
Screening for Metal-Chelating Activity in Potato Protein Hydrolysates Using Surface Plasmon Resonance and Peptidomics


 ,
,  ,
,  and
and 
Abstract
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Production of Hydrolysates
2.2.1. Enzymatic Hydrolysis
2.2.2. Determination of Protein Content Using DUMAS
2.2.3. Determination of the Degree of Hydrolysis
2.3. Fractionation
2.3.1. Ultrafiltration
2.3.2. SDS-PAGE
2.4. Peptidomics Analysis by nLC-MS/MS
2.4.1. LC-MS/MS Data Analysis
2.4.2. Processing of MaxQuant Data and Prediction of Antioxidant Properties
2.5. Surface Plasmon Resonance
2.5.1. Glycine Equivalence as Determined by OPA
2.5.2. SPR
2.6. Emulsion Production and Storage Experiment
2.7. Physical Stability of Emulsions
2.7.1. Droplet Size Distribution
2.7.2. Zeta Potential
2.8. Oxidative Stability
2.8.1. Oil Extraction and Peroxide Value
2.8.2. Determination of Tocopherol Content
2.8.3. Determination of Secondary, Volatile Oxidation Products Using Dynamic Headspace GC-MS
2.9. Statistical Analysis
3. Results and Discussion
3.1. Production of Hydrolysates
3.1.1. Enzymatic Hydrolysis
3.1.2. Size Fractionation
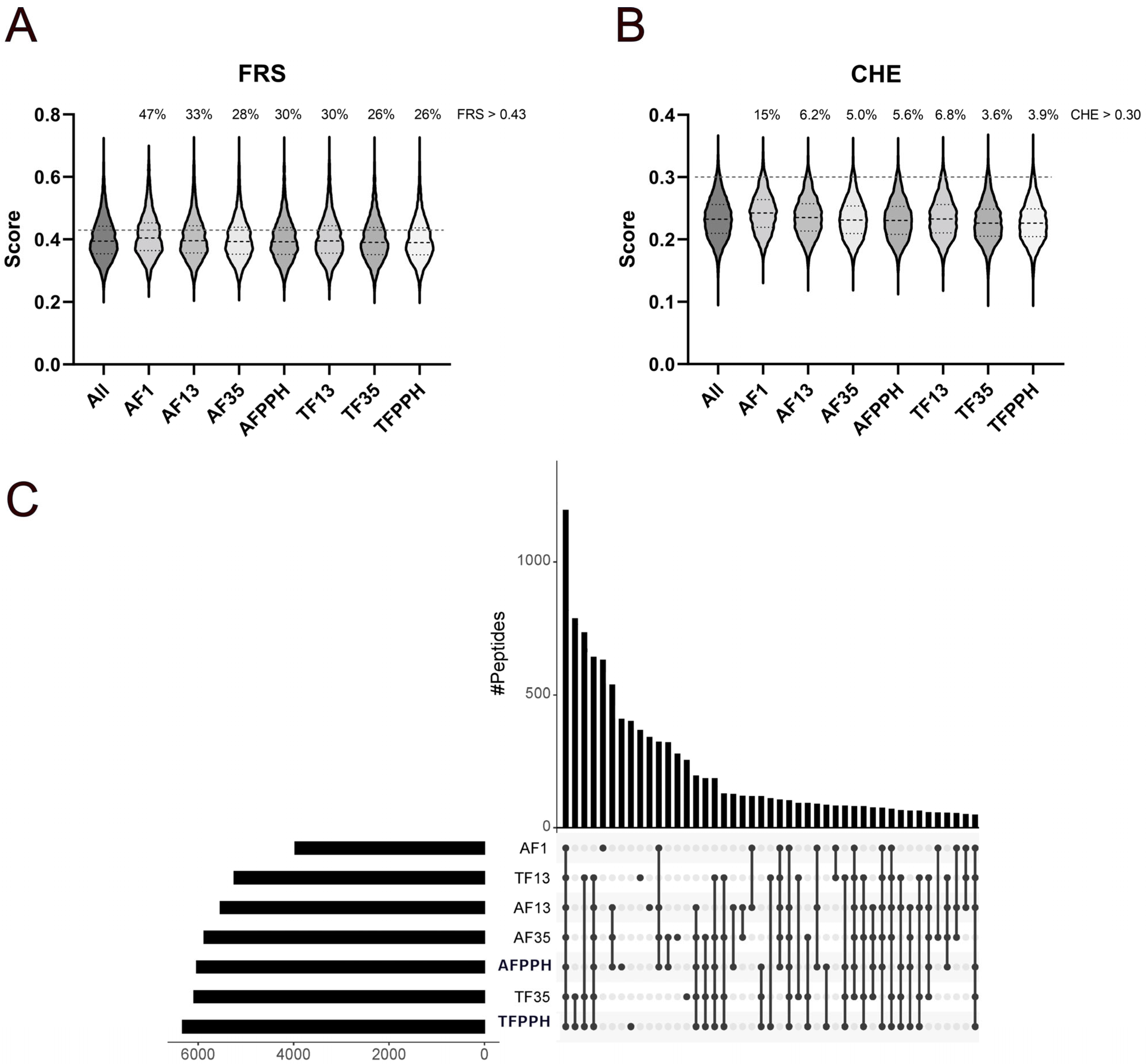
3.2. Peptidomics
Prediction of Peptide-Level Antioxidant Properties
3.3. Surface Plasmon Resonance (SPR)
3.4. Storage Experiment with Emulsions
3.4.1. Physical Stability of the Emulsions
Droplet Size Distribution
Zeta Potential
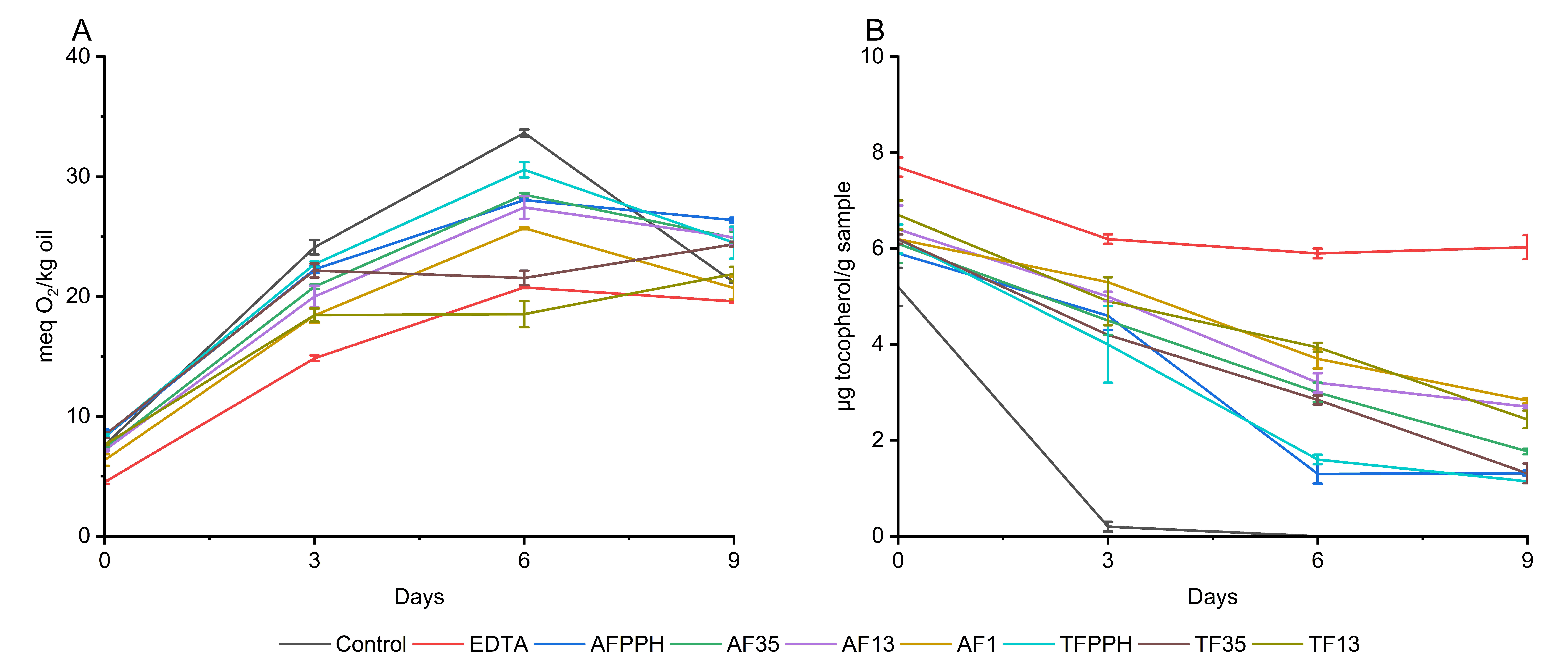
3.4.2. Oxidative Stability of Emulsions
Peroxide Value
Tocopherols
Development of Secondary Volatile Oxidation Products
3.5. Can SPR, Peptidomics, and Bioinformatics Be Used as Alternative Screening Methods?
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- McClements, D.J.; Decker, E.A. Lipids. In Fennema’s Food Chemistry, 5th ed.; CRC Press: Boca Raton, FL, USA, 2017. [Google Scholar] [CrossRef]
- Nenadis, N.; Tsimidou, M.Z. Assessing the activity of natural food antioxidants. In Oxidation in Foods and Beverages and Antioxidant Applications; Woodhead Publishing: Cambridge, UK, 2010; pp. 332–367. [Google Scholar] [CrossRef]
- Frankel, E.N. Lipid Oxidation; Woodhead Publishing Limited: Cambridge, UK, 2005. [Google Scholar] [CrossRef]
- Asioli, D.; Aschemann-Witzel, J.; Caputo, V.; Vecchio, R.; Annunziata, A.; Næs, T.; Varela, P. Making sense of the ‘clean label’ trends: A review of consumer food choice behavior and discussion of industry implications. Food Res. Int. 2017, 99, 58–71. [Google Scholar] [CrossRef]
- Mitterer-Daltoé, M.; Bordim, J.; Lise, C.; Breda, L.; Casagrande, M.; Lima, V. Consumer awareness of food antioxidants. Synthetic vs. Natural. Food Sci. Technol. 2020, 41, 208–212. [Google Scholar] [CrossRef]
- Elias, R.J.; Kellerby, S.S.; Decker, E.A. Antioxidant Activity of Proteins and Peptides. Crit. Rev. Food Sci. Nutr. 2008, 48, 430–441. [Google Scholar] [CrossRef] [PubMed]
- Pan, M.; Liu, K.; Yang, J.; Liu, S.; Wang, S.; Wang, S. Advances on Food-Derived Peptidic Antioxidants—A Review. Antioxidants 2020, 9, 799. [Google Scholar] [CrossRef]
- Sarmadi, B.H.; Ismail, A. Antioxidative peptides from food proteins: A review. Peptides 2010, 31, 1949–1956. [Google Scholar] [CrossRef]
- Mardani, M.; Badakné, K.; Farmani, J.; Aluko, R.E. Antioxidant peptides: Overview of production, properties, and applications in food systems. Compr. Rev. Food Sci. Food Saf. 2023, 22, 46–106. [Google Scholar] [CrossRef] [PubMed]
- Ranamukhaarachchi, S.; Meissner, L.; Moresoli, C. Production of antioxidant soy protein hydrolysates by sequential ultrafiltration and nanofiltration. J. Membr. Sci. 2013, 429, 81–87. [Google Scholar] [CrossRef]
- United Nations Environment Programme. Food Waste Index Report. 2021. Available online: https://wedocs.unep.org/bitstream/handle/20.500.11822/35280/FoodWaste.pdf (accessed on 29 January 2024).
- Zhang, H.; Xu, F.; Wu, Y.; Hu, H.H.; Dai, X.F. Progress of potato staple food research and industry development in China. J. Integr. Agric. 2017, 16, 2924–2932. [Google Scholar] [CrossRef]
- Hussain, M.; Qayum, A.; Xiuxiu, Z.; Liu, L.; Hussain, K.; Yue, P.; Yue, S.; Koko, M.Y.; Hussain, A.; Li, X. Potato protein: An emerging source of high quality and allergy free protein, and its possible future based products. Food Res. Int. 2021, 148, 110583. [Google Scholar] [CrossRef]
- Yesiltas, B.; García-Moreno, P.J.; Gregersen, S.; Olsen, T.H.; Jones, N.C.; Hoffmann, S.V.; Marcatili, P.; Overgaard, M.T.; Hansen, E.B.; Jacobsen, C. Antioxidant peptides derived from potato, seaweed, microbial and spinach proteins: Oxidative stability of 5% fish oil-in-water emulsions. Food Chem. 2022, 385, 13269. [Google Scholar] [CrossRef]
- Canabady-Rochelle, L.L.S.; Selmeczi, K.; Collin, S.; Pasc, A.; Muhr, L.; Boschi-Muller, S. SPR screening of metal chelating peptides in a hydrolysate for their antioxidant properties. Food Chem. 2018, 239, 478–485. [Google Scholar] [CrossRef] [PubMed]
- El Hajj, S.; Irankunda, R.; Echavarría, J.A.C.; Arnoux, P.; Paris, C.; Stefan, L.; Gaucher, C.; Boschi-Muller, S.; Canabady-Rochelle, L. Metal-chelating activity of soy and pea protein hydrolysates obtained after different enzymatic treatments from protein isolates. Food Chem. 2023, 405, 134788. [Google Scholar] [CrossRef] [PubMed]
- Pearson, R.G. Hard and soft acids and bases, HSAB, part 1: Fundamental principles. J. Chem. Educ. 1968, 45, 581. [Google Scholar] [CrossRef]
- Olsen, T.H.; Yesiltas, B.; Marin, F.I.; Pertseva, M.; García-Moreno, P.J.; Gregersen, S.; Overgaard, M.T.; Jacobsen, C.; Lund, O.; Hansen, E.B.; et al. AnOxPePred: Using deep learning for the prediction of antioxidative properties of peptides. Sci. Rep. 2020, 10, 21471. [Google Scholar] [CrossRef] [PubMed]
- Jafarpour, A.; Gregersen, S.; Gomes, R.M.; Marcatili, P.; Olsen, T.H.; Jacobsen, C.; Overgaard, M.T.; Sørensen, A.-D.M. Biofunctionality of Enzymatically Derived Peptides from Codfish (Gadus morhua) Frame: Bulk In Vitro Properties, Quantitative Proteomics, and Bioinformatic Prediction. Mar. Drugs 2020, 18, 599. [Google Scholar] [CrossRef] [PubMed]
- Gregersen Echers, S.; Abdul-Khalek, N.; Mikkelsen, R.K.; Holdt, S.L.; Jacobsen, C.; Hansen, E.B.; Olsen, T.H.; Sejberg, J.J.; Overgaard, M.T. Is Gigartina a potential source of food protein and functional peptide-based ingredients? Evaluating an industrial, pilot-scale extract by proteomics and bioinformatics. Future Foods 2022, 6, 100189. [Google Scholar] [CrossRef]
- Gregersen Echers, S.; Jafarpour, A.; Yesiltas, B.; García-Moreno, P.J.; Greve-Poulsen, M.; Hansen, D.K.; Jacobsen, C.; Overgaard, M.T.; Hansen, E.B. Targeted hydrolysis of native potato protein: A novel workflow for obtaining hydrolysates with improved interfacial properties. Food Hydrocoll. 2023, 137, 108299. [Google Scholar] [CrossRef]
- Merz, M.; Eisele, T.; Berends, P.; Appel, D.; Rabe, S.; Blank, I.; Stressler, T.; Fischer, L. Flavourzyme, an Enzyme Preparation with Industrial Relevance: Automated Nine-Step Purification and Partial Characterization of Eight Enzymes. J. Agric. Food Chem. 2015, 63, 5682–5693. [Google Scholar] [CrossRef]
- Kamnerdpetch, C.; Weiss, M.; Kasper, C.; Scheper, T. An improvement of potato pulp protein hydrolyzation process by the combination of protease enzyme systems. Enzym. Microb. Technol. 2007, 40, 508–514. [Google Scholar] [CrossRef]
- García-Moreno, P.J.; Yang, J.; Gregersen, S.; Jones, N.C.; Berton-Carabin, C.C.; Sagis, L.M.; Hoffmann, S.V.; Marcatili, P.; Overgaard, M.T.; Hansen, E.B.; et al. The structure, viscoelasticity and charge of potato peptides adsorbed at the oil-water interface determine the physicochemical stability of fish oil-in-water emulsions. Food Hydrocoll. 2021, 115, 106605. [Google Scholar] [CrossRef]
- Gregersen, S.; Kongsted, A.-S.H.; Nielsen, R.B.; Hansen, S.S.; Lau, F.A.; Rasmussen, J.B.; Holdt, S.L.; Jacobsen, C. Enzymatic extraction improves intracellular protein recovery from the industrial carrageenan seaweed Eucheuma denticulatum revealed by quantitative, subcellular protein profiling: A high potential source of functional food ingredients. Food Chem. X 2021, 12, 100137. [Google Scholar] [CrossRef] [PubMed]
- Knecht, S.; Ricklin, D.; Eberle, A.N.; Ernst, B. Oligohis-tags: Mechanisms of binding to Ni2+-NTA surfaces. J. Mol. Recognit. 2009, 22, 270–279. [Google Scholar] [CrossRef] [PubMed]
- Bligh, E.; Dyer, W. A rapid method of total lipid extraction and purification. Can. J. Biochem. Physiol. 1959, 37, 911–917. [Google Scholar] [CrossRef] [PubMed]
- Shantha, N.C.; Decker, E.A. Rapid, sensitive, iron-based spectrophotometric methods for determination of peroxide values of food lipids. J. AOAC Int. 1994, 77, 421–424. [Google Scholar] [CrossRef] [PubMed]
- Olsen, J.V.; Ong, S.E.; Mann, M. Trypsin cleaves exclusively C-terminal to arginine and lysine residues. Mol. Cell. Proteom. 2004, 3, 608–614. [Google Scholar] [CrossRef] [PubMed]
- Miedzianka, J.; Pȩksa, A.; Pokora, M.; Rytel, E.; Tajner-Czopek, A.; Kita, A. Improving the properties of fodder potato protein concentrate by enzymatic hydrolysis. Food Chem. 2014, 159, 512–518. [Google Scholar] [CrossRef] [PubMed]
- Ugolini, L.; Cinti, S.; Righetti, L.; Stefan, A.; Matteo, R.; D’avino, L.; Lazzeri, L. Production of an enzymatic protein hydrolyzate from defatted sunflower seed meal for potential application as a plant biostimulant. Ind. Crops Prod. 2015, 75, 15–23. [Google Scholar] [CrossRef]
- Rezvankhah, A.; Yarmand, M.S.; Ghanbarzadeh, B.; Mirzaee, H. Characterization of bioactive peptides produced from green lentil (Lens culinaris) seed protein concentrate using Alcalase and Flavourzyme in single and sequential hydrolysis. J. Food Process Preserv. 2021, 45, e15932. [Google Scholar] [CrossRef]
- Zhang, M.N.; Huang, G.R.; Jiang, J.X. Iron binding capacity of dephytinised soy protein isolate hydrolysate as influenced by the degree of hydrolysis and enzyme type. J. Food Sci. Technol. 2014, 51, 994–999. [Google Scholar] [CrossRef]
- Netto, F.M.; Desobry, S.A.; Labuza, T.P. Effect of water content on the glass transition, caking and stickiness of protein hydrolysates. Int. J. Food Prop. 2009, 1, 141–161. [Google Scholar] [CrossRef]
- Wallack, D.A.; King, C.J. Sticking and Agglomeration of Hygroscopic, Amorphous Carbohydrate and Food Powders. Biotechnol. Prog. 1988, 4, 31–35. [Google Scholar] [CrossRef]
- Wang, Y.; Li, D.; Wang, L.J.; Adhikari, B. The effect of addition of flaxseed gum on the emulsion properties of soybean protein isolate (SPI). J. Food Eng. 2011, 104, 56–62. [Google Scholar] [CrossRef]
- Chai, T.T.; Tong, S.R.; Law, Y.C.; Ismail, N.I.M.; Manan, F.A.; Wong, F.C. Anti-oxidative, metal chelating and radical scavenging effects of protein hydrolysates from blue-spotted stingray. Trop. J. Pharm. Res. 2015, 14, 1349. [Google Scholar] [CrossRef]
- Cheng, Y.; Chen, J.; Xiong, Y.L. Chromatographic Separation and Tandem MS Identification of Active Peptides in Potato Protein Hydrolysate That Inhibit Autoxidation of Soybean Oil-in-Water Emulsions. J. Agric. Food Chem. 2010, 58, 8825–8832. [Google Scholar] [CrossRef]
- Yuan, H.; Lv, J.; Gong, J.; Xiao, G.; Zhu, R.; Li, L.; Qiu, J. Secondary structures and their effects on antioxidant capacity of antioxidant peptides in yogurt. Int. J. Food Prop. 2018, 21, 2167–2180. [Google Scholar] [CrossRef]
- Smit, B.A.; Engels, W.J.M.; Smit, G. Branched chain aldehydes: Production and breakdown pathways and relevance for flavour in foods. Appl. Microbiol. Biotechnol. 2009, 81, 987–999. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Sample Name | Enzyme(s) | Protein Content of Hydrolysates (%) | Protein Yield (%) | DH (%) |
|---|---|---|---|---|
| Alc | Alcalase | 69 ± 0.01 a | 31.9 ± 0.6 ab | 20.7 ± 0.7 b |
| AlcFla | Alcalase and Flavourzyme | 88 ± 0.06 b | 40.2 ± 4.2 b | 38.8 ± 3.6 c |
| Try | Trypsin | 60 ± 0.00 a | 27.2 ± 0.6 a | 13.1 ± 1.0 a |
| TryFla | Trypsin and Flavourzyme | 86 ± 0.07 b | 39.0 ± 4.1 b | 38.1 ± 3.0 c |
| AF1 | AF13 | AF35 | AFPPH | TF13 | TF35 | TFPPH | ||
|---|---|---|---|---|---|---|---|---|
| Peptide IDs | 3968 | 5535 | 5872 | 6032 | 5241 | 6085 | 6325 | |
| Length | Mean | 6.36 | 7.75 | 8.67 | 8.76 | 8.17 | 9.66 | 9.79 |
| Weighted mean | 4.80 | 6.57 | 7.77 | 7.79 | 7.91 | 9.50 | 9.51 | |
| Charge 1 | Mean | −0.41 | −0.68 | −0.82 | −0.82 | −0.72 | −1.01 | −1.02 |
| Weighted mean | −0.30 | −0.64 | −0.83 | −0.78 | −0.64 | −0.98 | −1.02 | |
| Amino acid composition (relative molar abundance) | ||||||||
| Ala | 4.2% | 3.7% | 4.0% | 3.8% | 4.1% | 5.0% | 4.8% | |
| Arg | 0.7% | 1.4% | 1.6% | 1.5% | 1.6% | 1.9% | 1.6% | |
| Asn | 2.1% | 3.5% | 3.8% | 3.8% | 4.2% | 4.5% | 4.4% | |
| Asp | 4.0% | 7.6% | 8.4% | 7.8% | 5.5% | 8.0% | 7.9% | |
| Cys | 0.2% | 0.3% | 0.3% | 0.6% | 0.1% | 0.3% | 0.4% | |
| Gln | 1.4% | 1.9% | 2.0% | 2.1% | 1.3% | 1.6% | 1.5% | |
| Glu | 2.7% | 4.6% | 5.8% | 5.5% | 5.8% | 6.6% | 6.9% | |
| Gly | 8.2% | 9.6% | 9.9% | 9.7% | 11.3% | 12.4% | 11.2% | |
| His | 0.6% | 0.7% | 0.6% | 0.7% | 0.7% | 0.6% | 0.6% | |
| Ile | 1.3% | 3.0% | 4.2% | 3.8% | 4.2% | 4.9% | 4.9% | |
| Leu | 23.5% | 18.3% | 15.4% | 15.4% | 15.1% | 11.3% | 11.3% | |
| Lys | 0.7% | 1.4% | 2.1% | 2.4% | 1.7% | 2.5% | 2.7% | |
| Met | 0.8% | 1.0% | 0.9% | 0.8% | 1.1% | 0.9% | 1.2% | |
| Phe | 12.8% | 7.8% | 6.4% | 6.4% | 6.5% | 4.2% | 4.6% | |
| Pro | 14.2% | 12.4% | 11.9% | 12.1% | 12.9% | 10.6% | 11.0% | |
| Ser | 2.9% | 3.4% | 3.8% | 4.6% | 4.8% | 5.7% | 5.8% | |
| Thr | 3.3% | 4.6% | 5.3% | 5.4% | 5.5% | 6.7% | 6.4% | |
| Trp | 3.3% | 1.6% | 1.1% | 1.3% | 1.1% | 0.7% | 0.7% | |
| Tyr | 3.6% | 3.7% | 3.0% | 3.3% | 2.7% | 2.7% | 2.8% | |
| Val | 9.4% | 9.6% | 9.4% | 8.8% | 9.8% | 9.2% | 9.3% | |
| Sample | Peptide Concentration (mM Eq. Gly for 1 mg/mL PPH) | KD (mM Eq. Gly) | SE (KD) |
|---|---|---|---|
| AF1 | 3.53 | 0.72 | 0.43 |
| AF13 | 2.78 | 1.41 | 0.87 |
| AF35 | 2.41 | 2.27 | 0.58 |
| AFPPH | 2.41 | 6.81 | 2.10 |
| TF13 | 2.82 | 4.85 | 2.40 |
| TF35 | 2.27 | 9.19 | 7.80 |
| TFPPH | 2.27 | 8.16 | 7.50 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bjørlie, M.; Hartmann, J.C.; Rasmussen, L.H.; Yesiltas, B.; Sørensen, A.-D.M.; Gregersen Echers, S.; Jacobsen, C. Screening for Metal-Chelating Activity in Potato Protein Hydrolysates Using Surface Plasmon Resonance and Peptidomics. Antioxidants 2024, 13, 346. https://doi.org/10.3390/antiox13030346
Bjørlie M, Hartmann JC, Rasmussen LH, Yesiltas B, Sørensen A-DM, Gregersen Echers S, Jacobsen C. Screening for Metal-Chelating Activity in Potato Protein Hydrolysates Using Surface Plasmon Resonance and Peptidomics. Antioxidants. 2024; 13(3):346. https://doi.org/10.3390/antiox13030346
Chicago/Turabian StyleBjørlie, Mads, Julie Christina Hartmann, Line Hyrup Rasmussen, Betül Yesiltas, Ann-Dorit Moltke Sørensen, Simon Gregersen Echers, and Charlotte Jacobsen. 2024. "Screening for Metal-Chelating Activity in Potato Protein Hydrolysates Using Surface Plasmon Resonance and Peptidomics" Antioxidants 13, no. 3: 346. https://doi.org/10.3390/antiox13030346
APA StyleBjørlie, M., Hartmann, J. C., Rasmussen, L. H., Yesiltas, B., Sørensen, A.-D. M., Gregersen Echers, S., & Jacobsen, C. (2024). Screening for Metal-Chelating Activity in Potato Protein Hydrolysates Using Surface Plasmon Resonance and Peptidomics. Antioxidants, 13(3), 346. https://doi.org/10.3390/antiox13030346