Abstract
Acute kidney injury (AKI) and chronic kidney disease (CKD) are interconnected conditions, and CKD is projected to become the fifth leading global cause of death by 2040. New therapeutic approaches are needed. Mitochondrial dysfunction and oxidative stress have emerged as drivers of kidney injury in acute and chronic settings, promoting the AKI-to-CKD transition. In this work, we review the role of mitochondrial dysfunction and oxidative stress in AKI and CKD progression and discuss novel therapeutic approaches. Specifically, evidence for mitochondrial dysfunction in diverse models of AKI (nephrotoxicity, cytokine storm, and ischemia-reperfusion injury) and CKD (diabetic kidney disease, glomerulopathies) is discussed; the clinical implications of novel information on the key role of mitochondria-related transcriptional regulators peroxisome proliferator-activated receptor gamma coactivator 1-alpha, transcription factor EB (PGC-1α, TFEB), and carnitine palmitoyl-transferase 1A (CPT1A) in kidney disease are addressed; the current status of the clinical development of therapeutic approaches targeting mitochondria are updated; and barriers to the clinical development of mitochondria-targeted interventions are discussed, including the lack of clinical diagnostic tests that allow us to categorize the baseline renal mitochondrial dysfunction/mitochondrial oxidative stress and to monitor its response to therapeutic intervention. Finally, key milestones for further research are proposed.
1. AKI and CKD Progression
Chronic kidney disease (CKD) is one of the fastest growing causes of death worldwide, set to become the fifth leading global cause of death by 2040 [1]. Patients with CKD have an increased risk of acute kidney injury (AKI), and AKI may accelerate CKD progression [2]. A better understanding of the molecular mechanisms of kidney injury may allow the development of novel therapeutic strategies. In this regard, recent trials have reported the nephroprotective and cardioprotective impact of sodium-glucose cotransporter-2 (SGLT2) inhibitors, a family of drugs that decrease the transport of molecules in kidney proximal tubules, thus potentially decreasing energy consumption [3]. The kidneys receive 20% of the cardiac output, and proximal tubular cells display a high oxygen consumption rate since they reabsorb the largest portion of the glomerular ultrafiltrate of about 180 L per day. They are also the primary targets in AKI and CKD due to their vulnerability to injury induced by hypoxia, drug-related toxicity, uremic toxins, metabolic disorders, and senescence [4]. In AKI, initial tubular cell death and inflammation is followed by subsequent regeneration [5,6]. The failure of regeneration may lead to the AKI-to-CKD transition [2]. Here, we review the role of mitochondrial dysfunction and oxidative stress in AKI and CKD progression and discuss novel therapeutic approaches.
2. Mitochondrial Function in Healthy Kidneys
Mitochondria are intracellular organelles essential to produce adenosine triphosphate (ATP); maintain redox and iron homeostasis; control cell death, inflammation, and intracellular calcium; traffic phospholipids; regulate danger signaling; and synthesize 1,25-(OH)2-vitamin D. Therefore, mitochondrial dysfunction can lead to tissue damage and organ failure including AKI and CKD [7,8,9]. Given its critical functions, the homeostasis of mitochondria number and function is key to kidney health. Mitochondria are dynamic and continuously adapt to evolving cell requirements through biogenesis (generation of new mitochondria driven by peroxisome proliferator-activated receptor gamma coactivator 1-alpha (PGC-1α), mitophagy (clearance of damaged mitochondria), fusion, and fission. Disruption of these processes may cause mitochondrial dysfunction.
Kidney tubules are rich in mitochondria because of the high ATP demands required for the absorption of high volumes of ultrafiltrate and solutes. Indeed, the kidney is the second organ with the most mitochondria after the heart; therefore, healthy mitochondria are crucial for normal kidney function [10,11]. Kidney tubules primarily rely on fatty acid β-oxidation (FAO) and mitochondrial oxidative phosphorylation (OXPHOS), likely as an adaptation to generate large amounts of ATP through the electron transport chain (ETC), which is the major source reactive of oxygen species (ROS) in the mitochondria [9] (Figure 1). Electrons released by the ETC react with oxygen to form superoxide anion (O2−), which is converted to hydrogen peroxide (H2O2) by superoxidase dismutase (SOD). H2O2 can be reduced to water by antioxidant enzymes such as catalase and glutathione peroxidases. In this regard, a balance between mitochondrial ROS (mtROS) production and scavenging is critical for a correct mitochondrial function [12]. Increased mtROS production and/or deficient antioxidant defenses may induce mitochondrial dysfunction, leading to cell death, inflammation, and AKI [12]. Tubular cell death induced by different mechanisms, such as cisplatin, calcium oxalate crystals, or ischemia/reperfusion, has been associated to mtROS production and mitochondrial permeability transition pore (MPTP) opening [6,13,14,15,16,17]. The mtROS can activate pro-inflammatory genes, NF-ĸB signaling, and inflammasome proteins [18,19]. Indeed, spontaneous kidney inflammation is observed in PGC-1α-deficient mice [20]. Mitochondrial FAO is the preferred pathway to produce acetyl-CoA as a substrate for the tricarboxylic acid (TCA) cycle in kidney tubules, and FAO dysfunction results in ATP depletion, lipid accumulation, inflammation, and subsequent fibrosis [21,22,23,24]. The mtROS can also affect FAO, as antioxidant strategies restored the expression of proteins involved in FAO in experimental polycystic kidney disease [25]. Antioxidant strategies also improved FAO, lipid accumulation, mtROS-associated lipid peroxidation, and kidney function in murine cisplatin-induced AKI (cisplatin-AKI) [26].
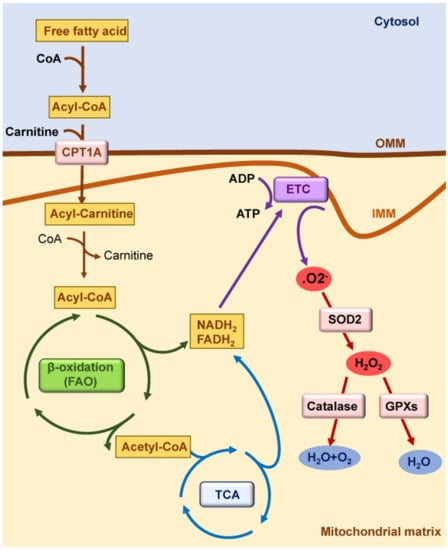
Figure 1.
Mitochondrial function in healthy kidneys. Mitochondrial fatty acid β-oxidation (FAO) is the preferred pathway to generate ATP in kidney tubules. Fatty acids are converted in acyl-CoA in cytosol; they are conjugated with carnitine in the outer mitochondrial membrane (OMM) by carnitine palmitoyl-transferase 1A (CPT1A) to cross the inner mitochondrial membrane (IMM). In the mitochondrial matrix, acyl-carnitine is reconverted to acyl-CoA, and it enters the tricarboxylic acid (TCA) cycle. Reduced nicotinamide adenine dinucleotide (NADH2) and reduced flavin adenine dinucleotide (FADH2) generated by FAO and by the TCA cycle deposit their electrons into the electron transport chain (ETC). Electrons released by the ETC react with oxygen to form superoxide anion (O2−), which is converted to hydrogen peroxide (H2O2) by superoxidase dismutase 2 (SOD2). H2O2 can be reduced to water by antioxidant enzymes such as catalase and glutathione peroxidases (GPXs).
3. Mitochondrial Dysfunction and Oxidative Stress in AKI
The key causes of AKI include nephrotoxicity, cytokine storm, and ischemia-reperfusion injury (IRI) [27,28]. Treatments aiming to reduce mitochondrial oxidative stress were protective in preclinical AKI triggered by these causes (Table 1).
3.1. Nephrotoxic AKI
Drug-induced nephrotoxicity accounts for up to 60% of hospital-acquired AKI [29]. Several preclinical models have been used to address pathogenic mechanisms in nephrotoxic AKI.
Folic acid-induced AKI (FA-AKI) is a classical model of AKI characterized by tubular cell death, interstitial leukocyte infiltration, and subsequent tubular regeneration and CKD transition; it has been reported in humans [30,31]. Mitochondrial alterations occur at early stages and during AKI-to-CKD progression [21,32,33]. Mitochondrial biogenesis appears to be depressed, along with decreased expression of PGC-1α and its transcriptional regulatory activity [33,34]. Furthermore, FA-AKI is more severe in PGC-1α-deficient mice or those exposed to a mitochondrial complex I inhibitor [34,35]. N-acetylcysteine (NAC) is an antioxidant in clinical use frequently prescribed to prevent radiocontrast nephrotoxicity despite the conflicting results of clinical trials. NAC has been reported to prevent mitochondrial dysfunction in preclinical Huntington’s disease and myocardial infarction [36,37]. In the kidney, NAC pre-treatment in murine FA-AKI prevented mitochondrial and kidney function, but delayed NAC administration increased the severity of FA-AKI [21,32,38]. Variability in intervention timing may be one of the factors underlying the inconclusive results from clinical studies.
The use of the antineoplastic agent cisplatin is limited by nephrotoxicity, especially by AKI that may become irreversible (AKI-to-CKD transition). Preclinical studies support a role for mitochondrial dysfunction in cisplatin-AKI [39,40,41]. Cisplatin nephrotoxicity is characterized by mtROS production, reduced mitochondrial membrane potential, mitochondrial swelling, and loss of mitochondrial function [42]. As in FA-AKI, mitochondrial biogenesis is compromised and PGC-1α expression is decreased in murine cisplatin-AKI [43]. Successful strategies to improve mitochondrial function and to limit preclinical cisplatin nephrotoxicity in mice included SOD mimetics or mitochondrial-targeted antioxidants [42,44,45,46].
Contrast-induced AKI (CI-AKI) is a common cause of AKI in hospitalized patients. During CI-AKI, mtROS increases in association with inflammation and cell death [47,48]. Mitophagy was protective in murine CI-AKI as it removes damaged mitochondria, and rapamycin-induced mitophagy reduced cell injury in cultured human tubular cells [47,49]. The antioxidant compound tetramethylpyrazine preserved kidney function and reduced kidney oxidative stress, inflammation, and aberrant mitochondrial dynamics in CI-AKI in rats [48].
3.2. Cytokine Storm
Cytokine storm is a life-threatening organ dysfunction resulting from a systemic inflammatory response to bacterial or viral (e.g., SARS-CoV-2) infection and constitutes the leading cause of AKI in the intensive care setting [50,51,52]. The term sepsis is usually restricted to patients with active bacterial infection; preclinical models frequently involve the administration of sterile bacterial lipopolysaccharide (LPS) or bacteriemia resulting from cecal ligation and puncture (CLP). Cytokine storm-induced AKI (cytokine storm-AKI) is characterized by variable levels of tubular cell death, interstitial inflammatory cell infiltration, and mitochondrial swelling and dysfunction [53,54].
Biopsies from patients with sepsis-associated AKI showed signs of oxidative stress and mitochondrial injury as assessed by the upregulation of oxidative stress markers, mitochondrial DNA (mtDNA) damage, and reduced expression of mitochondrial markers [55]. Moreover, urine mtDNA correlated with mitochondrial dysfunction and AKI severity in sepsis patients [56]. Sepsis causes a metabolic reprogramming in immune cells characterized by reduced mitochondrial OXPHOS and ATP production and increased aerobic glycolysis. This shift promotes a pro-inflammatory phenotype necessary to frame an inflammatory response, but its persistence may promote an exacerbated inflammation, boosting tissue injury [57]. Although there is no clear evidence that tubular cells undergo this metabolic change during sepsis, some data suggest that it may occur [57]. In murine CLP, a change in the metabolite milieu was consistent with reduced flux through the TCA cycle and increased glycolysis in the kidney [58]. Moreover, in murine LPS-induced AKI (LPS-AKI), genes encoding OXPHOS elements were downregulated [59]. Mitochondrial dysfunction in cytokine storm-AKI has been associated with renal failure, which can lead to compensatory quality control mechanisms, as observed in biopsies from sepsis-associated AKI patients, in which genes involved in antioxidant defense, such as SIRT1, in mitochondrial biogenesis, as mitochondrial transcription factor A (TFAM) and PGC-1α and in mitophagy as PTEN-induced kinase 1 (PINK1) and Parkin RBR E3 ubiquitin-protein ligase (PARKIN), were downregulated [55]. This may be interpreted as a scenario of limited generation of new mitochondria and limited removal of damaged mitochondria.
Different strategies against mtROS have been tested in preclinical cytokine storm-AKI. Mitochondria-target ceria nanoparticles have ROS-scavenging activity, improved kidney function, and reduced kidney inflammation and kidney mtROS in murine LPS-AKI [60]. Moreover, both MitoQ, a coenzyme Q10 (CoQ10) analogue, and MitoTEMPO, a SOD mimetic, protected from murine cytokine storm-AKI induced by LPS or CLP, respectively [61,62].
3.3. Ischemia-Reperfusion Injury (IRI)
Kidney IRI is a frequent cause of AKI (IRI-AKI) after major surgery and following kidney transplantation. Hypoxia promotes a switch from aerobic to anaerobic metabolism that decreases ATP levels. This is accompanied by a rapid depolarization of mitochondrial membranes and increased intracellular and mitochondrial Ca2+ [63,64]. During hypoxia, only a few cells die [64]; however, during reperfusion, multiple mechanisms increase the severity of injury, including oxidative stress resulting from oxygen availability, depletion of antioxidants, and the Ca2+-dependent activation of calpains as pH is normalized. Increased mtROS and mitochondrial calcium content lead to MPTP opening and the activation of different pathways of cell death [16]. In addition, mtROS also promote renal inflammation and NLRP3 activation, as in other AKI models [65,66]. The mtROS directly decrease the expression of the mitochondrial transcriptional factor TFAM in tubular cells undergoing hypoxia/reoxygenation and in murine IRI-AKI; in turn, decreased TFAM levels mediate mtROS-induced mitochondrial dysfunction and inflammation in tubular cells, although the effect of TFAM downregulation over cell death was not analyzed [15]. However, it is unknown whether mtROS directly promote kidney inflammation or whether inflammation is mediated by damage-associated molecular patterns (DAMPs) released by dying tubular cells [67].
Pre-treatment with elamipretide (also called Szeto–Schiller 31 (SS-31), MTP-131, and Bendavia) reduced tubular injury and favored kidney regeneration in IRI-AKI in rat, mice, and pigs [68,69,70]. Additionally, CoQ10 nanoparticles and the plastoquinone analogue SkQR1 reduced oxidative stress and tissue injury in IRI-AKI in rats, and MitoQ favored the cold preservation of porcine kidneys [71,72,73]. Taraxasterol, a natural product with antioxidant properties that reduce mtROS in hypoxia/reperfusion-stimulated tubular cells, was also protective in murine IRI-AKI [74]. Administration of isolated healthy mitochondria protected from IRI-AKI in rats and in swine, likely through the uptake and incorporation into cells [75].
Mitochondrial fragmentation, mitophagy, and biogenesis have also been targeted therapeutically. Dynamin-related protein 1 (Drp1) promotes mitochondrial fragmentation, generating mtROS and mitochondrial DAMPs. A specific inhibitor of Drp1, mitochondrial division inhibitor-1 (mdivi-1), attenuated tubular injury and tubular cell death in tubular cells and in murine IRI-AKI [76,77]. Mitophagy protects from kidney IRI by removing damaged mitochondria, and mitophagy activation with an AMP-activated protein kinase (AMPK) activator protected from murine IRI-AKI [78,79,80]. Mitochondrial biogenesis is also reduced in IRI-AKI, and PGC-1α overexpression protected from murine IRI-AKI [20,81]. Moreover, the long-acting beta2-adrenergic agonist formoterol stimulated mitochondrial biogenesis and improved kidney function after murine IRI-AKI [82].

Table 1.
Drugs targeting mitochondrial ROS that have shown beneficial effect in experimental AKI.
Table 1.
Drugs targeting mitochondrial ROS that have shown beneficial effect in experimental AKI.
| Model | Type of Drug | Drug | Ref. |
|---|---|---|---|
| FA-AKI | Antioxidant | NAC pre-treatment | [21,32] |
| Cisplatin | CoQ10 analogue | MitoQ | [45] |
| SOD mimetics | TEMPOL | [44] | |
| GC4419 | [42] | ||
| Mito-CP | [45] | ||
| MitoTEMPO | [46] | ||
| CI-AKI | Antioxidant | Tetramethylpyrazine | [48] |
| s-AKI | CoQ10 analogue | MitoQ | [61] |
| SOD mimetics | MitoTEMPO | [62] | |
| Antioxidant | Mitochondria-targeted ceria nanoparticles | [60] | |
| CoQ10 analogue | SkQR1 | [71] | |
| Mitochondria-targeted TPP CoQ10 nanoparticles | [72] | ||
| SOD mimetics | MitoTEMPO | [15] | |
| SS-peptide | Elamipretide * | [68,69] | |
| Isolated healthy mitochondria | [75] | ||
| Reperfusion in experimental atherosclerotic renal artery stenosis in pigs | SS-peptide | Elamipretide * | [70] |
| Cold preservation of porcine kidneys | CoQ10 analogue | MitoQ | [73] |
Abbreviations: FA-AKI: folic acid-AKI; CI-AKI: contrast induce-AKI; s-AKI: sepsis-induced AKI; IRI-AKI: ischemia-reperfusion injury AKI; NAC: N-acetylcysteine; SOD: superoxide dismutase; CoQ10: coenzyme Q10. * Elamipretide is also called SS-31, MTP-131, and Bendavia.
4. Mitochondrial Dysfunction and Oxidative Stress in CKD
There is evidence of mitochondrial dysfunction and related oxidative stress in CKD, as suggested by data in two of the most common causes of CKD, diabetic kidney disease (DKD) and glomerulonephritis.
4.1. Diabetic Kidney Disease
DKD is associated with mitochondrial dysfunction in kidney cells including endothelial cells [83] and podocytes [84]. Indeed, abnormal mitochondrial bioenergetics and dynamics precede CKD progression in rat type 1 diabetes (T1D) and altered mitochondrial morphology in tubules of patients with DKD support the idea that mitochondrial injury is an early event in DKD [85,86]. Indeed, in diabetic db/db mice, serum and urine metabolomics revealed differential concentrations of metabolites relevant to energy production by mitochondria, including the TCA cycle, lipid metabolism, glycolysis, and amino acid turnover [87].
The mtROS production in response to chronic hyperglycemia may initiate diverse pathogenic pathways, as was observed in mesangial cells, tubular cells, and in DKD kidneys in rodents [88,89]. Indeed, elamipretide or CoQ10 reduced kidney injury in experimental type 1 (streptozotocin) and type 2 (db/db mice) diabetes (Table 2) [90,91,92]. Post-translational modifications of mitochondrial proteins may also alter mitochondria function and biogenesis [93,94]. Increased nitrotyrosine staining in the kidneys of streptozotocin-induced T1D in ApoE-/- mice was associated with reduced manganese SOD activity [95]. In Madin–Darby canine kidney tubular cells, high glucose culture conditions increased the phosphorylation and oxidation of mitochondrial proteins [96].
A deficiency in the apoptosis-inducing factor (AIF) could contribute to mitochondrial dysfunction and DKD in streptozotocin-induced T1D. In patients with diabetic nephropathy, tubular AIF was decreased and correlated with declining AMP-activated protein kinase (GFR), while AIF overexpression in primary proximal tubule epithelial cells restored OXPHOS capacity under high glucose conditions [97].
The mtDNA released by damaged cells has been proposed as a biomarker of mitochondrial dysfunction. Circulating mtDNA levels were higher in patients with DKD than in healthy subjects or diabetic patients without DKD [98]. These findings were not confirmed by another study that found lower circulating mtDNA in DKD patients than in diabetic patients without DKD [99]. The reason for the discrepancy is unclear and may depend on patient characteristics or comorbidities. However, even when mtDNA was found decreased in plasma, it was increased in urine in DKD patients and in mice with streptozotocin-induced T1D [99]. The mtDNA may play a pathogenic role, as mtDNA infusion induced kidney injury and inflammation in healthy mice.
Altogether, these results suggest that mitochondrial injury may contribute to DKD and mtDNA may be a marker of mitochondrial disfunction in DKD and have a pro-inflammatory role; however, clinical evidence is incomplete. Indeed, the molecular mechanisms of the kidney and cardioprotective effect of SGLT2 inhibitors is not yet fully understood, but there is evidence that they may improve mitochondrial function [100].
4.2. Glomerular Disease
In addition to tubular epithelial cells, another epithelial cell type, podocytes, may be marred by mitochondrial dysfunction. Podocyte injury is a key feature of proteinuric glomerular disease, which may also progress to tubulointerstitial injury through proteinuria-induced tubular cell injury. Podocyte injury usually causes a morphological pattern of focal segmental glomerulosclerosis (FSGS) [101]. In humans, several mutations in the nuclear or mitochondrial DNA involving COQ2, COQ6, and PDSS2 and MTTL1, respectively, lead to glomerular disease phenotypes compatible with FSGS and nephrotic syndrome. Moreover, the genetic knockdown of specific mitochondrial genes in animals, such as Pdss2, Tsc1, Mtorc1, Rock1, and Atg5, resulted in proteinuria, glomerulosclerosis, and foot process effacement [102]. The spectrum of mitochondrial functions affected by these genetic defects or manipulations includes mitochondrial tRNA, biosynthesis of CoQ10, protein synthesis control, mitochondrial fission, and mitophagy.
Dysfunction of the energy metabolism and mitochondria may underly several glomerular diseases through deficits in ATP supply, ionic disbalance, unwanted or excessive apoptosis, oxidative stress, and inflammation. Unlike tubular cells, whose energy requirements almost fully rely on OXPHOS, energy generation may shift to glycolysis in injured podocytes during DKD and FSGS [103]. Additionally, a non-glycolytic pathway was uncovered in podocytes under hyperglycemic conditions [104,105]. Under physiological conditions, a deficiency in PGC-1α, Drp1, or TFAM negatively impacted OXPHOS progression in podocytes. However, any defective mitochondrial biogenesis, fission, or mtDNA transcription caused by the deficiency in these factors did not result in mitochondria-associated pathological phenotypes in mice. Thus, podocyte health is maintained when dysfunctional mitochondria lead to mitochondrial respiration deficits. This may be explained by the fact that anaerobic glycolysis was the primary metabolic energy source in podocytes [106]. Nevertheless, a thorough assessment of the impact of mitochondria-associated genetic defects on acquired glomerular injury in vivo is still lacking. Both elamipretide and the SOD mimetic MitoTEMPO were beneficial in high-fat diet-induced glomerulopathy in mice; however, there is limited information regarding their effects on other forms of glomerular injury [107,108] (Table 2). In any case, podocyte injury and albuminuria cause injury of proximal tubular cells, as albuminuria indicates that the capacity of proximal tubular cells to reabsorb filtered albumin has been exceeded. Excessive albumin reabsorption leads to tubular cell injury, inflammatory responses, and loss of the capacity to synthesize Klotho, an antiaging and kidney-protective protein of tubular cell origin [109]. In this regard, Pgc1α-KO mice display spontaneous kidney inflammation as evidence of subclinical CKD, while specific proximal tubular TFAM deficiency in mice favors kidney fibrosis through tuna leakage and activation of Sting-dependent innate immune inflammation [20,110].
Table 2.
Drugs targeting mitochondrial ROS that have shown beneficial effects in experimental CKD.
Table 2.
Drugs targeting mitochondrial ROS that have shown beneficial effects in experimental CKD.
| Model | Type of Drug | Drug | Ref. |
|---|---|---|---|
| STZ-induced diabetic nephropathy | SS-peptide | Elamipretide * | [90] |
| Type 2 diabetes (db/db mice) | SS-peptide | Elamipretide * | [91] |
| CoQ10 analogue | CoQ10 | [92] | |
| High-fat diet-induced glomerulopathy | SS-peptide | Elamipretide * | [107] |
| SOD mimetics | MitoTEMPO | [108] |
Abbreviations: STZ: streptozotocin; CoQ10: coenzyme Q10. * Elamipretide is also called SS-31, MTP-131, and Bendavia.
5. Novel Mitochondria-Related Therapeutic Targets in Kidney Disease
In recent years, accumulated evidence suggests a key role of mitochondria-related transcriptional regulators peroxisome proliferator-activated receptor gamma coactivator 1-alpha and transcription factor EB (PGC-1α, TFEB, respectively) and of carnitine palmitoyl-transferase 1A (CPT1A) in kidney disease (Figure 2).
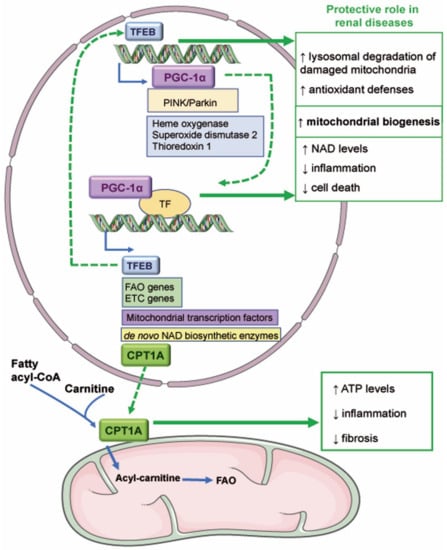
Figure 2.
Novel mitochondria-related therapeutic targets in kidney disease. The transcriptional activity of TFEB promotes the expression of genes involved in mitochondrial biogenesis such as PGC-1α and in antioxidant defenses. TFEB could be protective in AKI since it favors the degradation of damaged mitochondria and mitochondrial biogenesis. PGC-1α regulates the expression of TFEB, fatty acid β-oxidation (FAO), and electron transport chain (ETC) genes, mitochondrial transcription factors, and de novo NAD biosynthetic enzymes. Various reports have demonstrated that PGC-1α reduces renal inflammation and cell death and favors NAD synthesis and mitochondrial biogenesis. PGC-1α may also mediate the expression of carnitine palmitoyl-transferase 1A (CPT1A), which mediates the transport of fatty acids into the mitochondrial matrix. Overexpression of CPT1A in murine renal tubules protected from preclinical kidney disease by increasing ATP levels and reducing inflammation and fibrosis. ↑ increase; ↓ decrease.
5.1. PGC-1α
PGC-1α is the master regulator of mitochondrial biogenesis, a process that coordinates the transcriptional machinery leading to increased mitochondrial mass, allowing tissue adaptation to high energy demands [20,111]. Thus, PGC-1α expression is expected to increase in metabolically demanding tissues such as the kidney [112]. However, PGC-1α levels fall dramatically in kidney diseases, including AKI and CKD [20]. In this regard, PGC-1α downregulation is observed in human AKI of different causes and in experimental AKI models induced by sepsis, IRI, cisplatin, or folic acid, where it is associated with mitochondrial impairment and reduced mitochondrial biogenesis [33,34,59,81,113,114]. Indeed, PGC-1α was the transcription regulator with the most reduced transcriptional activity in FA-AKI [34]. Inflammatory mediators such as TWEAK or TNF-α decreased PGC-1α levels both in vivo and in vitro [33,59]. In fact, neutralizing anti-TWEAK or anti-TNF-α antibodies prevented the downregulation of PGC-1α and its mitochondrial biogenesis-associated target genes in FA-AKI and in sepsis-associated AKI, respectively [33,115]. Additionally, TGF-β1-driven SMAD3 binding overlaps with the active enhancer histone tail modification H3K4me1 of the PGC-1α promoter sequence, blocking the progression of its transcription machinery [116].
By contrast, PGC-1α overexpression increased mitochondrial abundance and protected from AKI induced by sepsis, IRI, and cisplatin in mice [81,114]. PGC-1α overexpression also increases mitochondrial biogenesis in cultured tubular cells [33]. Moreover, PGC-1α enhances the NAD biosynthesis pathway. NAD biosynthesis has emerged as a guardian against the age-related decline in health and mitochondrial function [81] and, potentially, against AKI [117]. PGC-1α also has anti-inflammatory activity since PGC-1α-deficient mice displayed subclinical kidney pro-inflammation and developed more severe FA-AKI [34]. Treatment with the AMPK activator AICAR (5-aminoimidazole-4-carboxamide-1-β-d-ribofuranoside) and the antioxidant ALCAR (acetyl-l-carnitine) increased PGC-1α expression and decreased the severity of cisplatin-AKI by reducing mitochondrial fragmentation, a mechanism that involves Sirt3 activation [43]. Other activators of PGC-1α that also increased mitochondrial biogenesis in preclinical AKI were the Sirt1 inducer SRT1720 in IRI and phosphodiesterase (PDE) inhibitors, which increase cGMP in FA-AKI [118,119].
PGC-1α has been extensively studied in DKD, where PGC-1α activators were nephroprotective in preclinical T1D and type 2 diabetes (T2D) [20]. Thus, pharmacological pyruvate kinase M2 (PKM2) activation and GTPase Rap1b reversed mitochondrial dysfunction in mice with streptozotocin-induced T1D by inducing PGC-1α expression [84,120]. A protective PGC-1α contribution to mitochondrial homeostasis was also observed in glomeruli of T2D BTBR ob/ob mice treated with honokiol, a Sirt3 inducer [121]. In this murine model, podocyte-specific overexpression of Tug1 improved mitochondrial bioenergetics, restoring the expression of PGC-1α and its target genes [122]. PGC-1α overexpression was also protective in tubular cells cultured in high-glucose conditions [123]. In cultured podocytes, Sirt1 agonists such as BT175 and resveratrol protected against high-glucose-mediated mitochondrial injury and reduced oxidative stress through increasing PGC-1α expression [124,125]. However, forced PGC-1α overexpression in podocytes caused collapsing glomerulopathy [126]. CKD is associated to kidney fibrosis; TGF-β1, a key driver of fibrosis, also downregulates PGC-1α expression, leading to lipid accumulation and impaired FAO. Indeed, PGC-1α overexpression in primary murine proximal tubular cells normalized the expression of FAO enzymes after TGF-β1 stimulation [116]. These data support a nephroprotective role of PGC-1α through the improvement of the mitochondrial function in kidney diseases [127]. However, excess PGC-1α expression may not be desirable, at least in some conditions [126].
5.2. Transcription Factor EB (TFEB)
TFEB is a master regulator of the autophagy-lysosomal pathway that removes misfolded protein aggregates or damaged organelles. Under baseline conditions, TFEB is retained phosphorylated in the cytosol. Under adverse conditions, TFEB is dephosphorylated and migrates to the nucleus where it promotes the expression of genes of the coordinated lysosomal expression and regulation (CLEAR) network [128]. The TFEB pathway controls mitochondria quality at three levels: mitophagy, mitochondrial biogenesis, and ROS scavenging [129]. TFEB promotes mitophagy by inducing general autophagy upon PINK/PARKIN-induced activation of TFEB, leading to further lysosomal degradation of damaged mitochondria [129,130]. TFEB promotes mitochondrial biogenesis by promoting PGC-1α expression, which, in turn, promotes TFEB expression [129]. Additionally, mtROS promote TFEB activation, and TFEB increases the expression of antioxidant genes such as heme oxygenase 1, SOD2, and thioredoxin 1 [129].
Although TFEB might protect from AKI, direct evidence (i.e., TFEB-deficient tubular cells) is lacking. In PGC-1α-KO mice, kidney TFEB is downregulated, and the number of lysosomes is lower than in WT mice; this was reversed in transgenic mice overexpressing PGC-1α [114]. ZLN005, an activator of PGC-1α expression, alleviates kidney injury in cisplatin-induced AKI; in cultured tubular cells, ZLN005-induced PGC-1α expression has also a protective role partially due to TFEB activation [131]. Moreover, trehalose induces TFEB expression and autophagy and reduces tubular injury and mitochondrial dysfunction induced by cisplatin both in mice and human proximal tubular cells [132]. However, trehalose promoted cell death and inflammation in rat kidney epithelial cells [133]. During murine IRI-AKI, TFEB is activated and upregulates genes of the CLEAR network, promoting autophagy. Indeed, treatment with urolithin A protected from IRI-AKI and promoted TFEB activation [134]. Additionally, the necroptotic protein RIPK3 reduced TFEB expression and autophagy in cytokine storm-AKI, but mitochondrial function was not explored [135].
Regarding CKD, TFEB and its target genes were upregulated in mice with adenine-induced CKD and in patients with IgA nephropathy, but its role was not addressed in interventional studies [133]. However, TFEB was downregulated in human DKD, sub-totally nephrectomized rats, and other forms of kidney fibrosis; this could be mediated by TFEB deacetylation by HDAC6 [136]. Thus, further characterization of the time course of TFEB expression in different models of CKD and the identification of the cell types with upregulated or downregulated TFEB expression are needed to reconcile the current discrepant information.
5.3. CPT1A
Experimental and clinical evidence has illustrated the significance of the drastic reduction of FAO coupled with intracellular lipid deposition in proximal tubules in the pathogenesis of both AKI and CKD [137]. CPT1A is the predominant renal isoform of the mitochondrial outer membrane fatty acid shuttling enzyme. It catalyzes the FAO rate-limiting step, i.e., the conversion of long-chain acyl-CoAs into acylcarnitine that is transported into the mitochondrial matrix to undergo FAO [138]. In CKD patients, decreased CPT1A levels are associated to an increased accumulation of short- and middle- chain acylcarnitines, reflecting impaired FAO [139,140]. Both genetic deletion of CPT1A and its pharmacological inhibition with etomoxir uncoupled mitochondrial function and increased the severity of tubular injury and interstitial fibrosis [116,141]. However, the accumulation of fatty acids and lipotoxicity do not appear to be the main drivers of kidney injury associated to impaired FAO. Although the inhibition of CD36-, FATP2-, or KIM-1-mediated fatty acid uptake decreased experimental tubulointerstitial inflammation and fibrosis, increased lipid overload resulting from tubular CD36 overexpression was not sufficient to drive spontaneous renal fibrogenesis [142,143].
Several factors may suppress CPT1A expression in kidney injury. Reduced PGC-1α leads to transcriptional repression of CPT1A and other fatty acid uptake- and oxidation-related genes. The Krüppel-like factor 15 (KLF15) is another transcriptional regulator of CPT1A and Acaa2 that is downregulated in different models of kidney injury, leading to a reduced FAO [144]. Post-transcriptional targeting of CPT1A by miR-33, miR-150, and miR-495 has also been reported in renal epithelial cells under a profibrotic insult [145,146].
Conversely, increasing FAO through genetic or pharmacologic interventions protects tubules from injury. Inducible FAO gain of function through CPT1A overexpression in murine kidney tubules protected from fibrosis in three models of CKD. It also decreased the kidney inflammation and epithelial cell dedifferentiation and preserved structural mitochondrial integrity and ATP generation [139]. There is also more indirect evidence supporting a protective role of FAO restoration: enhanced tubular expression of PGC-1α (a strong inducer of CPT1A) and treatment with fenofibrate (a PPAR-α agonist), C75 (a CPT1A activator and fatty acid synthase blocker), or AICAR (an AMPK activator, which induces CPT1A expression and reduces the levels of its physiological inhibitor, malonyl-CoA) mitigated the chemical or surgical induction of kidney fibrosis and associated functional decline [34,147,148]. Although a protective effect of FAO has been also suggested in AKI, it merits further investigation [59,81,149].
Despite the lack of complete knowledge of the metabolic profile of the renal cell populations involved in fibrogenesis and of whether enhanced FAO is safe in the long term, this strategy has gained momentum as a potential therapeutic approach. The future development of specific, non-toxic CPT1 activators may pave the way for clinical development.
6. Therapeutic Modulation of Mitochondrial Dysfunction in Kidney Diseases
Mitochondria-targeted therapies aim to enhance mitochondrial function or to dampen the cellular consequences of mitochondrial dysfunction, including apoptosis, MPTP opening, altered mitochondrial dynamics and mitophagy, or ROS production [20,150,151]. Inhibiting apoptosis-associated mitochondrial pathways was protective in experimental AKI, as exemplified by the genetic deletion of Bak in IRI-AKI. However, other strategies to prevent apoptosis in AKI were not successful; recently, other forms of cell death different than apoptosis, such as ferroptosis and necroptosis, have gained relevance [6,152]. The inhibition of cyclophilin D (CypD) with cyclosporine A or sanglifehrin suppresses MPTP opening and mitochondrial swelling, improving IRI-AKI. However, the therapeutic use of cyclosporine A is limited by its nephrotoxicity [16,153,154,155]. The modulation of mitochondrial dynamics with the mdivi-1, which blocks Drp-1-induced mitochondrial fission, protected from IRI- and cisplatin-AKI [76] but increased the severity of kidney fibrosis [156], evidencing the complexity of interfering with mitochondrial division in injured and regenerating cells [151].
SS peptides, such as elamipretide, are cell-permeable tetrapeptides that bind to cardiolipin on the inner mitochondrial membrane (IMM) and promote ATP synthesis, reduce electron leak and ROS production, and inhibit cardiolipin peroxidation [157]. Elamipretide was renoprotective and restored the mitochondrial structure in preclinical IRI-AKI, experimental DKD, or glomerulopathy (Table 1 and Table 2) [68,69,70,90,91,107]. Additionally, the safety and efficacy of elamipretide in kidney patients was tested in clinical trials (Table 3). A phase 1 trial evaluated the safety of elamipretide in patients with impaired kidney function; however, the results have not been published more than 7 years after its completion (NCT02436447). A phase 2 trial (n = 308) evaluated the safety and efficacy of elamipretide in patients with congestive heart failure (NCT02914665), having the impact on kidney function as a secondary outcome. However, no results have been posted. Elamipretide improved kidney function and decreased blood pressure 3 months after angioplasty of the renal artery in a small (n = 14) phase 2 trial in patients with atherosclerotic renal artery stenosis [158]. Interestingly, despite elamipretide attenuating renal hypoxia 24 h after contrast imaging and renal artery stent revascularization, it increased peripheral venous levels of G1 cell cycle arrest markers IGFBP-7*TIMP-2 at 24 h after stenting [158]. These biomarkers are associated with AKI, despite the authors’ optimistic interpretation of these findings. In a further small trial (n = 24) in heart failure, no temporal change in kidney function was observed associated with elamipretide [159]. The current clinical development program of elamipretide is focused on cardiology, neurology, and ophthalmology, and kidney disease is no longer pursued.
Mitochonic acid (MA-5) also binds to the IMM and increases cellular ATP levels independently of OXPHOS and ETC, promoting survival in fibroblasts isolated from patients with mitochondrial disease. In mice, MA-5 stabilized mitochondrial function, increased ATP content and improved renal function in cisplatin-AKI and IRI-AKI [160]; however, information on clinical trial development is not available on clinicaltrials.gov.
CoQ10 is a component of the ETC. CoQ10 or its analogues are nephroprotective by reducing oxidative stress in diverse forms of kidney injury and are undergoing clinical trials (Table 1, Table 2 and Table 3). CoQ10 protected from DKD in db/db mice and murine IRI-AKI. Indeed, the selective delivery of CoQ10 to mitochondria increased its efficacy compared with free CoQ10 in experimental IRI-AKI [71,72,92]. CoQ10 was safe and well tolerated in hemodialysis patients and decreased oxidative stress in plasma (NCT00908297). Additionally, an ongoing clinical trial (enrolment n = 100) is exploring whether pre-treatment with CoQ10 as a dietary supplement before cardiac surgery reduces the incidence of AKI (NCT04445779), while another trial (enrolment n = 84) is testing CoQ10 as a dietary supplement to improve renal function after kidney transplantation (NCT04972552). Idebenone, another CoQ10 analog, is already approved in some countries to treat glaucoma and authorized by the EMA to treat Leber’s hereditary optic neuropathy, an inherited disease characterized by progressive loss of sight (https://www.ema.europa.eu/en/medicines/human/EPAR/raxone; accessed on 2 June 2022). Idebenone reduced glomerular injury in murine lupus but has not yet been tested clinically for kidney disease [161]. Among CoQ1 analogs, MitoQ (mitoquinone mesylate) has shown the most promising results. It is currently commercially available as a dietary supplement. In mice, it protected from DKD (db/db mice), from lupus nephritis, and from cisplatin- and LPS-AKI. Furthermore, MitoQ perfusion during cold ischemia before transplantation improved kidney function ex vivo in human and pig kidneys [45,61,162,163,164,165]. An ongoing, small (n = 18) clinical trial is exploring the effect of MitoQ as a dietary supplement on exercise capacity in patients with CKD and heart failure (NCT03960073) but will not study its effects on kidney function or markers of kidney injury. Another clinical trial is testing the effect of MitoQ as a nutritional supplement on vascular function and blood pressure reactivity in healthy adults (NCT04334135).
Other SOD mimetic antioxidant strategies tested in preclinical AKI, such as Mito-CP, MitoTEMPO, tempol, and GC441, are not listed in clinicaltrials.gov as being in clinical development (Table 1 and Table 2) [15,45,46,62,108].
Table 3.
Clinical trials related to kidney disease of drugs targeting mitochondria according to clinicaltrials.gov, accessed on 20 May 2022.
Table 3.
Clinical trials related to kidney disease of drugs targeting mitochondria according to clinicaltrials.gov, accessed on 20 May 2022.
| Drug (Family of Drugs) | Clinicaltrials.Gov Identifier (Phase) | Title | Disease or Condition | Status |
|---|---|---|---|---|
| Elamipretide * (SS-peptide) | NCT02436447 (Phase 1) | A Phase 1 Study Investigating the Safety and Pharmacokinetics of Repeat-dose Intravenous Infusion of MTP-131 in Subjects with Impaired Renal Function | Normal and impaired renal function | C |
| NCT01755858 (Phase 1, 2) [158] | Effects of Intravenous Bendavia™ on Reperfusion Injury in Patients Undergoing Angioplasty of the Renal Artery (EVOLVE) | Renal artery obstruction, hypertension, renovascular ischemia reperfusion injury | T | |
| NCT02914665 (Phase 2) | A Phase 2 Study to Evaluate the Cardiac and Renal Effects of Short Term Treatment With Elamipretide in Patients Hospitalized With Congestion Due to Heart Failure | Heart failure | C | |
| CoQ10 (ETC component) | NCT00307996 (Phase 4) | The Effect of CoQ10 Administration on Hemodialysis Patients | Chronic renal failure hemodialysis | C |
| NCT00908297 (Phase, not applicable) [166] | Safety and Tolerability of Coenzyme Q10 in Hemodialysis Patients | Cardiovascular disease, ESRD, atherosclerosis oxidative stress | C | |
| NCT01408680 (Phase, not applicable) | Assessing the Effect of the Dietary Supplement Coenzyme Q10 on Biomarkers of Oxidative Stress, Systemic Inflammation, and Endothelial Function in Hemodialysis Patients | ESRD receiving thrice weekly hemodialysis | C | |
| NCT03579693 (Phase 2) | Cross-over Randomized Controlled Trial of Coenzyme Q10 or Nicotinamide Riboside in Chronic Kidney Disease | CKD, sarcopenia, frailty | C | |
| NCT04445779 (Phase, not applicable) | Q10 Preloading Before Cardiac Surgery for Kidney Failure Reduction | AKI | R | |
| NCT04972552 (Phase, not applicable) | Watermelon/UBIQuinone Study (WUBI-Q Trial) | Kidney transplantation | R | |
| MitoQ (CoQ10 analogue) | NCT02364648 (Phase 4) | Mitochondrial Oxidative Stress and Vascular Health in Chronic Kidney Disease | CKD | U |
| NCT03960073 (Phase, not applicable) | Chronic Kidney Disease and Heart Failure with Preserved Ejection Fraction: The Role of Mitochondrial Dysfunction | Chronic renal insufficiency, heart failure with preserved ejection fraction | R | |
| NCT04334135 (Phase, not applicable) | The Influence of Mitochondrial-Derived Reactive Oxygen Species on Racial Disparities in Neurovascular Function (MAVHS) | Racial disparities, blood pressure, cardiovascular risk factor, renal function | R |
Abbreviations: CoQ10: coenzyme Q10; ETC: electronic transporter chain; ESRD: end-stage renal disease; CKD: chronic kidney disease. * Elamipretide is also called SS-31, MTP-131, and Bendavia. Status: C: completed, R: Recruiting, T: terminated.
7. Summary and Future Perspectives
In summary, there is increasing preclinical evidence supporting a key role of mitochondria in AKI, the AKI-to-CKD transition, and CKD. However, this has not permeated to the clinic and there are gaps of knowledge regarding the cause-and-effect relationship between mitochondrial injury and kidney injury and the clinical relevance of preclinical studies. The clinical development of some promising agent has stalled (i.e., elamipretide). Others (CoQ10 or its analog MitoQ) are being tested in clinical trials as dietary supplements, which will limit the clinical impact of the results. However, most interventions mainly addressed AKI, while clinical trials exploring kidney protection in CKD are not available and will be difficult to perform. Recent data have identified a key role of mitochondria-related transcription factors (PGC-1α, TFEB) and the fatty acid translocation enzyme CPT1A in kidney disease, unveiling a new set of mitochondria-related therapeutic targets. Another unmet need relates to the lack of clinical diagnostic tests that allow us to categorize the baseline kidney mitochondrial dysfunction/mitochondrial oxidative stress to monitor a patient’s response to therapeutic intervention. The development of such companion diagnostics’ tests should be prioritized to enroll patients most likely to benefit from mitochondria-targeted interventions in clinical trials, to evaluate the pharmacologic effect, to identify responders and non-responders in early phase clinical trials, and to optimize protocols in pivotal trials.
Funding
FIS/Fondos FEDER (PI18/01366, PI19/00588, PI19/00815, DTS18/00032, PI21/00251, PI20/000140 ERA-PerMed-JTC2018 KIDNEY ATTACK AC18/00064 and PERSTIGAN AC18/00071, SPACKDc PMP21/00109, ISCIII-RETIC REDinREN RD016/0009), Sociedad Española de Nefrología, FRIAT, Comunidad de Madrid en Biomedicina B2017/BMD-3686 CIFRA2-CM; Instituto de Salud Carlos III (ISCIII) RICORS2040 (RD21/0005/0001), Financiado por la Unión Europea—NextGenerationEU and Mecanismo para la Recuperación y la Resiliencia (MRR) and Salary support: AC18/00064 to M.F.-B., ISCIII PFIS to A.M.L.-D., Ramon y Cajal program to A.B.S. and M.D.S.N., MICIU to J.G.-M., FEBS Long-Term Fellowship to V.M.
Conflicts of Interest
A.O. has received grants from Sanofi and consultancy or speaker fees or travel support from Advicciene, Astellas, Astrazeneca, Amicus, Amgen, Fresenius Medical Care, GSK, Bayer, Sanofi-Genzyme, Menarini, Mundipharma, Kyowa Kirin, Alexion, Freeline, Idorsia, Chiesi, Otsuka, Novo-Nordisk, and Vifor Fresenius Medical Care Renal Pharma and is Director of the Catedra Mundipharma-UAM of diabetic kidney disease and the Catedra Astrazeneca-UAM of chronic kidney disease and electrolytes. The rest of the authors have nothing to disclose.
References
- Ortiz, A.; Asociación Información Enfermedades Renales Genéticas (AIRG-E); European Kidney Patients’ Federation; Federación Nacional de Asociaciones para la Lucha Contra las Enfermedades del Riñón (ALCER); Fundación Renal Íñigo Álvarez de Toledo (FRIAT); Red de Investigación Renal (REDINREN); Resultados en Salud 2040 (RICORS2040); Sociedad Española de Nefrología (SENEFRO) Council, Sociedad Española de Trasplante (SET) Council; Organización Nacional de Trasplantes (ONT). RICORS2040: The need for collaborative research in chronic kidney disease. Clin. Kidney J. 2022, 15, 372–387. [Google Scholar] [CrossRef]
- Ruiz-Ortega, M.; Rayego-Mateos, S.; Lamas, S.; Ortiz, A.; Rodrigues-Diez, R.R. Targeting the progression of chronic kidney disease. Nat. Rev. Nephrol. 2020, 16, 269–288. [Google Scholar] [CrossRef]
- Fernandez-Fernandez, B.; Sarafidis, P.; Kanbay, M.; Navarro-González, J.F.; Soler, M.J.; Górriz, J.L.; Ortiz, A. SGLT2 inhibitors for non-diabetic kidney disease: Drugs to treat CKD that also improve glycaemia. Clin. Kidney J. 2020, 13, 728–733. [Google Scholar] [CrossRef]
- Chevalier, R.L. The proximal tubule is the primary target of injury and progression of kidney disease: Role of the glomerulotubular junction. Am. J. Physiol. Renal Physiol. 2016, 311, F145–F161. [Google Scholar] [CrossRef]
- Martin-Sanchez, D.; Poveda, J.; Fontecha-Barriuso, M.; Ruiz-Andres, O.; Sanchez-Niño, M.D.; Ruiz-Ortega, M.; Ortiz, A.; Sanz, A.B. Targeting of regulated necrosis in kidney disease. Nefrologia 2018, 38, 125–135. [Google Scholar] [CrossRef]
- Maremonti, F.; Meyer, C.; Linkermann, A. Mechanisms and Models of Kidney Tubular Necrosis and Nephron Loss. J. Am. Soc. Nephrol. 2022, 33, 472–486. [Google Scholar] [CrossRef]
- Suliman, H.B.; Piantadosi, C.A. Mitochondrial Quality Control as a Therapeutic Target. Pharmacol. Rev. 2016, 68, 20–48. [Google Scholar] [CrossRef]
- Ishimoto, Y.; Inagi, R. Mitochondria: A therapeutic target in acute kidney injury. Nephrol. Dial Transplant. 2016, 31, 1062–1069. [Google Scholar] [CrossRef]
- Bhargava, P.; Schnellmann, R.G. Mitochondrial energetics in the kidney. Nat. Rev. Nephrol. 2017, 13, 629–646. [Google Scholar] [CrossRef]
- Emma, F.; Montini, G.; Parikh, S.M.; Salviati, L. Mitochondrial dysfunction in inherited renal disease and acute kidney injury. Nat. Rev. Nephrol. 2016, 12, 267–280. [Google Scholar] [CrossRef]
- Lan, R.; Geng, H.; Singha, P.K.; Saikumar, P.; Bottinger, E.P.; Weinberg, J.M.; Venkatachalam, M.A. Mitochondrial Pathology and Glycolytic Shift during Proximal Tubule Atrophy after Ischemic AKI. J. Am. Soc. Nephrol. 2016, 27, 3356–3367. [Google Scholar] [CrossRef]
- Tang, C.; Cai, J.; Yin, X.M.; Weinberg, J.M.; Venkatachalam, M.A.; Dong, Z. Mitochondrial quality control in kidney injury and repair. Nat. Rev. Nephrol. 2021, 17, 299–318. [Google Scholar] [CrossRef]
- Choi, Y.M.; Kim, H.K.; Shim, W.; Anwar, M.A.; Kwon, J.W.; Kwon, H.K.; Kim, H.J.; Jeong, H.; Kim, H.M.; Hwang, D.; et al. Mechanism of Cisplatin-Induced Cytotoxicity Is Correlated to Impaired Metabolism Due to Mitochondrial ROS Generation. PLoS ONE 2015, 10, e0135083. [Google Scholar] [CrossRef]
- Mao, R.W.; He, S.P.; Lan, J.G.; Zhu, W.Z. Honokiol ameliorates cisplatin-induced acute kidney injury via inhibition of mitochondrial fission. Br. J. Pharmacol. 2022, 179, 3886–3904. [Google Scholar] [CrossRef]
- Zhao, M.; Wang, Y.; Li, L.; Liu, S.; Wang, C.; Yuan, Y.; Yang, G.; Chen, Y.; Cheng, J.; Lu, Y.; et al. Mitochondrial ROS promote mitochondrial dysfunction and inflammation in ischemic acute kidney injury by disrupting TFAM-mediated mtDNA maintenance. Theranostics 2021, 11, 1845–1863. [Google Scholar] [CrossRef] [PubMed]
- Linkermann, A.; Bräsen, J.H.; Darding, M.; Jin, M.K.; Sanz, A.B.; Heller, J.O.; De Zen, F.; Weinlich, R.; Ortiz, A.; Walczak, H.; et al. Two independent pathways of regulated necrosis mediate ischemia-reperfusion injury. Proc. Natl. Acad. Sci. USA 2013, 110, 12024–12029. [Google Scholar] [CrossRef]
- Mulay, S.R.; Honarpisheh, M.M.; Foresto-Neto, O.; Shi, C.; Desai, J.; Zhao, Z.B.; Marschner, J.A.; Popper, B.; Buhl, E.M.; Boor, P.; et al. Mitochondria Permeability Transition versus Necroptosis in Oxalate-Induced AKI. J. Am. Soc. Nephrol. 2019, 30, 1857–1869. [Google Scholar] [CrossRef]
- Aranda-Rivera, A.K.; Cruz-Gregorio, A.; Aparicio-Trejo, O.E.; Pedraza-Chaverri, J. Mitochondrial Redox Signaling and Oxidative Stress in Kidney Diseases. Biomolecules 2021, 11, 1144. [Google Scholar] [CrossRef]
- Bulua, A.C.; Simon, A.; Maddipati, R.; Pelletier, M.; Park, H.; Kim, K.Y.; Sack, M.N.; Kastner, D.L.; Siegel, R.M. Mitochondrial reactive oxygen species promote production of proinflammatory cytokines and are elevated in TNFR1-associated periodic syndrome (TRAPS). J. Exp. Med. 2011, 208, 519–533. [Google Scholar] [CrossRef]
- Fontecha-Barriuso, M.; Martin-Sanchez, D.; Martinez-Moreno, J.M.; Monsalve, M.; Ramos, A.M.; Sanchez-Niño, M.D.; Ruiz-Ortega, M.; Ortiz, A.; Sanz, A.B. The Role of PGC-1α and Mitochondrial Biogenesis in Kidney Diseases. Biomolecules 2020, 10, 347. [Google Scholar] [CrossRef]
- Aparicio-Trejo, O.E.; Avila-Rojas, S.H.; Tapia, E.; Rojas-Morales, P.; León-Contreras, J.C.; Martínez-Klimova, E.; Hernández-Pando, R.; Sánchez-Lozada, L.G.; Pedraza-Chaverri, J. Chronic impairment of mitochondrial bioenergetics and β-oxidation promotes experimental AKI-to-CKD transition induced by folic acid. Free Radic. Biol. Med. 2020, 154, 18–32. [Google Scholar] [CrossRef] [PubMed]
- Jang, H.S.; Noh, M.R.; Jung, E.M.; Kim, W.Y.; Southekal, S.; Guda, C.; Foster, K.W.; Oupicky, D.; Ferrer, F.A.; Padanilam, B.J. Proximal tubule cyclophilin D regulates fatty acid oxidation in cisplatin-induced acute kidney injury. Kidney Int. 2020, 97, 327–339. [Google Scholar] [CrossRef] [PubMed]
- Jang, H.S.; Noh, M.R.; Kim, J.; Padanilam, B.J. Defective Mitochondrial Fatty Acid Oxidation and Lipotoxicity in Kidney Diseases. Front. Med. 2020, 7, 65. [Google Scholar] [CrossRef]
- Li, J.; Yang, Y.; Li, Q.; Wei, S.; Zhou, Y.; Yu, W.; Xue, L.; Zhou, L.; Shen, L.; Lu, G.; et al. STAT6 contributes to renal fibrosis by modulating PPARα-mediated tubular fatty acid oxidation. Cell Death Dis. 2022, 13, 66. [Google Scholar] [CrossRef]
- Daneshgar, N.; Baguley, A.W.; Liang, P.I.; Wu, F.; Chu, Y.; Kinter, M.T.; Benavides, G.A.; Johnson, M.S.; Darley-Usmar, V.; Zhang, J.; et al. Metabolic derangement in polycystic kidney disease mouse models is ameliorated by mitochondrial-targeted antioxidants. Commun. Biol. 2021, 4, 1200. [Google Scholar] [CrossRef]
- Li, M.; Li, C.M.; Ye, Z.C.; Huang, J.; Li, Y.; Lai, W.; Peng, H.; Lou, T.Q. Sirt3 modulates fatty acid oxidation and attenuates cisplatin-induced AKI in mice. J. Cell Mol. Med. 2020, 24, 5109–5121. [Google Scholar] [CrossRef]
- Iavecchia, L.; Cereza García, G.; Sabaté Gallego, M.; Vidal Guitart, X.; Ramos Terrades, N.; de la Torre, J.; Segarra Medrano, A.; Agustí Escasany, A. Drug-related acute renal failure in hospitalised patients. Nefrologia 2015, 35, 523–532. [Google Scholar] [CrossRef]
- Rodrigo, E.; Suberviola, B.; Albines, Z.; Castellanos, Á.; Heras, M.; Rodriguez-Borregán, J.C.; Piñera, C.; Serrano, M.; Arias, M. A comparison of acute kidney injury classification systems in sepsis. Nefrologia 2016, 36, 530–534. [Google Scholar] [CrossRef][Green Version]
- Kwiatkowska, E.; Domański, L.; Dziedziejko, V.; Kajdy, A.; Stefańska, K.; Kwiatkowski, S. The Mechanism of Drug Nephrotoxicity and the Methods for Preventing Kidney Damage. Int. J. Mol. Sci. 2021, 22, 16109. [Google Scholar] [CrossRef]
- Metz-Kurschel, U.; Kurschel, E.; Wagner, K.; Aulbert, E.; Graben, N.; Philipp, T. Folate nephropathy occurring during cytotoxic chemotherapy with high-dose folinic acid and 5-fluorouracil. Renal Fail. 1990, 12, 93–97. [Google Scholar] [CrossRef]
- Martin-Sanchez, D.; Ruiz-Andres, O.; Poveda, J.; Carrasco, S.; Cannata-Ortiz, P.; Sanchez-Niño, M.D.; Ruiz Ortega, M.; Egido, J.; Linkermann, A.; Ortiz, A.; et al. Ferroptosis, but Not Necroptosis, Is Important in Nephrotoxic Folic Acid-Induced AKI. J. Am. Soc. Nephrol. 2017, 28, 218–229. [Google Scholar] [CrossRef] [PubMed]
- Aparicio-Trejo, O.E.; Reyes-Fermín, L.M.; Briones-Herrera, A.; Tapia, E.; León-Contreras, J.C.; Hernández-Pando, R.; Sánchez-Lozada, L.G.; Pedraza-Chaverri, J. Protective effects of N-acetyl-cysteine in mitochondria bioenergetics, oxidative stress, dynamics and S-glutathionylation alterations in acute kidney damage induced by folic acid. Free Radic. Biol. Med. 2019, 130, 379–396. [Google Scholar] [CrossRef] [PubMed]
- Ruiz-Andres, O.; Suarez-Alvarez, B.; Sanchez-Ramos, C.; Monsalve, M.; Sanchez-Nino, M.D.; Ruiz-Ortega, M.; Egido, J.; Ortiz, A.; Sanz, A.B. The inflammatory cytokine TWEAK decreases PGC-1alpha expression and mitochondrial function in acute kidney injury. Kidney Int. 2016, 89, 399–410. [Google Scholar] [CrossRef] [PubMed]
- Fontecha-Barriuso, M.; Martín-Sánchez, D.; Martinez-Moreno, J.M.; Carrasco, S.; Ruiz-Andrés, O.; Monsalve, M.; Sanchez-Ramos, C.; Gómez, M.J.; Ruiz-Ortega, M.; Sánchez-Niño, M.D.; et al. PGC-1α deficiency causes spontaneous kidney inflammation and increases the severity of nephrotoxic AKI. J. Pathol. 2019, 249, 65–78. [Google Scholar] [CrossRef]
- Zhang, W.; Yang, Y.; Gao, H.; Zhang, Y.; Jia, Z.; Huang, S. Inhibition of Mitochondrial Complex I Aggravates Folic Acid-Induced Acute Kidney Injury. Kidney Blood Press. Res. 2019, 44, 1002–1013. [Google Scholar] [CrossRef]
- Basha, R.H.; Priscilla, D.H. An in vivo and in vitro study on the protective effects of N-acetylcysteine on mitochondrial dysfunction in isoproterenol treated myocardial infarcted rats. Exp. Toxicol. Pathol. 2013, 65, 7–14. [Google Scholar] [CrossRef]
- Titeler, M.; Lyon, R.A.; Kuhar, M.J.; Frost, J.F.; Dannals, R.F.; Leonhardt, S.; Bullock, A.; Rydelek, L.T.; Price, D.L.; Struble, R.G. Mu opiate receptors are selectively labelled by [3H]carfentanil in human and rat brain. Eur. J. Pharmacol. 1989, 167, 221–228. [Google Scholar] [CrossRef]
- Wang, H.Z.; Peng, Z.Y.; Wen, X.Y.; Rimmelé, T.; Bishop, J.V.; Kellum, J.A. N-acetylcysteine is effective for prevention but not for treatment of folic acid-induced acute kidney injury in mice. Crit. Care Med. 2011, 39, 2487–2494. [Google Scholar] [CrossRef]
- Manohar, S.; Leung, N. Cisplatin nephrotoxicity: A review of the literature. J. Nephrol. 2018, 31, 15–25. [Google Scholar] [CrossRef]
- Yang, Y.; Liu, H.; Liu, F.; Dong, Z. Mitochondrial dysregulation and protection in cisplatin nephrotoxicity. Arch. Toxicol. 2014, 88, 1249–1256. [Google Scholar] [CrossRef]
- Mapuskar, K.A.; Steinbach, E.J.; Zaher, A.; Riley, D.P.; Beardsley, R.A.; Keene, J.L.; Holmlund, J.T.; Anderson, C.M.; Zepeda-Orozco, D.; Buatti, J.M.; et al. Mitochondrial Superoxide Dismutase in Cisplatin-Induced Kidney Injury. Antioxidants 2021, 10, 1329. [Google Scholar] [CrossRef]
- Mapuskar, K.A.; Wen, H.; Holanda, D.G.; Rastogi, P.; Steinbach, E.; Han, R.; Coleman, M.C.; Attanasio, M.; Riley, D.P.; Spitz, D.R.; et al. Persistent increase in mitochondrial superoxide mediates cisplatin-induced chronic kidney disease. Redox. Biol. 2019, 20, 98–106. [Google Scholar] [CrossRef]
- Morigi, M.; Perico, L.; Rota, C.; Longaretti, L.; Conti, S.; Rottoli, D.; Novelli, R.; Remuzzi, G.; Benigni, A. Sirtuin 3-dependent mitochondrial dynamic improvements protect against acute kidney injury. J. Clin. Investig. 2015, 125, 715–726. [Google Scholar] [CrossRef]
- Ahmed, L.A.; Shehata, N.I.; Abdelkader, N.F.; Khattab, M.M. Tempol, a superoxide dismutase mimetic agent, ameliorates cisplatin-induced nephrotoxicity through alleviation of mitochondrial dysfunction in mice. PLoS ONE 2014, 9, e108889. [Google Scholar] [CrossRef]
- Mukhopadhyay, P.; Horváth, B.; Zsengellér, Z.; Zielonka, J.; Tanchian, G.; Holovac, E.; Kechrid, M.; Patel, V.; Stillman, I.E.; Parikh, S.M.; et al. Mitochondrial-targeted antioxidants represent a promising approach for prevention of cisplatin-induced nephropathy. Free Radic. Biol. Med. 2012, 52, 497–506. [Google Scholar] [CrossRef]
- Kong, M.J.; Bak, S.H.; Han, K.H.; Kim, J.I.; Park, J.W.; Park, K.M. Fragmentation of kidney epithelial cell primary cilia occurs by cisplatin and these cilia fragments are excreted into the urine. Redox Biol. 2019, 20, 38–45. [Google Scholar] [CrossRef]
- Lin, Q.; Li, S.; Jiang, N.; Shao, X.; Zhang, M.; Jin, H.; Zhang, Z.; Shen, J.; Zhou, Y.; Zhou, W.; et al. PINK1-parkin pathway of mitophagy protects against contrast-induced acute kidney injury via decreasing mitochondrial ROS and NLRP3 inflammasome activation. Redox Biol. 2019, 26, 101254. [Google Scholar] [CrossRef]
- Gong, X.; Duan, Y.; Zheng, J.; Ye, Z.; Hei, T.K. Tetramethylpyrazine Prevents Contrast-Induced Nephropathy via Modulating Tubular Cell Mitophagy and Suppressing Mitochondrial Fragmentation, CCL2/CCR2-Mediated Inflammation, and Intestinal Injury. Oxid. Med. Cell Longev. 2019, 2019, 7096912. [Google Scholar] [CrossRef]
- Lei, R.; Zhao, F.; Tang, C.Y.; Luo, M.; Yang, S.K.; Cheng, W.; Li, X.W.; Duan, S.B. Mitophagy Plays a Protective Role in Iodinated Contrast-Induced Acute Renal Tubular Epithelial Cells Injury. Cell Physiol. Biochem. 2018, 46, 975–985. [Google Scholar] [CrossRef]
- Uchino, S.; Kellum, J.A.; Bellomo, R.; Doig, G.S.; Morimatsu, H.; Morgera, S.; Schetz, M.; Tan, I.; Bouman, C.; Macedo, E.; et al. Acute renal failure in critically ill patients: A multinational, multicenter study. JAMA 2005, 294, 813–818. [Google Scholar] [CrossRef]
- Singer, M.; Deutschman, C.S.; Seymour, C.W.; Shankar-Hari, M.; Annane, D.; Bauer, M.; Bellomo, R.; Bernard, G.R.; Chiche, J.D.; Coopersmith, C.M.; et al. The Third International Consensus Definitions for Sepsis and Septic Shock (Sepsis-3). JAMA 2016, 315, 801–810. [Google Scholar] [CrossRef] [PubMed]
- Roushani, J.; Thomas, D.; Oliver, M.J.; Ip, J.; Tang, Y.; Yeung, A.; Taji, L.; Cooper, R.; Magner, P.O.; Garg, A.X.; et al. Acute kidney injury requiring renal replacement therapy in people with COVID-19 disease in Ontario, Canada: A prospective analysis of risk factors and outcomes. Clin. Kidney J. 2022, 15, 507–516. [Google Scholar] [CrossRef] [PubMed]
- Lerolle, N.; Nochy, D.; Guérot, E.; Bruneval, P.; Fagon, J.Y.; Diehl, J.L.; Hill, G. Histopathology of septic shock induced acute kidney injury: Apoptosis and leukocytic infiltration. Intensive Care Med. 2010, 36, 471–478. [Google Scholar] [CrossRef] [PubMed]
- Parikh, S.M.; Yang, Y.; He, L.; Tang, C.; Zhan, M.; Dong, Z. Mitochondrial function and disturbances in the septic kidney. Semin. Nephrol. 2015, 35, 108–119. [Google Scholar] [CrossRef]
- Van der Slikke, E.C.; Star, B.S.; van Meurs, M.; Henning, R.H.; Moser, J.; Bouma, H.R. Sepsis is associated with mitochondrial DNA damage and a reduced mitochondrial mass in the kidney of patients with sepsis-AKI. Crit. Care 2021, 25, 36. [Google Scholar] [CrossRef]
- Hu, Q.; Ren, J.; Ren, H.; Wu, J.; Wu, X.; Liu, S.; Wang, G.; Gu, G.; Guo, K.; Li, J. Urinary Mitochondrial DNA Identifies Renal Dysfunction and Mitochondrial Damage in Sepsis-Induced Acute Kidney Injury. Oxid. Med. Cell Longev. 2018, 2018, 8074936. [Google Scholar] [CrossRef]
- Toro, J.; Manrique-Caballero, C.L.; Gómez, H. Metabolic Reprogramming and Host Tolerance: A Novel Concept to Understand Sepsis-Associated AKI. J. Clin. Med. 2021, 10, 4184. [Google Scholar] [CrossRef]
- Waltz, P.; Carchman, E.; Gomez, H.; Zuckerbraun, B. Sepsis results in an altered renal metabolic and osmolyte profile. J. Surg. Res. 2016, 202, 8–12. [Google Scholar] [CrossRef]
- Tran, M.; Tam, D.; Bardia, A.; Bhasin, M.; Rowe, G.C.; Kher, A.; Zsengeller, Z.K.; Akhavan-Sharif, M.R.; Khankin, E.V.; Saintgeniez, M.; et al. PGC-1α promotes recovery after acute kidney injury during systemic inflammation in mice. J. Clin. Investig. 2011, 121, 4003–4014. [Google Scholar] [CrossRef]
- Yu, H.; Jin, F.; Liu, D.; Shu, G.; Wang, X.; Qi, J.; Sun, M.; Yang, P.; Jiang, S.; Ying, X.; et al. ROS-responsive nano-drug delivery system combining mitochondria-targeting ceria nanoparticles with atorvastatin for acute kidney injury. Theranostics 2020, 10, 2342–2357. [Google Scholar] [CrossRef]
- Liang, N.N.; Zhao, Y.; Guo, Y.Y.; Zhang, Z.H.; Gao, L.; Yu, D.X.; Xu, D.X.; Xu, S. Mitochondria-derived reactive oxygen species are involved in renal cell ferroptosis during lipopolysaccharide-induced acute kidney injury. Int. Immunopharmacol. 2022, 107, 108687. [Google Scholar] [CrossRef]
- Patil, N.K.; Parajuli, N.; MacMillan-Crow, L.A.; Mayeux, P.R. Inactivation of renal mitochondrial respiratory complexes and manganese superoxide dismutase during sepsis: Mitochondria-targeted antioxidant mitigates injury. Am. J. Physiol. Renal Physiol. 2014, 306, F734–F743. [Google Scholar] [CrossRef]
- Hall, A.M.; Rhodes, G.J.; Sandoval, R.M.; Corridon, P.R.; Molitoris, B.A. In vivo multiphoton imaging of mitochondrial structure and function during acute kidney injury. Kidney Int. 2013, 83, 72–83. [Google Scholar] [CrossRef]
- Salvadori, M.; Rosso, G.; Bertoni, E. Update on ischemia-reperfusion injury in kidney transplantation: Pathogenesis and treatment. World J. Transplant. 2015, 5, 52–67. [Google Scholar] [CrossRef]
- Liu, D.; Xu, M.; Ding, L.H.; Lv, L.L.; Liu, H.; Ma, K.L.; Zhang, A.H.; Crowley, S.D.; Liu, B.C. Activation of the Nlrp3 inflammasome by mitochondrial reactive oxygen species: A novel mechanism of albumin-induced tubulointerstitial inflammation. Int. J. Biochem. Cell Biol. 2014, 57, 7–19. [Google Scholar] [CrossRef]
- Ding, W.; Guo, H.; Xu, C.; Wang, B.; Zhang, M.; Ding, F. Mitochondrial reactive oxygen species-mediated NLRP3 inflammasome activation contributes to aldosterone-induced renal tubular cells injury. Oncotarget 2016, 7, 17479–17491. [Google Scholar] [CrossRef]
- Meissner, M.; Viehmann, S.F.; Kurts, C. DAMPening sterile inflammation of the kidney. Kidney Int. 2019, 95, 489–491. [Google Scholar] [CrossRef]
- Birk, A.V.; Liu, S.; Soong, Y.; Mills, W.; Singh, P.; Warren, J.D.; Seshan, S.V.; Pardee, J.D.; Szeto, H.H. The mitochondrial-targeted compound SS-31 re-energizes ischemic mitochondria by interacting with cardiolipin. J. Am. Soc. Nephrol. 2013, 24, 1250–1261. [Google Scholar] [CrossRef]
- Liu, S.; Soong, Y.; Seshan, S.V.; Szeto, H.H. Novel cardiolipin therapeutic protects endothelial mitochondria during renal ischemia and mitigates microvascular rarefaction, inflammation, and fibrosis. Am. J. Physiol. Renal Physiol. 2014, 306, F970–F980. [Google Scholar] [CrossRef]
- Eirin, A.; Li, Z.; Zhang, X.; Krier, J.D.; Woollard, J.R.; Zhu, X.Y.; Tang, H.; Herrmann, S.M.; Lerman, A.; Textor, S.C.; et al. A mitochondrial permeability transition pore inhibitor improves renal outcomes after revascularization in experimental atherosclerotic renal artery stenosis. Hypertension 2012, 60, 1242–1249. [Google Scholar] [CrossRef]
- Plotnikov, E.Y.; Chupyrkina, A.A.; Jankauskas, S.S.; Pevzner, I.B.; Silachev, D.N.; Skulachev, V.P.; Zorov, D.B. Mechanisms of nephroprotective effect of mitochondria-targeted antioxidants under rhabdomyolysis and ischemia/reperfusion. Biochim. Biophys. Acta 2011, 1812, 77–86. [Google Scholar] [CrossRef]
- Liu, Z.; Li, Y.; Li, C.; Yu, L.; Chang, Y.; Qu, M. Delivery of coenzyme Q10 with mitochondria-targeted nanocarrier attenuates renal ischemia-reperfusion injury in mice. Mater. Sci. Eng. C Mater. Biol. Appl. 2021, 131, 112536. [Google Scholar] [CrossRef]
- Parajuli, N.; Campbell, L.H.; Marine, A.; Brockbank, K.G.; Macmillan-Crow, L.A. MitoQ blunts mitochondrial and renal damage during cold preservation of porcine kidneys. PLoS ONE 2012, 7, e48590. [Google Scholar] [CrossRef]
- Li, C.; Zheng, Z.; Xie, Y.; Zhu, N.; Bao, J.; Yu, Q.; Zhou, Z.; Liu, J. Protective effect of taraxasterol on ischemia/reperfusion-induced acute kidney injury via inhibition of oxidative stress, inflammation, and apoptosis. Int. Immunopharmacol. 2020, 89, 107169. [Google Scholar] [CrossRef]
- Pabla, N.; Bajwa, A. Role of Mitochondrial Therapy for Ischemic-Reperfusion Injury and Acute Kidney Injury. Nephron 2022, 146, 253–258. [Google Scholar] [CrossRef]
- Brooks, C.; Wei, Q.; Cho, S.G.; Dong, Z. Regulation of mitochondrial dynamics in acute kidney injury in cell culture and rodent models. J. Clin. Investig. 2009, 119, 1275–1285. [Google Scholar] [CrossRef]
- Zhao, W.; Sui, M.; Chen, R.; Lu, H.; Zhu, Y.; Zhang, L.; Zeng, L. SIRT3 protects kidneys from ischemia-reperfusion injury by modulating the DRP1 pathway to induce mitochondrial autophagy. Life Sci. 2021, 286, 120005. [Google Scholar] [CrossRef] [PubMed]
- Tang, C.; Han, H.; Yan, M.; Zhu, S.; Liu, J.; Liu, Z.; He, L.; Tan, J.; Liu, Y.; Liu, H.; et al. PINK1-PRKN/PARK2 pathway of mitophagy is activated to protect against renal ischemia-reperfusion injury. Autophagy 2018, 14, 880–897. [Google Scholar] [CrossRef] [PubMed]
- Livingston, M.J.; Wang, J.; Zhou, J.; Wu, G.; Ganley, I.G.; Hill, J.A.; Yin, X.M.; Dong, Z. Clearance of damaged mitochondria via mitophagy is important to the protective effect of ischemic preconditioning in kidneys. Autophagy 2019, 15, 2142–2162. [Google Scholar] [CrossRef] [PubMed]
- Ma, H.; Guo, X.; Cui, S.; Wu, Y.; Zhang, Y.; Shen, X.; Xie, C.; Li, J. Dephosphorylation of AMP-activated protein kinase exacerbates ischemia/reperfusion-induced acute kidney injury via mitochondrial dysfunction. Kidney Int. 2022, 101, 315–330. [Google Scholar] [CrossRef]
- Tran, M.T.; Zsengeller, Z.K.; Berg, A.H.; Khankin, E.V.; Bhasin, M.K.; Kim, W.; Clish, C.B.; Stillman, I.E.; Karumanchi, S.A.; Rhee, E.P.; et al. PGC1α drives NAD biosynthesis linking oxidative metabolism to renal protection. Nature 2016, 531, 528–532. [Google Scholar] [CrossRef]
- Jesinkey, S.R.; Funk, J.A.; Stallons, L.J.; Wills, L.P.; Megyesi, J.K.; Beeson, C.C.; Schnellmann, R.G. Formoterol restores mitochondrial and renal function after ischemia-reperfusion injury. J. Am. Soc. Nephrol. 2014, 25, 1157–1162. [Google Scholar] [CrossRef]
- Qi, H.; Casalena, G.; Shi, S.; Yu, L.; Ebefors, K.; Sun, Y.; Zhang, W.; D’Agati, V.; Schlondorff, D.; Haraldsson, B.; et al. Glomerular Endothelial Mitochondrial Dysfunction Is Essential and Characteristic of Diabetic Kidney Disease Susceptibility. Diabetes 2017, 66, 763–778. [Google Scholar] [CrossRef]
- Qi, W.; Keenan, H.A.; Li, Q.; Ishikado, A.; Kannt, A.; Sadowski, T.; Yorek, M.A.; Wu, I.H.; Lockhart, S.; Coppey, L.J.; et al. Pyruvate kinase M2 activation may protect against the progression of diabetic glomerular pathology and mitochondrial dysfunction. Nat. Med. 2017, 23, 753–762. [Google Scholar] [CrossRef]
- Coughlan, M.T.; Nguyen, T.V.; Penfold, S.A.; Higgins, G.C.; Thallas-Bonke, V.; Tan, S.M.; Van Bergen, N.J.; Sourris, K.C.; Harcourt, B.E.; Thorburn, D.R.; et al. Mapping time-course mitochondrial adaptations in the kidney in experimental diabetes. Clin. Sci. 2016, 130, 711–720. [Google Scholar] [CrossRef]
- Zhan, M.; Usman, I.; Yu, J.; Ruan, L.; Bian, X.; Yang, J.; Yang, S.; Sun, L.; Kanwar, Y.S. Perturbations in mitochondrial dynamics by p66Shc lead to renal tubular oxidative injury in human diabetic nephropathy. Clin. Sci. 2018, 132, 1297–1314. [Google Scholar] [CrossRef]
- Li, M.; Wang, X.; Aa, J.; Qin, W.; Zha, W.; Ge, Y.; Liu, L.; Zheng, T.; Cao, B.; Shi, J.; et al. GC/TOFMS analysis of metabolites in serum and urine reveals metabolic perturbation of TCA cycle in db/db mice involved in diabetic nephropathy. Am. J. Physiol. Renal Physiol. 2013, 304, F1317–F1324. [Google Scholar] [CrossRef]
- Coughlan, M.T.; Thorburn, D.R.; Penfold, S.A.; Laskowski, A.; Harcourt, B.E.; Sourris, K.C.; Tan, A.L.; Fukami, K.; Thallas-Bonke, V.; Nawroth, P.P.; et al. RAGE-induced cytosolic ROS promote mitochondrial superoxide generation in diabetes. J. Am. Soc. Nephrol. 2009, 20, 742–752. [Google Scholar] [CrossRef]
- Han, Y.; Xu, X.; Tang, C.; Gao, P.; Chen, X.; Xiong, X.; Yang, M.; Yang, S.; Zhu, X.; Yuan, S.; et al. Reactive oxygen species promote tubular injury in diabetic nephropathy: The role of the mitochondrial ros-txnip-nlrp3 biological axis. Redox Biol. 2018, 16, 32–46. [Google Scholar] [CrossRef]
- Hou, Y.; Li, S.; Wu, M.; Wei, J.; Ren, Y.; Du, C.; Wu, H.; Han, C.; Duan, H.; Shi, Y. Mitochondria-targeted peptide SS-31 attenuates renal injury via an antioxidant effect in diabetic nephropathy. Am. J. Physiol. Renal Physiol. 2016, 310, F547–F559. [Google Scholar] [CrossRef]
- Miyamoto, S.; Zhang, G.; Hall, D.; Oates, P.J.; Maity, S.; Madesh, M.; Han, X.; Sharma, K. Restoring mitochondrial superoxide levels with elamipretide (MTP-131) protects. J. Biol. Chem. 2020, 295, 7249–7260. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Zhu, H.; Wang, X.; Gao, Q.; Li, Z.; Huang, H. CoQ10 ameliorates mitochondrial dysfunction in diabetic nephropathy through mitophagy. J. Endocrinol. 2019, 240, 445–465. [Google Scholar] [CrossRef] [PubMed]
- Lechado Terradas, A.; Zittlau, K.I.; Macek, B.; Fraiberg, M.; Elazar, Z.; Kahle, P.J. Regulation of mitochondrial cargo-selective autophagy by posttranslational modifications. J. Biol. Chem. 2021, 297, 101339. [Google Scholar] [CrossRef] [PubMed]
- Peng, Y.; Liu, H.; Liu, J.; Long, J. Post-translational modifications on mitochondrial metabolic enzymes in cancer. Free Radic. Biol. Med. 2022, 179, 11–23. [Google Scholar] [CrossRef] [PubMed]
- Xu, S.; Jiang, B.; Maitland, K.A.; Bayat, H.; Gu, J.; Nadler, J.L.; Corda, S.; Lavielle, G.; Verbeuren, T.J.; Zuccollo, A.; et al. The thromboxane receptor antagonist S18886 attenuates renal oxidant stress and proteinuria in diabetic apolipoprotein E-deficient mice. Diabetes 2006, 55, 110–119. [Google Scholar] [CrossRef] [PubMed]
- Aluksanasuwan, S.; Plumworasawat, S.; Malaitad, T.; Chaiyarit, S.; Thongboonkerd, V. High glucose induces phosphorylation and oxidation of mitochondrial proteins in renal tubular cells: A proteomics approach. Sci. Rep. 2020, 10, 5843. [Google Scholar] [CrossRef] [PubMed]
- Coughlan, M.T.; Higgins, G.C.; Nguyen, T.V.; Penfold, S.A.; Thallas-Bonke, V.; Tan, S.M.; Ramm, G.; van Bergen, N.J.; Henstridge, D.C.; Sourris, K.C.; et al. Deficiency in Apoptosis-Inducing Factor Recapitulates Chronic Kidney Disease via Aberrant Mitochondrial Homeostasis. Diabetes 2016, 65, 1085–1098. [Google Scholar] [CrossRef]
- Malik, A.N.; Shahni, R.; Iqbal, M.M. Increased peripheral blood mitochondrial DNA in type 2 diabetic patients with nephropathy. Diabetes Res. Clin. Pract. 2009, 86, e22–e24. [Google Scholar] [CrossRef]
- Cao, H.; Wu, J.; Luo, J.; Chen, X.; Yang, J.; Fang, L. Urinary mitochondrial DNA: A potential early biomarker of diabetic nephropathy. Diabetes Metab. Res. Rev. 2019, 35, e3131. [Google Scholar] [CrossRef]
- Afsar, B.; Hornum, M.; Afsar, R.E.; Ertuglu, L.A.; Ortiz, A.; Covic, A.; van Raalte, D.H.; Cherney, D.Z.I.; Kanbay, M. Mitochondrion-driven nephroprotective mechanisms of novel glucose lowering medications. Mitochondrion 2021, 58, 72–82. [Google Scholar] [CrossRef]
- Jefferson, J.A.; Shankland, S.J. The pathogenesis of focal segmental glomerulosclerosis. Adv. Chronic Kidney Dis. 2014, 21, 408–416. [Google Scholar] [CrossRef]
- Imasawa, T.; Rossignol, R. Podocyte energy metabolism and glomerular diseases. Int. J. Biochem. Cell Biol. 2013, 45, 2109–2118. [Google Scholar] [CrossRef]
- Abe, Y.; Sakairi, T.; Kajiyama, H.; Shrivastav, S.; Beeson, C.; Kopp, J.B. Bioenergetic characterization of mouse podocytes. Am. J. Physiol. Cell Physiol. 2010, 299, C464–C476. [Google Scholar] [CrossRef]
- Kaufman, J.; Salomonsen, J.; Skjødt, K.; Thorpe, D. Size polymorphism of chicken major histocompatibility complex-encoded B-G molecules is due to length variation in the cytoplasmic heptad repeat region. Proc. Natl. Acad. Sci. USA 1990, 87, 8277–8281. [Google Scholar] [CrossRef]
- Imasawa, T.; Obre, E.; Bellance, N.; Lavie, J.; Rigothier, C.; Delmas, Y.; Combe, C.; Lacombe, D.; Benard, G.; Claverol, S.; et al. High glucose repatterns human podocyte energy metabolism during differentiation and diabetic nephropathy. FASEB J. 2017, 31, 294–307. [Google Scholar] [CrossRef]
- Brinkkoetter, P.T.; Bork, T.; Salou, S.; Liang, W.; Mizi, A.; Özel, C.; Koehler, S.; Hagmann, H.H.; Ising, C.; Kuczkowski, A.; et al. Anaerobic Glycolysis Maintains the Glomerular Filtration Barrier Independent of Mitochondrial Metabolism and Dynamics. Cell Rep. 2019, 27, 1551–1566.e5. [Google Scholar] [CrossRef]
- Szeto, H.H.; Liu, S.; Soong, Y.; Alam, N.; Prusky, G.T.; Seshan, S.V. Protection of mitochondria prevents high-fat diet-induced glomerulopathy and proximal tubular injury. Kidney Int. 2016, 90, 997–1011. [Google Scholar] [CrossRef]
- Noh, M.R.; Kong, M.J.; Han, S.J.; Kim, J.I.; Park, K.M. Isocitrate dehydrogenase 2 deficiency aggravates prolonged high-fat diet intake-induced hypertension. Redox Biol. 2020, 34, 101548. [Google Scholar] [CrossRef]
- Fernandez-Fernandez, B.; Izquierdo, M.C.; Valiño-Rivas, L.; Nastou, D.; Sanz, A.B.; Ortiz, A.; Sanchez-Niño, M.D. Albumin downregulates Klotho in tubular cells. Nephrol. Dial Transplant. 2018, 33, 1712–1722. [Google Scholar] [CrossRef]
- Chung, K.W.; Dhillon, P.; Huang, S.; Sheng, X.; Shrestha, R.; Qiu, C.; Kaufman, B.A.; Park, J.; Pei, L.; Baur, J.; et al. Mitochondrial Damage and Activation of the STING Pathway Lead to Renal Inflammation and Fibrosis. Cell Metab. 2019, 30, 784–799.e5. [Google Scholar] [CrossRef]
- Li, P.A.; Hou, X.; Hao, S. Mitochondrial biogenesis in neurodegeneration. J. Neurosci. Res. 2017, 95, 2025–2029. [Google Scholar] [CrossRef] [PubMed]
- Villena, J.A. New insights into PGC-1 coactivators: Redefining their role in the regulation of mitochondrial function and beyond. FEBS J. 2015, 282, 647–672. [Google Scholar] [CrossRef] [PubMed]
- Stallons, L.J.; Whitaker, R.M.; Schnellmann, R.G. Suppressed mitochondrial biogenesis in folic acid-induced acute kidney injury and early fibrosis. Toxicol. Lett. 2014, 224, 326–332. [Google Scholar] [CrossRef] [PubMed]
- Lynch, M.R.; Tran, M.T.; Ralto, K.M.; Zsengeller, Z.K.; Raman, V.; Bhasin, S.S.; Sun, N.; Chen, X.; Brown, D.; Rovira, I.I.; et al. TFEB-driven lysosomal biogenesis is pivotal for PGC1α-dependent renal stress resistance. JCI Insight 2019, 5, e126749. [Google Scholar] [CrossRef]
- Smith, J.A.; Stallons, L.J.; Collier, J.B.; Chavin, K.D.; Schnellmann, R.G. Suppression of mitochondrial biogenesis through toll-like receptor 4-dependent mitogen-activated protein kinase kinase/extracellular signal-regulated kinase signaling in endotoxin-induced acute kidney injury. J. Pharmacol. Exp. Ther. 2015, 352, 346–357. [Google Scholar] [CrossRef]
- Kang, H.M.; Ahn, S.H.; Choi, P.; Ko, Y.A.; Han, S.H.; Chinga, F.; Park, A.S.; Tao, J.; Sharma, K.; Pullman, J.; et al. Defective fatty acid oxidation in renal tubular epithelial cells has a key role in kidney fibrosis development. Nat. Med. 2015, 21, 37–46. [Google Scholar] [CrossRef]
- Fontecha-Barriuso, M.; Lopez-Diaz, A.M.; Carriazo, S.; Ortiz, A.; Sanz, A.B. Nicotinamide and acute kidney injury. Clin. Kidney J. 2021, 14, 2453–2462. [Google Scholar] [CrossRef]
- Whitaker, R.M.; Wills, L.P.; Stallons, L.J.; Schnellmann, R.G. cGMP-selective phosphodiesterase inhibitors stimulate mitochondrial biogenesis and promote recovery from acute kidney injury. J. Pharmacol. Exp. Ther. 2013, 347, 626–634. [Google Scholar] [CrossRef]
- Khader, A.; Yang, W.L.; Kuncewitch, M.; Jacob, A.; Prince, J.M.; Asirvatham, J.R.; Nicastro, J.; Coppa, G.F.; Wang, P. Sirtuin 1 activation stimulates mitochondrial biogenesis and attenuates renal injury after ischemia-reperfusion. Transplantation 2014, 98, 148–156. [Google Scholar] [CrossRef]
- Xiao, L.; Zhu, X.; Yang, S.; Liu, F.; Zhou, Z.; Zhan, M.; Xie, P.; Zhang, D.; Li, J.; Song, P.; et al. Rap1 ameliorates renal tubular injury in diabetic nephropathy. Diabetes 2014, 63, 1366–1380. [Google Scholar] [CrossRef]
- Locatelli, M.; Zoja, C.; Zanchi, C.; Corna, D.; Villa, S.; Bolognini, S.; Novelli, R.; Perico, L.; Remuzzi, G.; Benigni, A.; et al. Manipulating Sirtuin 3 pathway ameliorates renal damage in experimental diabetes. Sci. Rep. 2020, 10, 8418. [Google Scholar] [CrossRef]
- Long, J.; Badal, S.S.; Ye, Z.; Wang, Y.; Ayanga, B.A.; Galvan, D.L.; Green, N.H.; Chang, B.H.; Overbeek, P.A.; Danesh, F.R. Long noncoding RNA Tug1 regulates mitochondrial bioenergetics in diabetic nephropathy. J. Clin. Invest. 2016, 126, 4205–4218. [Google Scholar] [CrossRef]
- Yuan, S.; Liu, X.; Zhu, X.; Qu, Z.; Gong, Z.; Li, J.; Xiao, L.; Yang, Y.; Liu, H.; Sun, L.; et al. The Role of TLR4 on PGC-1. Oxid. Med. Cell Longev. 2018, 2018, 6296802. [Google Scholar] [CrossRef]
- Hong, Q.; Zhang, L.; Das, B.; Li, Z.; Liu, B.; Cai, G.; Chen, X.; Chuang, P.Y.; He, J.C.; Lee, K. Increased podocyte Sirtuin-1 function attenuates diabetic kidney injury. Kidney Int. 2018, 93, 1330–1343. [Google Scholar] [CrossRef]
- Zhang, T.; Chi, Y.; Kang, Y.; Lu, H.; Niu, H.; Liu, W.; Li, Y. Resveratrol ameliorates podocyte damage in diabetic mice via SIRT1/PGC-1α mediated attenuation of mitochondrial oxidative stress. J. Cell Physiol. 2019, 234, 5033–5043. [Google Scholar] [CrossRef]
- Li, S.Y.; Park, J.; Qiu, C.; Han, S.H.; Palmer, M.B.; Arany, Z.; Susztak, K. Increasing the level of peroxisome proliferator-activated receptor γ coactivator-1α in podocytes results in collapsing glomerulopathy. JCI Insight 2017, 2, e92930. [Google Scholar] [CrossRef]
- Ruiz-Andres, O.; Sanchez-Niño, M.D.; Moreno, J.A.; Ruiz-Ortega, M.; Ramos, A.M.; Sanz, A.B.; Ortiz, A. Downregulation of kidney protective factors by inflammation: Role of transcription factors and epigenetic mechanisms. Am. J. Physiol. Renal Physiol. 2016, 311, F1329–F1340. [Google Scholar] [CrossRef]
- Puertollano, R.; Ferguson, S.M.; Brugarolas, J.; Ballabio, A. The complex relationship between TFEB transcription factor phosphorylation and subcellular localization. EMBO J. 2018, 37, e98804. [Google Scholar] [CrossRef]
- Wang, S.; Chen, Y.; Li, X.; Zhang, W.; Liu, Z.; Wu, M.; Pan, Q.; Liu, H. Emerging role of transcription factor EB in mitochondrial quality control. BioMed. Pharmacother. 2020, 128, 110272. [Google Scholar] [CrossRef]
- Ivankovic, D.; Chau, K.Y.; Schapira, A.H.; Gegg, M.E. Mitochondrial and lysosomal biogenesis are activated following PINK1/parkin-mediated mitophagy. J. Neurochem. 2016, 136, 388–402. [Google Scholar] [CrossRef]
- Yuan, L.; Yuan, Y.; Liu, F.; Li, L.; Liu, J.; Chen, Y.; Cheng, J.; Lu, Y. PGC-1α alleviates mitochondrial dysfunction via TFEB-mediated autophagy in cisplatin-induced acute kidney injury. Aging 2021, 13, 8421–8439. [Google Scholar] [CrossRef] [PubMed]
- Zhu, L.; Yuan, Y.; Yuan, L.; Li, L.; Liu, F.; Liu, J.; Chen, Y.; Lu, Y.; Cheng, J. Activation of TFEB-mediated autophagy by trehalose attenuates mitochondrial dysfunction in cisplatin-induced acute kidney injury. Theranostics 2020, 10, 5829–5844. [Google Scholar] [CrossRef] [PubMed]
- Yuan, H.; Zheng, C.; Zhu, L.; Song, Z.; Dai, L.; Hu, Q.; Wang, L.; Chen, Y.; Xiong, J. Contribution of TFEB-mediated autophagy to tubulointerstitial fibrosis in mice with adenine-induced chronic kidney disease. BioMed. Pharmacother. 2021, 133, 110949. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Huang, H.; Jin, Y.; Shen, K.; Chen, X.; Xu, Z.; Jin, B.; Pan, H. Role of TFEB in autophagic modulation of ischemia reperfusion injury in mice kidney and protection by urolithin A. Food Chem. Toxicol. 2019, 131, 110591. [Google Scholar] [CrossRef]
- Li, R.; Zhao, X.; Zhang, S.; Dong, W.; Zhang, L.; Chen, Y.; Li, Z.; Yang, H.; Huang, Y.; Xie, Z.; et al. RIP3 impedes transcription factor EB to suppress autophagic degradation in septic acute kidney injury. Cell Death Dis. 2021, 12, 593. [Google Scholar] [CrossRef]
- Brijmohan, A.S.; Batchu, S.N.; Majumder, S.; Alghamdi, T.A.; Thieme, K.; McGaugh, S.; Liu, Y.; Advani, S.L.; Bowskill, B.B.; Kabir, M.G.; et al. HDAC6 Inhibition Promotes Transcription Factor EB Activation and Is Protective in Experimental Kidney Disease. Front. Pharmacol. 2018, 9, 34. [Google Scholar] [CrossRef]
- Simon, N.; Hertig, A. Alteration of Fatty Acid Oxidation in Tubular Epithelial Cells: From Acute Kidney Injury to Renal Fibrogenesis. Front. Med. 2015, 2, 52. [Google Scholar] [CrossRef]
- Schlaepfer, I.R.; Joshi, M. CPT1A-mediated Fat Oxidation, Mechanisms, and Therapeutic Potential. Endocrinology 2020, 161, bqz046. [Google Scholar] [CrossRef]
- Miguel, V.; Tituaña, J.; Herrero, J.I.; Herrero, L.; Serra, D.; Cuevas, P.; Barbas, C.; Puyol, D.R.; Márquez-Expósito, L.; Ruiz-Ortega, M.; et al. Renal tubule Cpt1a overexpression protects from kidney fibrosis by restoring mitochondrial homeostasis. J. Clin. Investig. 2021, 131, e140695. [Google Scholar] [CrossRef]
- Afshinnia, F.; Rajendiran, T.M.; Soni, T.; Byun, J.; Wernisch, S.; Sas, K.M.; Hawkins, J.; Bellovich, K.; Gipson, D.; Michailidis, G.; et al. Impaired β-Oxidation and Altered Complex Lipid Fatty Acid Partitioning with Advancing CKD. J. Am. Soc. Nephrol. 2018, 29, 295–306. [Google Scholar] [CrossRef]
- Yuan, Q.; Lv, Y.; Ding, H.; Ke, Q.; Shi, C.; Luo, J.; Jiang, L.; Yang, J.; Zhou, Y. CPT1α maintains phenotype of tubules via mitochondrial respiration during kidney injury and repair. Cell Death Dis. 2021, 12, 792. [Google Scholar] [CrossRef]
- Mori, Y.; Ajay, A.K.; Chang, J.H.; Mou, S.; Zhao, H.; Kishi, S.; Li, J.; Brooks, C.R.; Xiao, S.; Woo, H.M.; et al. KIM-1 mediates fatty acid uptake by renal tubular cells to promote progressive diabetic kidney disease. Cell Metab. 2021, 33, 1042–1061.e7. [Google Scholar] [CrossRef]
- Chen, Y.; Yan, Q.; Lv, M.; Song, K.; Dai, Y.; Huang, Y.; Zhang, L.; Zhang, C.; Gao, H. Involvement of FATP2-mediated tubular lipid metabolic reprogramming in renal fibrogenesis. Cell Death Dis. 2020, 11, 994. [Google Scholar] [CrossRef]
- Piret, S.E.; Attallah, A.A.; Gu, X.; Guo, Y.; Gujarati, N.A.; Henein, J.; Zollman, A.; Hato, T.; Ma’ayan, A.; Revelo, M.P.; et al. Loss of proximal tubular transcription factor Krüppel-like factor 15 exacerbates kidney injury through loss of fatty acid oxidation. Kidney Int. 2021, 100, 1250–1267. [Google Scholar] [CrossRef]
- Price, N.L.; Miguel, V.; Ding, W.; Singh, A.K.; Malik, S.; Rotllan, N.; Moshnikova, A.; Toczek, J.; Zeiss, C.; Sadeghi, M.M.; et al. Genetic deficiency or pharmacological inhibition of miR-33 protects from kidney fibrosis. JCI Insight 2019, 4, e131102. [Google Scholar] [CrossRef]
- Miguel, V.; Ramos, R.; García-Bermejo, L.; Rodríguez-Puyol, D.; Lamas, S. The program of renal fibrogenesis is controlled by microRNAs regulating oxidative metabolism. Redox Biol. 2021, 40, 101851. [Google Scholar] [CrossRef]
- Han, S.H.; Wu, M.Y.; Nam, B.Y.; Park, J.T.; Yoo, T.H.; Kang, S.W.; Park, J.; Chinga, F.; Li, S.Y.; Susztak, K. PGC-1α Protects from Notch-Induced Kidney Fibrosis Development. J. Am. Soc. Nephrol. 2017, 28, 3312–3322. [Google Scholar] [CrossRef]
- Chen, K.H.; Hsu, H.H.; Lee, C.C.; Yen, T.H.; Ko, Y.C.; Yang, C.W.; Hung, C.C. The AMPK agonist AICAR inhibits TGF-β1 induced activation of kidney myofibroblasts. PLoS ONE 2014, 9, e106554. [Google Scholar] [CrossRef]
- Idrovo, J.P.; Yang, W.L.; Nicastro, J.; Coppa, G.F.; Wang, P. Stimulation of carnitine palmitoyltransferase 1 improves renal function and attenuates tissue damage after ischemia/reperfusion. J. Surg. Res. 2012, 177, 157–164. [Google Scholar] [CrossRef]
- Zhang, X.; Agborbesong, E.; Li, X. The Role of Mitochondria in Acute Kidney Injury and Chronic Kidney Disease and Its Therapeutic Potential. Int. J. Mol. Sci. 2021, 22, 11253. [Google Scholar] [CrossRef]
- Tábara, L.C.; Poveda, J.; Martin-Cleary, C.; Selgas, R.; Ortiz, A.; Sanchez-Niño, M.D. Mitochondria-targeted therapies for acute kidney injury. Expert Rev. Mol. Med. 2014, 16, e13. [Google Scholar] [CrossRef] [PubMed]
- Wei, Q.; Dong, G.; Chen, J.K.; Ramesh, G.; Dong, Z. Bax and Bak have critical roles in ischemic acute kidney injury in global and proximal tubule-specific knockout mouse models. Kidney Int. 2013, 84, 138–148. [Google Scholar] [CrossRef] [PubMed]
- Singh, D.; Chander, V.; Chopra, K. Cyclosporine protects against ischemia/reperfusion injury in rat kidneys. Toxicology 2005, 207, 339–347. [Google Scholar] [CrossRef] [PubMed]
- González-Guerrero, C.; Ocaña-Salceda, C.; Berzal, S.; Carrasco, S.; Fernández-Fernández, B.; Cannata-Ortiz, P.; Egido, J.; Ortiz, A.; Ramos, A.M. Calcineurin inhibitors recruit protein kinases JAK2 and JNK, TLR signaling and the UPR to activate NF-κB-mediated inflammatory responses in kidney tubular cells. Toxicol. Appl. Pharmacol. 2013, 272, 825–841. [Google Scholar] [CrossRef]
- Claus, M.; Herro, R.; Wolf, D.; Buscher, K.; Rudloff, S.; Huynh-Do, U.; Burkly, L.; Croft, M.; Sidler, D. The TWEAK/Fn14 pathway is required for calcineurin inhibitor toxicity of the kidneys. Am. J. Transplant. 2018, 18, 1636–1645. [Google Scholar] [CrossRef]
- Li, S.; Lin, Q.; Shao, X.; Zhu, X.; Wu, J.; Wu, B.; Zhang, M.; Zhou, W.; Zhou, Y.; Jin, H.; et al. Drp1-regulated PARK2-dependent mitophagy protects against renal fibrosis in unilateral ureteral obstruction. Free Radic. Biol. Med. 2020, 152, 632–649. [Google Scholar] [CrossRef]
- Birk, A.V.; Chao, W.M.; Bracken, C.; Warren, J.D.; Szeto, H.H. Targeting mitochondrial cardiolipin and the cytochrome c/cardiolipin complex to promote electron transport and optimize mitochondrial ATP synthesis. Br. J. Pharmacol. 2014, 171, 2017–2028. [Google Scholar] [CrossRef]
- Saad, A.; Herrmann, S.M.S.; Eirin, A.; Ferguson, C.M.; Glockner, J.F.; Bjarnason, H.; McKusick, M.A.; Misra, S.; Lerman, L.O.; Textor, S.C. Phase 2a Clinical Trial of Mitochondrial Protection (Elamipretide) During Stent Revascularization in Patients With Atherosclerotic Renal Artery Stenosis. Circ. Cardiovasc. Interv. 2017, 10, e005487. [Google Scholar] [CrossRef]
- Daubert, M.A.; Yow, E.; Dunn, G.; Marchev, S.; Barnhart, H.; Douglas, P.S.; O’Connor, C.; Goldstein, S.; Udelson, J.E.; Sabbah, H.N. Novel Mitochondria-Targeting Peptide in Heart Failure Treatment: A Randomized, Placebo-Controlled Trial of Elamipretide. Circ. Heart Fail. 2017, 10, e004389. [Google Scholar] [CrossRef]
- Suzuki, T.; Yamaguchi, H.; Kikusato, M.; Hashizume, O.; Nagatoishi, S.; Matsuo, A.; Sato, T.; Kudo, T.; Matsuhashi, T.; Murayama, K.; et al. Mitochonic Acid 5 Binds Mitochondria and Ameliorates Renal Tubular and Cardiac Myocyte Damage. J. Am. Soc. Nephrol. 2016, 27, 1925–1932. [Google Scholar] [CrossRef]
- Blanco, L.P.; Pedersen, H.L.; Wang, X.; Lightfoot, Y.L.; Seto, N.; Carmona-Rivera, C.; Yu, Z.X.; Hoffmann, V.; Yuen, P.S.T.; Kaplan, M.J. Improved Mitochondrial Metabolism and Reduced Inflammation Following Attenuation of Murine Lupus With Coenzyme Q10 Analog Idebenone. Arthritis Rheumatol. 2020, 72, 454–464. [Google Scholar] [CrossRef] [PubMed]
- Fortner, K.A.; Blanco, L.P.; Buskiewicz, I.; Huang, N.; Gibson, P.C.; Cook, D.L.; Pedersen, H.L.; Yuen, P.S.T.; Murphy, M.P.; Perl, A.; et al. Targeting mitochondrial oxidative stress with MitoQ reduces NET formation and kidney disease in lupus-prone MRL. Lupus Sci. Med. 2020, 7, e000387. [Google Scholar] [CrossRef] [PubMed]
- Xiao, L.; Xu, X.; Zhang, F.; Wang, M.; Xu, Y.; Tang, D.; Wang, J.; Qin, Y.; Liu, Y.; Tang, C.; et al. The mitochondria-targeted antioxidant MitoQ ameliorated tubular injury mediated by mitophagy in diabetic kidney disease via Nrf2/PINK1. Redox Biol. 2017, 11, 297–311. [Google Scholar] [CrossRef]
- Hamed, M.; Logan, A.; Gruszczyk, A.V.; Beach, T.E.; James, A.M.; Dare, A.J.; Barlow, A.; Martin, J.; Georgakopoulos, N.; Gane, A.M.; et al. Mitochondria-targeted antioxidant MitoQ ameliorates ischaemia-reperfusion injury in kidney transplantation models. Br. J. Surg. 2021, 108, 1072–1081. [Google Scholar] [CrossRef]
- Correction: Targeting mitochondrial oxidative stress with MitoQ reduces NET formation and kidney disease in lupus-prone MRL-lpr mice. Lupus Sci. Med. 2020, 7, e000387corr1. [CrossRef] [PubMed]
- Yeung, C.K.; Billings, F.T.; Claessens, A.J.; Roshanravan, B.; Linke, L.; Sundell, M.B.; Ahmad, S.; Shao, B.; Shen, D.D.; Ikizler, T.A.; et al. Coenzyme Q10 dose-escalation study in hemodialysis patients: Safety, tolerability, and effect on oxidative stress. BMC Nephrol. 2015, 16, 183. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).