
The Key Role of GSH in Keeping the Redox Balance in Mammalian Cells: Mechanisms and Significance of GSH in Detoxification via Formation of Conjugates

Abstract
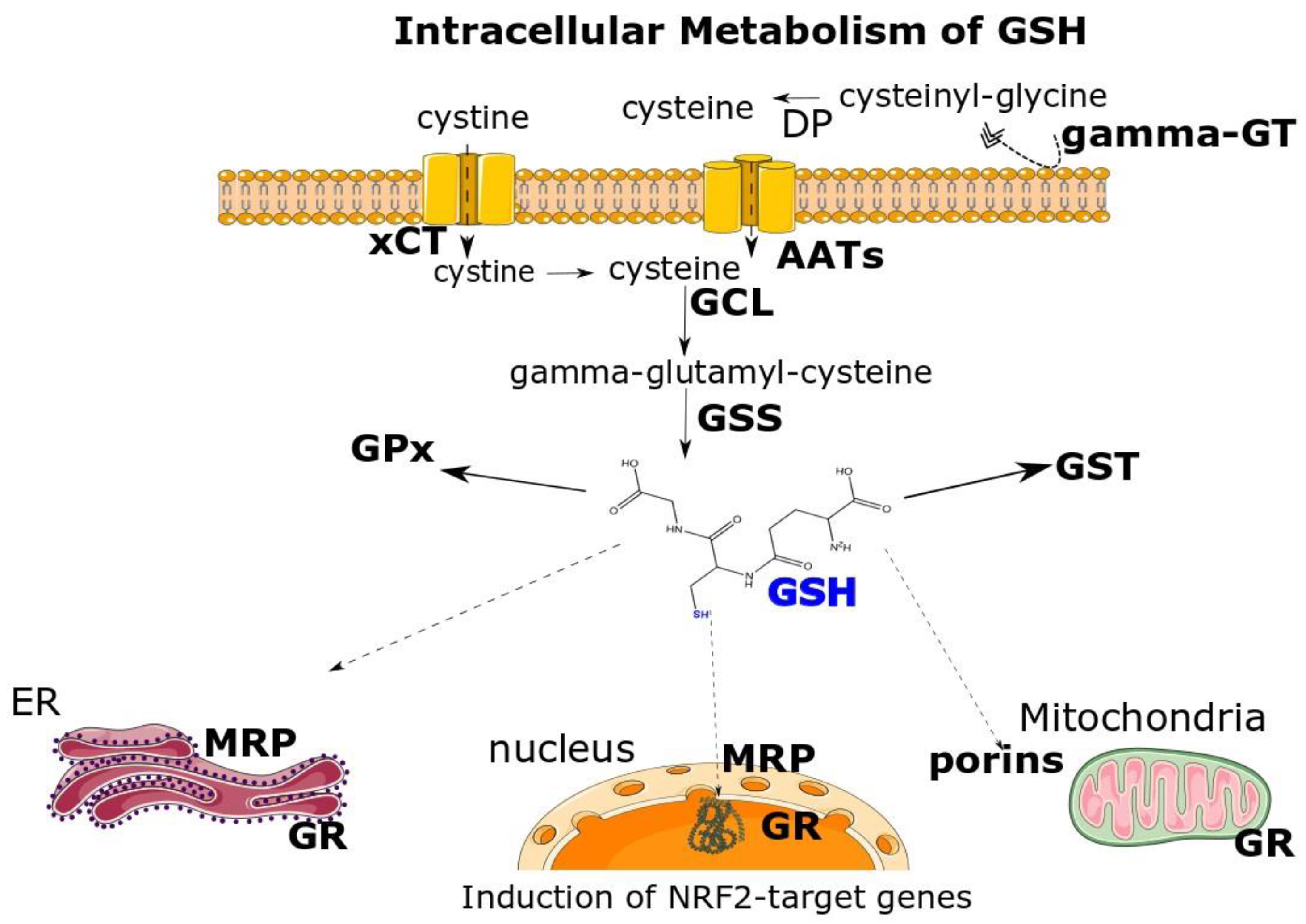
:1. Structure, Biosynthesis of GSH and Key Enzymes Involved in GSH Homeostasis
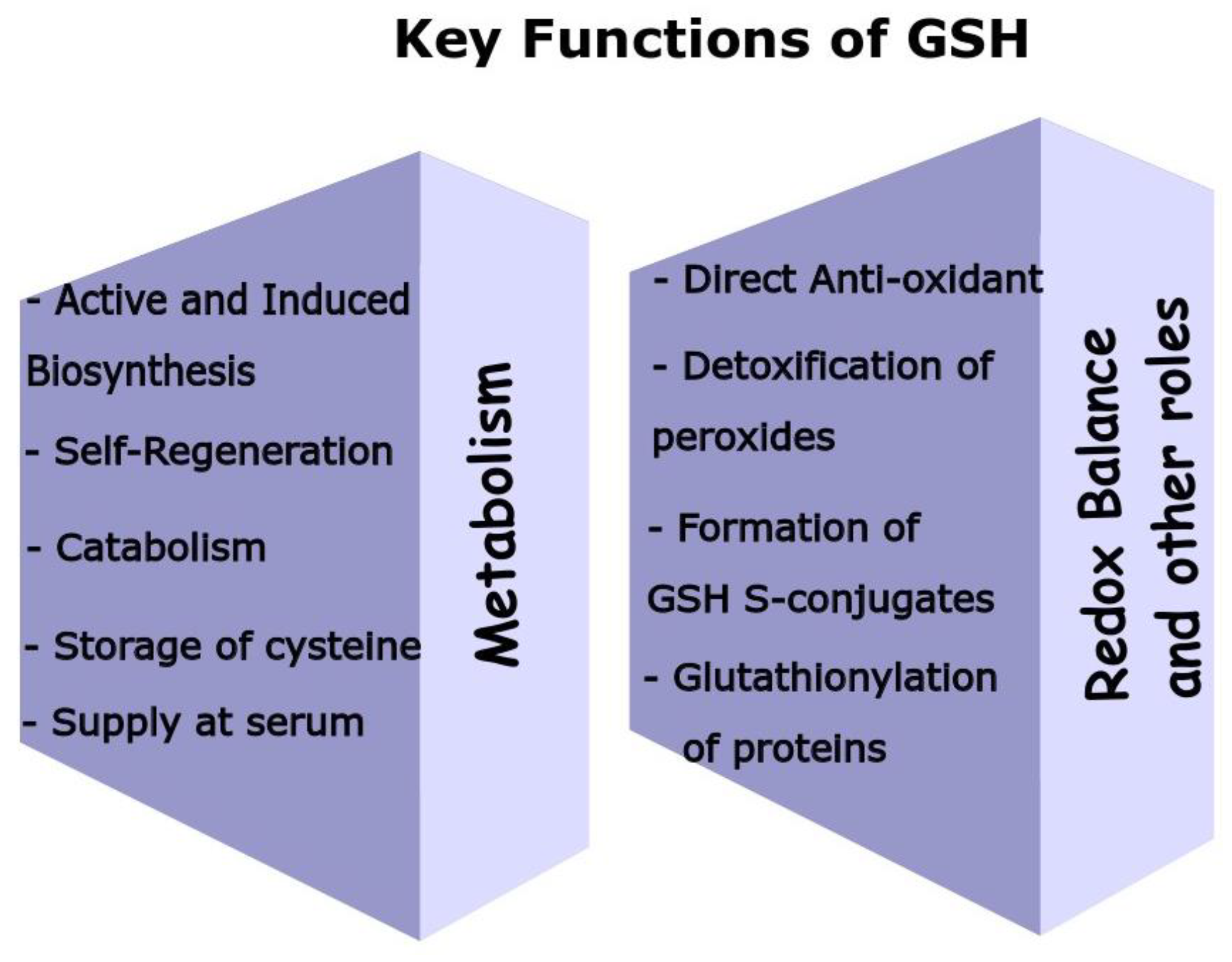
2. The Biological Functions of GSH
2.1. Antioxidant Role
2.2. Cell Redox Status
2.3. Formation of GSH S-Conjugates
3. GSH Conjugates: Mechanisms and Biological Significance
3.1. Extrinsic Compounds (Xenobiotics)—The Case of Poisons/Toxins

{kind=link}
{kind=link}
{kind=link}
| Xenobiotic Compound (Reactant) | GSH-S Conjugate (Product) | Chemical Group(s) Involved | Tissues/Organ | Biological Significance | Spontaneous (Direct) and/or GST-Dependent |
|---|---|---|---|---|---|
| Benzenes and their Metabolites | |||||
| 3,4-Bromobenzene oxide [79] | Bromobenzene–GSH adducts (4-glutathionyl conjugate, 3-glutathionyl conjugate) | Epoxide | Liver | Mainly bromobenzene detoxification | GST |
| 1,4 Benzoquinone | Hydroquinone–GSH conjugates | C-C double bond | Liver, bone marrow | Not detoxification; they are labile to a cycle of formation of myelotoxic, multiple GSH S-adducts | Spontaneous |
| Naphthalene [86] | Glutathionyl–naphtalene adduct | Epoxide | Liver, lung, nasal mucosa | Probably detoxification | Spontaneous, GST [87] |
| Quinones | |||||
| Bisphenol A-3,4-quinone (BPAQ) [91] | BPAQ–GSH adduct | C-C double bond | Liver [93] | Not known yet (detoxification of BPAQ or more toxic) | Spontaneous |
| Polychlorinated biphenyl (PCB) quinones [94] | Glutathionylated conjugated hydroquinone | C-C double bond | Liver | As for BPA | Spontaneous |
| Quercetin o-quinone [96] | Glutathionyl–quercetin adducts | C-C double bond | Liver [103], aortic endothelial cells [97] | Possible protective role, as these conjugates are excreted from cells [97] | Spontaneous |
| Other chemical groups | |||||
| Aflatoxin | Aflatoxin B1(AFB1)–GSH conjugates | Epoxide | Kidneys, liver | Detoxification [98] | GST |
| Isothiocyanates | Isothiocyanate–GSH adduct | Isothiocyanate group | Liver, kidney [104] | Unstable conjugates, release isothiocyanates [102] | GST, spontaneous |
| Formaldehyde | BiGF 2 | Carbonyl | Liver and possibly in other tissues | Masking, detoxification [100] | Spontaneous |
3.2. Extrinsic Compounds (Xenobiotics)—The Case of Medicines (Drugs)
| Xenobiotic Compound (Reactant) | GSH-S Conjugate (Product) | Chemical Group(s) Involved | Tissues/Organ | Biological Significance | Spontaneous (Direct) and/or GST-Dependent |
|---|---|---|---|---|---|
| Painkillers | |||||
| NAPQI [112] | APAP-GSH conjugate | C-C double bond, imine double bond | Main liver but also kidneys and others | Detoxification of APAP | Both |
| Anticancer drugs | |||||
| Cisplatin | Diglutathionyl- platinum adducts [115] | Pt-O | Cell extracts (leukemia), liver, lungs, heart, kidneys, brain, pancreas [116] | Quench the action of cisplatin, excretion of cisplatin, the adducts still inhibit translation of mRNAs | Spontaneous [115] |
| Landomycin E [123] | Landomycin E-GSH adduct | C-C double bond | In vitro (cell extracts) | Potent anti-neoplasmatic action for GSH depletion | Spontaneous |
| Immunosuppressive | |||||
| Azathioprine [126] | 6-mercaptopurine | Thiolysis | Liver | Bioactivation | GSTA [140] |
| Cardiovascular disease drugs | |||||
| Simvastatin | 5′,6′-dihydroxy-4′a-glutathione adduct of simvastatin [128] | C-C double bond | Bile | Probable detoxification, since it favors the formation of degradation products excreted in bile | Probably GST-dependent |
| Antidepressants | |||||
| Diazepine [129] | GSH–adduct of Tricyclic Diazepine | High electron density of tricyclic core | In vitro | Probable detoxification | Spontaneous |
| 3′-oxohexobarbital [130] | Cyclohexenone–glutathione adduct | C-C double bond | Liver, bile | Detoxification through excretion in bile | Probably by GST |
| Anticoagulant | |||||
| Ticlopidine [138] | Multiples ticlopidine–GSH adducts | C-C double bond and epoxide | Liver | Not known, perhaps detoxification | GST, unknown if spontaneous |
| Addictive drugs | |||||
| Cocaine [139] | Cocaine–GSH adducts | Aryl moiety | Liver | Not known, possibly detoxification | GST |
3.3. Endogenous Compounds
| Endogenous Compound (Reactant) | GSH-S Conjugate (Product) | Chemical Group(s) Involved | Tissues/Organ | Biological Significance | Spontaneous (Direct) and/or GST-Dependent |
|---|---|---|---|---|---|
| Vitamins | |||||
| Menadione [142] | Menadione-GSH conjugate | C-C double bond | Liver | Detoxification through excretion [142] | Spontaneous [143] |
| Hormones | |||||
| Estrogen | catechol estrogen quinone–GSH conjugates [175] | C-C double bond | Liver, breast | Decrease in carcinogenic potential | GST |
| Neurotransmitters | |||||
| Dopamine | 5-S-glutathionyl-catecholamine conjugates [153] | C-C double bond | Brain | Possibly neurotoxic through interconversions [154] | Spontaneous (in presence of tyrosinase) [153] |
| Eicosanoids | |||||
| Prostaglandins | PGA1-GSH adduct [158] | C-C double bond | RBCs, liver | Cytotoxic [161], novel biochemical properties [162,163] | Spontaneous, GST |
| Leukotrienes | LTA4-GSH adduct (LTC4) | C-C double bond | Hematopoietic cells | Synthesis [166] | GST- LTC4Synthase |
| 4-hydroxynonetal (lipid peroxidation product) | 3-glutathionyl-4-hydroxynonanal | C-C double bond [176] | Liver [177] | Detoxification | Spontaneous, GSTA4 |
3.4. Catabolism and Excretion of GSH S-Conjugates
4. GSH–Hematin Adducts: Characterization, Mechanism of Formation and Possible Implications in Heme Associated Pathologies
5. Can the GSH S-Conjugates Be Considered Novel Metabolic Signatures in Disease States in the Upcoming Field of Metabolomics?
6. Conclusions
Funding
Conflicts of Interest
Abbreviations
References
- Rey-Pailhade, D. Sur un corps d’origine organique hydrogénant le soufre á froid. Hebd. Séances Acad. Sci. 1888, 106, 1683–1684. [Google Scholar]
- Rey-Pailhade, D. Sur un nouveau principe immédiat organique. le philothion. Bull. Soc. Hist Nat. Toulouse 1888, 173–180. [Google Scholar]
- Fruton, J.S. Contrasts in Scientific Style. Emil Fischer and Franz Hofmeister: Their Research Groups and Their Theory of Protein Structure. Proc. Am. Philos. Soc. JSTOR 1985, 129, 313–370. [Google Scholar]
- Hopkins, F.G. On an Autoxidisable Constituent of the Cell. Biochem. J. 1921, 15, 286–305. [Google Scholar] [CrossRef]
- Hunter, G.; Eagles, B.A. Glutathione. A critical Study. J. Biol. Chem. 1927, 72, 147–166. [Google Scholar] [CrossRef]
- Robert, D.; Simoni, R.L.H. Martha Vaughan. The Discovery of Glutathione by F. Gowland Hopkins and the Beginning of Biochemistry at Cambridge University. J. Biol. Chem. 2002, 277, 27–28. [Google Scholar] [CrossRef]
- Meister, A. On the discovery of glutathione. Trends Biochem. Sci. 1988, 13, 185–188. [Google Scholar] [CrossRef] [PubMed]
- Kretzschmar, M. Regulation of hepatic glutathione metabolism and its role in hepatotoxicity. Exp. Toxicol. Pathol. Off. J. Ges. Fur. Toxikol. Pathol. 1996, 48, 439–446. [Google Scholar] [CrossRef]
- Franco, R.; Navarro, G.; Martinez-Pinilla, E. Antioxidant Defense Mechanisms in Erythrocytes and in the Central Nervous System. Antioxidants 2019, 8, 46. [Google Scholar] [CrossRef] [PubMed]
- Couto, N.; Wood, J.; Barber, J. The role of glutathione reductase and related enzymes on cellular redox homoeostasis network. Free Radic. Biol. Med. 2016, 95, 27–42. [Google Scholar] [CrossRef]
- Kowaltowski, A.J.; de Souza-Pinto, N.C.; Castilho, R.F.; Vercesi, A.E. Mitochondria and reactive oxygen species. Free Radic. Biol. Med. 2009, 47, 333–343. [Google Scholar] [CrossRef] [PubMed]
- Couto, N.; Malys, N.; Gaskell, S.J.; Barber, J. Partition and turnover of glutathione reductase from Saccharomyces cerevisiae: A proteomic approach. J. Proteome Res. 2013, 12, 2885–2894. [Google Scholar] [CrossRef]
- Chiang, H.S.; Maric, M. Lysosomal thiol reductase negatively regulates autophagy by altering glutathione synthesis and oxidation. Free Radic. Biol. Med. 2011, 51, 688–699. [Google Scholar] [CrossRef] [PubMed]
- Rahman, I.; Kode, A.; Biswas, S.K. Assay for quantitative determination of glutathione and glutathione disulfide levels using enzymatic recycling method. Nat. Protoc. 2006, 1, 3159–3165. [Google Scholar] [CrossRef] [PubMed]
- Bass, R.; Ruddock, L.W.; Klappa, P.; Freedman, R.B. A major fraction of endoplasmic reticulum-located glutathione is present as mixed disulfides with protein. J. Biol. Chem. 2004, 279, 5257–5262. [Google Scholar] [CrossRef]
- Griffith, O.W. Biologic and pharmacologic regulation of mammalian glutathione synthesis. Free Radic. Biol. Med. 1999, 27, 922–935. [Google Scholar] [CrossRef] [PubMed]
- Crook, T.R.; Souhami, R.L.; Whyman, G.D.; McLean, A.E. Glutathione depletion as a determinant of sensitivity of human leukemia cells to cyclophosphamide. Cancer Res. 1986, 46, 5035–5038. [Google Scholar]
- Meierjohann, S.; Walter, R.D.; Muller, S. Glutathione synthetase from Plasmodium falciparum. Biochem. J. 2002, 363, 833–838. [Google Scholar] [CrossRef] [PubMed]
- Zuhra, K.; Augsburger, F.; Majtan, T.; Szabo, C. Cystathionine-beta-Synthase: Molecular Regulation and Pharmacological Inhibition. Biomolecules 2020, 10, 697. [Google Scholar] [CrossRef]
- Mani, M.; Khaghani, S.; Gol Mohammadi, T.; Zamani, Z.; Azadmanesh, K.; Meshkani, R.; Pasalar, P.; Mostafavi, E. Activation of Nrf2-Antioxidant Response Element Mediated Glutamate Cysteine Ligase Expression in Hepatoma Cell line by Homocysteine. Hepat. Mon. 2013, 13, e8394. [Google Scholar] [CrossRef]
- Balla, G.; Jacob, H.S.; Eaton, J.W.; Belcher, J.D.; Vercellotti, G.M. Hemin: A possible physiological mediator of low density lipoprotein oxidation and endothelial injury. Arterioscler. Thromb. A J. Vasc. Biol. 1991, 11, 1700–1711. [Google Scholar] [CrossRef]
- Kumar, S.; Bandyopadhyay, U. Free heme toxicity and its detoxification systems in human. Toxicol. Lett. 2005, 157, 175–188. [Google Scholar] [CrossRef] [PubMed]
- Georgiou-Siafis, S.K.; Tsiftsoglou, A.S. Activation of KEAP1/NRF2 stress signaling involved in the molecular basis of hemin-induced cytotoxicity in human pro-erythroid K562 cells. Biochem. Pharmacol. 2020, 175, 113900. [Google Scholar] [CrossRef]
- Suzuki, T.; Yamamoto, M. Molecular basis of the Keap1-Nrf2 system. Free Radic. Biol. Med. 2015, 88, 93–100. [Google Scholar] [CrossRef]
- Atamna, H.; Ginsburg, H. Heme degradation in the presence of glutathione. A proposed mechanism to account for the high levels of non-heme iron found in the membranes of hemoglobinopathic red blood cells. J. Biol. Chem. 1995, 270, 24876–24883. [Google Scholar] [CrossRef]
- Laird, M.D.; Wakade, C.; Alleyne, C.H., Jr.; Dhandapani, K.M. Hemin-induced necroptosis involves glutathione depletion in mouse astrocytes. Free Radic. Biol. Med. 2008, 45, 1103–1114. [Google Scholar] [CrossRef] [PubMed]
- Richie, J.P., Jr.; Abraham, P.; Leutzinger, Y. Long-term stability of blood glutathione and cysteine in humans. Clin. Chem. 1996, 42, 1100–1105. [Google Scholar] [CrossRef] [PubMed]
- Perez, L.M.; Hooshmand, B.; Mangialasche, F.; Mecocci, P.; Smith, A.D.; Refsum, H.; Inzitari, M.; Fratiglioni, L.; Rizzuto, D.; Calderon-Larranaga, A. Glutathione Serum Levels and Rate of Multimorbidity Development in Older Adults. J. Gerontol. Ser. A Biol. Sci. Med. Sci. 2020, 75, 1089–1094. [Google Scholar] [CrossRef]
- van‘t Erve, T.J.; Wagner, B.A.; Ryckman, K.K.; Raife, T.J.; Buettner, G.R. The concentration of glutathione in human erythrocytes is a heritable trait. Free Radic. Biol. Med. 2013, 65, 742–749. [Google Scholar] [CrossRef]
- Lu, S.C. Glutathione synthesis. Biochim. Biophys. Acta 2013, 1830, 3143–3153. [Google Scholar] [CrossRef]
- Rahman, I.; MacNee, W. Lung glutathione and oxidative stress: Implications in cigarette smoke-induced airway disease. Am. J. Physiol. 1999, 277, L1067–L1088. [Google Scholar] [CrossRef]
- Dringen, R.; Hamprecht, B. Glutathione restoration as indicator for cellular metabolism of astroglial cells. Dev. Neurosci. 1998, 20, 401–407. [Google Scholar] [CrossRef]
- Vazquez-Meza, H.; Vilchis-Landeros, M.M.; Vazquez-Carrada, M.; Uribe-Ramirez, D.; Matuz-Mares, D. Cellular Compartmentalization, Glutathione Transport and Its Relevance in Some Pathologies. Antioxidants 2023, 12, 834. [Google Scholar] [CrossRef]
- Giustarini, D.; Milzani, A.; Dalle-Donne, I.; Rossi, R. Red blood cells as a physiological source of glutathione for extracellular fluids. Blood Cells Mol. Dis. 2008, 40, 174–179. [Google Scholar] [CrossRef]
- Montero, D.; Tachibana, C.; Rahr Winther, J.; Appenzeller-Herzog, C. Intracellular glutathione pools are heterogeneously concentrated. Redox Biol. 2013, 1, 508–513. [Google Scholar] [CrossRef]
- Ballatori, N.; Hammond, C.L.; Cunningham, J.B.; Krance, S.M.; Marchan, R. Molecular mechanisms of reduced glutathione transport: Role of the MRP/CFTR/ABCC and OATP/SLC21A families of membrane proteins. Toxicol. Appl. Pharmacol. 2005, 204, 238–255. [Google Scholar] [CrossRef]
- Bachhawat, A.K.; Thakur, A.; Kaur, J.; Zulkifli, M. Glutathione transporters. Biochim Biophys Acta 2013, 1830, 3154–3164. [Google Scholar] [CrossRef]
- Mari, M.; de Gregorio, E.; de Dios, C.; Roca-Agujetas, V.; Cucarull, B.; Tutusaus, A.; Morales, A.; Colell, A. Mitochondrial Glutathione: Recent Insights and Role in Disease. Antioxidants 2020, 9, 909. [Google Scholar] [CrossRef] [PubMed]
- Ballatori, N.; Rebbeor, J.F. Roles of MRP2 and oatp1 in hepatocellular export of reduced glutathione. Semin. Liver Dis. 1998, 18, 377–387. [Google Scholar] [CrossRef] [PubMed]
- Hanigan, M.H. Gamma-glutamyl transpeptidase: Redox regulation and drug resistance. Adv. Cancer Res. 2014, 122, 103–141. [Google Scholar] [CrossRef] [PubMed]
- Pizzorno, J. Glutathione! Integr. Med. A Clin. J. 2014, 13, 8–12. [Google Scholar]
- Averill-Bates, D.A. The antioxidant glutathione. Vitam. Horm. 2023, 121, 109–141. [Google Scholar] [CrossRef] [PubMed]
- Rushworth, G.F.; Megson, I.L. Existing and potential therapeutic uses for N-acetylcysteine: The need for conversion to intracellular glutathione for antioxidant benefits. Pharmacol. Ther. 2014, 141, 150–159. [Google Scholar] [CrossRef]
- Jones, C.M.; Lawrence, A.; Wardman, P.; Burkitt, M.J. Electron paramagnetic resonance spin trapping investigation into the kinetics of glutathione oxidation by the superoxide radical: Re-evaluation of the rate constant. Free Radic. Biol. Med. 2002, 32, 982–990. [Google Scholar] [CrossRef] [PubMed]
- Kirsch, M.; Lehnig, M.; Korth, H.G.; Sustmann, R.; de Groot, H. Inhibition of peroxynitrite-induced nitration of tyrosine by glutathione in the presence of carbon dioxide through both radical repair and peroxynitrate formation. Chemistry 2001, 7, 3313–3320. [Google Scholar] [CrossRef]
- Gibson, K.R.; Neilson, I.L.; Barrett, F.; Winterburn, T.J.; Sharma, S.; MacRury, S.M.; Megson, I.L. Evaluation of the antioxidant properties of N-acetylcysteine in human platelets: Prerequisite for bioconversion to glutathione for antioxidant and antiplatelet activity. J. Cardiovasc. Pharmacol. 2009, 54, 319–326. [Google Scholar] [CrossRef]
- Winterbourn, C.C.; Metodiewa, D. Reactivity of biologically important thiol compounds with superoxide and hydrogen peroxide. Free Radic. Biol. Med. 1999, 27, 322–328. [Google Scholar] [CrossRef] [PubMed]
- Pirie, N.W.; Pinhey, K.G. The Titration Curve of Glutathione. J. Biol. Chem. 1929, 84, 321–333. [Google Scholar] [CrossRef]
- Deponte, M. The Incomplete Glutathione Puzzle: Just Guessing at Numbers and Figures? Antioxid. Redox Signal. 2017, 27, 1130–1161. [Google Scholar] [CrossRef]
- Stone, J.R.; Yang, S. Hydrogen peroxide: A signaling messenger. Antioxid. Redox Signal. 2006, 8, 243–270. [Google Scholar] [CrossRef]
- Ribas, V.; Garcia-Ruiz, C.; Fernandez-Checa, J.C. Glutathione and mitochondria. Front. Pharmacol. 2014, 5, 151. [Google Scholar] [CrossRef]
- Hollins, D.L.; Suliman, H.B.; Piantadosi, C.A.; Carraway, M.S. Glutathione regulates susceptibility to oxidant-induced mitochondrial DNA damage in human lymphocytes. Free Radic. Biol. Med. 2006, 40, 1220–1226. [Google Scholar] [CrossRef]
- Cichoz-Lach, H.; Michalak, A. Oxidative stress as a crucial factor in liver diseases. World J. Gastroenterol. 2014, 20, 8082–8091. [Google Scholar] [CrossRef] [PubMed]
- Deponte, M. Glutathione catalysis and the reaction mechanisms of glutathione-dependent enzymes. Biochim. Biophys. Acta 2013, 1830, 3217–3266. [Google Scholar] [CrossRef]
- Guo, Q.; Packer, L. Ascorbate-dependent recycling of the vitamin E homologue Trolox by dihydrolipoate and glutathione in murine skin homogenates. Free Radic. Biol. Med. 2000, 29, 368–374. [Google Scholar] [CrossRef] [PubMed]
- Orlowski, M.; Meister, A. The γ-Glutamyl Cycle: A Possible Transport System for Amino Acids. Proc. Natl. Acad. Sci. USA 1970, 67, 1248–1255. [Google Scholar] [CrossRef] [PubMed]
- Poole, L.B. The basics of thiols and cysteines in redox biology and chemistry. Free Radic. Biol. Med. 2015, 80, 148–157. [Google Scholar] [CrossRef]
- Higdon, A.N.; Benavides, G.A.; Chacko, B.K.; Ouyang, X.; Johnson, M.S.; Landar, A.; Zhang, J.; Darley-Usmar, V.M. Hemin causes mitochondrial dysfunction in endothelial cells through promoting lipid peroxidation: The protective role of autophagy. Am. J. Physiol. Heart Circ. Physiol. 2012, 302, H1394–H1409. [Google Scholar] [CrossRef]
- Solar, I.; Muller-Eberhard, U.; Shviro, Y.; Shaklai, N. Long-term intercalation of residual hemin in erythrocyte membranes distorts the cell. Biochim. Biophys. Acta 1991, 1062, 51–58. [Google Scholar] [CrossRef] [PubMed]
- Dinkova-Kostova, A.T.; Kostov, R.V.; Canning, P. Keap1, the cysteine-based mammalian intracellular sensor for electrophiles and oxidants. Arch. Biochem. Biophys. 2017, 617, 84–93. [Google Scholar] [CrossRef] [PubMed]
- Aquilano, K.; Baldelli, S.; Ciriolo, M.R. Glutathione: New roles in redox signaling for an old antioxidant. Front. Pharmacol. 2014, 5, 196. [Google Scholar] [CrossRef]
- Jones, D.P.; Liang, Y. Measuring the poise of thiol/disulfide couples in vivo. Free Radic. Biol. Med. 2009, 47, 1329–1338. [Google Scholar] [CrossRef] [PubMed]
- Jones, D.P. Redox potential of GSH/GSSG couple: Assay and biological significance. Methods Enzymol. 2002, 348, 93–112. [Google Scholar] [CrossRef]
- Sen, C.K.; Packer, L. Antioxidant and redox regulation of gene transcription. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 1996, 10, 709–720. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, H.; Sugiyama, Y. Excretion of GSSG and glutathione conjugates mediated by MRP1 and cMOAT/MRP2. Semin. Liver Dis. 1998, 18, 359–376. [Google Scholar] [CrossRef] [PubMed]
- Musaogullari, A.; Chai, Y.C. Redox Regulation by Protein S-Glutathionylation: From Molecular Mechanisms to Implications in Health and Disease. Int. J. Mol. Sci. 2020, 21, 8113. [Google Scholar] [CrossRef]
- Yang, J.; Zhang, H.; Gong, W.; Liu, Z.; Wu, H.; Hu, W.; Chen, X.; Wang, L.; Wu, S.; Chen, C.; et al. S-Glutathionylation of human inducible Hsp70 reveals a regulatory mechanism involving the C-terminal alpha-helical lid. J. Biol. Chem. 2020, 295, 8302–8324. [Google Scholar] [CrossRef]
- Mohr, S.; Hallak, H.; de Boitte, A.; Lapetina, E.G.; Brune, B. Nitric oxide-induced S-glutathionylation and inactivation of glyceraldehyde-3-phosphate dehydrogenase. J. Biol. Chem. 1999, 274, 9427–9430. [Google Scholar] [CrossRef]
- Hwang, C.; Sinskey, A.J.; Lodish, H.F. Oxidized redox state of glutathione in the endoplasmic reticulum. Science 1992, 257, 1496–1502. [Google Scholar] [CrossRef]
- Tsunoda, S.; Avezov, E.; Zyryanova, A.; Konno, T.; Mendes-Silva, L.; Pinho Melo, E.; Harding, H.P.; Ron, D. Intact protein folding in the glutathione-depleted endoplasmic reticulum implicates alternative protein thiol reductants. Elife 2014, 3, e03421. [Google Scholar] [CrossRef]
- Georgiou-Siafis, S.K.; Samiotaki, M.K.; Demopoulos, V.J.; Panayotou, G.; Tsiftsoglou, A.S. Formation of novel N-acetylcysteine-hemin adducts abrogates hemin-induced cytotoxicity and suppresses the NRF2-driven stress response in human pro-erythroid K562 cells. Eur. J. Pharmacol. 2020, 880, 173077. [Google Scholar] [CrossRef] [PubMed]
- Georgiou-Siafis, S.K.; Samiotaki, M.K.; Demopoulos, V.J.; Panayotou, G.; Tsiftsoglou, A.S. Glutathione-Hemin/Hematin Adduct Formation to Disintegrate Cytotoxic Oxidant Hemin/Hematin in Human K562 Cells and Red Blood Cells’ Hemolysates: Impact of Glutathione on the Hemolytic Disorders and Homeostasis. Antioxidants 2022, 11, 1959. [Google Scholar] [CrossRef] [PubMed]
- Almazroo, O.A.; Miah, M.K.; Venkataramanan, R. Drug Metabolism in the Liver. Clin. Liver Dis. 2017, 21, 1–20. [Google Scholar] [CrossRef]
- Angelucci, F.; Baiocco, P.; Brunori, M.; Gourlay, L.; Morea, V.; Bellelli, A. Insights into the catalytic mechanism of glutathione S-transferase: The lesson from Schistosoma haematobium. Structure 2005, 13, 1241–1246. [Google Scholar] [CrossRef]
- Yu, S.J. Substrate specificity of glutathione S-transferases from the fall armyworm. Pestic. Biochem. Physiol. 2002, 74, 41–51. [Google Scholar] [CrossRef]
- Ji, X.; Johnson, W.W.; Sesay, M.A.; Dickert, L.; Prasad, S.M.; Ammon, H.L.; Armstrong, R.N.; Gilliland, G.L. Structure and function of the xenobiotic substrate binding site of a glutathione S-transferase as revealed by X-ray crystallographic analysis of product complexes with the diastereomers of 9-(S-glutathionyl)-10-hydroxy-9,10-dihydrophenanthrene. Biochemistry 1994, 33, 1043–1052. [Google Scholar] [CrossRef]
- Schultzen, O.; Naunyn, B. The behavior of benzene-derived hydrocarbons in the animal organism. du Bois-Reymond’s Arch Anat Physiol. 1867, 349. [Google Scholar]
- Snyder, R.; Kocsis, J.J. Current concepts of chronic benzene toxicity. CRC Crit. Rev. Toxicol. 1975, 3, 265–288. [Google Scholar] [CrossRef]
- Jollow, D.J.; Mitchell, J.R.; Zampaglione, N.; Gillette, J.R. Bromobenzene-induced liver necrosis. Protective role of glutathione and evidence for 3,4-bromobenzene oxide as the hepatotoxic metabolite. Pharmacology 1974, 11, 151–169. [Google Scholar] [CrossRef]
- Fakjian, N.; Buckpitt, A.R. Metabolism of bromobenzene to glutathione adducts in lung slices from mice treated with pneumotoxicants. Biochem. Pharmacol. 1984, 33, 1479–1486. [Google Scholar] [CrossRef]
- Lau, S.S.; Zannoni, V.G. Hepatic microsomal epoxidation of bromobenzene to phenols and its toxicological implication. Toxicol. Appl. Pharmacol. 1979, 50, 309–318. [Google Scholar] [CrossRef] [PubMed]
- Monks, T.J.; Butterworth, M.; Lau, S.S. The fate of benzene-oxide. Chem.-Biol. Interact. 2010, 184, 201–206. [Google Scholar] [CrossRef] [PubMed]
- Lau, S.S.; Hill, B.A.; Highet, R.J.; Monks, T.J. Sequential oxidation and glutathione addition to 1,4-benzoquinone: Correlation of toxicity with increased glutathione substitution. Mol. Pharmacol. 1988, 34, 829–836. [Google Scholar] [PubMed]
- Hill, B.A.; Kleiner, H.E.; Ryan, E.A.; Dulik, D.M.; Monks, T.J.; Lau, S.S. Identification of multi-S-substituted conjugates of hydroquinone by HPLC-coulometric electrode array analysis and mass spectroscopy. Chem. Res. Toxicol. 1993, 6, 459–469. [Google Scholar] [CrossRef] [PubMed]
- Bratton, S.B.; Lau, S.S.; Monks, T.J. Identification of quinol thioethers in bone marrow of hydroquinone/phenol-treated rats and mice and their potential role in benzene-mediated hematotoxicity. Chem. Res. Toxicol. 1997, 10, 859–865. [Google Scholar] [CrossRef]
- Phimister, A.J.; Lee, M.G.; Morin, D.; Buckpitt, A.R.; Plopper, C.G. Glutathione depletion is a major determinant of inhaled naphthalene respiratory toxicity and naphthalene metabolism in mice. Toxicol. Sci. Off. J. Soc. Toxicol. 2004, 82, 268–278. [Google Scholar] [CrossRef]
- Buckpitt, A.; Buonarati, M.; Avey, L.B.; Chang, A.M.; Morin, D.; Plopper, C.G. Relationship of cytochrome P450 activity to Clara cell cytotoxicity. II. Comparison of stereoselectivity of naphthalene epoxidation in lung and nasal mucosa of mouse, hamster, rat and rhesus monkey. J. Pharmacol. Exp. Ther. 1992, 261, 364–372. [Google Scholar]
- Shultz, M.A.; Choudary, P.V.; Buckpitt, A.R. Role of murine cytochrome P-450 2F2 in metabolic activation of naphthalene and metabolism of other xenobiotics. J. Pharmacol. Exp. Ther. 1999, 290, 281–288. [Google Scholar]
- Wu, Q.; Fang, J.; Li, S.; Wei, J.; Yang, Z.; Zhao, H.; Zhao, C.; Cai, Z. Interaction of bisphenol A 3,4-quinone metabolite with glutathione and ribonucleosides/deoxyribonucleosides in vitro. J. Hazard. Mater. 2017, 323, 195–202. [Google Scholar] [CrossRef]
- Hafezi, S.A.; Abdel-Rahman, W.M. The Endocrine Disruptor Bisphenol A (BPA) Exerts a Wide Range of Effects in Carcinogenesis and Response to Therapy. Curr. Mol. Pharmacol. 2019, 12, 230–238. [Google Scholar] [CrossRef]
- Stack, D.E.; Conrad, J.A.; Mahmud, B. Structural Identification and Kinetic Analysis of the in Vitro Products Formed by Reaction of Bisphenol A-3,4-quinone with N-Acetylcysteine and Glutathione. Chem. Res. Toxicol. 2018, 31, 81–87. [Google Scholar] [CrossRef]
- Qiu, S.X.; Yang, R.Z.; Gross, M.L. Synthesis and liquid chromatography/tandem mass spectrometric characterization of the adducts of bisphenol A o-quinone with glutathione and nucleotide monophosphates. Chem. Res. Toxicol. 2004, 17, 1038–1046. [Google Scholar] [CrossRef]
- Schmidt, J.; Kotnik, P.; Trontelj, J.; Knez, Z.; Masic, L.P. Bioactivation of bisphenol A and its analogs (BPF, BPAF, BPZ and DMBPA) in human liver microsomes. Toxicol. Vitr. Int. J. Publ. Assoc. BIBRA 2013, 27, 1267–1276. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.; Wagner, B.A.; Witmer, J.R.; Lehmler, H.J.; Buettner, G.R. Nonenzymatic displacement of chlorine and formation of free radicals upon the reaction of glutathione with PCB quinones. Proc. Natl. Acad. Sci. USA 2009, 106, 9725–9730. [Google Scholar] [CrossRef] [PubMed]
- Rao, D.N.; Takahashi, N.; Mason, R.P. Characterization of a glutathione conjugate of the 1,4-benzosemiquinone-free radical formed in rat hepatocytes. J. Biol. Chem. 1988, 263, 17981–17986. [Google Scholar] [CrossRef]
- Boersma, M.G.; Vervoort, J.; Szymusiak, H.; Lemanska, K.; Tyrakowska, B.; Cenas, N.; Segura-Aguilar, J.; Rietjens, I.M. Regioselectivity and reversibility of the glutathione conjugation of quercetin quinone methide. Chem. Res. Toxicol. 2000, 13, 185–191. [Google Scholar] [CrossRef]
- Li, C.; Zhang, W.J.; Choi, J.; Frei, B. Quercetin affects glutathione levels and redox ratio in human aortic endothelial cells not through oxidation but formation and cellular export of quercetin-glutathione conjugates and upregulation of glutamate-cysteine ligase. Redox Biol. 2016, 9, 220–228. [Google Scholar] [CrossRef]
- Allameh, A.; Farahani, M.; Zarghi, A. Kinetic studies of aflatoxin B1-glutathione conjugate formation in liver and kidneys of adult and weanling rats. Mech. Ageing Dev. 2000, 115, 73–83. [Google Scholar] [CrossRef]
- Jowsey, I.R.; Jiang, Q.; Itoh, K.; Yamamoto, M.; Hayes, J.D. Expression of the aflatoxin B1-8,9-epoxide-metabolizing murine glutathione S-transferase A3 subunit is regulated by the Nrf2 transcription factor through an antioxidant response element. Mol. Pharmacol. 2003, 64, 1018–1028. [Google Scholar] [CrossRef] [PubMed]
- Bateman, R.; Rauh, D.; Shokat, K.M. Glutathione traps formaldehyde by formation of a bicyclo[4.4.1]undecane adduct. Org. Biomol. Chem. 2007, 5, 3363–3367. [Google Scholar] [CrossRef]
- Navarro, S.L.; Li, F.; Lampe, J.W. Mechanisms of action of isothiocyanates in cancer chemoprevention: An update. Food Funct. 2011, 2, 579–587. [Google Scholar] [CrossRef] [PubMed]
- Bruggeman, I.M.; Temmink, J.H.; van Bladeren, P.J. Glutathione- and cysteine-mediated cytotoxicity of allyl and benzyl isothiocyanate. Toxicol. Appl. Pharmacol. 1986, 83, 349–359. [Google Scholar] [CrossRef] [PubMed]
- Omar, K.; Grant, M.H.; Henderson, C.; Watson, D.G. The complex degradation and metabolism of quercetin in rat hepatocyte incubations. Xenobiotica Fate Foreign Compd. Biol. Syst. 2014, 44, 1074–1082. [Google Scholar] [CrossRef]
- Brusewitz, G.; Cameron, B.D.; Chasseaud, L.F.; Gorler, K.; Hawkins, D.R.; Koch, H.; Mennicke, W.H. The metabolism of benzyl isothiocyanate and its cysteine conjugate. Biochem. J. 1977, 162, 99–107. [Google Scholar] [CrossRef]
- Hodgman, M.J.; Garrard, A.R. A review of acetaminophen poisoning. Crit. Care Clin. 2012, 28, 499–516. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, J.R.; Jollow, D.J.; Potter, W.Z.; Davis, D.C.; Gillette, J.R.; Brodie, B.B. Acetaminophen-induced hepatic necrosis. I. Role of Drug Metabolism. J. Pharmacol. Exp. Ther. 1973, 187, 185. [Google Scholar]
- Davis, D.C.; Potter, W.Z.; Jollow, D.J.; Mitchell, J.R. Species differences in hepatic glutathione depletion, covalent binding and hepatic necrosis after acetaminophen. Life Sci. 1974, 14, 2099–2109. [Google Scholar] [CrossRef]
- Hinson, J.A.; Mitchell, J.R.; Jollow, D.J. Microsomal N-hydroxylation of p-Chloroacetanilide. Mol. Pharmacol. 1975, 11, 462. [Google Scholar]
- Mitchell, J.R.; Jollow, D.J.; Potter, W.Z.; Gillette, J.R.; Brodie, B.B. Acetaminophen-induced hepatic necrosis. IV. Protective role of glutathione. J. Pharmacol. Exp. Ther. 1973, 187, 211–217. [Google Scholar]
- Albano, E.; Rundgren, M.; Harvison, P.J.; Nelson, S.D.; Moldéus, P. Mechanisms of N-acetyl-p-benzoquinone imine cytotoxicity. Mol. Pharmacol. 1985, 28, 306–311. [Google Scholar]
- Allameh, A.; Alikhani, N. Acetaminophen-glutathione conjugate formation in a coupled cytochrome P-450-glutathione S-transferase assay system mediated by subcellular preparations from adult and weanling rat tissues. Toxicol. Vitr. Int. J. Publ. Assoc. BIBRA 2002, 16, 637–641. [Google Scholar] [CrossRef]
- Coles, B.; Wilson, I.; Wardman, P.; Hinson, J.A.; Nelson, S.D.; Ketterer, B. The spontaneous and enzymatic reaction of N-acetyl-p-benzoquinonimine with glutathione: A stopped-flow kinetic study. Arch. Biochem. Biophys. 1988, 264, 253–260. [Google Scholar] [CrossRef] [PubMed]
- Potega, A. Glutathione-Mediated Conjugation of Anticancer Drugs: An Overview of Reaction Mechanisms and Biological Significance for Drug Detoxification and Bioactivation. Molecules 2022, 27, 5252. [Google Scholar] [CrossRef]
- Fuertes, M.A.; Castilla, J.; Alonso, C.; Perez, J.M. Novel concepts in the development of platinum antitumor drugs. Curr. Med. Chem. Anti-Cancer Agents 2002, 2, 539–551. [Google Scholar] [CrossRef] [PubMed]
- Ishikawa, T.; Ali-Osman, F. Glutathione-associated cis-diamminedichloroplatinum(II) metabolism and ATP-dependent efflux from leukemia cells. Molecular characterization of glutathione-platinum complex and its biological significance. J. Biol. Chem. 1993, 268, 20116–20125. [Google Scholar] [CrossRef] [PubMed]
- Nagar, R.; Khan, A.R.; Poonia, A.; Mishra, P.K.; Singh, S. Metabolism of cisplatin in the organs of Rattus norvegicus: Role of Glutathione S-transferase P1. Eur. J. Drug Metab. Pharmacokinet. 2015, 40, 45–51. [Google Scholar] [CrossRef]
- Kasherman, Y.; Sturup, S.; Gibson, D. Is glutathione the major cellular target of cisplatin? A study of the interactions of cisplatin with cancer cell extracts. J. Med. Chem. 2009, 52, 4319–4328. [Google Scholar] [CrossRef]
- Karpusas, M.; Axarli, I.; Chiniadis, L.; Papakyriakou, A.; Bethanis, K.; Scopelitou, K.; Clonis, Y.D.; Labrou, N.E. The interaction of the chemotherapeutic drug chlorambucil with human glutathione transferase A1-1: Kinetic and structural analysis. PLoS ONE 2013, 8, e56337. [Google Scholar] [CrossRef]
- Perry, C.S.; Liu, X.; Lund, L.G.; Whitman, C.P.; Kehrer, J.P. Differential toxicities of cyclophosphamide and its glutathione metabolites to A549 cells. Toxicol. Vitr. Int. J. Publ. Assoc. BIBRA 1995, 9, 21–26. [Google Scholar] [CrossRef]
- Sharma, M.; Tomasz, M. Conjugation of glutathione and other thiols with bioreductively activated mitomycin C. Effect of thiols on the reductive activation rate. Chem. Res. Toxicol. 1994, 7, 390–400. [Google Scholar] [CrossRef]
- Jackson, K.D.; Durandis, R.; Vergne, M.J. Role of Cytochrome P450 Enzymes in the Metabolic Activation of Tyrosine Kinase Inhibitors. Int. J. Mol. Sci. 2018, 19, 2376. [Google Scholar] [CrossRef]
- Panchuk, R.R.; Lehka, L.V.; Terenzi, A.; Matselyukh, B.P.; Rohr, J.; Jha, A.K.; Downey, T.; Kril, I.J.; Herbacek, I.; van Schoonhoven, S.; et al. Rapid generation of hydrogen peroxide contributes to the complex cell death induction by the angucycline antibiotic landomycin E. Free Radic. Biol. Med. 2017, 106, 134–147. [Google Scholar] [CrossRef]
- Terenzi, A.; La Franca, M.; van Schoonhoven, S.; Panchuk, R.; Martinez, A.; Heffeter, P.; Gober, R.; Pirker, C.; Vician, P.; Kowol, C.R.; et al. Landomycins as glutathione-depleting agents and natural fluorescent probes for cellular Michael adduct-dependent quinone metabolism. Commun. Chem. 2021, 4, 162. [Google Scholar] [CrossRef] [PubMed]
- IARC Working Group on the Evaluation of Carcinogenic Risks to Humans. Pharmaceuticals. Lyon (FR): International Agency for Research on Cancer; (IARC Monographs on the Evaluation of Carcinogenic Risks to Humans, No. 100A.) Azathioprine. Available online: https://www.ncbi.nlm.nih.gov/books/NBK304317/ (accessed on 25 August 2023).
- Lennard, L. The clinical pharmacology of 6-mercaptopurine. Eur. J. Clin. Pharmacol. 1992, 43, 329–339. [Google Scholar] [CrossRef]
- Chalmers, A.H. Studies on the mechanism of formation of 5-mercapto-1-methyl-4-nitroimidazole, a metabolite of the immunosuppressive drug azathioprine. Biochem. Pharmacol. 1974, 23, 1891–1901. [Google Scholar] [CrossRef]
- Tiede, I.; Fritz, G.; Strand, S.; Poppe, D.; Dvorsky, R.; Strand, D.; Lehr, H.A.; Wirtz, S.; Becker, C.; Atreya, R.; et al. CD28-dependent Rac1 activation is the molecular target of azathioprine in primary human CD4+ T lymphocytes. J. Clin. Investig. 2003, 111, 1133–1145. [Google Scholar] [CrossRef]
- Subramanian, R.; Fang, X.; Prueksaritanont, T. Structural characterization of in vivo rat glutathione adducts and a hydroxylated metabolite of simvastatin hydroxy acid. Drug Metab. Dispos. Biol. Fate Chem. 2002, 30, 225–230. [Google Scholar] [CrossRef]
- Huang, C.; Fischer, C.; Machacek, M.R.; Bogen, S.; Biftu, T.; Huang, X.; Reutershan, M.H.; Otte, R.; Hong, Q.; Wu, Z.; et al. Diminishing GSH-Adduct Formation of Tricyclic Diazepine-based Mutant IDH1 Inhibitors. ACS Med. Chem. Lett. 2022, 13, 734–741. [Google Scholar] [CrossRef]
- Takenoshita, R.; Toki, S. [New aspects of hexobarbital metabolism: Stereoselective metabolism, new metabolic pathway via GSH conjugation, and 3-hydroxyhexobarbital dehydrogenases]. Yakugaku Zasshi J. Pharm. Soc. Jpn. 2004, 124, 857–871. [Google Scholar] [CrossRef] [PubMed]
- Gallagher, R.E. Arsenic--new life for an old potion. N. Engl. J. Med. 1998, 339, 1389–1391. [Google Scholar] [CrossRef] [PubMed]
- Leslie, E.M. Arsenic-glutathione conjugate transport by the human multidrug resistance proteins (MRPs/ABCCs). J. Inorg. Biochem. 2012, 108, 141–149. [Google Scholar] [CrossRef]
- Jiang, Y.; Shen, X.; Zhi, F.; Wen, Z.; Gao, Y.; Xu, J.; Yang, B.; Bai, Y. An overview of arsenic trioxide-involved combined treatment algorithms for leukemia: Basic concepts and clinical implications. Cell Death Discov. 2023, 9, 266. [Google Scholar] [CrossRef] [PubMed]
- Parker, L.J.; Bocedi, A.; Ascher, D.B.; Aitken, J.B.; Harris, H.H.; Lo Bello, M.; Ricci, G.; Morton, C.J.; Parker, M.W. Glutathione transferase P1-1 as an arsenic drug-sequestering enzyme. Protein Sci. A Publ. Protein Soc. 2017, 26, 317–326. [Google Scholar] [CrossRef] [PubMed]
- Lassila, T.; Rousu, T.; Mattila, S.; Chesne, C.; Pelkonen, O.; Turpeinen, M.; Tolonen, A. Formation of GSH-trapped reactive metabolites in human liver microsomes, S9 fraction, HepaRG-cells, and human hepatocytes. J. Pharm. Biomed. Anal. 2015, 115, 345–351. [Google Scholar] [CrossRef] [PubMed]
- Gilliland, R.A.; Möller, C.; DeCaprio, A.P. LC-MS/MS based detection and characterization of covalent glutathione modifications formed by reactive drug of abuse metabolites. Xenobiotica Fate Foreign Compd. Biol. Syst. 2019, 49, 778–790. [Google Scholar] [CrossRef]
- Gan, J.; Ruan, Q.; He, B.; Zhu, M.; Shyu, W.C.; Humphreys, W.G. In vitro screening of 50 highly prescribed drugs for thiol adduct formation--comparison of potential for drug-induced toxicity and extent of adduct formation. Chem. Res. Toxicol. 2009, 22, 690–698. [Google Scholar] [CrossRef]
- Ruan, Q.; Zhu, M. Investigation of bioactivation of ticlopidine using linear ion trap/orbitrap mass spectrometry and an improved mass defect filtering technique. Chem. Res. Toxicol. 2010, 23, 909–917. [Google Scholar] [CrossRef]
- Schneider, K.J.; DeCaprio, A.P. Covalent thiol adducts arising from reactive intermediates of cocaine biotransformation. Chem. Res. Toxicol. 2013, 26, 1755–1764. [Google Scholar] [CrossRef]
- Moden, O.; Mannervik, B. Glutathione transferases in the bioactivation of azathioprine. Adv. Cancer Res. 2014, 122, 199–244. [Google Scholar] [CrossRef]
- Hassan, G.S. Menadione. In Profiles of Drug Substances, Excipients and Related Methodology; Elsevier: Amsterdam, The Netherlands, 2013; Volume 38, pp. 227–313. [Google Scholar] [CrossRef]
- Di Monte, D.; Ross, D.; Bellomo, G.; Eklow, L.; Orrenius, S. Alterations in intracellular thiol homeostasis during the metabolism of menadione by isolated rat hepatocytes. Arch. Biochem. Biophys. 1984, 235, 334–342. [Google Scholar] [CrossRef]
- Nickerson, W.J.; Falcone, G.; Strauss, G. Studies on Quinone-Thioethers. I. Mechanism of Formation and Properties of Thiodione. Biochemistry 1963, 2, 537–543. [Google Scholar] [CrossRef]
- Ross, D.; Thor, H.; Orrenius, S.; Moldeus, P. Interaction of menadione (2-methyl-1,4-naphthoquinone) with glutathione. Chem.-Biol. Interact. 1985, 55, 177–184. [Google Scholar] [CrossRef] [PubMed]
- Shearer, M.J.; Newman, P. Recent trends in the metabolism and cell biology of vitamin K with special reference to vitamin K cycling and MK-4 biosynthesis. J. Lipid Res. 2014, 55, 345–362. [Google Scholar] [CrossRef] [PubMed]
- Haenen, H.E.; Spenkelink, A.; Teunissen, C.; Temmink, J.H.; Koeman, J.H.; van Bladeren, P.J. Transport and metabolism of glutathione conjugates of menadione and ethacrynic acid in confluent monolayers of rat renal proximal tubular cells. Toxicology 1996, 112, 117–130. [Google Scholar] [CrossRef] [PubMed]
- Cavalieri, E.; Frenkel, K.; Liehr, J.G.; Rogan, E.; Roy, D. Estrogens as endogenous genotoxic agents--DNA adducts and mutations. JNCI Monogr. 2000, 2000, 75–93. [Google Scholar] [CrossRef] [PubMed]
- Liehr, J.G. Is estradiol a genotoxic mutagenic carcinogen? Endocr. Rev. 2000, 21, 40–54. [Google Scholar] [CrossRef]
- Cavalieri, E.L.; Stack, D.E.; Devanesan, P.D.; Todorovic, R.; Dwivedy, I.; Higginbotham, S.; Johansson, S.L.; Patil, K.D.; Gross, M.L.; Gooden, J.K.; et al. Molecular origin of cancer: Catechol estrogen-3,4-quinones as endogenous tumor initiators. Proc. Natl. Acad. Sci. USA 1997, 94, 10937–10942. [Google Scholar] [CrossRef]
- Tsutsui, T.; Suzuki, N.; Fukuda, S.; Sato, M.; Maizumi, H.; McLachlan, J.A.; Barrett, J.C. 17beta-Estradiol-induced cell transformation and aneuploidy of Syrian hamster embryo cells in culture. Carcinogenesis 1987, 8, 1715–1719. [Google Scholar] [CrossRef]
- Cao, K.; Stack, D.E.; Ramanathan, R.; Gross, M.L.; Rogan, E.G.; Cavalieri, E.L. Synthesis and structure elucidation of estrogen quinones conjugated with cysteine, N-acetylcysteine, and glutathione. Chem. Res. Toxicol. 1998, 11, 909–916. [Google Scholar] [CrossRef]
- Rogan, E.G.; Badawi, A.F.; Devanesan, P.D.; Meza, J.L.; Edney, J.A.; West, W.W.; Higginbotham, S.M.; Cavalieri, E.L. Relative imbalances in estrogen metabolism and conjugation in breast tissue of women with carcinoma: Potential biomarkers of susceptibility to cancer. Carcinogenesis 2003, 24, 697–702. [Google Scholar] [CrossRef]
- Spencer, J.P.; Jenner, P.; Daniel, S.E.; Lees, A.J.; Marsden, D.C.; Halliwell, B. Conjugates of catecholamines with cysteine and GSH in Parkinson’s disease: Possible mechanisms of formation involving reactive oxygen species. J. Neurochem. 1998, 71, 2112–2122. [Google Scholar] [CrossRef]
- Zhang, F.; Dryhurst, G. Reactions of Cysteine and Cysteinyl Derivatives with Dopamine-o-quinone and Further Insights into the Oxidation Chemistry of 5-S-Cysteinyldopamine: Potential Relevance to Idiopathic Parkinson′s Disease. Bioorg. Chem. 1995, 23, 193–216. [Google Scholar] [CrossRef]
- Norman, A.W. Chapter 8–Eicosanoids. Available online: https://www.semanticscholar.org/paper/Chapter-8-%E2%80%93-Eicosanoids-Norman/f862586410a346b09933ea4d212b23130eb1b5ed (accessed on 29 September 2023).
- Murphy, R.C.; Zarini, S. Glutathione adducts of oxyeicosanoids. Prostaglandins Other Lipid Mediat. 2002, 68–69, 471–482. [Google Scholar] [CrossRef]
- Golub, M.S.; Zia, P.K.; Horton, R. Metabolism of prostaglandins A1 and A2 by human whole blood. Prostaglandins 1974, 8, 13–20. [Google Scholar] [CrossRef]
- Cagen, L.M.; Fales, H.M.; Pisano, J.J. Formation of glutathione conjugates of prostaglandin A1 in human red blood cells. J. Biol. Chem. 1976, 251, 6550–6554. [Google Scholar] [CrossRef] [PubMed]
- Cagen, L.M.; Pisano, J.J.; Ketley, J.N.; Habig, W.H.; Jakoby, W.B. The conjugation of prostaglandin A1 and glutathione catalyzed by homogeneous glutathione s-transferases from human and rat liver. Biochim. Biophys. Acta 1975, 398, 205–208. [Google Scholar] [CrossRef]
- Gross, K.B.; Gillis, C.N. The formation of prostaglandin A1-glutathione adduct in the lung. Biochim. Biophys. Acta 1976, 450, 266–268. [Google Scholar] [CrossRef] [PubMed]
- Parker, J.; Ankel, H. Formation of a prostaglandin A2-glutathione conjugate in L1210 mouse leukemia cells. Biochem. Pharmacol. 1992, 43, 1053–1060. [Google Scholar] [CrossRef] [PubMed]
- Cagen, L.M.; Pisano, J.J. The glutathione conjugate of prostaglandin A1 is a better substrate than prostaglandin E for partially purified avian prostaglandin E 9-ketoreductase. Biochim. Biophys. Acta 1979, 573, 547–551. [Google Scholar] [CrossRef]
- Heasley, L.E.; Brunton, L.L. Prostaglandin A1 metabolism and inhibition of cyclic AMP extrusion by avian erythrocytes. J. Biol. Chem. 1985, 260, 11514–11519. [Google Scholar] [CrossRef] [PubMed]
- Dahlen, S.E.; Hedqvist, P.; Hammarstrom, S.; Samuelsson, B. Leukotrienes are potent constrictors of human bronchi. Nature 1980, 288, 484–486. [Google Scholar] [CrossRef]
- Marom, Z.; Shelhamer, J.H.; Bach, M.K.; Morton, D.R.; Kaliner, M. Slow-reacting substances, leukotrienes C4 and D4, increase the release of mucus from human airways in vitro. Am. Rev. Respir. Dis. 1982, 126, 449–451. [Google Scholar]
- Lam, B.K. Leukotriene C(4) synthase. Prostaglandins Leukot. Essent. Fat. Acids 2003, 69, 111–116. [Google Scholar] [CrossRef]
- Martinez Molina, D.; Wetterholm, A.; Kohl, A.; McCarthy, A.A.; Niegowski, D.; Ohlson, E.; Hammarberg, T.; Eshaghi, S.; Haeggstrom, J.Z.; Nordlund, P. Structural basis for synthesis of inflammatory mediators by human leukotriene C4 synthase. Nature 2007, 448, 613–616. [Google Scholar] [CrossRef]
- Esterbauer, H.; Schaur, R.J.; Zollner, H. Chemistry and biochemistry of 4-hydroxynonenal, malonaldehyde and related aldehydes. Free Radic. Biol. Med. 1991, 11, 81–128. [Google Scholar] [CrossRef]
- Poli, G.; Schaur, R.J.; Siems, W.G.; Leonarduzzi, G. 4-hydroxynonenal: A membrane lipid oxidation product of medicinal interest. Med. Res. Rev. 2008, 28, 569–631. [Google Scholar] [CrossRef]
- Shoeb, M.; Ansari, N.H.; Srivastava, S.K.; Ramana, K.V. 4-Hydroxynonenal in the pathogenesis and progression of human diseases. Curr. Med. Chem. 2014, 21, 230–237. [Google Scholar] [CrossRef] [PubMed]
- Balogh, L.M.; Atkins, W.M. Interactions of glutathione transferases with 4-hydroxynonenal. Drug Metab. Rev. 2011, 43, 165–178. [Google Scholar] [CrossRef]
- Falletti, O.; Cadet, J.; Favier, A.; Douki, T. Trapping of 4-hydroxynonenal by glutathione efficiently prevents formation of DNA adducts in human cells. Free Radic. Biol. Med. 2007, 42, 1258–1269. [Google Scholar] [CrossRef] [PubMed]
- De Maria, F.; Pedersen, J.Z.; Caccuri, A.M.; Antonini, G.; Turella, P.; Stella, L.; Lo Bello, M.; Federici, G.; Ricci, G. The specific interaction of dinitrosyl-diglutathionyl-iron complex, a natural NO carrier, with the glutathione transferase superfamily: Suggestion for an evolutionary pressure in the direction of the storage of nitric oxide. J. Biol. Chem. 2003, 278, 42283–42293. [Google Scholar] [CrossRef]
- Bocedi, A.; Fabrini, R.; Lo Bello, M.; Caccuri, A.M.; Federici, G.; Mannervik, B.; Cornish-Bowden, A.; Ricci, G. Evolution of Negative Cooperativity in Glutathione Transferase Enabled Preservation of Enzyme Function. J. Biol. Chem. 2016, 291, 26739–26749. [Google Scholar] [CrossRef] [PubMed]
- Cavalieri, E.L.; Rogan, E.G. Depurinating estrogen-DNA adducts, generators of cancer initiation: Their minimization leads to cancer prevention. Clin. Transl. Med. 2016, 5, 12. [Google Scholar] [CrossRef] [PubMed]
- Balestri, F.; Barracco, V.; Renzone, G.; Tuccinardi, T.; Pomelli, C.S.; Cappiello, M.; Lessi, M.; Rotondo, R.; Bellina, F.; Scaloni, A.; et al. Stereoselectivity of Aldose Reductase in the Reduction of Glutathionyl-Hydroxynonanal Adduct. Antioxidants 2019, 8, 502. [Google Scholar] [CrossRef]
- Warnke, M.M.; Wanigasekara, E.; Singhal, S.S.; Singhal, J.; Awasthi, S.; Armstrong, D.W. The determination of glutathione-4-hydroxynonenal (GSHNE), E-4-hydroxynonenal (HNE), and E-1-hydroxynon-2-en-4-one (HNO) in mouse liver tissue by LC-ESI-MS. Anal. Bioanal. Chem. 2008, 392, 1325–1333. [Google Scholar] [CrossRef] [PubMed]
- Hanna, P.E.; Anders, M.W. The mercapturic acid pathway. Crit. Rev. Toxicol. 2019, 49, 819–929. [Google Scholar] [CrossRef] [PubMed]
- Veiga-da-Cunha, M.; Tyteca, D.; Stroobant, V.; Courtoy, P.J.; Opperdoes, F.R.; Van Schaftingen, E. Molecular identification of NAT8 as the enzyme that acetylates cysteine S-conjugates to mercapturic acids. J. Biol. Chem. 2010, 285, 18888–18898. [Google Scholar] [CrossRef]
- Scholl, P.F.; Musser, S.M.; Groopman, J.D. Synthesis and characterization of aflatoxin B1 mercapturic acids and their identification in rat urine. Chem. Res. Toxicol. 1997, 10, 1144–1151. [Google Scholar] [CrossRef]
- Pombrio, J.M.; Giangreco, A.; Li, L.; Wempe, M.F.; Anders, M.W.; Sweet, D.H.; Pritchard, J.B.; Ballatori, N. Mercapturic acids (N-acetylcysteine S-conjugates) as endogenous substrates for the renal organic anion transporter-1. Mol. Pharmacol. 2001, 60, 1091–1099. [Google Scholar] [CrossRef]
- Goncalves-Dias, C.; Morello, J.; Semedo, V.; Correia, M.J.; Coelho, N.R.; Monteiro, E.C.; Antunes, A.M.M.; Pereira, S.A. The mercapturomic profile of health and non-communicable diseases. High-Throughput 2019, 8, 10. [Google Scholar] [CrossRef]
- Tate, S.S.; Meister, A. gamma-Glutamyl transpeptidase from kidney. Methods Enzymol. 1985, 113, 400–419. [Google Scholar] [CrossRef]
- Mikov, M. The metabolism of drugs by the gut flora. Eur. J. Drug Metab. Pharmacokinet. 1994, 19, 201–207. [Google Scholar] [CrossRef]
- Ma, L.; Landsiedel, R.; Kuhlow, A.; Engst, W.; Seidel, A.; Glatt, H. Detection of Mercapturic Acids and Nucleoside Adducts in Blood, Urine and Feces of Rats Treated with Metabolites of Methylpyrene. Polycycl. Aromat. Compd. 2000, 21, 135–149. [Google Scholar] [CrossRef]
- Cooper, A.J.; Pinto, J.T. Cysteine S-conjugate beta-lyases. Amino Acids 2006, 30, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Cooper, A.J.L.; Hanigan, M.H. Enzymes Involved in Processing Glutathione Conjugates. Compr. Toxicol. 2010, 323–366. [Google Scholar] [CrossRef]
- Anders, M.W. Glutathione-dependent bioactivation of haloalkanes and haloalkenes. Drug Metab. Rev. 2004, 36, 583–594. [Google Scholar] [CrossRef] [PubMed]
- Tsiftsoglou, A.S.; Tsamadou, A.I.; Papadopoulou, L.C. Heme as key regulator of major mammalian cellular functions: Molecular, cellular, and pharmacological aspects. Pharmacol. Ther. 2006, 111, 327–345. [Google Scholar] [CrossRef]
- Chou, A.C.; Fitch, C.D. Mechanism of hemolysis induced by ferriprotoporphyrin IX. J. Clin. Investig. 1981, 68, 672–677. [Google Scholar] [CrossRef] [PubMed]
- Shviro, Y.; Shaklai, N. Glutathione as a scavenger of free hemin. A mechanism of preventing red cell membrane damage. Biochem. Pharmacol. 1987, 36, 3801–3807. [Google Scholar] [CrossRef]
- Sahini, V.E.; Dumitrescu, M.; Volanschi, E.; Birla, L.; Diaconu, C. Spectral and interferometrical study of the interaction of haemin with glutathione. Biophys. Chem. 1996, 58, 245–253. [Google Scholar] [CrossRef]
- Raftos, J.E.; Whillier, S.; Kuchel, P.W. Glutathione synthesis and turnover in the human erythrocyte: Alignment of a model based on detailed enzyme kinetics with experimental data. J. Biol. Chem. 2010, 285, 23557–23567. [Google Scholar] [CrossRef]
- Dass, P.D.; Bermes, E.W., Jr.; Holmes, E.W. Renal and hepatic output of glutathione in plasma and whole blood. Biochim. Biophys. Acta 1992, 1156, 99–102. [Google Scholar] [CrossRef]
- Moller, M.N.; Orrico, F.; Villar, S.F.; Lopez, A.C.; Silva, N.; Donze, M.; Thomson, L.; Denicola, A. Oxidants and Antioxidants in the Redox Biochemistry of Human Red Blood Cells. ACS Omega 2023, 8, 147–168. [Google Scholar] [CrossRef]
- Kinoshita, A.; Nakayama, Y.; Kitayama, T.; Tomita, M. Simulation study of methemoglobin reduction in erythrocytes. Differential contributions of two pathways to tolerance to oxidative stress. FEBS J. 2007, 274, 1449–1458. [Google Scholar] [CrossRef]
- Njalsson, R.; Ristoff, E.; Carlsson, K.; Winkler, A.; Larsson, A.; Norgren, S. Genotype, enzyme activity, glutathione level, and clinical phenotype in patients with glutathione synthetase deficiency. Hum. Genet. 2005, 116, 384–389. [Google Scholar] [CrossRef]
- Hirono, A.; Iyori, H.; Sekine, I.; Ueyama, J.; Chiba, H.; Kanno, H.; Fujii, H.; Miwa, S. Three cases of hereditary nonspherocytic hemolytic anemia associated with red blood cell glutathione deficiency. Blood 1996, 87, 2071–2074. [Google Scholar] [CrossRef]
- Rossi, R.; Milzani, A.; Dalle-Donne, I.; Giannerini, F.; Giustarini, D.; Lusini, L.; Colombo, R.; Di Simplicio, P. Different metabolizing ability of thiol reactants in human and rat blood: Biochemical and pharmacological implications. J. Biol. Chem. 2001, 276, 7004–7010. [Google Scholar] [CrossRef]
- Cortese-Krott, M.M. The Reactive Species Interactome in Red Blood Cells: Oxidants, Antioxidants, and Molecular Targets. Antioxidants 2023, 12, 1736. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.-T.; Weiss, L. The Role of the Sinus Wall in the Passage of Erythrocytes Through the Spleen. Blood 1973, 41, 529–537. [Google Scholar] [CrossRef]
- Hultquist, D.E.; Passon, P.G. Catalysis of methaemoglobin reduction by erythrocyte cytochrome B5 and cytochrome B5 reductase. Nat. New Biol. 1971, 229, 252–254. [Google Scholar] [CrossRef]
- Mansouri, A.; Lurie, A.A. Concise review: Methemoglobinemia. Am. J. Hematol. 1993, 42, 7–12. [Google Scholar] [CrossRef]
- Werner, C.; Doenst, T.; Schwarzer, M. Chapter 4–Metabolic Pathways and Cycles. In The Scientist’s Guide to Cardiac Metabolism; Schwarzer, M., Doenst, T., Eds.; Academic Press: Boston, MA, USA, 2016; pp. 39–55. [Google Scholar] [CrossRef]
- Capoluongo, E.; Giardina, B.; Minucci, A. Glucose-6-Phosphate Dehydrogenase (G6PD) Deficiency. In Brenner’s Encyclopedia of Genetics; Elsevier: Amsterdam, The Netherlands, 2013; pp. 340–342. [Google Scholar] [CrossRef]
- Kassa, T.; Jana, S.; Meng, F.; Alayash, A.I. Differential heme release from various hemoglobin redox states and the upregulation of cellular heme oxygenase-1. FEBS Open Bio 2016, 6, 876–884. [Google Scholar] [CrossRef] [PubMed]
- Ponka, P.; Sheftel, A.D.; English, A.M.; Scott Bohle, D.; Garcia-Santos, D. Do Mammalian Cells Really Need to Export and Import Heme? Trends Biochem. Sci. 2017, 42, 395–406. [Google Scholar] [CrossRef] [PubMed]
- Cannon, J.B.; Kuo, F.S.; Pasternack, R.F.; Wong, N.M.; Muller-Eberhard, U. Kinetics of the interaction of hemin liposomes with heme binding proteins. Biochemistry 1984, 23, 3715–3721. [Google Scholar] [CrossRef] [PubMed]
- Tipping, E.; Ketterer, B.; Christodoulides, L. Interactions of small molecules with phospholipid bilayers. Binding to egg phosphatidylcholine of some organic anions (bromosulphophthalein, oestrone sulphate, haem and bilirubin) that bind to ligandin and aminoazo-dye-binding protein A. Biochem. J. 1979, 180, 327–337. [Google Scholar] [CrossRef] [PubMed]
- Chiu, D.T.; Huang, T.Y.; Hung, I.J.; Wei, J.S.; Liu, T.Z.; Stern, A. Hemin-induced membrane sulfhydryl oxidation: Possible involvement of thiyl radicals. Free Radic. Res. 1997, 27, 55–62. [Google Scholar] [CrossRef]
- Rifkind, J.M.; Nagababu, E. Hemoglobin redox reactions and red blood cell aging. Antioxid. Redox Signal. 2013, 18, 2274–2283. [Google Scholar] [CrossRef] [PubMed]
- Porto, B.N.; Alves, L.S.; Fernandez, P.L.; Dutra, T.P.; Figueiredo, R.T.; Graca-Souza, A.V.; Bozza, M.T. Heme induces neutrophil migration and reactive oxygen species generation through signaling pathways characteristic of chemotactic receptors. J. Biol. Chem. 2007, 282, 24430–24436. [Google Scholar] [CrossRef] [PubMed]
- Figueiredo, R.T.; Fernandez, P.L.; Mourao-Sa, D.S.; Porto, B.N.; Dutra, F.F.; Alves, L.S.; Oliveira, M.F.; Oliveira, P.L.; Graca-Souza, A.V.; Bozza, M.T. Characterization of heme as activator of Toll-like receptor 4. J. Biol. Chem. 2007, 282, 20221–20229. [Google Scholar] [CrossRef] [PubMed]
- Wagener, F.A.; Feldman, E.; de Witte, T.; Abraham, N.G. Heme induces the expression of adhesion molecules ICAM-1, VCAM-1, and E selectin in vascular endothelial cells. Exp. Biol. Med. 1997, 216, 456–463. [Google Scholar] [CrossRef] [PubMed]
- Neely, S.M.; Gardner, D.V.; Reynolds, N.; Green, D.; Ts’ao, C.H. Mechanism and characteristics of platelet activation by haematin. Br. J. Haematol. 1984, 58, 305–316. [Google Scholar] [CrossRef]
- Ilesanmi, O.O. Pathological basis of symptoms and crises in sickle cell disorder: Implications for counseling and psychotherapy. Hematol. Rep. 2010, 2, e2. [Google Scholar] [CrossRef]
- Baldwin, C.; Pandey, P.J.; Olarewaju, O. Hemolytic Anemia [Updated 2023 Jul 24]; StatPearls Publishing LLC.: Tampa, FL, USA, 2023. [Google Scholar]
- Marcus, C.J.; Habig, W.H.; Jakoby, W.B. Glutathione transferase from human erythrocytes. Nonidentity with the enzymes from liver. Arch. Biochem. Biophys. 1978, 188, 287–293. [Google Scholar] [CrossRef]
- Harvey, J.; Beutler, E. Binding of heme by glutathione S-transferase: A possible role of the erythrocyte enzyme. Blood 1982, 60, 1227–1230. [Google Scholar] [CrossRef]
- Aft, R.L.; Mueller, G.C. Degradation and covalent cross-linking of glutathione reductase by hemin. Life Sci. 1985, 36, 2153–2161. [Google Scholar] [CrossRef]
- O’Keeffe, R.; Latunde-Dada, G.O.; Chen, Y.L.; Kong, X.L.; Cilibrizzi, A.; Hider, R.C. Glutathione and the intracellular labile heme pool. Biometals Int. J. Role Met. Ions Biol. Biochem. Med. 2021, 34, 221–228. [Google Scholar] [CrossRef] [PubMed]
- Gouveia, Z.; Carlos, A.R.; Yuan, X.; Aires-da-Silva, F.; Stocker, R.; Maghzal, G.J.; Leal, S.S.; Gomes, C.M.; Todorovic, S.; Iranzo, O.; et al. Characterization of plasma labile heme in hemolytic conditions. FEBS J. 2017, 284, 3278–3301. [Google Scholar] [CrossRef]
- Carter, P. Spectrophotometric determination of serum iron at the submicrogram level with a new reagent (ferrozine). Anal. Biochem. 1971, 40, 450–458. [Google Scholar] [CrossRef]
- Mlejnek, P. Direct Interaction between N-Acetylcysteine and Cytotoxic Electrophile-An Overlooked In Vitro Mechanism of Protection. Antioxidants 2022, 11, 1485. [Google Scholar] [CrossRef]
- Kozlova, E.; Sherstyukova, E.; Sergunova, V.; Kozlov, A.; Gudkova, O.; Inozemtsev, V.; Chernysh, A. The Toxic Influence of Excess Free Iron on Red Blood Cells in the Biophysical Experiment: An In Vitro Study. J. Toxicol. 2022, 2022, 7113958. [Google Scholar] [CrossRef] [PubMed]
- Unno, M.; Matsui, T.; Ikeda-Saito, M. Structure and catalytic mechanism of heme oxygenase. Nat. Prod. Rep. 2007, 24, 553–570. [Google Scholar] [CrossRef] [PubMed]
- Guengerich, F.P. Destruction of heme and hemoproteins mediated by liver microsomal reduced nicotinamide adenine dinucleotide phosphate-cytochrome P-450 reductase. Biochemistry 1978, 17, 3633–3639. [Google Scholar] [CrossRef]
- Tahoun, M.; Engeser, M.; Svolacchia, L.; Sander, P.M.; Müller, C.E. Molecular Taphonomy of Heme: Chemical Degradation of Hemin under Presumed Fossilization Conditions. Molecules 2023, 28, 4887. [Google Scholar] [CrossRef]
- Maitra, D.; Byun, J.; Andreana, P.R.; Abdulhamid, I.; Diamond, M.P.; Saed, G.M.; Pennathur, S.; Abu-Soud, H.M. Reaction of hemoglobin with HOCl: Mechanism of heme destruction and free iron release. Free Radic. Biol. Med. 2011, 51, 374–386. [Google Scholar] [CrossRef]
- Aich, A.; Freundlich, M.; Vekilov, P.G. The free heme concentration in healthy human erythrocytes. Blood Cells Mol. Dis. 2015, 55, 402–409. [Google Scholar] [CrossRef] [PubMed]
- Bhagavan, N.V.; Ha, C.E. Chapter 27–Metabolism of Iron and Heme. In Essentials of Medical Biochemistry; Elsevier: Amsterdam, The Netherlands, 2015. [Google Scholar]
- Stocker, R.; Yamamoto, Y.; McDonagh, A.F.; Glazer, A.N.; Ames, B.N. Bilirubin is an antioxidant of possible physiological importance. Science 1987, 235, 1043–1046. [Google Scholar] [CrossRef] [PubMed]
- Soto Conti, C.P. Bilirubin: The toxic mechanisms of an antioxidant molecule. Arch. Argent. Pediatr. 2021, 119, e18–e25. [Google Scholar] [CrossRef]
- Bocedi, A.; Fabrini, R.; Lai, O.; Alfieri, L.; Roncoroni, C.; Noce, A.; Pedersen, J.Z.; Ricci, G. Erythrocyte glutathione transferase: A general probe for chemical contaminations in mammals. Cell Death Discov. 2016, 2, 16029. [Google Scholar] [CrossRef] [PubMed]
- Bocedi, A.; Noce, A.; Marrone, G.; Noce, G.; Cattani, G.; Gambardella, G.; Di Lauro, M.; Di Daniele, N.; Ricci, G. Glutathione Transferase P1-1 an Enzyme Useful in Biomedicine and as Biomarker in Clinical Practice and in Environmental Pollution. Nutrients 2019, 11, 1741. [Google Scholar] [CrossRef]
- Kopacz, A.; Kloska, D.; Cysewski, D.; Kraszewska, I.; Przepiorska, K.; Lenartowicz, M.; Loboda, A.; Grochot-Przeczek, A.; Nowak, W.; Jozkowicz, A.; et al. Co-administration of angiotensin II and simvastatin triggers kidney injury upon heme oxygenase-1 deficiency. Free Radic. Biol. Med. 2023, 205, 188–201. [Google Scholar] [CrossRef]
- Voskou, S.; Aslan, M.; Fanis, P.; Phylactides, M.; Kleanthous, M. Oxidative stress in beta-thalassaemia and sickle cell disease. Redox Biol. 2015, 6, 226–239. [Google Scholar] [CrossRef]
- Uzunova, V.V.; Pan, W.; Galkin, O.; Vekilov, P.G. Free heme and the polymerization of sickle cell hemoglobin. Biophys. J. 2010, 99, 1976–1985. [Google Scholar] [CrossRef]
- Nagababu, E.; Fabry, M.E.; Nagel, R.L.; Rifkind, J.M. Heme degradation and oxidative stress in murine models for hemoglobinopathies: Thalassemia, sickle cell disease and hemoglobin C disease. Blood Cells Mol. Dis. 2008, 41, 60–66. [Google Scholar] [CrossRef]
- Darghouth, D.; Koehl, B.; Madalinski, G.; Heilier, J.F.; Bovee, P.; Xu, Y.; Olivier, M.F.; Bartolucci, P.; Benkerrou, M.; Pissard, S.; et al. Pathophysiology of sickle cell disease is mirrored by the red blood cell metabolome. Blood 2011, 117, e57–e66. [Google Scholar] [CrossRef]
- Mussap, M. Special Issue on “The Application of Metabolomics in Clinical Practice: Challenges and Opportunities”. Metabolites 2022, 12, 296. [Google Scholar] [CrossRef]
- The Human Metabolome Database (HMDB). Available online: https://hmdb.ca/ (accessed on 1 September 2023).
- Lichtenberg, S.; Trifonova, O.P.; Maslov, D.L.; Balashova, E.E.; Lokhov, P.G. Metabolomic Laboratory-Developed Tests: Current Status and Perspectives. Metabolites 2021, 11, 423. [Google Scholar] [CrossRef] [PubMed]
- Odom, J.D.; Sutton, V.R. Metabolomics in Clinical Practice: Improving Diagnosis and Informing Management. Clin. Chem. 2021, 67, 1606–1617. [Google Scholar] [CrossRef]
- Considine, E.C. The Search for Clinically Useful Biomarkers of Complex Disease: A Data Analysis Perspective. Metabolites 2019, 9, 126. [Google Scholar] [CrossRef]
- Lokhov, P.G.; Maslov, D.L.; Lichtenberg, S.; Trifonova, O.P.; Balashova, E.E. Holistic Metabolomic Laboratory-Developed Test (LDT): Development and Use for the Diagnosis of Early-Stage Parkinson’s Disease. Metabolites 2020, 11, 14. [Google Scholar] [CrossRef] [PubMed]
- Hakki Kalkan, I.; Suher, M. The relationship between the level of glutathione, impairment of glucose metabolism and complications of diabetes mellitus. Pak. J. Med. Sci. 2013, 29, 938–942. [Google Scholar] [CrossRef] [PubMed]
- Morris, K. Glutathione may have major role in HIV-1 disease. Lancet 1997, 349, 781. [Google Scholar] [CrossRef]
- Bradford, S.; Cowan, J. From Traditional Drug Design to Catalytic Metallodrugs: A Brief History of the Use of Metals in Medicine. Metallodrugs 2014, 1, 10–23. [Google Scholar] [CrossRef]
- Kim-Shapiro, D.B.; Lee, J.; Gladwin, M.T. Storage lesion: Role of red blood cell breakdown. Transfusion 2011, 51, 844–851. [Google Scholar] [CrossRef]
- Benathan, M.; Labidi, F. Cysteine-dependent 5-S-cysteinyldopa formation and its regulation by glutathione in normal epidermal melanocytes. Arch. Dermatol. Res. 1996, 288, 697–702. [Google Scholar] [CrossRef]
- Banfalvi, T.; Gilde, K.; Boldizsar, M.; Fejos, Z.; Horvath, B.; Liszkay, G.; Beczassy, E.; Kremmer, T. Serum concentration of 5-S-cysteinyldopa in patients with melanoma. Eur. J. Clin. Investig. 2000, 30, 900–904. [Google Scholar] [CrossRef] [PubMed]
- Wakamatsu, K.; Kageshita, T.; Furue, M.; Hatta, N.; Kiyohara, Y.; Nakayama, J.; Ono, T.; Saida, T.; Takata, M.; Tsuchida, T.; et al. Evaluation of 5-S-cysteinyldopa as a marker of melanoma progression: 10 years’ experience. Melanoma Res. 2002, 12, 245–253. [Google Scholar] [CrossRef] [PubMed]
- Wakamatsu, K.; Fukushima, S.; Minagawa, A.; Omodaka, T.; Hida, T.; Hatta, N.; Takata, M.; Uhara, H.; Okuyama, R.; Ihn, H. Significance of 5-S-Cysteinyldopa as a Marker for Melanoma. Int. J. Mol. Sci. 2020, 21, 432. [Google Scholar] [CrossRef] [PubMed]
- Desideri, E.; Ciccarone, F.; Ciriolo, M.R. Targeting Glutathione Metabolism: Partner in Crime in Anticancer Therapy. Nutrients 2019, 11, 1926. [Google Scholar] [CrossRef] [PubMed]
- Celik, D.; Doruk, S.; Koseoglu, H.I.; Sahin, S.; Celikel, S.; Erkorkmaz, U. Cysteinyl leukotrienes in exhaled breath condensate of smoking asthmatics. Clin. Chem. Lab. Med. 2013, 51, 1069–1073. [Google Scholar] [CrossRef]
- Wennergren, G. Inflammatory mediators in blood and urine. Paediatr. Respir. Rev. 2000, 1, 259–265. [Google Scholar] [CrossRef] [PubMed]
- Eckert, E.; Schmid, K.; Schaller, B.; Hiddemann-Koca, K.; Drexler, H.; Goen, T. Mercapturic acids as metabolites of alkylating substances in urine samples of German inhabitants. Int. J. Hyg. Environ. Health 2011, 214, 196–204. [Google Scholar] [CrossRef]
- Goncalves-Dias, C.; Morello, J.; Correia, M.J.; Coelho, N.R.; Antunes, A.M.M.; Macedo, M.P.; Monteiro, E.C.; Soto, K.; Pereira, S.A. Mercapturate Pathway in the Tubulocentric Perspective of Diabetic Kidney Disease. Nephron 2019, 143, 17–23. [Google Scholar] [CrossRef]
- Allen, S.P.; Sampson, A.P.; Piper, P.J.; Chester, A.H.; Ohri, S.K.; Yacoub, M.H. Enhanced excretion of urinary leukotriene E4 in coronary artery disease and after coronary artery bypass surgery. Coron. Artery Dis. 1993, 4, 899–904. [Google Scholar] [CrossRef] [PubMed]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Georgiou-Siafis, S.K.; Tsiftsoglou, A.S. The Key Role of GSH in Keeping the Redox Balance in Mammalian Cells: Mechanisms and Significance of GSH in Detoxification via Formation of Conjugates. Antioxidants 2023, 12, 1953. https://doi.org/10.3390/antiox12111953
Georgiou-Siafis SK, Tsiftsoglou AS. The Key Role of GSH in Keeping the Redox Balance in Mammalian Cells: Mechanisms and Significance of GSH in Detoxification via Formation of Conjugates. Antioxidants. 2023; 12(11):1953. https://doi.org/10.3390/antiox12111953
Chicago/Turabian StyleGeorgiou-Siafis, Sofia K., and Asterios S. Tsiftsoglou. 2023. "The Key Role of GSH in Keeping the Redox Balance in Mammalian Cells: Mechanisms and Significance of GSH in Detoxification via Formation of Conjugates" Antioxidants 12, no. 11: 1953. https://doi.org/10.3390/antiox12111953