
Taurine Protects against Postischemic Brain Injury via the Antioxidant Activity of Taurine Chloramine

 ,
,
Abstract
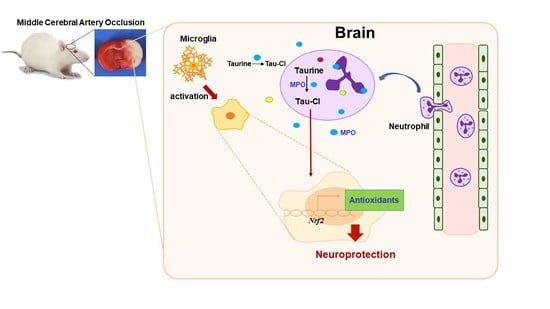
:
1. Introduction
2. Materials and Methods
2.1. Reagents and Tau-Cl Synthesis
2.2. MCAO Procedure
2.3. Intranasal Delivery
2.4. Infarct Volume Assessment
2.5. Modified Neurological Severity Scores
2.6. Rotarod Test
2.7. Hematoxylin and Eosin (H&E) Staining
2.8. Immunofluorescence Staining
2.9. BV2 Cell Culture
2.10. RNA Preparation and qRT-PCR
2.11. Western Blotting
2.12. Statistical Analysis
3. Results
3.1. Intranasally Delivered Tau-Cl Suppressed Infarct Formation in the Rat MCAO Model
3.2. Tau-Cl Improved Neurological Deficits and Motor Impairment in the Rat MCAO Model
3.3. Neutrophils Were Infiltrated into the Brain Parenchyma after Ischemic Insult
3.4. Tau-Cl Increased the Expression of Antioxidant Enzymes in BV2 Microglia
3.5. Tau-Cl Increased Antioxidant Enzyme Expression in a Rat MCAO Model
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Huxtable, R.J. Physiological actions of taurine. Physiol. Rev. 1992, 72, 101–163. [Google Scholar] [CrossRef] [Green Version]
- Schuller-Levis, G.B.; Park, E. Taurine: New implications for an old amino acid. FEMS Microbiol. Lett. 2003, 226, 195–202. [Google Scholar] [CrossRef]
- Kim, C.; Cha, Y.-N. Taurine chloramine produced from taurine under inflammation provides anti-inflammatory and cytoprotective effects. Amino Acids 2014, 46, 89–100. [Google Scholar] [CrossRef]
- Rea, M.A.; McBride, W.J.; Rohde, B.H. Levels of glutamate, aspartate, GABA, and taurine in different regions of the cerebellum after x-irradiation-induced neuronal loss. Neurochem. Res. 1981, 6, 33–39. [Google Scholar] [CrossRef]
- Sturman, J.A. Nutritional taurine and central nervous system development. Ann. NY. Acad. Sci. 1986, 477, 196–213. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.-H.; Jiang, Z.-L.; Fan, X.-J.; Zhang, L.; Li, X.; Ke, K.-F. Neuroprotective effect of taurine against focal cerebral ischemia in rats possibly mediated by activation of both GABAA and glycine receptors. Neuropharmacol. 2007, 52, 1199–1209. [Google Scholar] [CrossRef]
- Sun, M.; Zhao, Y.; Gu, Y.; Xu, C. Protective functions of taurine against experimental stroke through depressing mitochondria-mediated cell death in rats. Amino Acids 2011, 40, 1419–1429. [Google Scholar] [CrossRef] [PubMed]
- Sun, M.; Zhao, Y.; Gu, Y.; Xu, C. Anti-inflammatory mechanism of taurine against ischemic stroke is related to down-regulation of PARP and NF-kappaB. Amino Acids 2012, 42, 1735–1747. [Google Scholar] [CrossRef] [PubMed]
- Menzie, J.; Prentice, H.; Wu, J.Y. Neuroprotective mechanisms of taurine against ischemic stroke. Brain Sci. 2013, 3, 877–907. [Google Scholar] [CrossRef] [Green Version]
- Su, Y.; Fan, W.; Ma, Z.; Wen, X.; Wang, W.; Wu, Q.; Huang, H. Taurine improves functional and histological outcomes and reduces inflammation in traumatic brain injury. Neuroscience 2014, 266, 56–65. [Google Scholar] [CrossRef]
- Taranukhin, A.G.; Taranukhina, E.Y.; Saransaari, P.; Djatchkova, I.M.; Pelto-Huikko, M.; Oja, S.S. Taurine reduces caspase-8 and caspase-9 expression induced by ischemia in the mouse hypothalamic nuclei. Amino Acids 2008, 34, 169–174. [Google Scholar] [CrossRef]
- Gao, X.; Yang, X.; Zhang, B. Neuroprotection of taurine against bilirubin-induced elevation of apoptosis and intracellular free calcium ion in vivo. Toxicol. Mech. Methods 2011, 21, 383–387. [Google Scholar] [CrossRef]
- Prentice, H.; Gharibani, P.M.; Ma, Z.; Alexandrescu, A.; Genova, R.; Chen, P.-C.; Modi, J.; Menzie, J.; Pan, C.; Tao, R.; et al. Neuroprotective functions through inhibition of ER stress by taurine or taurine combination treatments in a rat stroke model. Adv. Exp. Med. Biol. 2017, 975, 193–205. [Google Scholar] [CrossRef]
- Jin, R.; Xiao, A.Y.; Liu, S.; Wang, M.; Li, G. Taurine reduces tPA (Tissue-Type Plasminogen Activator)-induced hemorrhage and microvascular thrombosis after embolic stroke in rat. Stroke 2018, 49, 1708–1718. [Google Scholar] [CrossRef]
- Fukuda, K.; Hirai, Y.; Yoshida, H.; Nakajima, T.; Usui, T. Free amino acid content of lymphocytes and granulocytes compared. Clin. Chem. 1982, 28, 1758–1761. [Google Scholar] [CrossRef]
- Mårtensson, J. The effect of fasting on leukocyte and plasma glutathione and sulfur amino acid concentrations. Metabolism 1986, 35, 118–121. [Google Scholar] [CrossRef]
- Weiss, S.J.; Klein, R.; Slivka, A.; Wei, M. Chlorination of taurine by human neutrophils. Evidence for hypochlorous acid generation. J. Clin. Investig. 1982, 70, 598–607. [Google Scholar] [CrossRef]
- Kim, C.; Jang, J.S.; Cho, M.R.; Agarawal, S.R.; Cha, Y.N. Taurine chloramine induces heme oxygenase-1 expression via Nrf2 activation in murine macrophages. Int. Immunopharmacol. 2010, 10, 440–446. [Google Scholar] [CrossRef]
- Jang, J.S.; Piao, S.; Cha, Y.N.; Kim, C. Taurine chloramine activates Nrf2, increases HO-1 expression and protects cells from death caused by hydrogen peroxide. J. Clin. Biochem. Nutr. 2009, 45, 37–43. [Google Scholar] [CrossRef] [Green Version]
- Huie, R.E.; Padmaja, S. The reaction of NO with superoxide. Free Radic. Res. Commun. 1993, 18, 195–199. [Google Scholar] [CrossRef]
- Motterlini, R.; Green, C.J.; Foresti, R. Regulation of heme oxygenase-1 by redox signals involving nitric oxide. Antioxid. Redox Signal. 2002, 4, 615–624. [Google Scholar] [CrossRef]
- Siegel, D.; Yan, C.; Ross, D. NAD(P)H: Quinone oxidoreductase 1 (NQO1) in the sensitivity and resistance to antitumor quinones. Biochem. Pharmacol. 2012, 83, 1033–1040. [Google Scholar] [CrossRef] [Green Version]
- Chae, H.Z.; Robison, K.; Poole, L.B.; Church, G.; Storz, G.; Rhee, S.G. Cloning and sequencing of thiol-specific antioxidant from mammalian brain: Alkyl hydroperoxide reductase and thiol-specific antioxidant define a large family of antioxidant enzymes. Proc. Natl. Acad. Sci. USA 1994, 91, 7017–7021. [Google Scholar] [CrossRef] [Green Version]
- Rhee, S.G.; Woo, H.A. Multiple functions of peroxiredoxins: Peroxidases, sensors and regulators of the intracellular messenger H2O2, and protein chaperones. Antioxid. Redox Signal. 2011, 15, 781–794. [Google Scholar] [CrossRef]
- Moskowitz, M.A.; Lo, E.H.; Iadecola, C. The science of stroke: Mechanisms in search of treatments. Neuron 2010, 67, 181–198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chan, P.H. Reactive oxygen radicals in signaling and damage in the ischemic brain. J. Cereb. Blood Flow Metab. 2001, 21, 2–14. [Google Scholar] [CrossRef]
- Chamorro, A.; Dirnagl, U.; Urra, X.; Planas, A.M. Neuroprotection in acute stroke: Targeting excitotoxicity, oxidative and nitrosative stress, and inflammation. Lancet Neurol. 2016, 15, 869–881. [Google Scholar] [CrossRef]
- Guan, W.; Zhao, Y.; Xu, C. A combined treatment with taurine and intra-arterial thrombolysis in an embolic model of stroke in rats: Increased neuroprotective efficacy and extended therapeutic time window. Transl. Stroke Res. 2011, 2, 80–91. [Google Scholar] [CrossRef]
- Gharibani, P.M.; Modi, J.; Pan, C.; Menzie, J.; Ma, Z.; Chen, P.C.; Tao, R.; Prentice, H.; Wu, J.Y. The mechanism of taurine protection against endoplasmic reticulum stress in an animal stroke model of cerebral artery occlusion and stroke-related conditions in primary neuronal cell culture. Adv. Exp. Med. Biol. 2013, 776, 241–258. [Google Scholar] [CrossRef]
- Zhu, X.Y.; Ma, P.S.; Wu, W.; Zhou, R.; Hao, Y.J.; Niu, Y.; Sun, T.; Li, Y.X.; Yu, J.Q. Neuroprotective actions of taurine on hypoxic-ischemic brain damage in neonatal rats. Brain Res. Bull. 2016, 124, 295–305. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Yu, R.; Cao, L. Neuroprotection of taurine through inhibition of 12/15 lipoxygenase pathway in cerebral ischemia of rats. Neurol. Res. 2017, 39, 453–458. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.W.; Kim, C. Inhibition of LPS-induced NO production by taurine chloramine in macrophages is mediated through Ras-ERK-NF-kappaB. Biochem. Pharmacol. 2005, 70, 1352–1360. [Google Scholar] [CrossRef]
- Kim, I.-D.; Lee, H.; Kim, S.-W.; Lee, H.-K.; Choi, J.; Han, P.-L.; Lee, J.-K. Alarmin HMGB1 induces systemic and brain inflammatory exacerbation in post-stroke infection rat model. Cell Death Dis. 2018, 9, 426–439. [Google Scholar] [CrossRef]
- Kim, I.-D.; Shin, J.-H.; Kim, S.-W.; Choi, S.; Ahn, J.; Han, P.-L.; Park, J.-S.; Lee, J.-K. Intranasal delivery of HMGB1 siRNA confers target gene knockdown and robust neuroprotection in the postischemic brain. Mol. Ther. 2012, 20, 829–839. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.-B.; Lim, C.-M.; Yu, Y.-M.; Lee, J.-K. Induction and subcellular localization of high-mobility group box-1 (HMGB1) in the postischemic rat brain. J. Neurosci. Res. 2008, 86, 1125–1131. [Google Scholar] [CrossRef]
- Kim, C.; Marschal, C.; Penninger, J.; Dinauer, M.C. The hemopoietic Rho/Rac guanine-nucleotide exchange factor Vav1 regulates N-formyl-Methionyl-Leucyl-Phenylalanine-activated neutrophil functions. J. Immunol. 2003, 171, 4425–4430. [Google Scholar] [CrossRef] [PubMed]
- Segel, G.B.; Halterman, M.W.; Lichtman, M.A. The paradox of the neutrophil’s role in tissue injury. J. Leukoc. Biol. 2011, 89, 359–372. [Google Scholar] [CrossRef]
- Krapfenbauer, K.; Yoo, B.C.; Fountoulakis, M.; Mitrova, E.; Lubec, G. Expression patterns of antioxidant proteins in brains of patients with sporadic Creutzfeldt-Jacob disease. Electrophoresis 2002, 23, 2541–2547. [Google Scholar] [CrossRef]
- Krapfenbauer, K.; Engidawork, E.; Cairns, N.; Fountoulakis, M.; Lubec, G. Aberrant expression of peroxiredoxin subtypes in neurodegenerative disorders. Brain Res. 2003, 967, 152–160. [Google Scholar] [CrossRef]
- Marcinkiewicz, J.; Kontny, E. Taurine and inflammatory diseases. Amino Acids 2014, 46, 7–20. [Google Scholar] [CrossRef] [Green Version]
- Jin, M.-H.; Lee, Y.-H.; Kim, J.-M.; Sun, H.-N.; Moon, E.-Y.; Shong, M.H.; Kim, S.-U.; Lee, S.H.; Lee, T.-H.; Yu, D.-Y.; et al. Characterization of neural cell types expressing peroxiredoxins in mouse brain. Neurosci. Lett. 2005, 381, 252–257. [Google Scholar] [CrossRef] [PubMed]
- Abbas, K.; Breton, J.; Picot, C.R.; Quesniaux, V.; Bouton, C.; Drapier, J.-C. Signaling events leading to peroxiredoxin 5 up-regulation in immunostimulated macrophages. Free Radic. Biol. Med. 2009, 47, 794–802. [Google Scholar] [CrossRef] [PubMed]
- Verdrengh, M.; Tarkowski, A. Inhibition of septic arthritis by local administration of taurine chloramine, a product of activated neutrophils. J. Rheumatol. 2005, 32, 1513–1517. [Google Scholar]
- Wang, Y.; Cha, Y.N.; Kim, K.S.; Kim, C. Taurine chloramine inhibits osteoclastogenesis and splenic lymphocyte proliferation in mice with collagen-induced arthritis. Eur. J. Pharmacol. 2011, 668, 325–330. [Google Scholar] [CrossRef] [PubMed]
- Schaalan, M.F.; Ramadan, B.K.; Elwahab, A.H.A. Ameliorative effect of taurine-chloramine in azathioprine-induced testicular damage; a deeper insight into the mechanism of protection. BMC Complement. Altern. Med. 2018, 18, 255. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dall’igna, D.M.; Daluz, J.M.; Vuolo, F.; Michels, M.; Dal-Pizzol, F. Taurine chloramine decreases cell viability and cytokine production in blood and spleen lymphocytes from septic rats. An. Acad. Bras. Cienc. 2020, 92, e20191311. [Google Scholar] [CrossRef]
- Chen, V.C.-H.; Chiu, C.-C.; Chen, L.-J.; Hsu, T.-C.; Tzang, B.-S. Effects of taurine on striatal dopamine transporter expression and dopamine uptake in SHR rats. Behav. Brain Res. 2018, 348, 219–226. [Google Scholar] [CrossRef]
- Serdar, M.; Mordelt, A.; Müser, K.; Kempe, K.; Felderhoff-Müser, U.; Herz, J.; Bendix, I. Detrimental impact of energy drink compounds on developing oligodendrocytes and neurons. Cells 2019, 8, 1381. [Google Scholar] [CrossRef] [Green Version]
- Jickling, G.C.; Liu, D.; Ander, B.P.; Stamova, B.; Zhan, X.; Sharp, F.R. Targeting neutrophils in ischemic stroke: Translational insights from experimental studies. J. Cereb. Blood Flow Metab. 2015, 35, 888–901. [Google Scholar] [CrossRef] [Green Version]
- Lee, H.-K.; Kim, I.-D.; Lee, H.; Luo, L.; Kim, S.-W.; Lee, J.-K. Neuroprotective and anti-inflammatory effects of a dodecamer peptide harboring Ninjurin 1 cell adhesion motif in the postischemic brain. Mol. Neurobiol. 2018, 55, 6094–6111. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.W.; Lee, H.; Lee, H.K.; Kim, I.D.; Lee, J.K. Neutrophil extracellular trap induced by HMGB1 exacerbates damages in the ischemic brain. Acta Neuropathol. Com. 2019, 7, 94. [Google Scholar] [CrossRef] [Green Version]
- Breckwoldt, M.O.; Chen, J.W.; Stangenberg, L.; Aikawa, E.; Rodriguez, E.; Qiu, S.; Moskowitz, M.A.; Weissleder, R. Tracking the inflammatory response in stroke in vivo by sensing the enzyme myeloperoxidase. Proc. Natl. Acad. Sci. USA 2008, 105, 18584–18589. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maki, R.A.; Tyurin, V.A.; Lyon, R.C.; Hamilton, R.L.; DeKosky, S.T.; Kagan, V.E.; Reynolds, W.F. Aberrant expression of myeloperoxidase in astrocytes promotes phospholipid oxidation and memory deficits in a mouse model of Alzheimer disease. J. Biol. Chem. 2009, 284, 3158–3169. [Google Scholar] [CrossRef] [Green Version]
- Forghani, R.; Kim, H.J.; Wojtkiewicz, G.R.; Bure, L.; Wu, Y.; Hayase, M.; Wei, Y.; Zheng, Y.; Moskowitz, M.A.; Chen, J.W. Myeloperoxidase propagates damage and is a potential therapeutic target for subacute stroke. J. Cereb. Blood Flow Metab. 2015, 35, 485–493. [Google Scholar] [CrossRef]
- Yu, G.; Liang, Y.; Huang, Z.; Jones, D.W.; Pritchard, K.A., Jr.; Zhang, H. Inhibition of myeloperoxidase oxidant production by N-acetyl lysyltyrosylcysteine amide reduces brain damage in a murine model of stroke. J. Neuroinflamm. 2016, 13, 119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Re, G.; Azzimondi, G.; Lanzarini, C.; Bassein, L.; Vaona, I.; Guarnieri, C. Plasma lipoperoxidative markers in ischemic stroke suggest brain embolism. Eur. J. Emerg. Med. 1997, 4, 5–9. [Google Scholar] [PubMed]
- Cojocaru, I.M.; Cojocaru, M.; Iliescu, I.; Botnaru, L.; Gurban, C.V.; Sfrijan, F.; Tănăsescu, R. Plasma myeloperoxidase levels in patients with acute ischemic stroke. Rom. J. Intern. Med. 2010, 48, 101–104. [Google Scholar] [PubMed]
- Hoy, A.; Leininger-Muller, B.; Poirier, O.; Siest, G.; Gautier, M.; Elbaz, A.; Amarenco, P.; Visvikis, S. Myeloperoxidase polymorphisms in brain infarction. Association with infarct size and functional outcome. Atherosclerosis 2003, 167, 223–230. [Google Scholar] [CrossRef]
- Nagra, R.M.; Becher, B.; Tourtellotte, W.W.; Antel, J.P.; Gold, D.; Paladino, T.; Smith, R.A.; Nelson, J.R.; Reynolds, W.F. Immunohistochemical and genetic evidence of myeloperoxidase involvement in multiple sclerosis. J. Neuroimmunol. 1997, 78, 97–107. [Google Scholar] [CrossRef]
- Rodrigues, M.R.; Rodriguez, D.; Russo, M.; Campa, A. Macrophage activation includes high intracellular myeloperoxidase activity. Biochem. Biophys. Res. Commun. 2002, 292, 869–873. [Google Scholar] [CrossRef] [PubMed]
- Guégan, C.; Ceballos-Picot, I.; Chevalier, E.; Nicole, A.; Onténiente, B.; Sola, B. Reduction of ischemic damage in NGF-transgenic mice: Correlation with enhancement of antioxidant enzyme activities. Neurobiol. Dis. 1999, 6, 180–189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malik, I.; Shah, F.A.; Ali, T.; Tan, Z.; Alattar, A.; Ullah, N.; Khan, A.-U.; Alshaman, R.; Li, S. Potent natural antioxidant carveol attenuates MCAO-stress induced oxidative, neurodegeneration by regulating the Nrf-2 pathway. Front. Neurosci. 2020, 14, 659. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Liu, W.; Ding, S.; Xu, W.; Guan, Y.; Zhang, J.H.; Sun, X. Hyperbaric oxygen preconditioning induces tolerance against brain ischemia–reperfusion injury by upregulation of antioxidant enzymes in rats. Brain Res. 2008, 1210, 223–229. [Google Scholar] [CrossRef] [PubMed]
- Wicha, P.; Tocharus, J.; Janyou, A.; Jittiwat, J.; Changtam, C.; Suksamrarn, A.; Tocharus, C. Hexahydrocurcumin protects against cerebral ischemia/reperfusion injury, attenuates inflammation, and improves antioxidant defenses in a rat stroke model. PLoS ONE 2017, 12, e0189211. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Vehicle-Treated Group (n = 3) | Tau-Cl-Treated Group (n = 3) | |||
|---|---|---|---|---|
| Base | During Ischemia | Base | During Ischemia | |
| Rectal Temperature (°C) | 36.1 ± 0.3 * | 36.5 ± 0.5 | 36.0 ± 0.6 | 36.1 ± 0.2 |
| pH | 7.5 ± 0.1 | 7.4 ± 0.1 | 7.5 ± 0.1 | 7.4 ± 0.1 |
| PO₂ mmHg | 82.3 ± 4.6 | 89.0 ± 12.2 | 91.0 ± 10.6 | 74.7 ± 0.6 |
| PCO₂ mmHg | 34.3 ± 7.4 | 43.8 ± 3.0 | 36.9 ± 6.1 | 36.2 ± 4.7 |
| Glucose, mg/dL | 111.3 ± 12.1 | 115.7 ± 7.1 | 106.7 ± 3.1 | 107.3 ± 3.1 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Seol, S.-I.; Kim, H.J.; Choi, E.B.; Kang, I.S.; Lee, H.-K.; Lee, J.-K.; Kim, C. Taurine Protects against Postischemic Brain Injury via the Antioxidant Activity of Taurine Chloramine. Antioxidants 2021, 10, 372. https://doi.org/10.3390/antiox10030372
Seol S-I, Kim HJ, Choi EB, Kang IS, Lee H-K, Lee J-K, Kim C. Taurine Protects against Postischemic Brain Injury via the Antioxidant Activity of Taurine Chloramine. Antioxidants. 2021; 10(3):372. https://doi.org/10.3390/antiox10030372
Chicago/Turabian StyleSeol, Song-I, Hyun Jae Kim, Eun Bi Choi, In Soon Kang, Hye-Kyung Lee, Ja-Kyeong Lee, and Chaekyun Kim. 2021. "Taurine Protects against Postischemic Brain Injury via the Antioxidant Activity of Taurine Chloramine" Antioxidants 10, no. 3: 372. https://doi.org/10.3390/antiox10030372