
Antioxidant Therapeutics in Parkinson’s Disease: Current Challenges and Opportunities



 , , and
, , and
Abstract
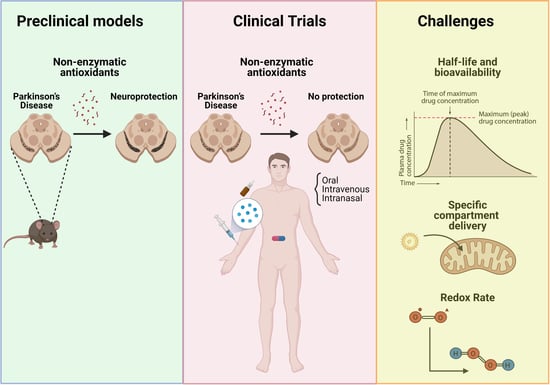
:
1. Introduction
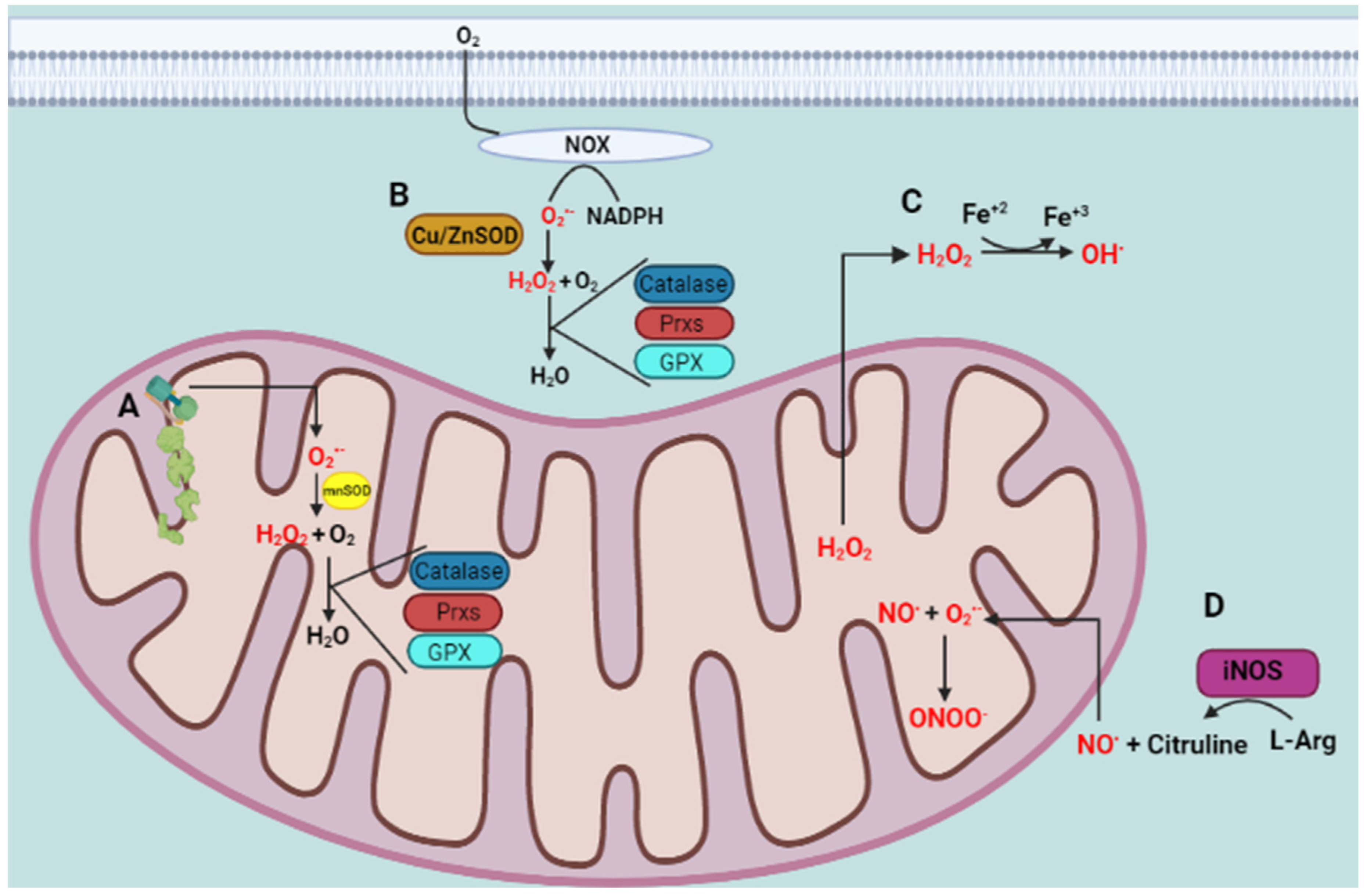
2. Oxidative Stress and Antioxidant Defense
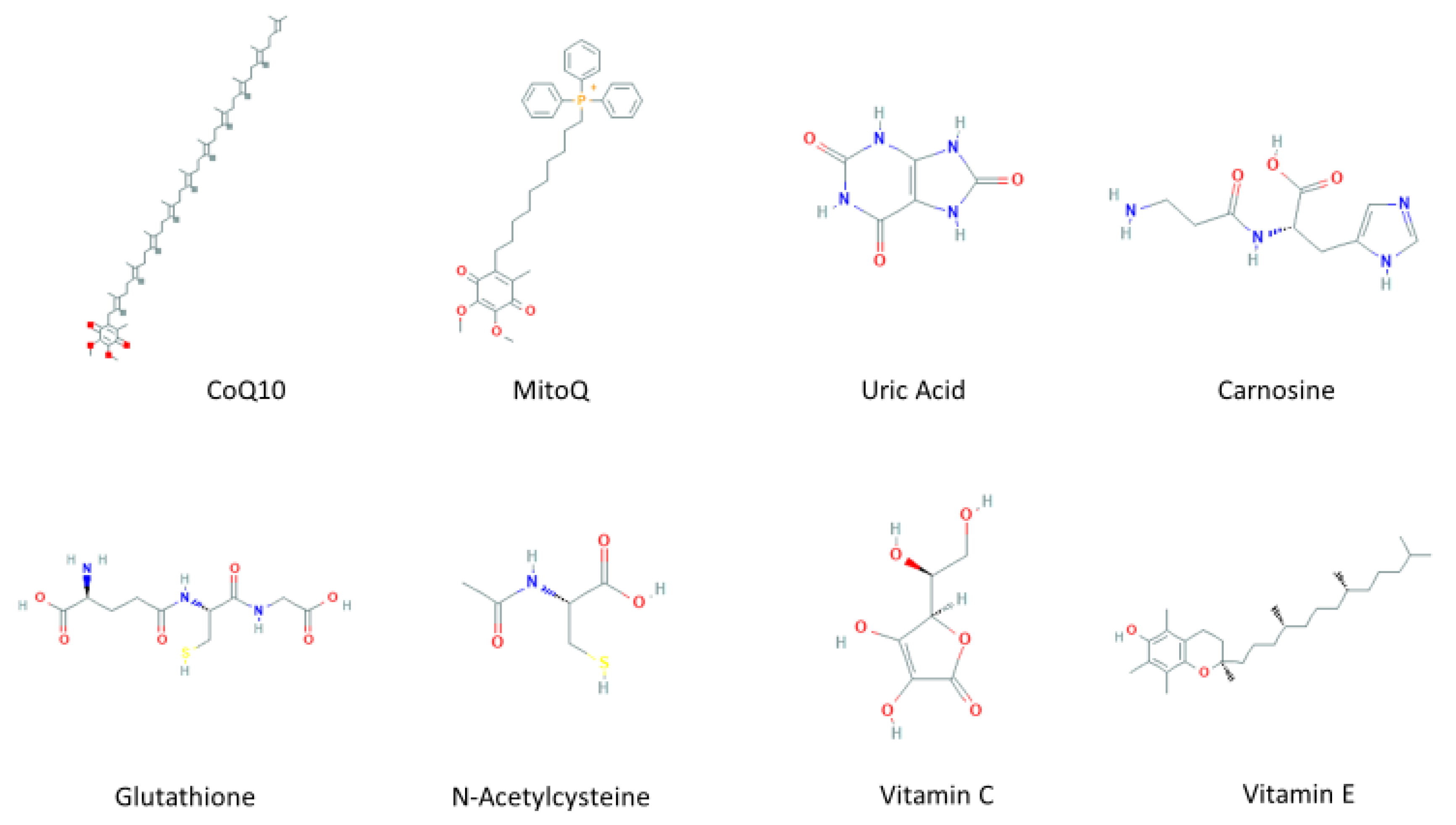
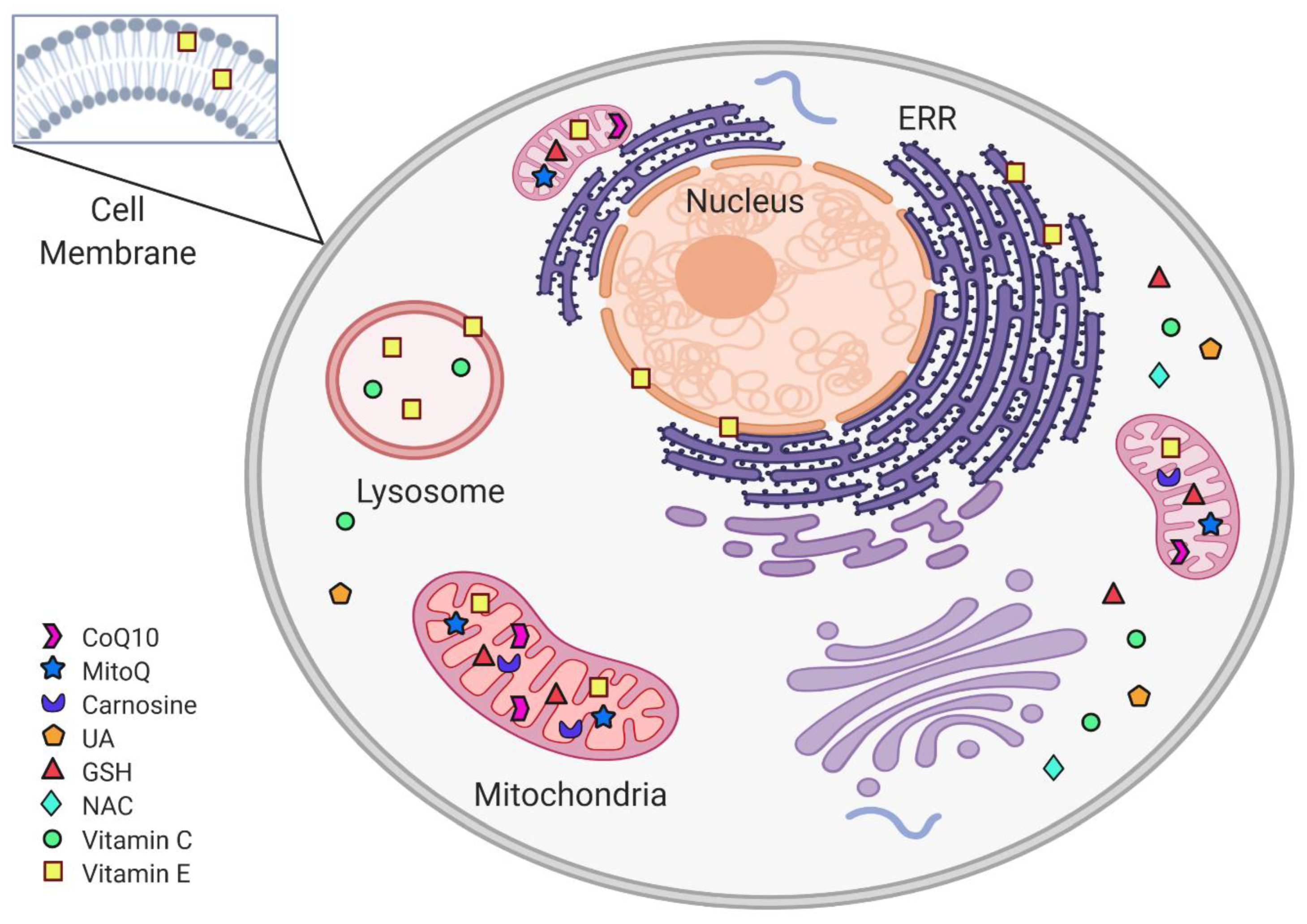
3. Mitochondria-Targeted Antioxidants Therapeutic Effect on PD
3.1. Coenzyme Q10
3.2. Mitochondria-Targeted Antioxidant (MitoQ)
3.3. Carnosine

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Antioxidant | Study Design | Clinical Trial | Subjects (n) | Primary Outcomes | Reference |
|---|---|---|---|---|---|
| Coenzyme Q10 | Pilot study | Not applicable | 10 | Safe and well-tolerated No significant effect on the clinical ratings | [67] |
| Pilot Study | Not applicable | 15 | Safe and well-tolerated No significant effect on the clinical ratings (UPDRS) | [68] | |
| Multicenter, randomized, parallel-group, placebo-controlled, double-blind, dosage-ranging | Phase II | 80 | Safe and well-tolerated at dosages of up to 1200 mg/d ↓ Disability developed, and the benefit was most significant in subjects receiving the highest dosage Slow the progressive deterioration of function in PD | [69] | |
| Randomized, placebo-controlled, double-blind clinical trial | Phase III | 397 + 201 | safe and well-tolerated No evidence of clinical benefit | [70] | |
| Randomized, double-blind, placebo-controlled, parallel-group pilot trials | Phase II | 28 + 20 | ↓ UPDRS score | [71] | |
| Meta-Analysis | Phase III | Does not slow functional decline or provide any symptomatic benefits | [72] | ||
| Double-blind, randomized, and placebo-controlled | Phase II | Total predict number: 84 | This study focuses on genetically stratified subgroups of Parkinson’s disease patients (PD) with enrichment of risk variants in mitochondrial genes, who might benefit from treatment with the “mitochondrial enhancer” coenzyme Q10 | [73] | |
| MitoQ | Double-blind, placebo-controlled study | Phase II | 128 | Did not slow the progression of PD | [80] |
| Carnosine | Pilot comparative clinical trial | Phase I | 36 + 20 | ↑ The efficiency of PD patients’ primary therapy. ↓ UPDRS score Restoration of SOD | [85] |
4. Inosine Therapeutic Effect on PD
| Antioxidant | Study Design | Clinical Trial | Subjects (n) | Primary Outcomes | Reference |
|---|---|---|---|---|---|
| Inosine | Prospective cohort study | NA | 4695 | ↑ serum levels of UA ↓ risk of PD | [94] |
| Double-blind randomized | NA | 800 | ↑ serum and CSF UA ↓ clinical decline in PD | [95] | |
| Randomized, double-blind, placebo-controlled, dose-ranging | Phase II | 75 | ↑serum and CSF urate levels in early PD | [98] | |
| Randomized, double-blind, placebo-controlled | Phase II | 75 | Dose-dependent and persistent elevation of plasma antioxidant capacity form oral inosine | [97] | |
| Placebo-controlled double-blind dose-ranging | Phase II b | 75 | ↑ serum and CSF urate in women compared to men | [98] |
5. Cysteine-Based Antioxidants Therapeutic Effect on PD
5.1. Glutathione
5.2. N-Acetylcysteine (NAC)
| Antioxidant | Study Design | Clinical Trial | Subjects (n) | Primary Outcomes | References |
|---|---|---|---|---|---|
| Glutathione | Pilot evaluation | Phase I | 9 | GSH has symptomatic effects and possibly retards the progression of the disease | [113] |
| Randomized, Double-Blind, Pilot Evaluation | Phase I | 11 + 10 | Safe and well-tolerated | [114] | |
| Randomized, double-blind | Phase I/IIa | 15 + 15 | These data support the safety and tolerability of intranasal GSH in this population Pharmacokinetic and dose-finding studies are warranted | [115,116] | |
| Double-blind placebo-controlled | Phase IIb | 45 | All cohorts improved over the intervention period, although neither treatment group was superior to the placebo | ||
| N-Acetylcysteine | Interventional Open label | Phase I | 6 + 2 | ↑GSH redox ratios ↑ Brain GSH | [128] |
| Single-center | Phase I | 12 | Orally ↑ CSF NAC concentrations | [132] | |
| Randomized open-label | NA (preliminary) | 65 | ↑ DAT binding in the caudate and putamen | [129] | |
| Randomized controlled trial | NA | 65 | ↑ DAT binding in patients with PD ↓ UPDRS score | [131] |
6. Vitamin Therapeutic Effect on PD
6.1. Vitamin C
6.2. Vitamin E
| Antioxidant | Study Design | Clinical Trial | Subjects (n) | Primary Outcomes | Reference |
|---|---|---|---|---|---|
| Vitamin C | Prospective cohort study | 76,890 | Do not reduce the risk of PD | [147] | |
| Comparative study | Phase I | 67 | ↑ Absorption and bioavailability of levodopa | [150] | |
| Randomized Double-blind Controlled Trial | Phase II | 76 | Extend the time necessary to start levodopa therapy to 2.5 years | [149] | |
| Deprenyl and Tocopherol | Placebo-Controlled | Phase I | 800 | There was no beneficial effect of tocopherol or any interaction between tocopherol and deprenyl | [159] |
| Double-blind, randomized clinical | Phase II | 199 + 191 | ↓ UPDRS score | [160] | |
| Prospective cohort study | 1032 | ↓ Parkinson’s disease risk (this result was not significant in a 4-y lag analysis) | [148] |
7. Conclusions
Funding
Acknowledgments
Conflicts of Interest
References
- de Rijk, M.C.; Launer, L.J.; Berger, K.; Breteler, M.M.; Dartigues, J.F.; Baldereschi, M.; Fratiglioni, L.; Lobo, A.; Martinez-Lage, J.; Trenkwalder, C.; et al. Prevalence of Parkinson’s disease in Europe: A collaborative study of population-based cohorts. Neurologic Diseases in the Elderly Research Group. Neurology 2000, 54, S21–S23. [Google Scholar] [PubMed]
- Rajput, A.H. Frequency and cause of Parkinson’s disease. Can. J. Neurol. Sci. 1992, 19, 103–107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Darweesh, S.K.; Koudstaal, P.J.; Stricker, B.H.; Hofman, A.; Ikram, M.A. Trends in the Incidence of Parkinson Disease in the General Population: The Rotterdam Study. Am. J. Epidemiol. 2016, 183, 1018–1026. [Google Scholar] [CrossRef] [Green Version]
- Braak, H.; Del Tredici, K.; Rüb, U.; de Vos, R.A.; Jansen Steur, E.N.; Braak, E. Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiol. Aging 2003, 24, 197–211. [Google Scholar] [CrossRef]
- Przedborski, S. Pathogenesis of nigral cell death in Parkinson’s disease. Parkinsonism Relat. Disord. 2005, 11 (Suppl. 1), S3–S7. [Google Scholar] [CrossRef] [PubMed]
- He, J.; Zhu, G.; Wang, G.; Zhang, F. Oxidative Stress and Neuroinflammation Potentiate Each Other to Promote Progression of Dopamine Neurodegeneration. Oxid. Med. Cell. Longev. 2020, 2020, 6137521. [Google Scholar] [CrossRef]
- Banati, R.B.; Daniel, S.E.; Blunt, S.B. Glial pathology but absence of apoptotic nigral neurons in long-standing Parkinson’s disease. Mov. Disord. 1998, 13, 221–227. [Google Scholar] [CrossRef]
- Damier, P.; Hirsch, E.C.; Zhang, P.; Agid, Y.; Javoy-Agid, F. Glutathione peroxidase, glial cells and Parkinson’s disease. Neuroscience 1993, 52, 1–6. [Google Scholar] [CrossRef]
- Mogi, M.; Harada, M.; Riederer, P.; Narabayashi, H.; Fujita, K.; Nagatsu, T. Tumor necrosis factor-alpha (TNF-alpha) increases both in the brain and in the cerebrospinal fluid from parkinsonian patients. Neurosci. Lett. 1994, 165, 208–210. [Google Scholar] [CrossRef]
- Knott, C.; Stern, G.; Wilkin, G.P. Inflammatory regulators in Parkinson’s disease: iNOS, lipocortin-1, and cyclooxygenases-1 and -2. Mol. Cell. Neurosci. 2000, 16, 724–739. [Google Scholar] [CrossRef]
- Reed, T.T. Lipid peroxidation and neurodegenerative disease. Free Radic. Biol. Med. 2011, 51, 1302–1319. [Google Scholar] [CrossRef] [PubMed]
- Ramalingam, M.; Kim, S.J. Reactive oxygen/nitrogen species and their functional correlations in neurodegenerative diseases. J. Neural Transm. 2012, 119, 891–910. [Google Scholar] [CrossRef] [PubMed]
- Yoritaka, A.; Hattori, N.; Uchida, K.; Tanaka, M.; Stadtman, E.R.; Mizuno, Y. Immunohistochemical detection of 4-hydroxynonenal protein adducts in Parkinson disease. Proc. Natl. Acad. Sci. USA 1996, 93, 2696–2701. [Google Scholar] [CrossRef] [Green Version]
- Khan, Z.; Ali, S.A. Oxidative stress-related biomarkers in Parkinson’s disease: A systematic review and meta-analysis. Iran. J. Neurol. 2018, 17, 137–144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alam, Z.I.; Daniel, S.E.; Lees, A.J.; Marsden, D.C.; Jenner, P.; Halliwell, B. A generalised increase in protein carbonyls in the brain in Parkinson’s but not incidental Lewy body disease. J. Neurochem. 1997, 69, 1326–1329. [Google Scholar] [CrossRef]
- Good, P.F.; Hsu, A.; Werner, P.; Perl, D.P.; Olanow, C.W. Protein nitration in Parkinson’s disease. J. Neuropathol. Exp. Neurol. 1998, 57, 338–342. [Google Scholar] [CrossRef]
- Klein, J.A.; Ackerman, S.L. Oxidative stress, cell cycle, and neurodegeneration. J. Clin. Investig. 2003, 111, 785–793. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Du, H.; Yan, S.S. Mitochondrial medicine for neurodegenerative diseases. Int. J. Biochem. Cell. Biol. 2010, 42, 560–572. [Google Scholar] [CrossRef] [Green Version]
- Kannan, K.; Jain, S.K. Oxidative stress and apoptosis. Pathophysiology 2000, 7, 153–163. [Google Scholar] [CrossRef]
- Giasson, B.I.; Duda, J.E.; Murray, I.V.; Chen, Q.; Souza, J.M.; Hurtig, H.I.; Ischiropoulos, H.; Trojanowski, J.Q.; Lee, V.M. Oxidative damage linked to neurodegeneration by selective alpha-synuclein nitration in synucleinopathy lesions. Science 2000, 290, 985–989. [Google Scholar] [CrossRef]
- Jenner, P.; Olanow, C.W. The pathogenesis of cell death in Parkinson’s disease. Neurology 2006, 66, S24–S36. [Google Scholar] [CrossRef]
- Nimse, S.B.; Pal, D. Free radicals, natural antioxidants, and their reaction mechanisms. RSC Adv. 2015, 5, 27986–28006. [Google Scholar] [CrossRef] [Green Version]
- Swallow, A.J. Radiation Chemistry of Organic Compounds. In Radiation Chemistry of Organic Compounds; Thomson, S., Ed.; Pergamon Press: Oxford, UK, 1960. [Google Scholar]
- Tanaka, M.; Vécsei, L. Monitoring the Redox Status in Multiple Sclerosis. Biomedicines 2020, 8, 406. [Google Scholar] [CrossRef]
- Quinn, M.T.; Ammons, M.C.; Deleo, F.R. The expanding role of NADPH oxidases in health and disease: No longer just agents of death and destruction. Clin. Sci. 2006, 111, 1–20. [Google Scholar] [CrossRef] [Green Version]
- Cairns, R.A.; Harris, I.S.; Mak, T.W. Regulation of cancer cell metabolism. Nat. Rev. Cancer 2011, 11, 85–95. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brookes, P.S.; Yoon, Y.; Robotham, J.L.; Anders, M.W.; Sheu, S.S. Calcium, ATP, and ROS: A mitochondrial love-hate triangle. Am. J. Physiol. Cell Physiol. 2004, 287, C817–C833. [Google Scholar] [CrossRef] [PubMed]
- Lambeth, J.D. NOX enzymes and the biology of reactive oxygen. Nat. Rev. Immunol. 2004, 4, 181–189. [Google Scholar] [CrossRef]
- McCord, J.M.; Fridovich, I. The utility of superoxide dismutase in studying free radical reactions. I. Radicals generated by the interaction of sulfite, dimethyl sulfoxide, and oxygen. J. Biol. Chem. 1969, 244, 6056–6063. [Google Scholar] [CrossRef]
- Hjalmarsson, K.; Marklund, S.L.; Engström, A.; Edlund, T. Isolation and sequence of complementary DNA encoding human extracellular superoxide dismutase. Proc. Natl. Acad. Sci. USA 1987, 84, 6340–6344. [Google Scholar] [CrossRef] [Green Version]
- Salvi, M.; Battaglia, V.; Brunati, A.M.; La Rocca, N.; Tibaldi, E.; Pietrangeli, P.; Marcocci, L.; Mondovì, B.; Rossi, C.A.; Toninello, A. Catalase takes part in rat liver mitochondria oxidative stress defense. J. Biol. Chem. 2007, 282, 24407–24415. [Google Scholar] [CrossRef] [Green Version]
- Netto, L.E.S.; Chae, H.Z.; Kang, S.W.; Rhee, S.G.; Stadtman, E.R. Removal of hydrogen peroxide by thiol-specific antioxidant enzyme (TSA) is involved with its antioxidant properties. TSA possesses thiol peroxidase activity. J. Biol. Chem. 1996, 271, 15315–15321. [Google Scholar] [CrossRef] [Green Version]
- Flohe, L.; Günzler, W.A.; Schock, H.H. Glutathione peroxidase: A selenoenzyme. FEBS Lett. 1973, 32, 132–134. [Google Scholar] [CrossRef] [Green Version]
- Lynch, R.E.; Fridovich, I. Permeation of the erythrocyte stroma by superoxide radical. J. Biol. Chem. 1978, 253, 4697–4699. [Google Scholar] [CrossRef]
- Ramasarma, T. Generation of H2O in biomembranes. Biochim. Biophys. Acta 1982, 694, 69–93. [Google Scholar] [CrossRef]
- Zigler, J.S.; Jernigan, H.M.; Garland, D.; Reddy, V.N. The effects of “oxygen radicals” generated in the medium on lenses in organ culture: Inhibition of damage by chelated iron. Arch. Biochem. Biophys. 1985, 241, 163–172. [Google Scholar] [CrossRef]
- Radi, R. Nitric oxide, oxidants, and protein tyrosine nitration. Proc. Natl. Acad. Sci. USA 2004, 101, 4003–4008. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huie, R.E.; Padmaja, S. The reaction of no with superoxide. Free Radic. Res. Commun. 1993, 18, 195–199. [Google Scholar] [CrossRef] [PubMed]
- Radi, R.; Rodriguez, M.; Castro, L.; Telleri, R. Inhibition of mitochondrial electron transport by peroxynitrite. Arch. Biochem. Biophys. 1994, 308, 89–95. [Google Scholar] [CrossRef]
- Botti, H.; Batthyány, C.; Trostchansky, A.; Radi, R.; Freeman, B.A.; Rubbo, H. Peroxynitrite-mediated alpha-tocopherol oxidation in low-density lipoprotein: A mechanistic approach. Free Radic. Biol. Med. 2004, 36, 152–162. [Google Scholar] [CrossRef] [PubMed]
- Kelso, G.F.; Porteous, C.M.; Coulter, C.V.; Hughes, G.; Porteous, W.K.; Ledgerwood, E.C.; Smith, R.A.; Murphy, M.P. Selective targeting of a redox-active ubiquinone to mitochondria within cells: Antioxidant and antiapoptotic properties. J. Biol. Chem. 2001, 276, 4588–4596. [Google Scholar] [CrossRef] [Green Version]
- Crane, F.L. Biochemical functions of coenzyme Q10. J. Am. Coll. Nutr. 2001, 20, 591–598. [Google Scholar] [CrossRef]
- Kohen, R.; Yamamoto, Y.; Cundy, K.C.; Ames, B.N. Antioxidant activity of carnosine, homocarnosine, and anserine present in muscle and brain. Proc. Natl. Acad. Sci. USA 1988, 85, 3175–3179. [Google Scholar] [CrossRef] [Green Version]
- Zhang, N.; Shu, H.Y.; Huang, T.; Zhang, Q.L.; Li, D.; Zhang, G.Q.; Peng, X.Y.; Liu, C.F.; Luo, W.F.; Hu, L.F. Nrf2 signaling contributes to the neuroprotective effects of urate against 6-OHDA toxicity. PLoS ONE 2014, 9, e100286. [Google Scholar] [CrossRef] [Green Version]
- Samuni, Y.; Goldstein, S.; Dean, O.M.; Berk, M. The chemistry and biological activities of N-acetylcysteine. Biochim. Biophys. Acta 2013, 1830, 4117–4129. [Google Scholar] [CrossRef]
- Beutler, E. Nutritional and metabolic aspects of glutathione. Annu. Rev. Nutr. 1989, 9, 287–302. [Google Scholar] [CrossRef] [PubMed]
- Sies, H.; Stahl, W. Vitamins E and C, beta-carotene, and other carotenoids as antioxidants. Am. J. Clin. Nutr. 1995, 62, 1315S–1321S. [Google Scholar] [CrossRef]
- Ernster, L.; Dallner, G. Biochemical, physiological and medical aspects of ubiquinone function. Biochim. Biophys. Acta 1995, 1271, 195–204. [Google Scholar] [CrossRef] [Green Version]
- Bhagavan, H.N.; Chopra, R.K. Coenzyme Q10: Absorption, tissue uptake, metabolism and pharmacokinetics. Free Radic. Res. 2006, 40, 445–453. [Google Scholar] [CrossRef]
- Kaikkonen, J.; Nyyssönen, K.; Tuomainen, T.P.; Ristonmaa, U.; Salonen, J.T. Determinants of plasma coenzyme Q10 in humans. FEBS Lett. 1999, 443, 163–166. [Google Scholar] [CrossRef] [Green Version]
- Chopra, R.K.; Goldman, R.; Sinatra, S.T.; Bhagavan, H.N. Relative bioavailability of coenzyme Q10 formulations in human subjects. Int. J. Vitam. Nutr. Res. 1998, 68, 109–113. [Google Scholar] [PubMed]
- López-Lluch, G.; Del Pozo-Cruz, J.; Sánchez-Cuesta, A.; Cortés-Rodríguez, A.B.; Navas, P. Bioavailability of coenzyme Q10 supplements depends on carrier lipids and solubilization. Nutrition 2019, 57, 133–140. [Google Scholar] [CrossRef] [PubMed]
- Weis, M.; Mortensen, S.A.; Rassing, M.R.; Møller-Sonnergaard, J.; Poulsen, G.; Rasmussen, S.N. Bioavailability of four oral coenzyme Q10 formulations in healthy volunteers. Mol. Asp. Med. 1994, 15, s273–s280. [Google Scholar] [CrossRef]
- Bhagavan, H.N.; Chopra, R.K. Plasma coenzyme Q10 response to oral ingestion of coenzyme Q10 formulations. Mitochondrion 2007, 7, S78–S88. [Google Scholar] [CrossRef]
- Mantle, D.; Hargreaves, I. Coenzyme Q10 and Degenerative Disorders Affecting Longevity: An Overview. Antioxidants 2019, 8, 44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matthews, R.T.; Yang, L.; Browne, S.; Baik, M.; Beal, M.F. Coenzyme Q10 administration increases brain mitochondrial concentrations and exerts neuroprotective effects. Proc. Natl. Acad. Sci. USA 1998, 95, 8892–8897. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eggens, I.; Elmberger, P.G.; Löw, P. Polyisoprenoid, cholesterol and ubiquinone levels in human hepatocellular carcinomas. Br. J. Exp. Pathol. 1989, 70, 83–92. [Google Scholar]
- Alkholy, U.M.; Abdalmonem, N.; Zaki, A.; Elkoumi, M.A.; Hashim, M.I.A.; Basset, M.A.A.; Salah, H.E. The antioxidant status of coenzyme Q10 and vitamin E in children with type 1 diabetes. J. Pediatr. 2019, 95, 224–230. [Google Scholar] [CrossRef]
- Compta, Y.; Giraldo, D.M.; Muñoz, E.; Antonelli, F.; Fernández, M.; Bravo, P.; Soto, M.; Cámara, A.; Torres, F.; Martí, M.J.; et al. Cerebrospinal fluid levels of coenzyme Q10 are reduced in multiple system atrophy. Parkinsonism Relat. Disord. 2018, 46, 16–23. [Google Scholar] [CrossRef] [PubMed]
- Mischley, L.K.; Allen, J.; Bradley, R. Coenzyme Q10 deficiency in patients with Parkinson’s disease. J. Neurol. Sci. 2012, 318, 72–75. [Google Scholar] [CrossRef] [Green Version]
- Hargreaves, I.P.; Lane, A.; Sleiman, P.M. The coenzyme Q10 status of the brain regions of Parkinson’s disease patients. Neurosci. Lett. 2008, 447, 17–19. [Google Scholar] [CrossRef] [PubMed]
- Sohmiya, M.; Tanaka, M.; Tak, N.W.; Yanagisawa, M.; Tanino, Y.; Suzuki, Y.; Okamoto, K.; Yamamoto, Y. Redox status of plasma coenzyme Q10 indicates elevated systemic oxidative stress in Parkinson’s disease. J. Neurol. Sci. 2004, 223, 161–166. [Google Scholar] [CrossRef]
- Shults, C.W.; Haas, R.H.; Passov, D.; Beal, M.F. Coenzyme Q10 levels correlate with the activities of complexes I and II/III in mitochondria from parkinsonian and nonparkinsonian subjects. Ann. Neurol. 1997, 42, 261–264. [Google Scholar] [CrossRef] [PubMed]
- Gille, G.; Hung, S.T.; Reichmann, H.; Rausch, W.D. Oxidative stress to dopaminergic neurons as models of Parkinson’s disease. Ann. N. Y. Acad. Sci. 2004, 1018, 533–540. [Google Scholar] [CrossRef] [PubMed]
- Sherer, T.B.; Betarbet, R.; Testa, C.M.; Seo, B.B.; Richardson, J.R.; Kim, J.H.; Miller, G.W.; Yagi, T.; Matsuno-Yagi, A.; Greenamyre, J.T. Mechanism of toxicity in rotenone models of Parkinson’s disease. J. Neurosci. 2003, 23, 10756–10764. [Google Scholar] [CrossRef]
- Horvath, T.L.; Diano, S.; Leranth, C.; Garcia-Segura, L.M.; Cowley, M.A.; Shanabrough, M.; Elsworth, J.D.; Sotonyi, P.; Roth, R.H.; Dietrich, E.H.; et al. Coenzyme Q induces nigral mitochondrial uncoupling and prevents dopamine cell loss in a primate model of Parkinson’s disease. Endocrinology 2003, 144, 2757–2760. [Google Scholar] [CrossRef] [Green Version]
- Strijks, E.; Kremer, H.P.; Horstink, M.W. Q10 therapy in patients with idiopathic Parkinson’s disease. Mol. Asp. Med. 1997, 18, S237–S240. [Google Scholar] [CrossRef]
- Shults, C.W.; Beal, M.F.; Fontaine, D.; Nakano, K.; Haas, R.H. Absorption, tolerability, and effects on mitochondrial activity of oral coenzyme Q10 in parkinsonian patients. Neurology 1998, 50, 793–795. [Google Scholar] [CrossRef] [PubMed]
- Shults, C.W.; Oakes, D.; Kieburtz, K.; Beal, M.F.; Haas, R.; Plumb, S.; Juncos, J.L.; Nutt, J.; Shoulson, I.; Carter, J.; et al. Effects of coenzyme Q10 in early Parkinson disease: Evidence of slowing of the functional decline. Arch. Neurol. 2002, 59, 1541–1550. [Google Scholar] [CrossRef] [PubMed]
- Beal, M.F.; Oakes, D.; Shoulson, I.; Henchcliffe, C.; Galpern, W.R.; Haas, R.; Juncos, J.L.; Nutt, J.G.; Voss, T.S.; Ravina, B.; et al. A randomized clinical trial of high-dosage coenzyme Q10 in early Parkinson disease: No evidence of benefit. JAMA Neurol. 2014, 71, 543–552. [Google Scholar] [CrossRef] [PubMed]
- Yoritaka, A.; Kawajiri, S.; Yamamoto, Y.; Nakahara, T.; Ando, M.; Hashimoto, K.; Nagase, M.; Saito, Y.; Hattori, N. Randomized, double-blind, placebo-controlled pilot trial of reduced coenzyme Q10 for Parkinson’s disease. Parkinsonism Relat. Disord. 2015, 21, 911–916. [Google Scholar] [CrossRef] [PubMed]
- Negida, A.; Menshawy, A.; El Ashal, G.; Elfouly, Y.; Hani, Y.; Hegazy, Y.; El Ghonimy, S.; Fouda, S.; Rashad, Y. Coenzyme Q10 for Patients with Parkinson’s Disease: A Systematic Review and Meta-Analysis. CNS Neurol. Disord. Drug Targets 2016, 15, 45–53. [Google Scholar] [CrossRef]
- Prasuhn, J.; Brüggemann, N.; Hessler, N.; Berg, D.; Gasser, T.; Brockmann, K.; Olbrich, D.; Ziegler, A.; König, I.R.; Klein, C.; et al. An omics-based strategy using coenzyme Q10 in patients with Parkinson’s disease: Concept evaluation in a double-blind randomized placebo-controlled parallel group trial. Neurol. Res. Pract. 2019, 1, 31. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Zhang, H.; Fawcett, J.P.; Tucker, I.G. Quantitation and metabolism of mitoquinone, a mitochondria-targeted antioxidant, in rat by liquid chromatography/tandem mass spectrometry. Rapid Commun. Mass Spectrom. 2007, 21, 1958–1964. [Google Scholar] [CrossRef]
- Smith, R.A.; Porteous, C.M.; Gane, A.M.; Murphy, M.P. Delivery of bioactive molecules to mitochondria in vivo. Proc. Natl. Acad. Sci. USA 2003, 100, 5407–5412. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murphy, M.P.; Smith, R.A. Targeting antioxidants to mitochondria by conjugation to lipophilic cations. Annu. Rev. Pharmacol. Toxicol. 2007, 47, 629–656. [Google Scholar] [CrossRef] [PubMed]
- James, A.M.; Cochemé, H.M.; Smith, R.A.; Murphy, M.P. Interactions of mitochondria-targeted and untargeted ubiquinones with the mitochondrial respiratory chain and reactive oxygen species. Implications for the use of exogenous ubiquinones as therapies and experimental tools. J. Biol. Chem. 2005, 280, 21295–21312. [Google Scholar] [CrossRef] [Green Version]
- Solesio, M.E.; Prime, T.A.; Logan, A.; Murphy, M.P.; Del Mar Arroyo-Jimenez, M.; Jordán, J.; Galindo, M.F. The mitochondria-targeted anti-oxidant MitoQ reduces aspects of mitochondrial fission in the 6-OHDA cell model of Parkinson’s disease. Biochim. Biophys. Acta 2013, 1832, 174–182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghosh, A.; Chandran, K.; Kalivendi, S.V.; Joseph, J.; Antholine, W.E.; Hillard, C.J.; Kanthasamy, A.; Kalyanaraman, B. Neuroprotection by a mitochondria-targeted drug in a Parkinson’s disease model. Free Radic. Biol. Med. 2010, 49, 1674–1684. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Snow, B.J.; Rolfe, F.L.; Lockhart, M.M.; Frampton, C.M.; O’Sullivan, J.D.; Fung, V.; Smith, R.A.; Murphy, M.P.; Taylor, K.M.; Group, P.S. A double-blind, placebo-controlled study to assess the mitochondria-targeted antioxidant MitoQ as a disease-modifying therapy in Parkinson’s disease. Mov. Disord. 2010, 25, 1670–1674. [Google Scholar] [CrossRef] [PubMed]
- Boldyrev, A.A.; Aldini, G.; Derave, W. Physiology and pathophysiology of carnosine. Physiol. Rev. 2013, 93, 1803–1845. [Google Scholar] [CrossRef] [PubMed]
- Kubota, M.; Kobayashi, N.; Sugizaki, T.; Shimoda, M.; Kawahara, M.; Tanaka, K.I. Carnosine suppresses neuronal cell death and inflammation induced by 6-hydroxydopamine in an in vitro model of Parkinson’s disease. PLoS ONE 2020, 15, e0240448. [Google Scholar] [CrossRef] [PubMed]
- Yuneva, A.O.; Kramarenko, G.G.; Vetreshchak, T.V.; Gallant, S.; Boldyrev, A.A. Effect of carnosine on Drosophila melanogaster lifespan. Bull. Exp. Biol. Med. 2002, 133, 559–561. [Google Scholar] [CrossRef] [PubMed]
- Bermúdez, M.L.; Seroogy, K.B.; Genter, M.B. Evaluation of Carnosine Intervention in the Thy1-aSyn Mouse Model of Parkinson’s Disease. Neuroscience 2019, 411, 270–278. [Google Scholar] [CrossRef] [PubMed]
- Boldyrev, A.; Fedorova, T.; Stepanova, M.; Dobrotvorskaya, I.; Kozlova, E.; Boldanova, N.; Bagyeva, G.; Ivanova-Smolenskaya, I.; Illarioshkin, S. Carnosine increases efficiency of DOPA therapy of Parkinson’s disease: A pilot study. Rejuvenation Res. 2008, 11, 821–827. [Google Scholar] [CrossRef] [PubMed]
- Gong, L.; Zhang, Q.L.; Zhang, N.; Hua, W.Y.; Huang, Y.X.; Di, P.W.; Huang, T.; Xu, X.S.; Liu, C.F.; Hu, L.F.; et al. Neuroprotection by urate on 6-OHDA-lesioned rat model of Parkinson’s disease: Linking to Akt/GSK3β signaling pathway. J. Neurochem. 2012, 123, 876–885. [Google Scholar] [CrossRef] [PubMed]
- Yeum, K.J.; Russell, R.M.; Krinsky, N.I.; Aldini, G. Biomarkers of antioxidant capacity in the hydrophilic and lipophilic compartments of human plasma. Arch. Biochem. Biophys. 2004, 430, 97–103. [Google Scholar] [CrossRef] [PubMed]
- Becker, B.F. Towards the physiological function of uric acid. Free Radic. Biol. Med. 1993, 14, 615–631. [Google Scholar] [CrossRef]
- Guerreiro, S.; Ponceau, A.; Toulorge, D.; Martin, E.; Alvarez-Fischer, D.; Hirsch, E.C.; Michel, P.P. Protection of midbrain dopaminergic neurons by the end-product of purine metabolism uric acid: Potentiation by low-level depolarization. J. Neurochem. 2009, 109, 1118–1128. [Google Scholar] [CrossRef]
- Hink, H.U.; Santanam, N.; Dikalov, S.; McCann, L.; Nguyen, A.D.; Parthasarathy, S.; Harrison, D.G.; Fukai, T. Peroxidase properties of extracellular superoxide dismutase: Role of uric acid in modulating in vivo activity. Arterioscler. Thromb. Vasc. Biol. 2002, 22, 1402–1408. [Google Scholar] [CrossRef] [Green Version]
- Davies, K.J.; Sevanian, A.; Muakkassah-Kelly, S.F.; Hochstein, P. Uric acid-iron ion complexes. A new aspect of the antioxidant functions of uric acid. Biochem. J. 1986, 235, 747–754. [Google Scholar] [CrossRef] [PubMed]
- Niklasson, F.; Hetta, J.; Degrell, I. Hypoxanthine, xanthine, urate and creatinine concentration gradients in cerebrospinal fluid. Ups. J. Med. Sci. 1988, 93, 225–232. [Google Scholar] [CrossRef] [PubMed]
- Cipriani, S.; Desjardins, C.A.; Burdett, T.C.; Xu, Y.; Xu, K.; Schwarzschild, M.A. Urate and its transgenic depletion modulate neuronal vulnerability in a cellular model of Parkinson’s disease. PLoS ONE 2012, 7, e37331. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Lau, L.M.; Koudstaal, P.J.; Hofman, A.; Breteler, M.M. Serum uric acid levels and the risk of Parkinson disease. Ann. Neurol. 2005, 58, 797–800. [Google Scholar] [CrossRef] [PubMed]
- Ascherio, A.; LeWitt, P.A.; Xu, K.; Eberly, S.; Watts, A.; Matson, W.R.; Marras, C.; Kieburtz, K.; Rudolph, A.; Bogdanov, M.B.; et al. Urate as a predictor of the rate of clinical decline in Parkinson disease. Arch. Neurol. 2009, 66, 1460–1468. [Google Scholar] [CrossRef] [Green Version]
- Zhu, T.G.; Wang, X.X.; Luo, W.F.; Zhang, Q.L.; Huang, T.T.; Xu, X.S.; Liu, C.F. Protective effects of urate against 6-OHDA-induced cell injury in PC12 cells through antioxidant action. Neurosci. Lett. 2012, 506, 175–179. [Google Scholar] [CrossRef]
- Bhattacharyya, S.; Bakshi, R.; Logan, R.; Ascherio, A.; Macklin, E.A.; Schwarzschild, M.A. Oral Inosine Persistently Elevates Plasma antioxidant capacity in Parkinson’s disease. Mov. Disord. 2016, 31, 417–421. [Google Scholar] [CrossRef]
- Schwarzschild, M.A.; Macklin, E.A.; Bakshi, R.; Battacharyya, S.; Logan, R.; Espay, A.J.; Hung, A.Y.; Bwala, G.; Goetz, C.G.; Russell, D.S.; et al. Sex differences by design and outcome in the Safety of Urate Elevation in PD (SURE-PD) trial. Neurology 2019, 93, e1328–e1338. [Google Scholar] [CrossRef]
- Fernández-Checa, J.C.; Kaplowitz, N.; García-Ruiz, C.; Colell, A. Mitochondrial glutathione: Importance and transport. Semin. Liver Dis. 1998, 18, 389–401. [Google Scholar] [CrossRef] [Green Version]
- Ursini, F.; Bindoli, A. The role of selenium peroxidases in the protection against oxidative damage of membranes. Chem. Phys. Lipids 1987, 44, 255–276. [Google Scholar] [CrossRef]
- Pompella, A.; Visvikis, A.; Paolicchi, A.; De Tata, V.; Casini, A.F. The changing faces of glutathione, a cellular protagonist. Biochem. Pharmacol. 2003, 66, 1499–1503. [Google Scholar] [CrossRef]
- Meister, A. Biosynthesis and functions of glutathione, an essential biofactor. J. Nutr. Sci. Vitaminol. 1992, 38, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Pizzorno, J. Glutathione! Integr. Med. 2014, 13, 8–12. [Google Scholar]
- Sian, J.; Dexter, D.T.; Lees, A.J.; Daniel, S.; Agid, Y.; Javoy-Agid, F.; Jenner, P.; Marsden, C.D. Alterations in glutathione levels in Parkinson’s disease and other neurodegenerative disorders affecting basal ganglia. Ann. Neurol. 1994, 36, 348–355. [Google Scholar] [CrossRef]
- Dexter, D.T.; Sian, J.; Rose, S.; Hindmarsh, J.G.; Mann, V.M.; Cooper, J.M.; Wells, F.R.; Daniel, S.E.; Lees, A.J.; Schapira, A.H. Indices of oxidative stress and mitochondrial function in individuals with incidental Lewy body disease. Ann. Neurol. 1994, 35, 38–44. [Google Scholar] [CrossRef] [PubMed]
- Chinta, S.J.; Kumar, M.J.; Hsu, M.; Rajagopalan, S.; Kaur, D.; Rane, A.; Nicholls, D.G.; Choi, J.; Andersen, J.K. Inducible alterations of glutathione levels in adult dopaminergic midbrain neurons result in nigrostriatal degeneration. J. Neurosci. 2007, 27, 13997–14006. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ibi, M.; Sawada, H.; Kume, T.; Katsuki, H.; Kaneko, S.; Shimohama, S.; Akaike, A. Depletion of intracellular glutathione increases susceptibility to nitric oxide in mesencephalic dopaminergic neurons. J. Neurochem. 1999, 73, 1696–1703. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wüllner, U.; Löschmann, P.A.; Schulz, J.B.; Schmid, A.; Dringen, R.; Eblen, F.; Turski, L.; Klockgether, T. Glutathione depletion potentiates MPTP and MPP+ toxicity in nigral dopaminergic neurones. Neuroreport 1996, 7, 921–923. [Google Scholar] [CrossRef]
- Pradhan, P.; Majhi, O.; Biswas, A.; Joshi, V.K.; Sinha, D. Enhanced accumulation of reduced glutathione by Scopoletin improves survivability of dopaminergic neurons in Parkinson’s model. Cell Death Dis. 2020, 11, 739. [Google Scholar] [CrossRef]
- Allen, J.; Bradley, R.D. Effects of oral glutathione supplementation on systemic oxidative stress biomarkers in human volunteers. J. Altern. Complement. Med. 2011, 17, 827–833. [Google Scholar] [CrossRef] [Green Version]
- Richie, J.P.; Nichenametla, S.; Neidig, W.; Calcagnotto, A.; Haley, J.S.; Schell, T.D.; Muscat, J.E. Randomized controlled trial of oral glutathione supplementation on body stores of glutathione. Eur. J. Nutr. 2015, 54, 251–263. [Google Scholar] [CrossRef]
- Witschi, A.; Reddy, S.; Stofer, B.; Lauterburg, B.H. The systemic availability of oral glutathione. Eur. J. Clin. Pharmacol. 1992, 43, 667–669. [Google Scholar] [CrossRef] [PubMed]
- Sechi, G.; Deledda, M.G.; Bua, G.; Satta, W.M.; Deiana, G.A.; Pes, G.M.; Rosati, G. Reduced intravenous glutathione in the treatment of early Parkinson’s disease. Prog. Neuropsychopharmacol. Biol. Psychiatry 1996, 20, 1159–1170. [Google Scholar] [CrossRef]
- Hauser, R.A.; Lyons, K.E.; McClain, T.; Carter, S.; Perlmutter, D. Randomized, double-blind, pilot evaluation of intravenous glutathione in Parkinson’s disease. Mov. Disord. 2009, 24, 979–983. [Google Scholar] [CrossRef] [PubMed]
- Mischley, L.K.; Vespignani, M.F.; Finnell, J.S. Safety survey of intranasal glutathione. J. Altern. Complement. Med. 2013, 19, 459–463. [Google Scholar] [CrossRef] [Green Version]
- Mischley, L.K.; Leverenz, J.B.; Lau, R.C.; Polissar, N.L.; Neradilek, M.B.; Samii, A.; Standish, L.J. A randomized, double-blind phase I/IIa study of intranasal glutathione in Parkinson’s disease. Mov. Disord. 2015, 30, 1696–1701. [Google Scholar] [CrossRef] [PubMed]
- Mischley, L.K.; Lau, R.C.; Shankland, E.G.; Wilbur, T.K.; Padowski, J.M. Phase IIb Study of Intranasal Glutathione in Parkinson’s Disease. J. Parkinsons Dis. 2017, 7, 289–299. [Google Scholar] [CrossRef] [Green Version]
- Smilkstein, M.J.; Knapp, G.L.; Kulig, K.W.; Rumack, B.H. Efficacy of oral N-acetylcysteine in the treatment of acetaminophen overdose. Analysis of the national multicenter study (1976 to 1985). N. Engl. J. Med. 1988, 319, 1557–1562. [Google Scholar] [CrossRef]
- Moldéus, P.; Cotgreave, I.A.; Berggren, M. Lung protection by a thiol-containing antioxidant: N-acetylcysteine. Respiration 1986, 50 (Suppl. 1), 31–42. [Google Scholar] [CrossRef]
- Staal, F.J.; Ela, S.W.; Roederer, M.; Anderson, M.T.; Herzenberg, L.A. Glutathione deficiency and human immunodeficiency virus infection. Lancet 1992, 339, 909–912. [Google Scholar] [CrossRef]
- Medina, S.; Martínez, M.; Hernanz, A. Antioxidants inhibit the human cortical neuron apoptosis induced by hydrogen peroxide, tumor necrosis factor alpha, dopamine and beta-amyloid peptide 1–42. Free Radic. Res. 2002, 36, 1179–1184. [Google Scholar] [CrossRef]
- Pocernich, C.B.; La Fontaine, M.; Butterfield, D.A. In-vivo glutathione elevation protects against hydroxyl free radical-induced protein oxidation in rat brain. Neurochem. Int. 2000, 36, 185–191. [Google Scholar] [CrossRef]
- Martínez Banaclocha, M. N-acetylcysteine elicited increase in complex I activity in synaptic mitochondria from aged mice: Implications for treatment of Parkinson’s disease. Brain Res. 2000, 859, 173–175. [Google Scholar] [CrossRef]
- Martínez Banaclocha, M.; Martínez, N. N-acetylcysteine elicited increase in cytochrome c oxidase activity in mice synaptic mitochondria. Brain Res. 1999, 842, 249–251. [Google Scholar] [CrossRef]
- Clark, J.; Clore, E.L.; Zheng, K.; Adame, A.; Masliah, E.; Simon, D.K. Oral N-acetyl-cysteine attenuates loss of dopaminergic terminals in alpha-synuclein overexpressing mice. PLoS ONE 2010, 5, e12333. [Google Scholar] [CrossRef] [Green Version]
- Holdiness, M.R. Clinical pharmacokinetics of N-acetylcysteine. Clin. Pharmacokinet. 1991, 20, 123–134. [Google Scholar] [CrossRef]
- Mahumane, G.D.; Kumar, P.; Pillay, V.; Choonara, Y.E. Repositioning. Pharmaceutics 2020, 12, 934. [Google Scholar] [CrossRef]
- Holmay, M.J.; Terpstra, M.; Coles, L.D.; Mishra, U.; Ahlskog, M.; Öz, G.; Cloyd, J.C.; Tuite, P.J. N-Acetylcysteine boosts brain and blood glutathione in Gaucher and Parkinson diseases. Clin. Neuropharmacol. 2013, 36, 103–106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Monti, D.A.; Zabrecky, G.; Kremens, D.; Liang, T.W.; Wintering, N.A.; Cai, J.; Wei, X.; Bazzan, A.J.; Zhong, L.; Bowen, B.; et al. N-Acetyl Cysteine May Support Dopamine Neurons in Parkinson’s Disease: Preliminary Clinical and Cell Line Data. PLoS ONE 2016, 11, e0157602. [Google Scholar] [CrossRef] [PubMed]
- Coles, L.D.; Tuite, P.J.; Öz, G.; Mishra, U.R.; Kartha, R.V.; Sullivan, K.M.; Cloyd, J.C.; Terpstra, M. Repeated-Dose Oral N-Acetylcysteine in Parkinson’s Disease: Pharmacokinetics and Effect on Brain Glutathione and Oxidative Stress. J. Clin. Pharmacol. 2018, 58, 158–167. [Google Scholar] [CrossRef] [PubMed]
- Monti, D.A.; Zabrecky, G.; Kremens, D.; Liang, T.W.; Wintering, N.A.; Bazzan, A.J.; Zhong, L.; Bowens, B.K.; Chervoneva, I.; Intenzo, C.; et al. N-Acetyl Cysteine Is Associated With Dopaminergic Improvement in Parkinson’s Disease. Clin. Pharmacol. Ther. 2019, 106, 884–890. [Google Scholar] [CrossRef] [PubMed]
- Katz, M.; Won, S.J.; Park, Y.; Orr, A.; Jones, D.P.; Swanson, R.A.; Glass, G.A. Cerebrospinal fluid concentrations of N-acetylcysteine after oral administration in Parkinson’s disease. Parkinsonism Relat. Disord. 2015, 21, 500–503. [Google Scholar] [CrossRef] [Green Version]
- Linster, C.L.; Van Schaftingen, E.; Vitamin, C. Biosynthesis, recycling and degradation in mammals. FEBS J. 2007, 274, 1–22. [Google Scholar] [CrossRef]
- Njus, D.; Kelley, P.M.; Tu, Y.J.; Schlegel, H.B. Ascorbic acid: The chemistry underlying its antioxidant properties. Free Radic. Biol. Med. 2020, 159, 37–43. [Google Scholar] [CrossRef]
- Packer, J.E.; Slater, T.F.; Willson, R.L. Direct observation of a free radical interaction between vitamin E and vitamin C. Nature 1979, 278, 737–738. [Google Scholar] [CrossRef]
- Tsukaguchi, H.; Tokui, T.; Mackenzie, B.; Berger, U.V.; Chen, X.Z.; Wang, Y.; Brubaker, R.F.; Hediger, M.A. A family of mammalian Na+-dependent L-ascorbic acid transporters. Nature 1999, 399, 70–75. [Google Scholar] [CrossRef] [PubMed]
- Frei, B.; Birlouez-Aragon, I.; Lykkesfeldt, J. Authors’ perspective: What is the optimum intake of vitamin C in humans? Crit. Rev. Food Sci. Nutr. 2012, 52, 815–829. [Google Scholar] [CrossRef]
- Hasselholt, S.; Tveden-Nyborg, P.; Lykkesfeldt, J. Distribution of vitamin C is tissue specific with early saturation of the brain and adrenal glands following differential oral dose regimens in guinea pigs. Br. J. Nutr. 2015, 113, 1539–1549. [Google Scholar] [CrossRef] [Green Version]
- Figueroa-Méndez, R.; Rivas-Arancibia, S. Vitamin C in Health and Disease: Its Role in the Metabolism of Cells and Redox State in the Brain. Front. Physiol. 2015, 6, 397. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stamford, J.A.; Kruk, Z.L.; Millar, J. Regional differences in extracellular ascorbic acid levels in the rat brain determined by high speed cyclic voltammetry. Brain Res. 1984, 299, 289–295. [Google Scholar] [CrossRef]
- Foy, C.J.; Passmore, A.P.; Vahidassr, M.D.; Young, I.S.; Lawson, J.T. Plasma chain-breaking antioxidants in Alzheimer’s disease, vascular dementia and Parkinson’s disease. QJM 1999, 92, 39–45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yapa, S.C. Detection of subclinical ascorbate deficiency in early Parkinson’s disease. Public Health 1992, 106, 393–395. [Google Scholar] [CrossRef]
- Ide, K.; Yamada, H.; Umegaki, K.; Mizuno, K.; Kawakami, N.; Hagiwara, Y.; Matsumoto, M.; Yoshida, H.; Kim, K.; Shiosaki, E.; et al. Lymphocyte vitamin C levels as potential biomarker for progression of Parkinson’s disease. Nutrition 2015, 31, 406–408. [Google Scholar] [CrossRef] [PubMed]
- Pardo, B.; Mena, M.A.; Fahn, S.; García de Yébenes, J. Ascorbic acid protects against levodopa-induced neurotoxicity on a catecholamine-rich human neuroblastoma cell line. Mov. Disord. 1993, 8, 278–284. [Google Scholar] [CrossRef] [PubMed]
- Man Anh, H.; Linh, D.M.; My Dung, V.; Thi Phuong Thao, D. Evaluating Dose- and Time-Dependent Effects of Vitamin C Treatment on a Parkinson’s Disease Fly Model. Parkinsons Dis. 2019, 2019, 9720546. [Google Scholar] [CrossRef] [Green Version]
- Bagga, V.; Dunnett, S.B.; Fricker-Gates, R.A. Ascorbic acid increases the number of dopamine neurons in vitro and in transplants to the 6-OHDA-lesioned rat brain. Cell Transplant. 2008, 17, 763–773. [Google Scholar] [CrossRef] [Green Version]
- Zhang, S.M.; Hernán, M.A.; Chen, H.; Spiegelman, D.; Willett, W.C.; Ascherio, A. Intakes of vitamins E and C, carotenoids, vitamin supplements, and PD risk. Neurology 2002, 59, 1161–1169. [Google Scholar] [CrossRef]
- Hughes, K.C.; Gao, X.; Kim, I.Y.; Rimm, E.B.; Wang, M.; Weisskopf, M.G.; Schwarzschild, M.A.; Ascherio, A. Intake of antioxidant vitamins and risk of Parkinson’s disease. Mov. Disord. 2016, 31, 1909–1914. [Google Scholar] [CrossRef] [Green Version]
- Fahn, S. A pilot trial of high-dose alpha-tocopherol and ascorbate in early Parkinson’s disease. Ann. Neurol. 1992, 32, S128–S132. [Google Scholar] [CrossRef]
- Nagayama, H.; Hamamoto, M.; Ueda, M.; Nito, C.; Yamaguchi, H.; Katayama, Y. The effect of ascorbic acid on the pharmacokinetics of levodopa in elderly patients with Parkinson disease. Clin. Neuropharmacol. 2004, 27, 270–273. [Google Scholar] [CrossRef]
- Burton, G.W.; Joyce, A.; Ingold, K.U. First proof that vitamin E is major lipid-soluble, chain-breaking antioxidant in human blood plasma. Lancet 1982, 2, 327. [Google Scholar] [CrossRef]
- Colombo, M.L. An update on vitamin E, tocopherol and tocotrienol-perspectives. Molecules 2010, 15, 2103–2113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bruno, R.S.; Leonard, S.W.; Park, S.I.; Zhao, Y.; Traber, M.G. Human vitamin E requirements assessed with the use of apples fortified with deuterium-labeled alpha-tocopheryl acetate. Am. J. Clin. Nutr. 2006, 83, 299–304. [Google Scholar] [CrossRef] [PubMed]
- Borel, P.; Preveraud, D.; Desmarchelier, C. Bioavailability of vitamin E in humans: An update. Nutr. Rev. 2013, 71, 319–331. [Google Scholar] [CrossRef] [PubMed]
- Odunze, I.N.; Klaidman, L.K.; Adams, J.D. MPTP toxicity in the mouse brain and vitamin E. Neurosci. Lett. 1990, 108, 346–349. [Google Scholar] [CrossRef]
- Martinovits, G.; Melamed, E.; Cohen, O.; Rosenthal, J.; Uzzan, A. Systemic administration of antioxidants does not protect mice against the dopaminergic neurotoxicity of 1-methyl-4-phenyl-1,2,5,6-tetrahydropyridine (MPTP). Neurosci. Lett. 1986, 69, 192–197. [Google Scholar] [CrossRef]
- Gong, L.; Daigneault, E.A.; Acuff, R.V.; Kostrzewa, R.M. Vitamin E supplements fail to protect mice from acute MPTP neurotoxicity. Neuroreport 1991, 2, 544–546. [Google Scholar] [CrossRef]
- Barc, S.; Page, G.; Barrier, L.; Huguet, F.; Fauconneau, B. Progressive alteration of neuronal dopamine transporter activity in a rat injured by an intranigral injection of MPP+. Brain Res. 2002, 941, 72–81. [Google Scholar] [CrossRef]
- Group, P.S. Effects of tocopherol and deprenyl on the progression of disability in early Parkinson’s disease. N. Engl. J. Med. 1993, 328, 176–183. [Google Scholar] [CrossRef]
- Marras, C.; Lang, A.E.; Eberly, S.W.; Oakes, D.; Fahn, S.; Schwid, S.R.; Hyson, C.; Shoulson, I. A comparison of treatment thresholds in two large Parkinson’s disease clinical trial cohorts. Mov. Disord. 2009, 24, 2370–2378. [Google Scholar] [CrossRef]
- Joutsa, J.; Gardberg, M.; Röyttä, M.; Kaasinen, V. Diagnostic accuracy of parkinsonism syndromes by general neurologists. Parkinsonism Relat. Disord. 2014, 20, 840–844. [Google Scholar] [CrossRef]
- Jankovic, J. Parkinson’s disease: Clinical features and diagnosis. J. Neurol. Neurosurg. Psychiatry 2008, 79, 368–376. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, Y.; Yao, Q.; Jiang, G.X.; Wang, G.; Cheng, Q. Identification of distinct blood-based biomarkers in early stage of Parkinson’s disease. Neurol. Sci. 2020, 41, 893–901. [Google Scholar] [CrossRef]
- Jin, H.; Kanthasamy, A.; Ghosh, A.; Anantharam, V.; Kalyanaraman, B.; Kanthasamy, A.G. Mitochondria-targeted antioxidants for treatment of Parkinson’s disease: Preclinical and clinical outcomes. Biochim. Biophys. Acta 2014, 1842, 1282–1294. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khalil, I.; Yehye, W.A.; Etxeberria, A.E.; Alhadi, A.A.; Dezfooli, S.M.; Julkapli, N.B.M.; Basirun, W.J.; Seyfoddin, A. Nanoantioxidants: Recent Trends in Antioxidant Delivery Applications. Antioxidants 2019, 9, 24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mohd Zaffarin, A.S.; Ng, S.F.; Ng, M.H.; Hassan, H.; Alias, E. Pharmacology and Pharmacokinetics of Vitamin E: Nanoformulations to Enhance Bioavailability. Int. J. Nanomed. 2020, 15, 9961–9974. [Google Scholar] [CrossRef] [PubMed]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Duarte-Jurado, A.P.; Gopar-Cuevas, Y.; Saucedo-Cardenas, O.; Loera-Arias, M.d.J.; Montes-de-Oca-Luna, R.; Garcia-Garcia, A.; Rodriguez-Rocha, H. Antioxidant Therapeutics in Parkinson’s Disease: Current Challenges and Opportunities. Antioxidants 2021, 10, 453. https://doi.org/10.3390/antiox10030453
Duarte-Jurado AP, Gopar-Cuevas Y, Saucedo-Cardenas O, Loera-Arias MdJ, Montes-de-Oca-Luna R, Garcia-Garcia A, Rodriguez-Rocha H. Antioxidant Therapeutics in Parkinson’s Disease: Current Challenges and Opportunities. Antioxidants. 2021; 10(3):453. https://doi.org/10.3390/antiox10030453
Chicago/Turabian StyleDuarte-Jurado, Ana Patricia, Yareth Gopar-Cuevas, Odila Saucedo-Cardenas, Maria de Jesus Loera-Arias, Roberto Montes-de-Oca-Luna, Aracely Garcia-Garcia, and Humberto Rodriguez-Rocha. 2021. "Antioxidant Therapeutics in Parkinson’s Disease: Current Challenges and Opportunities" Antioxidants 10, no. 3: 453. https://doi.org/10.3390/antiox10030453