

Butylated Hydroxytoluene (BHT) Protects SH-SY5Y Neuroblastoma Cells from Ferroptotic Cell Death: Insights from In Vitro and In Vivo Studies

Abstract
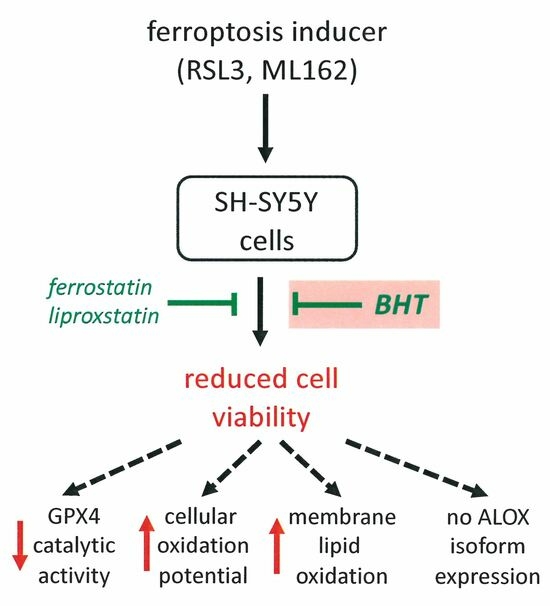
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Background
2. Materials and Methods
2.1. Chemicals and Devices
2.2. Cell-Viability Assay (CCK8)
2.3. Lipid Extraction, Hydrolysis, and HPLC Analysis
2.4. Measurements of the Cellular Oxidation Potential
2.5. RNA Extraction, Reverse Transcription and qRT-PCR of GPX4- and ALOX-Isoforms
2.6. In Vitro GPX4 Activity Assay
2.7. In Vivo Rat Ferroptosis Model
2.8. Numbers of Repetitions and Statistic Evaluation of the Experimental Raw Data
3. Results
3.1. Butylated Hydroxytoluene Does Not Impact the Viability of Human Neuroblastoma Cells

3.2. Butylated Hydroxytoluene Protects Human Neuroblastoma Cells from RSL-3-Induced Ferroptosis

3.3. Butylated Hydroxytoluene Also Protects SH-SY5Y Cells from ML162-Induced Ferroptosis

3.4. Butylated Hydroxytoluene Prevented RSL3-Induced Oxidation of Membrane Lipids


3.5. RSL3 Inhibits Intracellular GPX4 Activity but Does Not Modify GPX4 Gene Expression

3.6. RSL3 Does Not Inhibit Recombinant Selenium Containing GPX4

3.7. Lipoxygenase Isoforms Are Neither Expressed in Resting nor in RSL3-Treated SH-SY5Y Cells
3.8. Oral Application of BHT Normalizes the Expression of Ferroptosis-Related Genes in a Rat Alzheimer’s Disease Model

4. Discussion
4.1. BHT Does Not Impact the Viability of SH-SY5Y Cells under Baseline Conditions but Protects Cells from RSL3-Induced Ferroptosis
4.2. The Anti-Ferroptotic GPX4 Is High-Level Expressed in SH-SY5Y Neuroblastoma Cells but ALOX-Isoforms Are Absent
4.3. RSL3 Inhibits Cellular GPX4 Activity but Does Not Prevent the Catalytic Activity of the Selenocysteine-Containing Recombinant Enzyme
4.4. BHT Prevents Upregulation of Expression of Patho-Physiologically Relevant Genes in a Rat In Vivo Model of Alzheimer’s Disease
4.5. Degree of Novelty, Advancement of Science, and Limitations of Our Study
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Stockwell, B.R. Ferroptosis turns 10: Emerging mechanisms, physiological functions, and therapeutic applications. Cell 2022, 185, 2401–2421. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.; Stockwell, B.R.; Conrad, M. Ferroptosis: Mechanisms, biology and role in disease. Nat. Rev. Mol. Cell Biol. 2021, 22, 266–282. [Google Scholar] [CrossRef] [PubMed]
- Tang, D.; Chen, X.; Kang, R.; Kroemer, G. Ferroptosis: Molecular mechanisms and health implications. Cell Res. 2021, 31, 107–125. [Google Scholar] [CrossRef]
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Zaitsev, E.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantley, A.M.; Yang, W.S.; et al. Ferroptosis: An iron-dependent form of nonapoptotic cell death. Cell 2012, 149, 1060–1072. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Kang, R.; Tang, D. Signaling pathways and defense mechanisms of ferroptosis. FEBS J. 2022, 289, 7038–7050. [Google Scholar] [CrossRef]
- Rochette, L.; Dogon, G.; Rigal, E.; Zeller, M.; Cottin, Y.; Vergely, C. Lipid Peroxidation and Iron Metabolism: Two Corner Stones in the Homeostasis Control of Ferroptosis. Int. J. Mol. Sci. 2022, 24, 449. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Li, J.; Kang, R.; Klionsky, D.J.; Tang, D. Ferroptosis: Machinery and regulation. Autophagy 2021, 17, 2054–2081. [Google Scholar] [CrossRef]
- David, S.; Jhelum, P.; Ryan, F.; Jeong, S.Y.; Kroner, A. Dysregulation of Iron Homeostasis in the Central Nervous System and the Role of Ferroptosis in Neurodegenerative Disorders. Antioxid. Redox Signal. 2022, 37, 150–170. [Google Scholar] [CrossRef]
- Papanikolaou, G.; Pantopoulos, K. Iron metabolism and toxicity. Toxicol. Appl. Pharmacol. 2005, 202, 199–211. [Google Scholar] [CrossRef]
- Venkataramani, V. Iron Homeostasis and Metabolism: Two Sides of a Coin. Adv. Exp. Med. Biol. 2021, 1301, 25–40. [Google Scholar] [CrossRef]
- Griffiths, H.R.; Gao, D.; Pararasa, C. Redox regulation in metabolic programming and inflammation. Redox Biol. 2017, 12, 50–57. [Google Scholar] [CrossRef]
- Lee, B.W.L.; Ghode, P.; Ong, D.S.T. Redox regulation of cell state and fate. Redox Biol. 2019, 25, 101056. [Google Scholar] [CrossRef]
- Sies, H. Oxidative stress: A concept in redox biology and medicine. Redox Biol. 2015, 4, 180–183. [Google Scholar] [CrossRef]
- Forcina, G.C.; Dixon, S.J. GPX4 at the Crossroads of Lipid Homeostasis and Ferroptosis. Proteomics 2019, 19, 1800311. [Google Scholar] [CrossRef]
- Zheng, J.; Conrad, M. The Metabolic Underpinnings of Ferroptosis. Cell Metab. 2020, 32, 920–937. [Google Scholar] [CrossRef]
- Zhang, B.; Pan, C.; Feng, C.; Yan, C.; Yu, Y.; Chen, Z.; Guo, C.; Wang, X. Role of mitochondrial reactive oxygen species in homeostasis regulation. Redox Rep. 2022, 27, 45–52. [Google Scholar] [CrossRef]
- Yehye, W.A.; Rahman, N.A.; Ariffin, A.; Abd Hamid, S.B.; Alhadi, A.A.; Kadir, F.A.; Yaeghoobi, M. Understanding the chemistry behind the antioxidant activities of butylated hydroxytoluene (BHT): A review. Eur. J. Med. Chem. 2015, 101, 295–312. [Google Scholar] [CrossRef]
- Burton, G.W.; Doba, T.; Gabe, E.J.; Hughes, L.; Lee, F.L.; Prasad, L.; Ungold, K.U. Autoxidation of Biological Molecules. 4. Maximizing the Antioxidant Activity of Phenols. J. Am. Chem. Soc. 1985, 107, 7053–7065. [Google Scholar] [CrossRef]
- Snipes, W.; Person, S.; Keith, A.; Cupp, J. Butylated hydroxytoluene inactivated lipid-containing viruses. Science 1975, 188, 64–66. [Google Scholar] [CrossRef]
- Yamamoto, K.; Tajima, K.; Mizutani, T. The acute toxicity of butylated hydroxytoluene and its metabolites in mice. Toxicol. Lett. 1980, 6, 173–175. [Google Scholar] [CrossRef]
- Lanigan, R.S.; Yamarik, T.A. Final report on the safety assessment of BHT(1). Int. J. Toxicol. 2002, 21 (Suppl. S2), 19–94. [Google Scholar] [CrossRef] [PubMed]
- Williams, G.M.; Iatropoulos, M.J.; Whysner, J. Safety assessment of butylated hydroxyanisole and butylated hydroxytoluene as antioxidant food additives. Food Chem. Toxicol. 1999, 37, 1027–1038. [Google Scholar] [CrossRef] [PubMed]
- Bomhard, E.M.; Bremmer, J.N.; Herbold, B.A. Review of the mutagenicity/genotoxicity of butylated hydroxytoluene. Mutat. Res. 1992, 277, 187–200. [Google Scholar] [CrossRef] [PubMed]
- Botterweck, A.A.; Verhagen, H.; Goldbohm, R.A.; Kleinjans, J.; van den Brandt, P.A. Intake of butylated hydroxyanisole and butylated hydroxytoluene and stomach cancer risk: Results from analyses in the Netherlands Cohort Study. Food Chem. Toxicol. 2000, 38, 599–605. [Google Scholar] [CrossRef]
- Thompson, J.A.; Bolton, J.L.; Malkinson, A.M. Relationship between the Metabolism of Butylated Hydroxytoluene (Bht) and Lung-Tumor Promotion in Mice. Exp. Lung Res. 1991, 17, 439–453. [Google Scholar] [CrossRef] [PubMed]
- Bligh, E.G.; Dyer, W.J. A Rapid Method of Total Lipid Extraction and Purification. Can. J. Biochem. Phys. 1959, 37, 911–917. [Google Scholar] [CrossRef]
- Huang, L.S.; Kang, J.S.; Kim, M.R.; Sok, D.E. Oxygenation of arachidonoyl lysophospholipids by lipoxygenases from soybean, porcine leukocyte, or rabbit reticulocyte. J. Agric. Food Chem. 2008, 56, 1224–1232. [Google Scholar] [CrossRef]
- Akhtar, A.; Gupta, S.M.; Dwivedi, S.; Kumar, D.; Shaikh, M.F.; Negi, A. Preclinical Models for Alzheimer’s Disease: Past, Present, and Future Approaches. ACS Omega 2022, 7, 47504–47517. [Google Scholar] [CrossRef]
- Silva, S.S.L.; Tureck, L.V.V.; Souza, L.C.; Mello-Hortega, J.V.V.; Piumbini, A.L.; Teixeira, M.D.; Furtado-Alle, L.; Vital, M.A.B.F.; Souza, R.L.R. Animal model of Alzheimer’s disease induced by streptozotocin: New insights about cholinergic pathway. Brain Res. 2023, 1799, 148175. [Google Scholar] [CrossRef]
- Duan, L.; Qian, X.; Wang, Q.; Huang, L.; Ge, S. Experimental Periodontitis Deteriorates Cognitive Function and Impairs Insulin Signaling in a Streptozotocin-Induced Alzheimer’s Disease Rat Model. J. Alzheimer’s Dis. 2022, 88, 57–74. [Google Scholar] [CrossRef]
- Chen, Z.Y.; Zhang, Y. Animal models of Alzheimer’s disease: Applications, evaluation, and perspectives. Zool. Res. 2022, 43, 1026–1040. [Google Scholar] [CrossRef]
- Scarpellini, C.; Klejborowska, G.; Lanthier, C.; Hassannia, B.; Vanden Berghe, T.; Augustyns, K. Beyond ferrostatin-1: A comprehensive review of ferroptosis inhibitors. Trends Pharmacol. Sci. 2023, 44, 902–916. [Google Scholar] [CrossRef]
- Chen, H.; Qi, Q.; Wu, N.; Wang, Y.; Feng, Q.; Jin, R.; Jiang, L. Aspirin promotes RSL3-induced ferroptosis by suppressing mTOR/SREBP-1/SCD1-mediated lipogenesis in PIK3CA-mutant colorectal cancer. Redox Biol. 2022, 55, 102426. [Google Scholar] [CrossRef]
- Kawasaki, N.K.; Suhara, T.; Komai, K.; Shimada, B.K.; Yorichika, N.; Kobayashi, M.; Baba, Y.; Higa, J.K.; Matsui, T. The role of ferroptosis in cell-to-cell propagation of cell death initiated from focal injury in cardiomyocytes. Life Sci. 2023, 332, 122113. [Google Scholar] [CrossRef]
- Shintoku, R.; Takigawa, Y.; Yamada, K.; Kubota, C.; Yoshimoto, Y.; Takeuchi, T.; Koshiishi, I.; Torii, S. Lipoxygenase-mediated generation of lipid peroxides enhances ferroptosis induced by erastin and RSL3. Cancer Sci. 2017, 108, 2187–2194. [Google Scholar] [CrossRef]
- Chen, T.T.; Leng, J.F.; Tan, J.; Zhao, Y.J.; Xie, S.S.; Zhao, S.F.; Yan, X.; Zhu, L.; Luo, J.; Kong, L.; et al. Discovery of Novel Potent Covalent Glutathione Peroxidase 4 Inhibitors as Highly Selective Ferroptosis Inducers for the Treatment of Triple-Negative Breast Cancer. J. Med. Chem. 2023, 66, 10036–10059. [Google Scholar] [CrossRef]
- van Leyen, K.; Duvoisin, R.M.; Engelhardt, H.; Wiedmann, M. A function for lipoxygenase in programmed organelle degradation. Nature 1998, 395, 392–395. [Google Scholar] [CrossRef]
- Banthiya, S.; Pekarova, M.; Kuhn, H.; Heydeck, D. Secreted lipoxygenase from Pseudomonas aeruginosa exhibits biomembrane oxygenase activity and induces hemolysis in human red blood cells. Arch. Biochem. Biophys. 2015, 584, 116–124. [Google Scholar] [CrossRef]
- Kuhn, H.; Belkner, J.; Wiesner, R.; Brash, A.R. Oxygenation of biological membranes by the pure reticulocyte lipoxygenase. J. Biol. Chem. 1990, 265, 18351–18361. [Google Scholar] [CrossRef]
- Belkner, J.; Wiesner, R.; Rathman, J.; Barnett, J.; Sigal, E.; Kuhn, H. Oxygenation of lipoproteins by mammalian lipoxygenases. Eur. J. Biochem. 1993, 213, 251–261. [Google Scholar] [CrossRef]
- Escada-Rebelo, S.; Ramalho-Santos, J. Oxidative and Nitrosative Stress Detection in Human Sperm Using Fluorescent Probes. Methods Mol. Biol. 2023, 2566, 45–52. [Google Scholar] [CrossRef] [PubMed]
- Ursini, F.; Maiorino, M.; Roveri, A. Phospholipid hydroperoxide glutathione peroxidase (PHGPx): More than an antioxidant enzyme? Biomed. Environ. Sci. 1997, 10, 327–332. [Google Scholar] [PubMed]
- Cheff, D.M.; Huang, C.; Scholzen, K.C.; Gencheva, R.; Ronzetti, M.H.; Cheng, Q.; Hall, M.D.; Arner, E.S.J. The ferroptosis inducing compounds RSL3 and ML162 are not direct inhibitors of GPX4 but of TXNRD1. Redox Biol. 2023, 62, 102703. [Google Scholar] [CrossRef] [PubMed]
- Sui, X.B.; Zhang, R.N.; Liu, S.P.; Duan, T.; Zhai, L.J.; Zhang, M.M.; Han, X.; Xiang, Y.; Huang, X.; Lin, H.; et al. RSL3 Drives Ferroptosis Through GPX4 Inactivation and ROS Production in Colorectal Cancer. Front. Pharmacol. 2018, 9, 1371. [Google Scholar] [CrossRef] [PubMed]
- Sekhar, K.R.; Hanna, D.N.; Cyr, S.; Baechle, J.J.; Kuravi, S.; Balusu, R.; Rathmel, K.; Baregamian, N. Glutathione peroxidase 4 inhibition induces ferroptosis and mTOR pathway suppression in thyroid cancer. Sci. Rep. 2022, 12, 19396. [Google Scholar] [CrossRef]
- Moosmayer, D.; Hilpmann, A.; Hoffmann, J.; Schnirch, L.; Zimmermann, K.; Badock, V.; Furst, L.; Eaton, J.K.; Viswanathan, V.S.; Schreiber, S.L.; et al. Crystal structures of the selenoprotein glutathione peroxidase 4 in its apo form and in complex with the covalently bound inhibitor ML162. Acta Crystallogr. D Struct. Biol. 2021, 77 Pt 2, 237–248. [Google Scholar] [CrossRef] [PubMed]
- Probst, L.; Dachert, J.; Schenk, B.; Fulda, S. Lipoxygenase inhibitors protect acute lymphoblastic leukemia cells from ferroptotic cell death. Biochem. Pharmacol. 2017, 140, 41–52. [Google Scholar] [CrossRef]
- Liu, Y.; Wang, W.; Li, Y.; Xiao, Y.; Cheng, J.; Jia, J. The 5-Lipoxygenase Inhibitor Zileuton Confers Neuroprotection against Glutamate Oxidative Damage by Inhibiting Ferroptosis. Biol. Pharm. Bull. 2015, 38, 1234–1239. [Google Scholar] [CrossRef]
- Wenzel, S.E.; Tyurina, Y.Y.; Zhao, J.; St Croix, C.M.; Dar, H.H.; Mao, G.; Tyurin, V.A.; Anthonymuthu, T.S.; Kapralov, A.A.; Amoscato, A.A.; et al. PEBP1 Wardens Ferroptosis by Enabling Lipoxygenase Generation of Lipid Death Signals. Cell 2017, 171, 628–641.e26. [Google Scholar] [CrossRef]
- Khan, S.; Barve, K.H.; Kumar, M.S. Recent Advancements in Pathogenesis, Diagnostics and Treatment of Alzheimer’s Disease. Curr. Neuropharmacol. 2020, 18, 1106–1125. [Google Scholar] [CrossRef]
- Zhang, X.X.; Tian, Y.; Wang, Z.T.; Ma, Y.H.; Tan, L.; Yu, J.T. The Epidemiology of Alzheimer’s Disease Modifiable Risk Factors and Prevention. J. Prev. Alzheimer’s Dis. 2021, 8, 313–321. [Google Scholar] [CrossRef]
- Ma, H.; Dong, Y.; Chu, Y.; Guo, Y.; Li, L. The mechanisms of ferroptosis and its role in alzheimer’s disease. Front. Mol. Biosci. 2022, 9, 965064. [Google Scholar] [CrossRef] [PubMed]
- Yan, N.; Zhang, J. Iron Metabolism, Ferroptosis, and the Links With Alzheimer’s Disease. Front. Neurosci. 2019, 13, 1443. [Google Scholar] [CrossRef] [PubMed]
- Jafarzadeh, G.; Shakerian, S.; Farbood, Y.; Ghanbarzadeh, M. Effects of Eight Weeks of Resistance Exercises on Neurotrophins and Trk Receptors in Alzheimer Model Male Wistar Rats. Basic. Clin. Neurosci. 2021, 12, 349–359. [Google Scholar] [CrossRef]
- Plays, M.; Muller, S.; Rodriguez, R. Chemistry and biology of ferritin. Metallomics 2021, 13, mfab021. [Google Scholar] [CrossRef]
- Park, E.; Chung, S.W. ROS-mediated autophagy increases intracellular iron levels and ferroptosis by ferritin and transferrin receptor regulation. Cell Death Dis. 2019, 10, 822. [Google Scholar] [CrossRef]
- Doll, S.; Proneth, B.; Tyurina, Y.Y.; Panzilius, E.; Kobayashi, S.; Ingold, I.; Irmler, M.; Beckers, J.; Aichler, M.; Walch, A.; et al. ACSL4 dictates ferroptosis sensitivity by shaping cellular lipid composition. Nat. Chem. Biol. 2017, 13, 91–98. [Google Scholar] [CrossRef]
- Lane, C.A.; Hardy, J.; Schott, J.M. Alzheimer’s disease. Eur. J. Neurol. 2018, 25, 59–70. [Google Scholar] [CrossRef] [PubMed]
- Nieva-Echevarria, B.; Manzanos, M.J.; Goicoechea, E.; Guillen, M.D. 2,6-Di-Tert-Butyl-Hydroxytoluene and Its Metabolites in Foods. Compr. Rev. Food Sci. Food Saf. 2015, 14, 67–80. [Google Scholar] [CrossRef]
- Borchert, A.; Savaskan, N.E.; Kuhn, H. Regulation of expression of the phospholipid hydroperoxide/sperm nucleus glutathione peroxidase gene. Tissue-specific expression pattern and identification of functional cis- and trans-regulatory elements. J. Biol. Chem. 2003, 278, 2571–2580. [Google Scholar] [CrossRef]
- Borchert, A.; Kalms, J.; Roth, S.R.; Rademacher, M.; Schmidt, A.; Holzhutter, H.G.; Kuhn, H.; Scheerer, P. Crystal structure and functional characterization of selenocysteine-containing glutathione peroxidase 4 suggests an alternative mechanism of peroxide reduction. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2018, 1863, 1095–1107. [Google Scholar] [CrossRef]
- Schnurr, K.; Borchert, A.; Kuhn, H. Inverse regulation of lipid-peroxidizing and hydroperoxyl lipid-reducing enzymes by interleukins 4 and 13. FASEB J. 1999, 13, 143–154. [Google Scholar] [CrossRef]
- Weitzel, F.; Wendel, A. Selenoenzymes regulate the activity of leukocyte 5-lipoxygenase via the peroxide tone. J. Biol. Chem. 1993, 268, 6288–6292. [Google Scholar] [CrossRef]
- Kagan, V.E.; Mao, G.; Qu, F.; Angeli, J.P.; Doll, S.; Croix, C.S.; Hussain Dar, H.; Liu, B.; Tyurin, V.A.; Ritov, V.B.; et al. Oxidized arachidonic and adrenic PEs navigate cells to ferroptosis. Nat. Chem. Biol. 2017, 13, 81–90. [Google Scholar] [CrossRef]
- Funk, C.D.; Chen, X.S.; Johnson, E.N.; Zhao, L. Genes and their targeted disruption. Prostaglandins Other Lipid Mediat. 2002, 68–69, 303–312. [Google Scholar] [CrossRef] [PubMed]
- Vuckovic, A.M.; Bosello Travain, V.; Bordin, L.; Cozza, G.; Miotto, G.; Rossetto, M.; Toppo, S.; Venerando, R.; Zaccarin, M.; Maiorino, M.; et al. Inactivation of the glutathione peroxidase GPx4 by the ferroptosis-inducing molecule RSL3 requires the adaptor protein 14-3-3epsilon. FEBS Lett. 2020, 594, 611–624. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Forouhar, F.; Lin, A.J.; Wang, Q.; Polychronidou, V.; Soni, R.K.; Xia, X.; Stockwell, B.R. Small-molecule allosteric inhibitors of GPX4. Cell Chem. Biol. 2022, 29, 1680–1693.e9. [Google Scholar] [CrossRef] [PubMed]
- Gatt, A.; Lee, H.; Williams, G.; Thuret, S.; Ballard, C. Expression of neurogenic markers in Alzheimer’s disease: A systematic review and metatranscriptional analysis. Neurobiol. Aging 2019, 76, 166–180. [Google Scholar] [CrossRef] [PubMed]
- Sharma, V.K.; Mehta, V.; Singh, T.G. Alzheimer’s Disorder: Epigenetic Connection and Associated Risk Factors. Curr. Neuropharmacol. 2020, 18, 740–753. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Yang, H.X.; Lyu, M.; Zhou, L.H.; Zhang, Y.; Kang, C.S.; Wang, J.; Wang, Y. Association between behavioural risks and Alzheimer’s disease: Elucidated with an integrated analysis of gene expression patterns and molecular mechanisms. Neurosci. Biobehav. R. 2023, 150, 105207. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Faraji, P.; Borchert, A.; Ahmadian, S.; Kuhn, H. Butylated Hydroxytoluene (BHT) Protects SH-SY5Y Neuroblastoma Cells from Ferroptotic Cell Death: Insights from In Vitro and In Vivo Studies. Antioxidants 2024, 13, 242. https://doi.org/10.3390/antiox13020242
Faraji P, Borchert A, Ahmadian S, Kuhn H. Butylated Hydroxytoluene (BHT) Protects SH-SY5Y Neuroblastoma Cells from Ferroptotic Cell Death: Insights from In Vitro and In Vivo Studies. Antioxidants. 2024; 13(2):242. https://doi.org/10.3390/antiox13020242
Chicago/Turabian StyleFaraji, Parisa, Astrid Borchert, Shahin Ahmadian, and Hartmut Kuhn. 2024. "Butylated Hydroxytoluene (BHT) Protects SH-SY5Y Neuroblastoma Cells from Ferroptotic Cell Death: Insights from In Vitro and In Vivo Studies" Antioxidants 13, no. 2: 242. https://doi.org/10.3390/antiox13020242