] [PubMed]
- Reed, T.T.; Pierce, W.M.; Markesbery, W.R.; Butterfield, D.A. Proteomic identification of HNE-bound proteins in early Alzheimer disease: Insights into the role of lipid peroxidation in the progression of AD. Brain Res. 2009, 1274, 66–76. [Google Scholar] [CrossRef] [PubMed]
- Butterfield, D.A.; Bader Lange, M.L.; Sultana, R. Involvements of the lipid peroxidation product, HNE, in the pathogenesis and progression of Alzheimer’s disease. Biochim. Biophys. Acta 2010, 1801, 924–929. [Google Scholar] [CrossRef] [PubMed]
- Ruiperez, V.; Darios, F.; Davletov, B. Alpha-synuclein, lipids and Parkinson’s disease. Prog. Lipid Res. 2010, 49, 420–428. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Kosaras, B.; del Signore, S.J.; Cormier, K.; McKe, A.; Ratan, R.R.; Kowall, N.W.; Ryu, H. Modulation of lipid peroxidation and mitochondrial function improves neuropathology in Huntington’s disease mice. Acta Neuropathol. 2011, 121, 487–498. [Google Scholar] [CrossRef] [PubMed]
- Shichiri, M.; Yoshida, Y.; Ishida, N.; Hagihara, Y.; Iwahashi, H.; Tamai, H.; Niki, E. Alpha-tocopgerol suppresses lipid peroxidation and behavioral and cognitive impairments in the Ts65Dn mouse model of Down syndrome. Free Radic. Biol. Med. 2011, 50, 1801–1811. [Google Scholar] [CrossRef] [PubMed]
- Berlett, B.S.; Stadtman, E.R. Protein oxidation in aging, disease, and oxidative stress. J. Biol. Chem. 1997, 272, 20313–20316. [Google Scholar] [CrossRef] [PubMed]
- Pamplona, R.; Dalfó, E.; Ayala, V.; Bellmunt, M.J.; Prat, J.; Ferrer, I.; Portero-Otín, M. Proteins in human brain cortex are modified by oxidation, glycoxidation, and lipoxidation. Effects of Alzheimer disease and identification of lipoxidation targets. J. Biol. Chem. 2005, 280, 21522–21530. [Google Scholar] [CrossRef] [PubMed]
- Grimsrud, P.A.; Xie, H.; Griffin, T.J.; Bernlohr, D.A. Oxidative stress and covalent modification of protein with bioactive aldehydes. J. Biol. Chem. 2008, 283, 21837–21841. [Google Scholar] [CrossRef] [PubMed]
- Keller, J.N.; Schmitt, F.A.; Scheff, S.W.; Ding, Q.; Chen, Q.; Butterfield, D.A.; Markesbery, W.R. Evidence of increased oxidative damage in subjects with mild cognitive impairment. Neurology 2005, 64, 1152–1156. [Google Scholar] [CrossRef] [PubMed]
- Butterfield, D.A.; Poon, H.F.; St Clair, D.; Keller, J.N.; Pierce, W.M.; Klein, J.B.; Markesbery, W.R. Redox proteomics identification of oxidatively modified hippocampal proteins in mild cognitive impairment: Insights into the development of Alzheimer’s disease. Neurobiol. Dis. 2006, 22, 223–232. [Google Scholar] [CrossRef] [PubMed]
- Sultana, R.; Perluigi, M.; Butterfield, D.A. Lipid peroxidation triggers neurodegeneration: A redox proteomics view into the Alzheimer disease brain. Free Radic. Biol. Med. 2013, 62, 157–169. [Google Scholar] [CrossRef] [PubMed]
- Perluigi, M.; Sultana, R.; Cenini, G.; di Domenico, F.; Memo, M.; Pierce, W.M.; Coccia, R.; Butterfield, D.A. Redox proteomics identification of 4-hydroxynonenal modified brain proteins in Alzheimer’s disease: Role of lipid peroxidation in Alzheimer’s disease pathogenesis. Proteomics Clin. Appl. 2009, 3, 682–693. [Google Scholar] [CrossRef] [PubMed]
- Martínez, A.; Portero-Otin, M.; Pamplona, R.; Ferrer, I. Protein targets of oxidative damage in human neurodegenerative diseases with abnormal protein aggregates. Brain Pathol. 2010, 20, 281–297. [Google Scholar] [CrossRef] [PubMed]
- Reed, T. Lipid peroxidation and neurodegenerative disease. Free Radic. Biol. Med. 2011, 51, 1302–1319. [Google Scholar] [CrossRef] [PubMed]
- Siegel, S.J.; Bieschke, J.; Powers, E.T.; Kelly, J.W. The oxidative stress metabolite 4-hydroxynonenal promotes Alzheimer protofibril formation. Biochemistry 2007, 46, 1503–1510. [Google Scholar] [CrossRef] [PubMed]
- Federico, A.; Cardaioli, E.; da Pozzo, P.; Formichi, P.; Gallus, G.N.; Radi, E. Mitochondria, oxidative stress and neurodegeneration. J. Neurol. Sci. 2012, 322, 254–262. [Google Scholar] [CrossRef] [PubMed]
- Bhat, A.H.; Dar, K.B.; Anees, S.; Zargar, M.A.; Masood, A.; Sofi, M.A.; Ganie, S.A. Oxidative stress, mitochondrial dysfunction and neurodegenerative diseases; a mechanistic insight. Biomed. Pharmacother. 2015, 74, 101–110. [Google Scholar] [CrossRef] [PubMed]
- Online Mendelian Inheritance in Man (OMIM) database. Availalbe online: http://www.ncbi.nlm.nih.gov/omim (accessed on 17 February 2016).
- Terni, B.; Boada, J.; Portero-Otín, M.; Pamplona, R.; Ferrer, I. Mitochondrial ATP-synthase in the entorhinal cortex is a target of oxidative stress at stages I/II of Alzheimer’s disease pathology. Brain Pathol. 2010, 20, 222–233. [Google Scholar] [CrossRef] [PubMed]
- Sorolla, M.A.; Reverter-Branchat, G.; Tamarit, J.; Ferrer, I.; Ros, J.; Cabiscol, E. Proteomic and oxidative stress analysis in human brain samples of Huntington disease. Free Radic. Biol. Med. 2008, 45, 667–678. [Google Scholar] [CrossRef] [PubMed]
- Sorolla, M.A.; Rodríguez-Colman, M.J.; Tamarit, J.; Ortega, Z.; Lucas, J.J.; Ferrer, I.; Ros, J.; Cabiscol, E. Protein oxidation in Huntington disease affects energy production and vitamin B6 metabolism. Free Radic. Biol. Med. 2010, 49, 612–621. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.; Sullards, M.C.; Olzmann, J.A.; Rees, H.D.; Weintraub, S.T.; Bostwick, D.E.; Gearing, M.; Levey, A.I.; Chin, L.S.; Li, L. Oxidative damage of DJ-1 is linked to sporadic Parkinson and Alzheimer diseases. J. Biol. Chem. 2006, 281, 10816–10824. [Google Scholar] [CrossRef] [PubMed]
- Keeney, P.M.; Xie, J.; Capaldi, R.A.; Bennett, J.P. Parkinson’s disease brain mitochondrial complex I has oxidatively damaged subunits and is functionally impaired and misassembled. J. Neurosci. 2006, 26, 5256–5264. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.; Rees, H.D.; Weintraub, S.T.; Levey, A.I.; Chin, L.-S.; Li, L. Oxidative modifications and aggregation of Cu,Zn-superoxide dismutase associated with Alzheimer and Parkinson diseases. J. Biol. Chem. 2005, 280, 11648–11655. [Google Scholar] [CrossRef] [PubMed]
- Cabiscol, E.; Ros, J. Oxidative damage to proteins: Structural modifications and consequences in cell function. In Redox Proteomics: From Protein Modifications to Cellular Dysfunctions and Diseases; Dalle-Donne, I., Scaloni, A., Butterfield, D.A., Eds.; Wiley: Hoboken, NJ, USA, 2006; pp. 399–472. [Google Scholar]
- Chen, X.J.; Wang, X.; Kaufman, B.A.; Butow, R.A. Aconitase couples metabolic regulation to mitochondrial DNA maintenance. Science 2005, 307, 714–717. [Google Scholar] [CrossRef] [PubMed]
- Schagger, H.; Ohm, T.G. Human diseases with defects in oxidative phosphorylation. 2. F1F0 ATP-synthase defects in Alzheimer disease revealed by blue native polyacrylamide gel electrophoresis. Eur. J. Biochem. 1995, 227, 916–921. [Google Scholar] [CrossRef] [PubMed]
- Rhein, V.; Eckert, A. Effects of Alzheimer’s amyloid-beta and tau protein on mitochondrial function—Role of glucose metabolism and insulin signalling. Arch. Physiol. Biochem. 2007, 113, 131–141. [Google Scholar] [CrossRef] [PubMed]
- Sultana, R.; Poon, H.F.; Cai, J.; Pierce, W.M.; Merchant, M.; Klein, J.B.; Markesbery, W.R.; Butterfield, D.A. Identification of nitrated proteins in Alzheimer’s disease brain using a redox proteomics approach. Neurobiol. Dis. 2006, 22, 76–87. [Google Scholar] [CrossRef] [PubMed]
- Mattiazzi, M.; Vijayvergiya, C.; Gajewski, C.D.; DeVivo, D.C.; Lenaz, G.; Wiedmann, M.; Manfredi, G. The mtDNA T8993G (NARP) mutation results in an impairment of oxidative phosphorylation that can be improved by antioxidants. Hum. Molec. Genet. 2007, 13, 869–879. [Google Scholar] [CrossRef] [PubMed]
- Hansen, J.J.; Durr, A.; Cournu-Rebeix, I.; Georgopoulos, C.; Ang, D.; Nielsen, M.N.; Davoine, C.S.; Brice, A.; Fontaine, B.; Gregersen, N.; et al. Hereditary spastic paraplegia SPG13 is associated with a mutation in the gene encoding the mitochondrial chaperonin Hsp60. Am. J. Hum. Genet. 2002, 70, 1328–1332. [Google Scholar] [CrossRef] [PubMed]
- Tabrizi, S.J.; Cleeter, M.W.; Xuereb, J.; Taanman, J.W.; Cooper, J.M.; Schapira, A.H. Biochemical abnormalities and excitotoxicity in Huntington’s disease brain. Ann. Neurol. 1999, 45, 25–32. [Google Scholar] [CrossRef]
- Solans, A.; Zambrano, A.; Rodríguez, M.; Barrientos, A. Cytotoxicity of a mutant huntingtin fragment in yeast involves early alterations in mitochondrial OXPHOS complexes II and III. Hum. Mol. Genet. 2006, 15, 3063–3081. [Google Scholar] [CrossRef] [PubMed]
- Miyake, N.; Yano, S.; Sakai, C.; Hatakeyama, H.; Matsushima, Y.; Shiina, M.; Watanabe, Y.; Bartley, J.; Abdenut, J.E.; Wang, R.Y.; et al. Mitochondrial complex III deficiency caused by a homozygous UQCRC2 mutation presenting with neonatal-onset recurrent metabolic decompensation. Hum. Mutat. 2013, 34, 446–452. [Google Scholar] [CrossRef] [PubMed]
- Richarme, G.; Mihoub, M.; Dairou, J.; Bui, L.C.; Leger, T.; Lamouri, A. Parkinsonism-associated protein DJ-1/Park7 is a major protein deglycase that repairs methylglyoxal- and glyoxal-glycated cysteine, arginine, and lysine residues. J. Biol. Chem. 2015, 290, 1885–1897. [Google Scholar] [CrossRef] [PubMed]
- Sekito, A.; Koide-Yoshida, S.; Niki, T.; Taira, T.; Iguchi-Ariga, S.M.; Ariga, H. DJ-1 interacts with HIPK1 and affects H2O2-induced cell death. Free Radic. Res. 2006, 40, 155–165. [Google Scholar] [CrossRef] [PubMed]
- Björkblom, B.; Adilbayeva, A.; Maple-Grødem, J.; Piston, D.; Ökvist, M.; Xu, X.M.; Brede, C.; Larsen, J.P.; Møller, S.G. Parkinson disease protein DJ-1 binds metals and protects against metal-induced cytotoxicity. J. Biol. Chem. 2013, 288, 22809–22820. [Google Scholar] [CrossRef] [PubMed]
- Xiong, H.; Wang, D.; Chen, L.; Choo, Y.S.; Ma, H.; Tang, C.; Xia, K.; Jiang, W.; Ronai, Z.; Zhuang, X.; et al. Parkin, PINK1, and DJ-1 form a ubiquitin E3 ligase complex promoting unfolded protein degradation. J. Clin. Investig. 2009, 119, 650–660. [Google Scholar] [CrossRef] [PubMed]
- Tang, B.; Xiong, H.; Sun, P.; Zhang, Y.; Wang, D.; Hu, Z.; Zhu, Z.; Ma, H.; Pan, Q.; Xia, J.H.; et al. Association of PINK1 and DJ-1 confers digenic inheritance of early-onset Parkinson’s disease. Hum. Mol. Genet. 2006, 15, 1816–1825. [Google Scholar] [CrossRef] [PubMed]
- Junn, E.; Jang, W.H.; Zhao, X.; Jeong, B.S.; Mouradian, M.M. Mitochondrial localization of DJ-1 leads to enhanced neuroprotection. J. Neurosci. Res. 2009, 87, 123–129. [Google Scholar] [CrossRef] [PubMed]
- Rizzu, P.; Hinkle, D.A.; Zhukareva, V.; Bonifati, V.; Severijnen, L.A.; Martinez, D.; Ravid, R.; Kamphorst, W.; Eberwine, J.H.; Lee, V.M.; et al. DJ-1 colocalizes with tau inclusions: A link between Parkinsonism and dementia. Ann. Neurol. 2004, 55, 113–118. [Google Scholar] [CrossRef] [PubMed]
- Shults, C.W. Mitochondrial dysfunction and possible treatments in Parkinson’s disease—A review. Mitochondrion 2004, 4, 641–648. [Google Scholar] [CrossRef] [PubMed]
- Schumacker, P.T. Reactive oxygen species in cancer cells: Live by the sword, die by the sword. Cancer Cell 2006, 10, 175–176. [Google Scholar] [CrossRef] [PubMed]
- Fruehauf, J.P.; Meyskens, F.L., Jr. Reactive oxygen species: A breath of life or death? Clin. Cancer Res. 2007, 13, 789–794. [Google Scholar] [CrossRef] [PubMed]
- Kardeh, S.; Ashkani-Esfahani, S.; Alizadeh, A.M. Paradoxical action of reactive oxygen species in creation and therapy of cancer. Eur. J. Pharmacol. 2014, 735, 150–168. [Google Scholar] [CrossRef] [PubMed]
- Cojocneanu Petric, R.; Braicu, C.; Raduly, L.; Zanoaga, O.; Dragos, N.; Monroig, P.; Dumitrascu, D.; Berindan-Neagoe, I. Phytochemicals modulate carcinogenic signaling pathways in breast and hormone-related cancers. Onco Targets Ther. 2015, 8, 2053–2066. [Google Scholar] [CrossRef] [PubMed]
- Galasko, D.R.; Peskind, E.; Clark, C.M.; Quinn, J.F.; Ringman, J.M.; Jicha, G.A.; Cotman, C.; Cottrell, B.; Montine, T.J.; Thomas, R.G.; et al. Alzheimer’s Disease Cooperative Study. Antioxidants for Alzheimer disease: A randomized clinical trial with cerebrospinal fluid biomarker measures. Arch. Neurol. 2012, 69, 836–841. [Google Scholar] [PubMed]
- Gandhi, S.; Abramov, A.Y. Mechanism of oxidative stress in neurodegeneration. Oxid. Med. Cell Longev. 2012, 2012. [Google Scholar] [CrossRef] [PubMed]
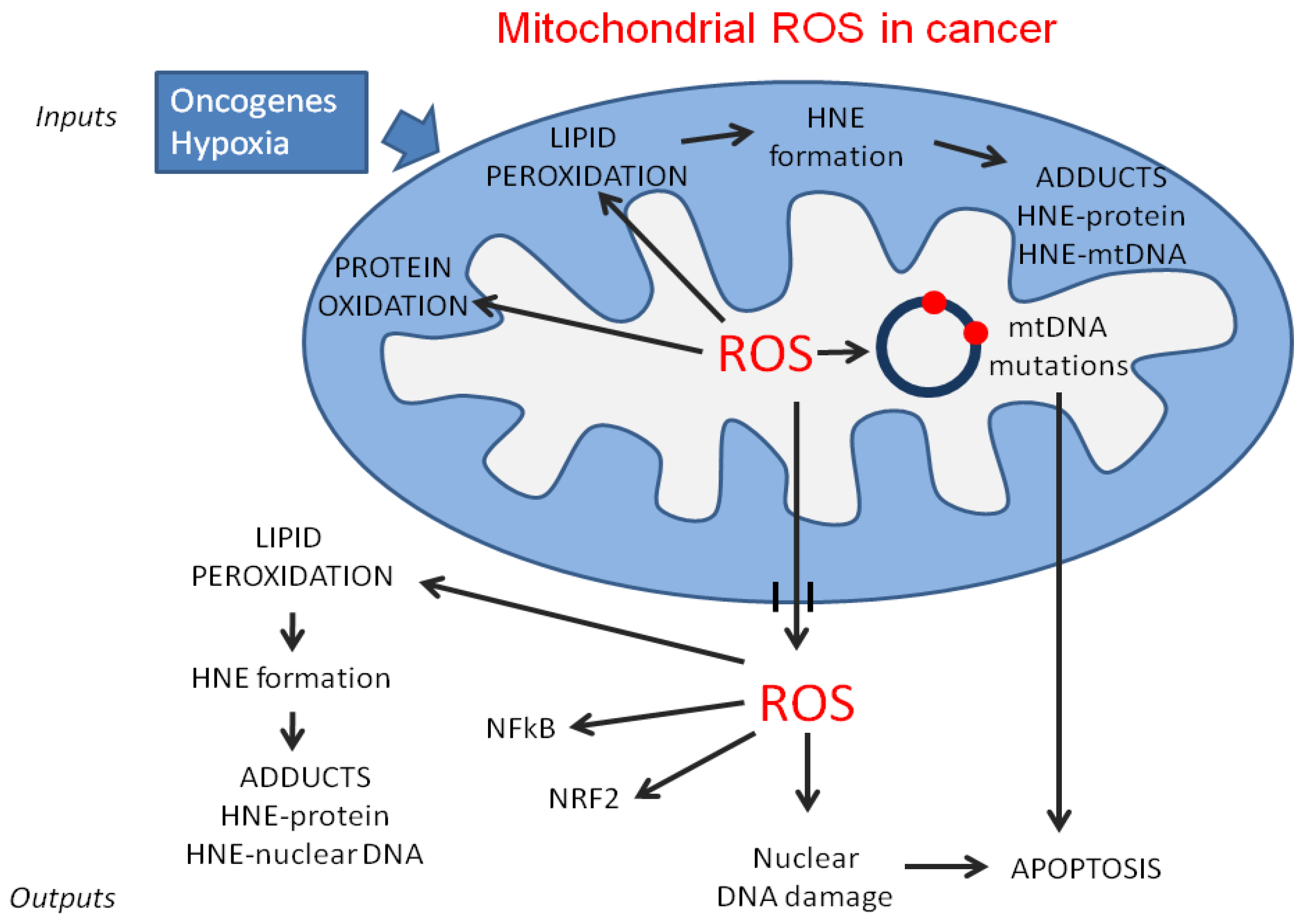

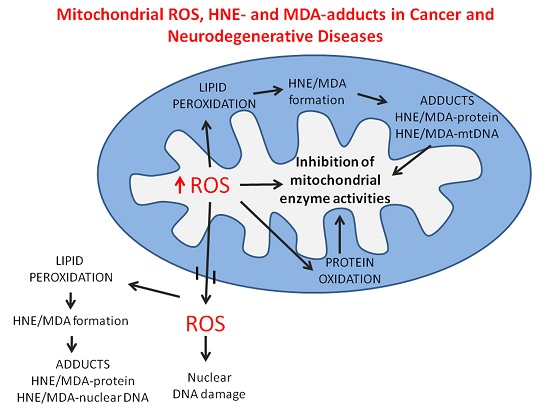{kind=link}
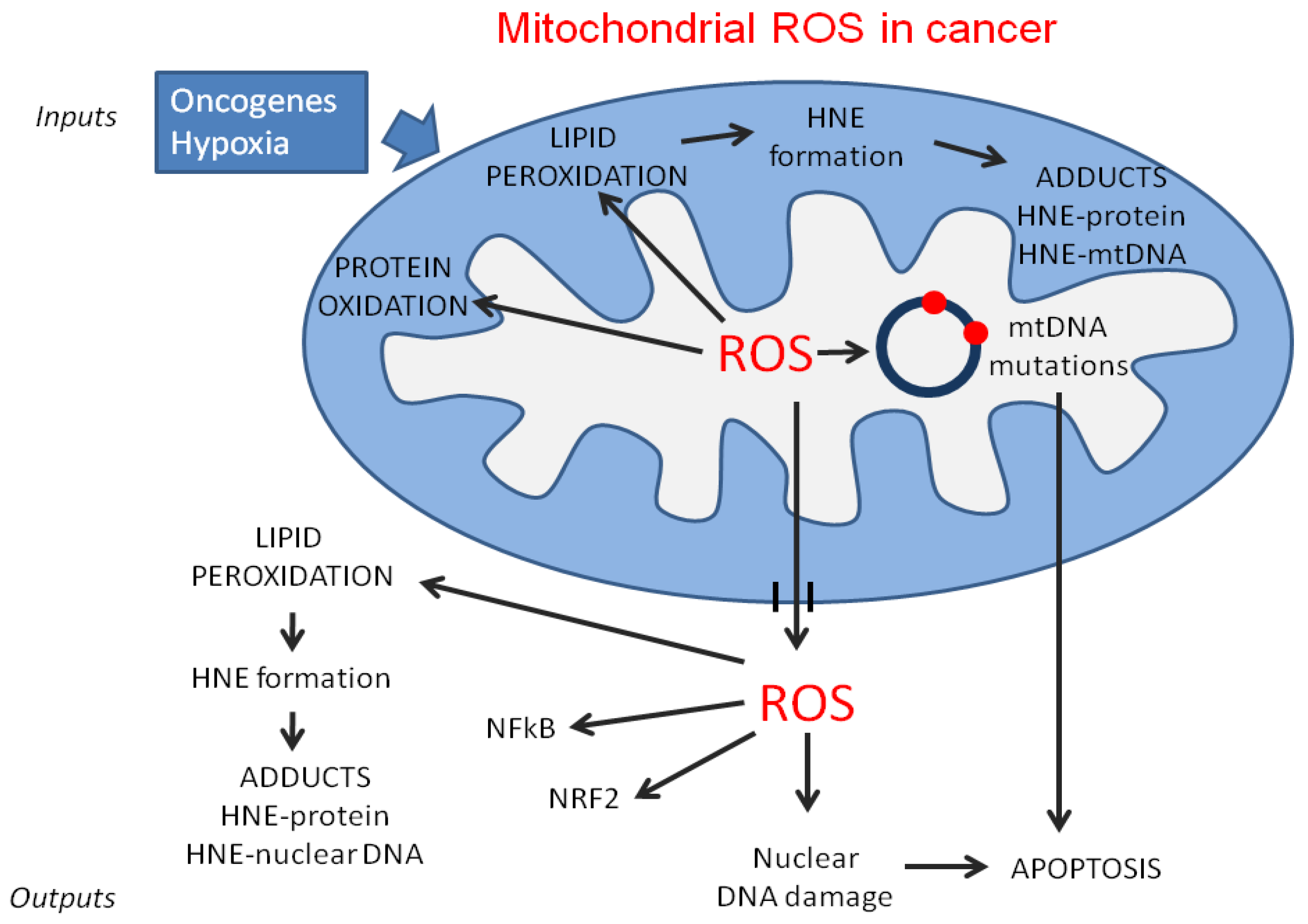{kind=link}
| Phospholipids | Percentage Content | |
|---|---|---|
| OMM | IMM | |
| Phosphatidylcholine | 54 | 40 |
| Phosphatidylethanolamine | 29 | 34 |
| Phosphatidylinositol | 13 | 5 |
| Phosphatidyserine | 2 | 3 |
| Cardiolipin | <1 | 18 |
| Others | <1 | 0 |
| Tissues/Cells | Phophatidyl− Choline | Phophatidyl− Ethanolamine | Phosphatldylserine+ Phosphatidylinositol | Cardiolipn | ||||
|---|---|---|---|---|---|---|---|---|
| MUFA | PUFA | MUFA | PUFA | MUFA | PUFA | MUFA | PUFA | |
| Normal liver | 11.41 | 37.11 | 7.08 | 43.11 | 6.84 | 30.18 | 18.04 | 55.46 |
| Nodules | 23.62 | 27.24 | 19.06 | 34.85 | 14.44 | 24.89 | 25.37 | 36.27 |
| Hepatoma | 29.68 | 25.68 | 22.57 | 37.78 | 22.12 | 25.64 | 28.54 | 23.72 |
| AH-130 Hepatoma | 25.33 | 26.28 | 18.54 | 47.21 | 17.83 | 28.23 | 22.17 | 28.75 |
| Cancer Model | Protein Targets | Mitochondrial Function | HNE Effect | Reference |
|---|---|---|---|---|
| PC12 pheochromocytoma cell line | cytochrome c oxidase aconitase | respiratory enzymes | inhibition | [81] |
| Kidney cancers | HNE-mitochondrial protein adducts | [101] | ||
| Skin carcinogenesis | HNE-mitochondrial protein adducts | - | - | [102] |
| Breast cancer cells | sirtuin 3 (SIRT3) HNE-SIRT3 adducts | NAD+-dependent deacetylase | inhibition | [103] |
| HeLa cervical adenocarcinoma cell line | thioredoxin reductase (TrxR) | Trx reduction | inhibition | [104] |
| RKO colorectal carcinoma cell line | - | cytochrome c release | induction | [105] |
| RAW 264.7 mouse monocytic/macrophagic leukemic cell line | - | cytochrome c release | induction | [106] |
| Protein | AD Stage | Function | Reference |
|---|---|---|---|
| HNE-modified proteins | |||
| Aconitate hydratase, mitochondrial (Aconitase 2, ACO2) | LAD | energy metabolism, mitochondrial function | [121] |
| ATP synthase (complex V) alpha subunit 1 (ATP5A1) | PAD, MCI, EAD, LAD | energy metabolism, ATP production | [109,110,121,128] |
| Translation elongation factor Tu (EF-Tu, TUFM) | MCI | protein synthesis | [109] |
| Malate dehydrogenase 2, mitochondrial (MDH2) | EAD | energy metabolism, gluconeogenesis | [110] |
| Mn Superoxide dysmutase, mitochondrial (SOD2) | EAD, LAD | antioxidant defense | [110,121] |
| MDA-modified proteins | |||
| Ubiquinol-cytochrome c reductase (complex III) core protein 1 (UQCRC1) | LAD | electron transport, ATP production | [116] |
| ATP synthase (complex V) beta subunit (ATP5B) | LAD | energy metabolism, ATP production | [116] |
| 60-kDa Heat shock protein (HSPD1, HSP60) | LAD | stress response | [116] |
| Glutamate dehydrogenase 1, mitochondrial (GDH1) | LAD | energy metabolism | [116] |
| Proteins with increased content of DPNH-reactive groups | |||
| Aconitate hydratase, mitochondrial (Aconitase 2, ACO2) | HD | energy metabolism, mitochondrial function | [129] |
| Citrate synthase, mitochondrial (CS) | HD | energy metabolism | [130] |
| Creatine kinase B, ubiquitous mitochondrial (CKMT1A) | HD | ATP production | [130] |
| Ubiquinol-cytochrome c reductase (complex III) core protein 2 (UQCRC2) | HD | electron transport, ATP production | [130] |
| ATP synthase (complex V) alpha subunit 1 (ATP5A1) | HD | energy metabolism, ATP production | [130] |
| DJ-1 (Parkinson protein 7, PARK7) | PD, AD | antioxidant defense | [131] |
| NADH-ubiquinone oxidoreductase (complex I) | PD | electron transport, ATP production | [132] |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Barrera, G.; Gentile, F.; Pizzimenti, S.; Canuto, R.A.; Daga, M.; Arcaro, A.; Cetrangolo, G.P.; Lepore, A.; Ferretti, C.; Dianzani, C.; et al. Mitochondrial Dysfunction in Cancer and Neurodegenerative Diseases: Spotlight on Fatty Acid Oxidation and Lipoperoxidation Products. Antioxidants 2016, 5, 7. https://doi.org/10.3390/antiox5010007
Barrera G, Gentile F, Pizzimenti S, Canuto RA, Daga M, Arcaro A, Cetrangolo GP, Lepore A, Ferretti C, Dianzani C, et al. Mitochondrial Dysfunction in Cancer and Neurodegenerative Diseases: Spotlight on Fatty Acid Oxidation and Lipoperoxidation Products. Antioxidants. 2016; 5(1):7. https://doi.org/10.3390/antiox5010007
Chicago/Turabian StyleBarrera, Giuseppina, Fabrizio Gentile, Stefania Pizzimenti, Rosa Angela Canuto, Martina Daga, Alessia Arcaro, Giovanni Paolo Cetrangolo, Alessio Lepore, Carlo Ferretti, Chiara Dianzani, and et al. 2016. "Mitochondrial Dysfunction in Cancer and Neurodegenerative Diseases: Spotlight on Fatty Acid Oxidation and Lipoperoxidation Products" Antioxidants 5, no. 1: 7. https://doi.org/10.3390/antiox5010007