
“Upstream Analysis”: An Integrated Promoter-Pathway Analysis Approach to Causal Interpretation of Microarray Data


Abstract
:1. Introduction
2. Experimental Section
2.1. Microarray Data, Differential Expression Analysis
2.2. Triclustering of Genes in Expression Data
2.3. Analysis of Enriched Transcription Factor Binding Sites

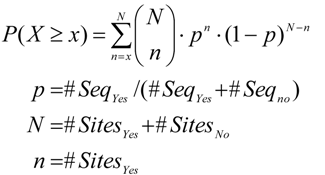
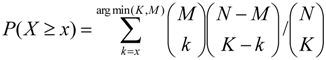

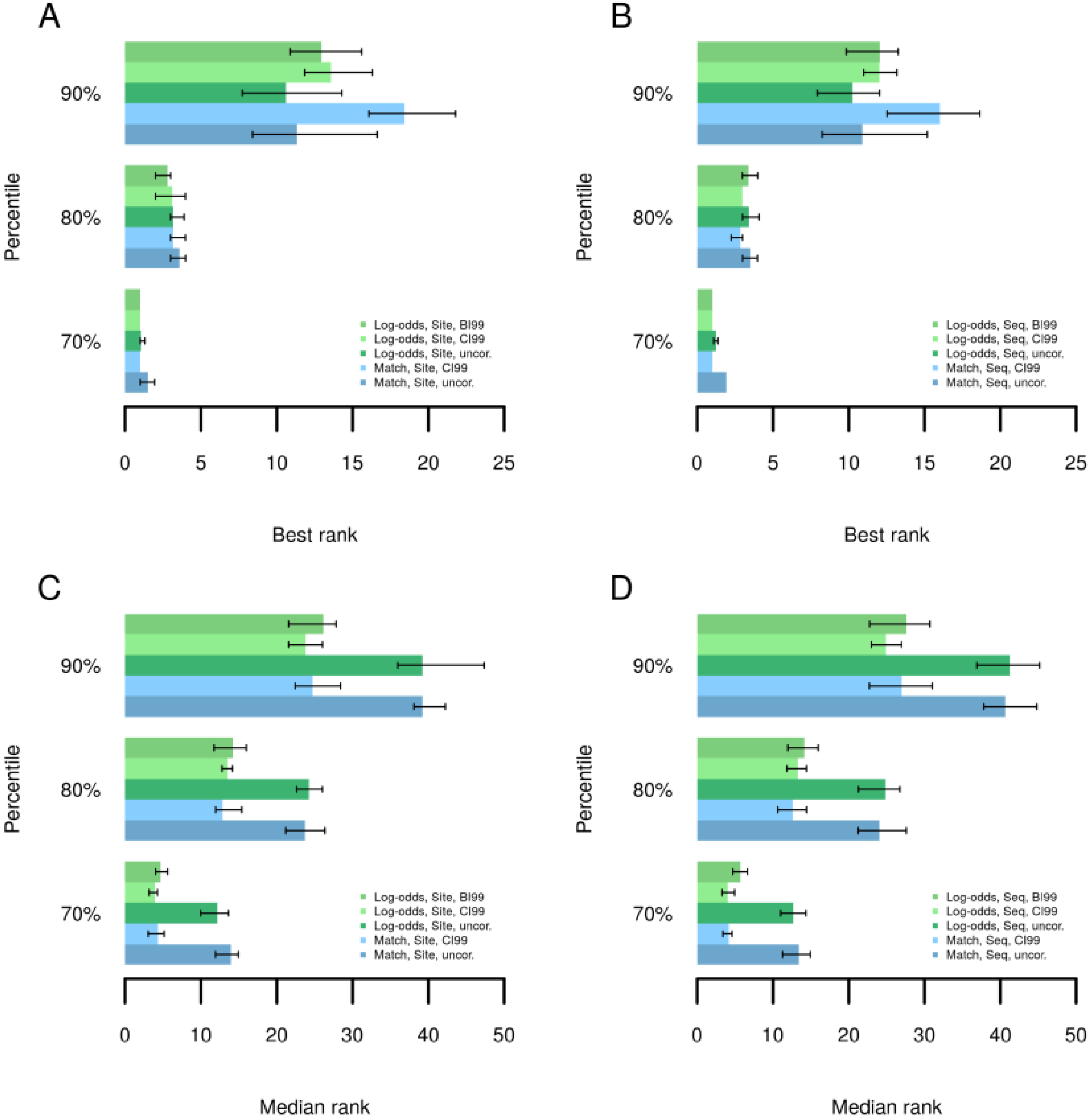
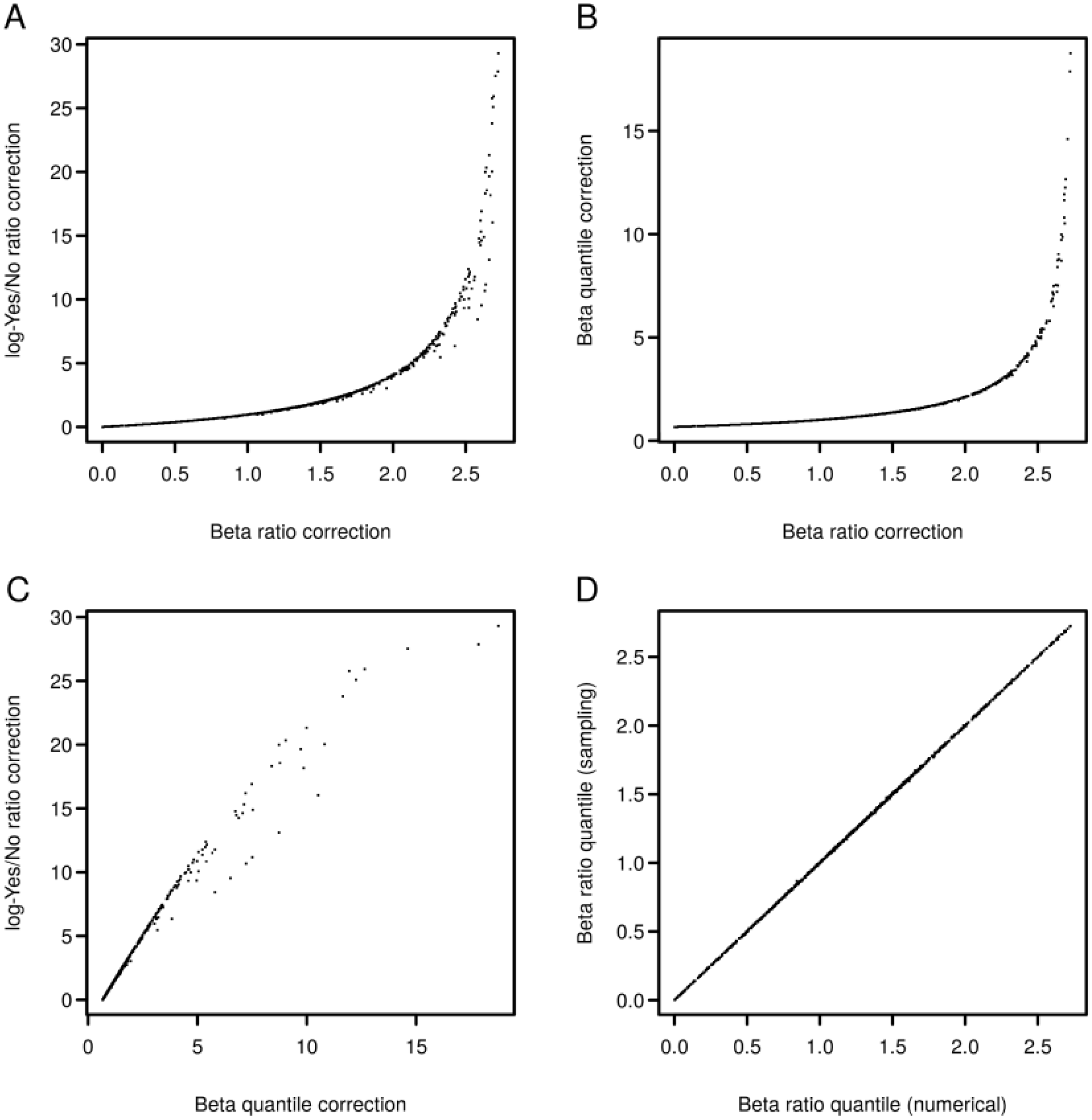
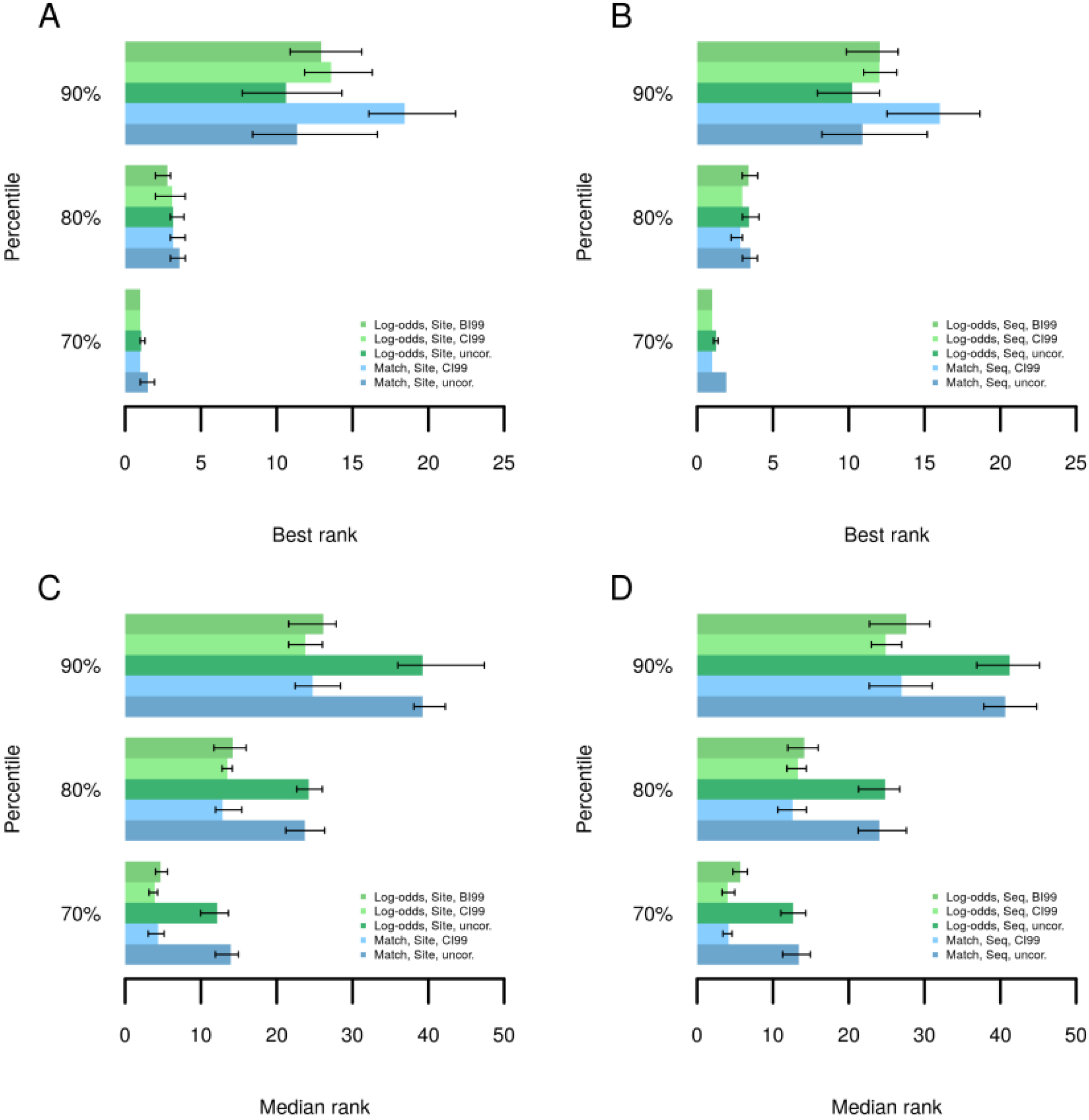
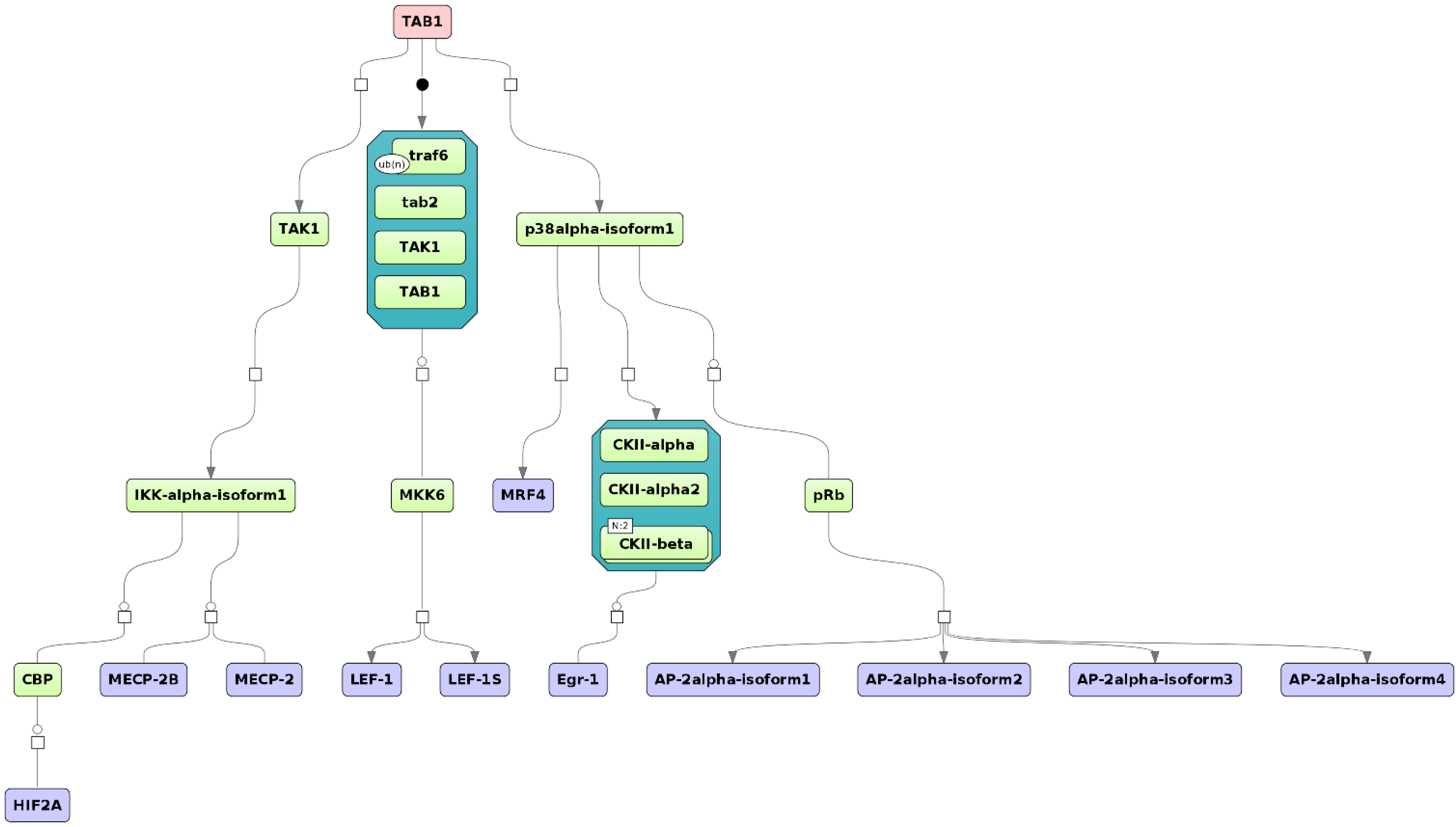
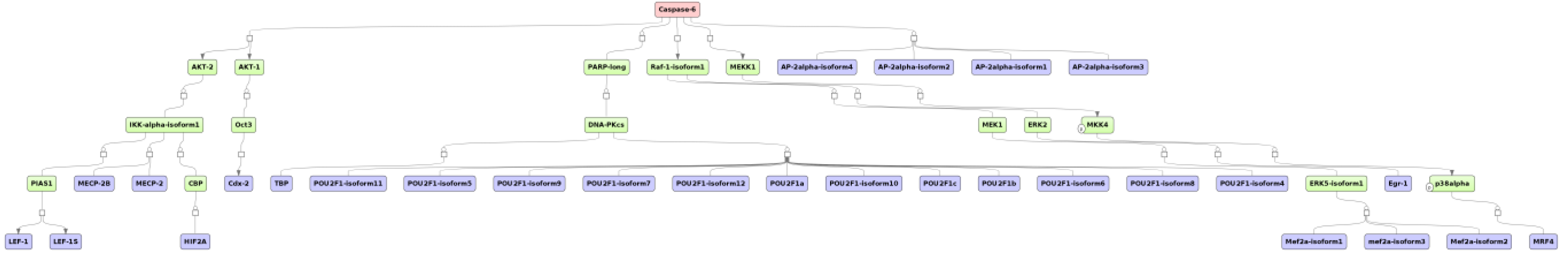
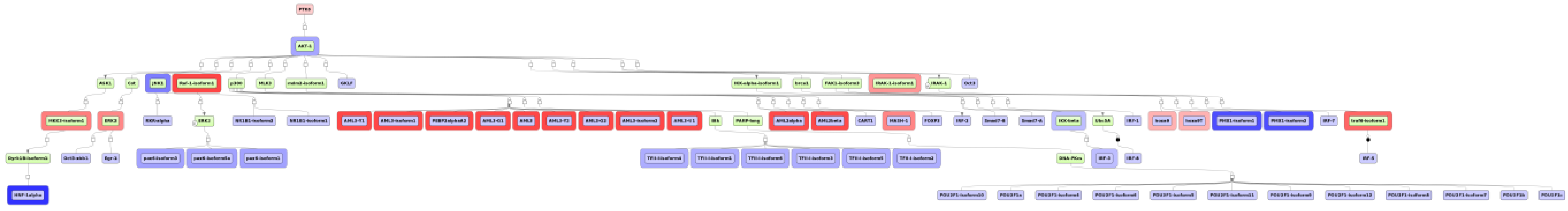
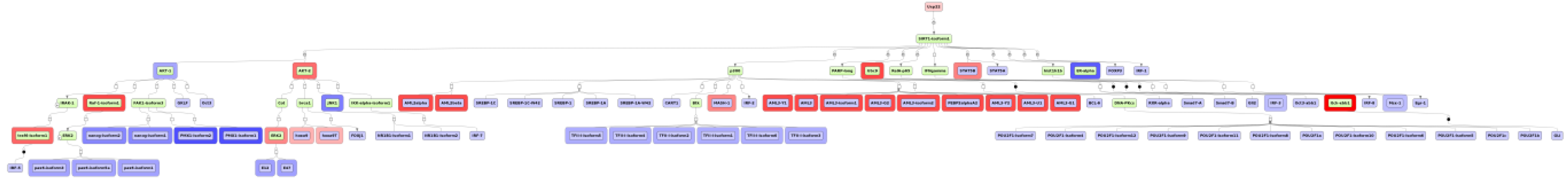
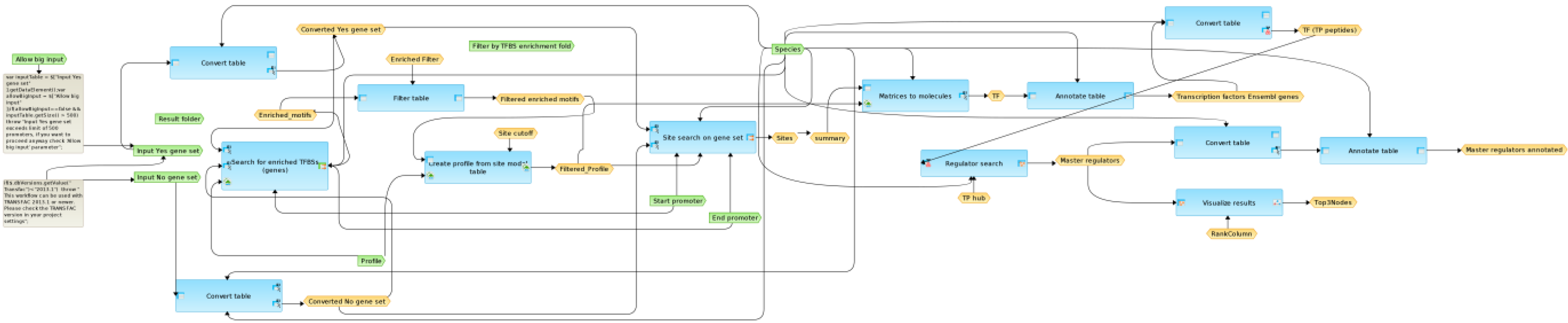
2.4. Finding Master Regulators in Networks
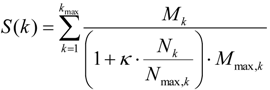
3. Results and Discussion
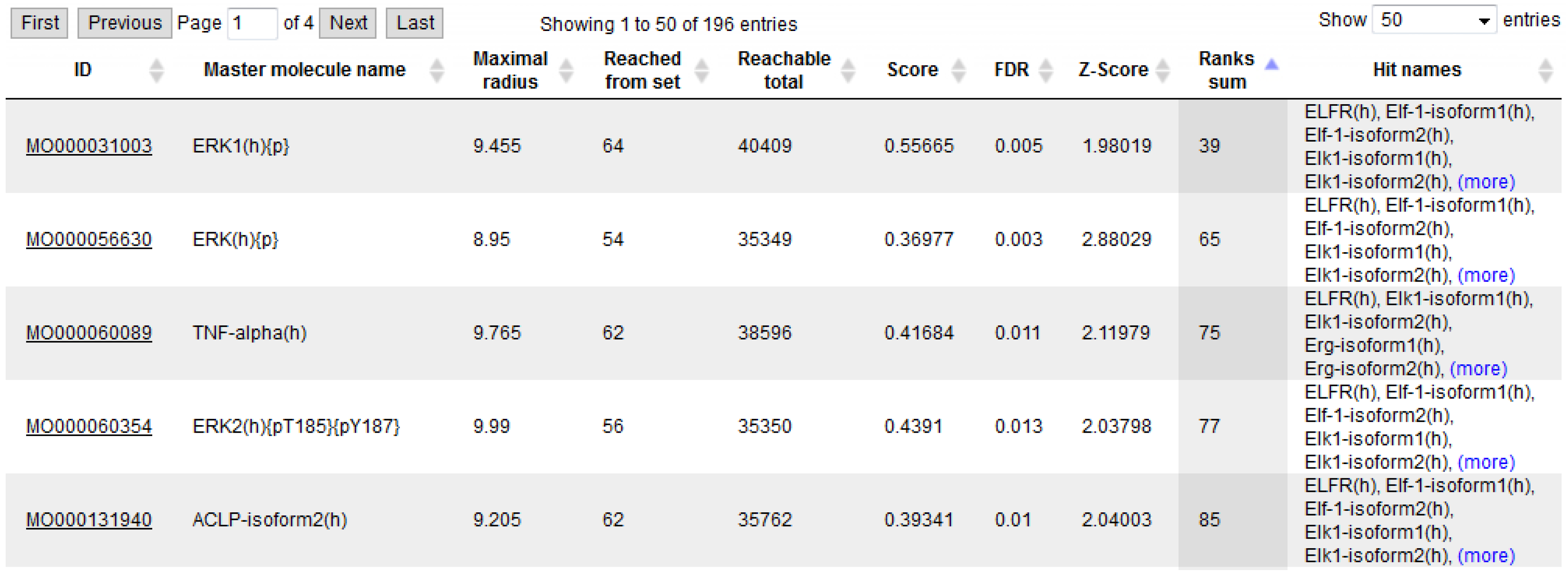
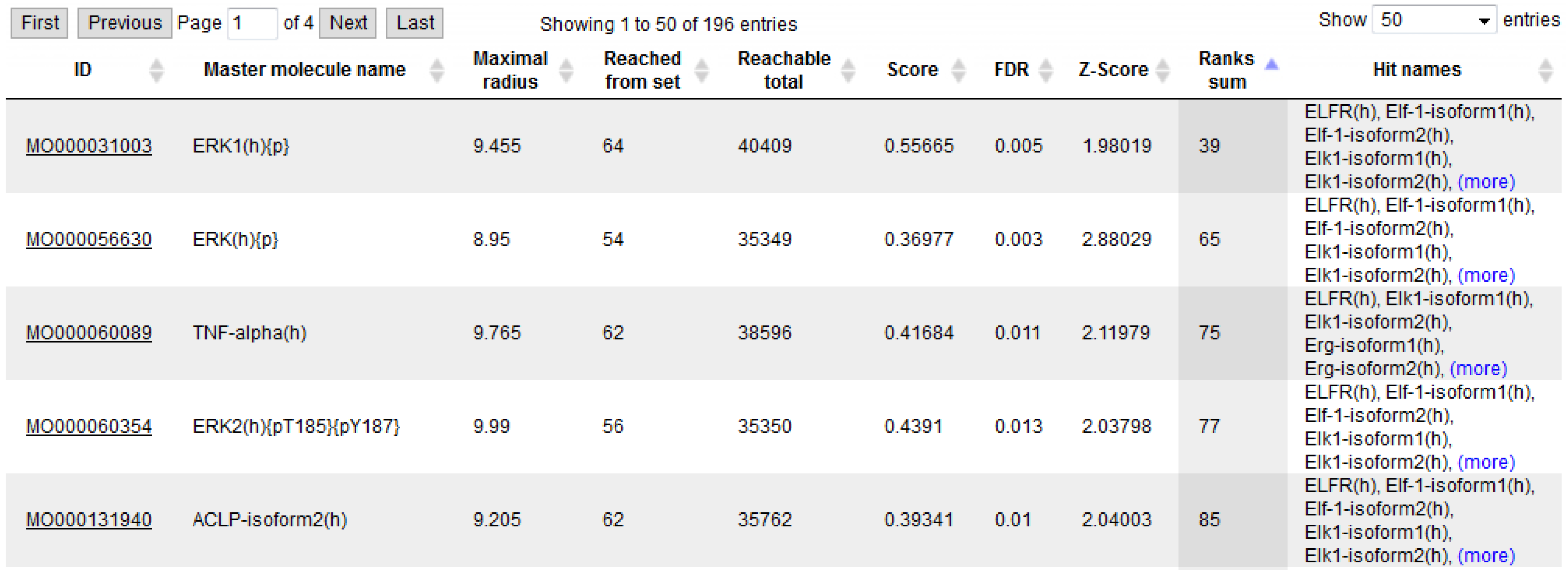
3.1. Integrated Promoter-Pathway Upstream Analysis: Proof of Principle
3.2. Triclustering Identifies Gene Clusters in Three-Dimensional Datasets


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Ensembl ID | Gene name | Cluster 3 | Cluster 3 | Cluster 4 | Cluster 4 |
|---|---|---|---|---|---|
| (log2) fold_change | adj. p_value | (log2) fold_change | adj. p_value | ||
| ENSMUSG00000015134 | Aldh1a3 | 0.436 | 0.04163 | 0.164 | 0.02642 |
| ENSMUSG00000022263 | Trio | 0.287 | 0.03742 | 0.158 | 0.03514 |
| ENSMUSG00000026470 | Stx6 | 0.444 | 0.03099 | 0.137 | 0.01988 |
| ENSMUSG00000029663 | Gngt1 | 0.582 | 0.03451 | 0.178 | 0.00599 |
| ENSMUSG00000024360 | Etf1 | −0.732 | 0.02545 | −0.393 | 0.00802 |
| ENSMUSG00000027962 | Vcam1 | −0.596 | 0.03890 | −0.151 | 0.03274 |
3.3. Promoter Analysis
| TFs | TFs | TFs | TFs | |||
|---|---|---|---|---|---|---|
| cluster 3 | cluster 4 | cluster 3 | cluster 4 | |||
| (liver) down | (lung) down | (liver) up | (lung) up | |||
| Cdx1 | Alx1 | Lhx1 | Ebf1 | Alx1 | Irf1 | Pou2f1 |
| Cdx2 | Alx4 | Lhx3 | Egr1 | Arid5a | Irf2 | Pou5f1 |
| Hoxc10 | Arid3a | Lhx5 | Egr2 | Ascl1 | Irf3 | Prdm1 |
| Mafb | Arid5a | Lmx1b | Egr3 | Cbfb | Irf4 | Prrx1 |
| Mef2a | Bcl6 | Nanog | Epas1 | Egr1 | Irf5 | Rara |
| Pou2f1 | Cnot3 | Nr2e1 | Hivep2 | Egr2 | Irf6 | Rfx2 |
| Pou3f1 | Egr2 | Otp | Lef1 | Foxc1 | Irf7 | Runx2 |
| Rfx1 | Foxa1 | Pbx1 | Mecp2 | Foxf1 | Irf8 | Runx3 |
| Rfx2 | Foxa2 | Pbx2 | Mtf1 | Foxg1 | Klf4 | Rxra |
| Rfx3 | Foxa3 | Pbx3 | Myf6 | Foxj2 | Lhx1 | Shox2 |
| Rfx4 | Foxc1 | Phox2b | Nr2f2 | Foxj3 | Lhx3 | Smad7 |
| Rfx5 | Foxd3 | Pknox1 | Rreb1 | Foxk1 | Lhx5 | Sox12 |
| Six6 | Foxf1 | Pou2f1 | Tcf12 | Foxp3 | Lhx8 | Sox14 |
| Sox21 | Foxf2 | Prdm1 | Tcf7 | Gfi1 | Lmx1b | Sox21 |
| Tbp | Foxh1 | Shox2 | Tfap2a | Gfi1b | Meis1 | Sox30 |
| Foxi1 | Sox12 | Zfp423 | Gtf2i | Meis3 | Sry | |
| Foxj1 | Sp5 | Zscan4f | Hdx | Msx1 | Tbx15 | |
| Foxk1 | Srebf1 | Hnf1a | Msx3 | Vsx1 | ||
| Foxp3 | Stat5a | Hnf1b | Nr2c2 | Zfp184 | ||
| Gfi1 | Stat5b | Hoxa4 | Nr2f2 | Zfp426 | ||
| Gli1 | Tcf3 | Hoxa9 | Pax6 | Zfp445 | ||
| Gli2 | Uncx | Hoxb4 | Phox2b | Zscan4f | ||
| Gtf2i | Vsx1 | Hoxc4 | Pknox2 | |||
| Hoxb4 | Zfp30 | |||||
| Hoxc4 | Zfp784 | |||||
| Hoxd8 | Zic1 | |||||
| Irf1 | Zscan4f | |||||
| Irf5 | ||||||
3.4. Find Master Regulators in Networks
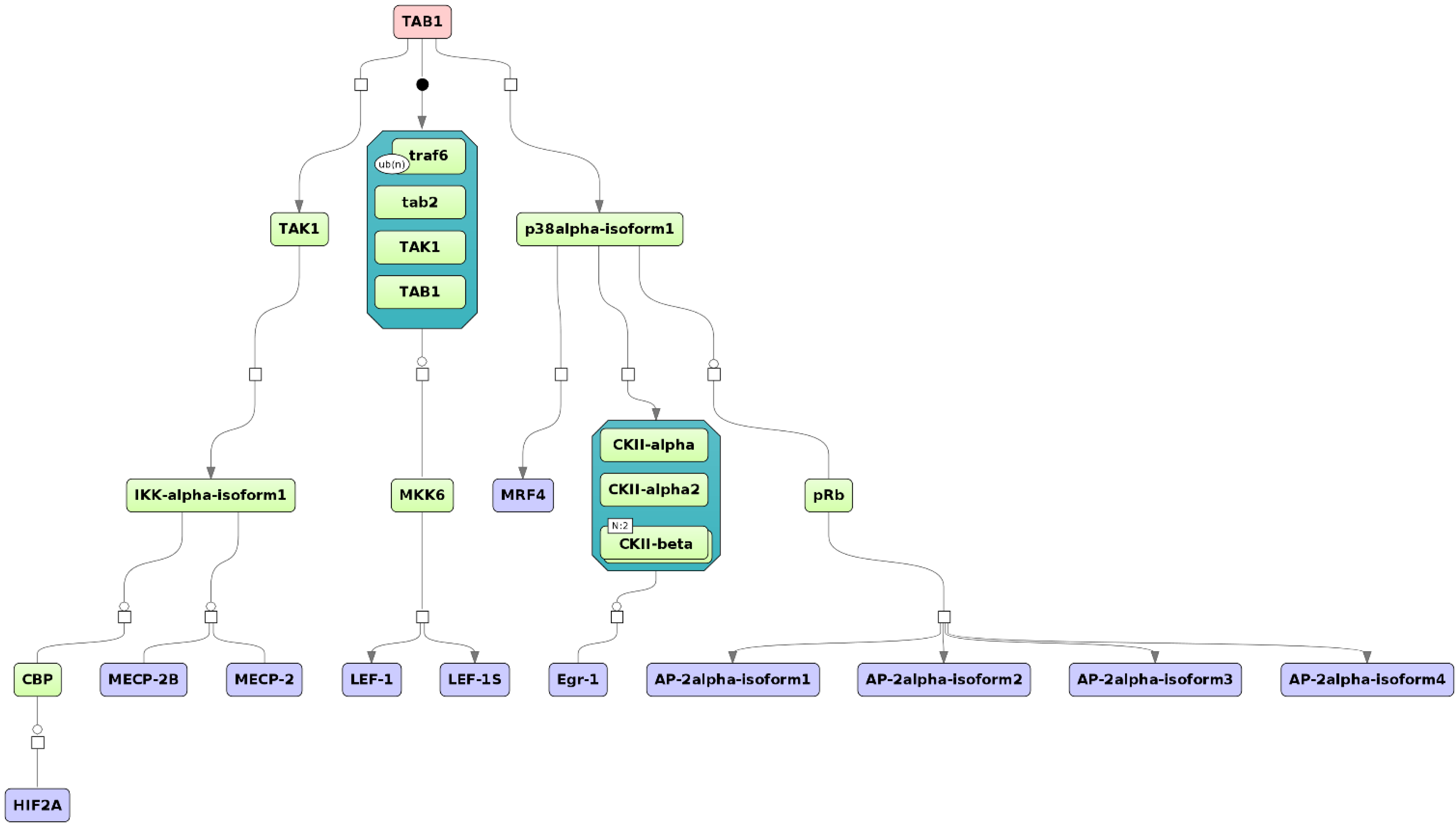
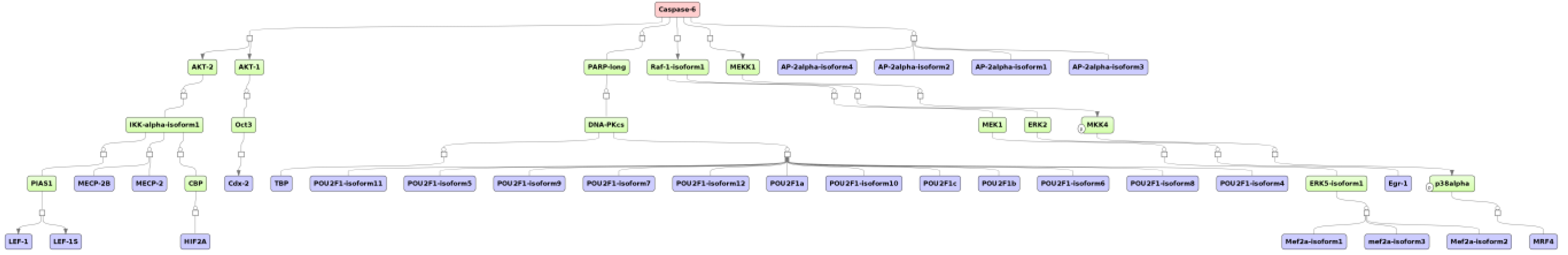

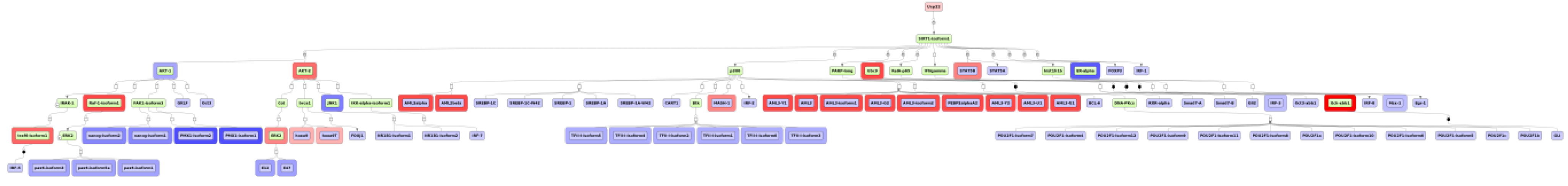
4. Conclusions
Supplementary Files
Supplementary File 1Acknowledgments
Author Contributions
Conflicts of Interest
References
- Kolesnikov, N.; Hastings, E.; Keays, M.; Melnichuk, O.; Tang, Y.A.; Williams, E.; Dylag, M.; Kurbatova, N.; Brandizi, M.; Burdett, T.; et al. ArrayExpress update—Simplifying data submissions. Nucleic Acids Res. 2015, 43, D1113–D1116. [Google Scholar] [CrossRef]
- Barrett, T.; Wilhite, S.E.; Ledoux, P.; Evangelista, C.; Kim, I.F.; Tomashevsky, M.; Marshall, K.A.; Phillippy, K.H.; Sherman, P.M.; Holko, M.; et al. NCBI GEO: archive for functional genomics data sets–update. Nucleic Acids Res. 2013, 41, D991–D995. [Google Scholar] [CrossRef]
- Petryszak, R.; Burdett, T.; Fiorelli, B.; Fonseca, N.A.; Gonzalez-Porta, M.; Hastings, E.; Huber, W.; Jupp, S.; Keays, M.; Kryvych, N.; et al. Expression Atlas update—A database of gene and transcript expression from microarray- and sequencing-based functional genomics experiments. Nucleic Acids Res. 2014, 42, D926–D932. [Google Scholar] [CrossRef] [PubMed]
- Smith, C.M.; Finger, J.H.; Hayamizu, T.F.; McCright, I.J.; Xu, J.; Berghout, J.; Campbell, J.; Corbani, L.E.; Forthofer, K.L.; Frost, P.J.; et al. The mouse Gene Expression Database (GXD): 2014 update. Nucleic Acids Res. 2014, 42, D818–D824. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.; MacLeod, I.; Su, A.I. BioGPS and MyGene.info: Organizing online, gene-entric information. Nucleic Acids Res. 2013, 41, D561–D565. [Google Scholar] [CrossRef] [PubMed]
- Gluz, O.; Hofmann, D.; Würstlein, R.; Liedtke, C.; Nitz, U.; Harbeck, N. Genomic profiling in luminal breast cancer. Breast Care (Basel) 2013, 8, 414–422. [Google Scholar] [CrossRef]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Goto, S.; Sato, Y.; Furumichi, M.; Tanabe, M. KEGG for integration and interpretation of large-scale molecular data sets. Nucleic Acids Res. 2012, 40, D109–D114. [Google Scholar] [CrossRef] [PubMed]
- Kel, A.; Voss, N.; Jauregui, R.; Kel-Margoulis, O.; Wingender, E. Beyond microarrays: find key transcription factors controlling signal transduction pathways. BMC Bioinformatics 2006, 7, S13. [Google Scholar] [CrossRef] [PubMed]
- Michael, H.; Hogan, J.; Kel, A.; Kel-Margoulis, O.; Schacherer, F.; Voss, N.; Wingender, E. Building a knowledge base for systems pathology. Brief. Bioinform. 2008, 9, 518–531. [Google Scholar] [CrossRef] [PubMed]
- Stegmaier, P.; Voss, N.; Meier, T.; Kel, A.; Wingender, E.; Borlak, J. Advanced computational biology methods identify molecular switches for malignancy in an EGF mouse model of liver cancer. PLoS ONE 2011, 6, e17738. [Google Scholar] [CrossRef] [PubMed]
- Heinemeyer, T.; Chen, X.; Karas, H.; Kel, A.E.; Kel, O.V.; Liebich, I.; Meinhardt, T.; Reuter, I.; Schacherer, F.; Wingender, E. Expanding the TRANSFAC database towards an expert system of regulatory molecular mechanisms. Nucleic Acids Res. 1999, 27, 318–322. [Google Scholar] [CrossRef] [PubMed]
- Krull, M.; Pistor, S.; Voss, N.; Kel, A.; Reuter, I.; Kronenberg, D.; Michael, H.; Schwarzer, K.; Potapov, A.; Choi, C.; Kel-Margoulis, O.; Wingender, E. TRANSPATH: an information resource for storing and visualizing signaling pathways and their pathological aberrations. Nucleic Acids Res. 2006, 34, D546–D551. [Google Scholar] [CrossRef] [PubMed]
- Sudakin, D.L.; Canoy, S.D.; Power, L. Naphthalene mothballs: emerging and recurring issues and their relevance to environmental health. Curr. Top. Toxicol. 2011, 7, 13–19. [Google Scholar] [PubMed]
- Santucci, K.; Shah, B. Association of naphthalene with acute hemolytic anemia. Acad. Emerg. Med. 2000, 7, 42–47. [Google Scholar] [CrossRef] [PubMed]
- Price, P.S.; Jayjock, M.A. Available data on naphthalene exposures: Strengths and limitations. Regul. Toxicol. Pharm. 2008, 51, S15–S21. [Google Scholar] [CrossRef]
- Bhar, A.; Haubrock, M.; Mukhopadhyay, A.; Maulik, U.; Bandyopadhyay, S.; Wingender, E. Coexpression and coregulation analysis of time-series gene expression data in estrogen-induced breast cancer cell. Algorithms Mol. Biol. 2013, 8, 9. [Google Scholar] [CrossRef] [PubMed]
- Demo workflows. Available online: http://www.genexplain.com/demo-workflows (accessed on 18 May 2015).
- Thomas, R.S.; Clewell, H.J., 3rd.; Allen, B.C.; Wesselkamper, S.C.; Wang, N.C.; Lambert, J.C.; Hess-Wilson, J.K.; Zhao, Q.J.; Andersen, M.E. Application of transcriptional benchmark dose values in quantitative cancer and noncancer risk assessment. Toxicol. Sci. 2011, 120, 194–205. [Google Scholar] [CrossRef] [PubMed]
- Thomas, R.S.; Bao, W.; Chu, T.M.; Bessarabova, M.; Nikolskaya, T.; Nikolsky, Y.; Andersen, M.E.; Wolfinger, R.D. Use of short-term transcriptional profiles to assess the long-term cancer-related safety of environmental and industrial chemicals. Toxicol. Sci. 2009, 112, 311–321. [Google Scholar] [CrossRef] [PubMed]
- Smyth, G.K. Limma: Linear models for microarray data. In Bioinformatics and Computational Biology Solutions Using R and Bioconductor; Gentleman, R., Carey, V., Dudoit, S., Irizarry, R., Huber, W., Eds.; Springer: New York, 2005; pp. 397–420. [Google Scholar]
- Bhar, A.; Haubrock, M.; Mukhopadhyay, A.; Wingender, E. Multiobjective Triclustering of Time-Series Transcriptome Data Reveals Key Genes of Biological Processes. BMC Bioinformatics 2015, in press. [Google Scholar]
- Wingender, E. The TRANSFAC project as an example of framework technology that supports the analysis of genomic regulation. Brief. Bioinform. 2008, 9, 326–332. [Google Scholar] [CrossRef] [PubMed]
- Morris, J.A.; Gardner, M.J. Calculating confidence intervals for relative risks (odds ratios) and standardised ratios and rates. Br. Med. J. (Clin. Res. Ed.) 1988, 296, 1313–1316. [Google Scholar] [CrossRef]
- Kellis, M.; Wold, B.; Snyder, M.P.; Bernstein, B.E.; Kundaje, A.; Marinov, G.K.; Ward, L.D.; Birney, E.; Crawford, G.E.; Dekker, J.; et al. Defining functional DNA elements in the human genome. Proc. Natl. Acad. Sci. USA 2014, 111, 6131–6138. [Google Scholar] [CrossRef] [PubMed]
- Kel, A.E.; Gössling, E.; Reuter, I.; Cheremushkin, E.; Kel-Margoulis, O.V.; Wingender, E. MATCH: A tool for searching transcription factor binding sites in DNA sequences. Nucleic Acids Res. 2003, 31, 3576–3579. [Google Scholar] [CrossRef] [PubMed]
- Kel, A. “Walking toxic pathways”—Changes in gene regulation circuits predict human toxicity of chemical compounds after repeated dose inhalation exposure. In Presented at the 20th EuroQSAR Symposium “Understanding Chemical-Biological Interactions”, St. Petersburg, Russia, 31 August –4 September 2014.
- Viemann, D.; Goebeler, M.; Schmid, S.; Klimmek, K.; Sorg, C.; Ludwig, S.; Roth, J. Transcriptional profiling of IKK2/NF-kappa B- and p38 MAP kinase-dependent gene expression in TNF-alpha-stimulated primary human endothelial cells. Blood 2004, 103, 3365–3373. [Google Scholar] [CrossRef] [PubMed]
- Zwang, Y.; Oren, M.; Yarden, Y. Consistency test of the cell cycle: roles for p53 and EGR1. Cancer Res. 2012, 72, 1051–1054. [Google Scholar] [CrossRef] [PubMed]
- Roh, Y.S.; Song, J.; Seki, E. TAK1 regulates hepatic cell survival and carcinogenesis. J. Gastroenterol. 2014, 49, 185–194. [Google Scholar] [CrossRef] [PubMed]
- Sakurai, H.; Nishi, A.; Sato, N.; Mizukami, J.; Miyoshi, H.; Sugita, T. TAK1-TAB1 fusion protein: a novel constitutively active mitogen-activated protein kinase kinase kinase that stimulates AP-1 and NF-kappaB signaling pathways. Biochem. Biophys. Res. Commun. 2002, 297, 1277–1281. [Google Scholar] [CrossRef] [PubMed]
- Inokuchi, S.; Aoyama, T.; Miura, K.; Osterreicher, CH.; Kodama, Y.; Miyai, K. Disruption of TAK1 in hepatocytes causes hepatic injury, inflammation, fibrosis, and carcinogenesis. Proc. Natl. Acad. Sci. USA 2010, 107, 844–849. [Google Scholar] [CrossRef] [PubMed]
- Wu, M.; Shi, L.; Cimic, A.; Romero, L.; Sui, G.; Lees, C.J.; Cline, J.M.; Seals, D.F.; Sirintrapun, J.S.; McCoy, T.P.; et al. Suppression of Tak1 promotes prostate tumorigenesis. Cancer Res. 2012, 72, 2833–2843. [Google Scholar] [CrossRef] [PubMed]
- Melisi, D.; Xia, Q.; Paradiso, G.; Ling, J.; Moccia, T.; Carbone, C.; Budillon, A.; Abbruzzese, J.L.; Chiao, P.J. Modulation of pancreatic cancer chemoresistance by inhibition of TAK1. J. Natl. Cancer Inst. 2011, 103, 1190–1204. [Google Scholar] [CrossRef] [PubMed]
- Srinivasula, S.M.; Fernandes-Alnemri, T.; Zangrilli, J.; Robertson, N.; Armstrong, R.C.; Wang, L.; Trapani, J.A.; Tomaselli, K.J.; Litwack, G.; Alnemri, E.S. The Ced-3/interleukin 1β converting enzyme-like homolog Mch6 and the lamin-cleaving enzyme Mch2α are substrates for the apoptotic mediator CPP32. J. Biol. Chem. 1996, 271, 27099–27106. [Google Scholar] [CrossRef] [PubMed]
- Chang, H.Y.; Yang, X. Proteases for cell suicide: functions and regulation of caspases. Microbiol. Mol. Biol. Rev. 2000, 64, 821–846. [Google Scholar] [CrossRef] [PubMed]
- Riedl, S.J.; Shi, Y. Molecular mechanisms of caspase regulation during apoptosis. Nat. Rev. Mol. Cell Biol. 2004, 5, 897–907. [Google Scholar] [CrossRef] [PubMed]
- Klaiman, G.; Champagne, N.; LeBlanc, A.C. Self-activation of Caspase-6 in vitro and in vivo: Caspase-6 activation does not induce cell death in HEK293T cells. Biochim. Biophys. Acta 2009, 1793, 592–601. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.F.; Ma, G.; Cao, X.; Huang, Z.L.; Zeng, M.S.; Wen, Z.S. Downregulated expression of PTK6 is correlated with poor survival in esophageal squamous cell carcinoma. Med. Oncol. 2014, 31, 317. [Google Scholar] [CrossRef] [PubMed]
- Zhao, C.; Chen, Y.; Zhang, W.; Zhang, J.; Xu, Y.; Li, W.; Chen, S.; Deng, A. Expression of protein tyrosine kinase 6 (PTK6) in nonsmall cell lung cancer and their clinical and prognostic significance. OncoTargets Ther. 2013, 6, 183–188. [Google Scholar]
- Lang, G.; Bonnet, J.; Umlauf, D.; Karmodiya, K.; Koffler, J.; Stierle, M.; Devys, D.; Tora, L. The tightly controlled deubiquitination activity of the human SAGA complex differentially modifies distinct gene regulatory elements. Mol. Cell. Biol. 2011, 31, 3734–3744. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Lang, G.; Ito, S.; Bonnet, J.; Metzger, E.; Sawatsubashi, S.; Suzuki, E.; Le Guezennec, X.; Stunnenberg, H.G.; Krasnov, A.; et al. A TFTC/STAGA module mediates histone H2A and H2B deubiquitination, coactivates nuclear receptors, and counteracts heterochromatin silencing. Mol. Cell. 2008, 29, 92–101. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.Y.; Varthi, M.; Sykes, S.M.; Phillips, C.; Warzecha, C.; Zhu, W.; Wyce, A.; Thorne, A.W.; Berger, S.L.; McMahon, S.B. The putative cancer stem cell marker USP22 is a subunit of the human SAGA complex required for activated transcription and cell-cycle progression. Mol. Cell. 2008, 29, 102–111. [Google Scholar] [CrossRef] [PubMed]
- Ning, J.; Zhang, J.; Liu, W.; Lang, Y.; Xue, Y.; Xu, S. Overexpression of ubiquitin-specific protease 22 predicts poor survival in patients with early-stage non-small cell lung cancer. Eur. J. Histochem. 2012, 56, e46. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Li, Y.P.; Chen, J.H.; Yuan, S.F.; Wang, L.; Zhang, J.L.; Yao, Q.; Li, N.L.; Bian, J.F.; Fan, J.; et al. Prognostic significance of USP22 as an oncogene in papillary thyroid carcinoma. Tumour Biol. 2013, 34, 1635–1639. [Google Scholar] [CrossRef] [PubMed]
- Piao, S.; Ma, J.; Wang, W.; Liu, Y.; Zhang, M.; Chen, H.; Guo, F.; Zhang, B.; Guo, F. Increased expression of USP22 is associated with disease progression and patient prognosis of salivary duct carcinoma. Oral Oncol. 2013, 49, 796–801. [Google Scholar] [CrossRef] [PubMed]
- Krämer, A.; Green, J.; Pollard, J.Jr; Tugendreich, S. Causal analysis approaches in Ingenuity Pathway Analysis. Bioinformatics 2014, 30, 523–530. [Google Scholar] [CrossRef] [PubMed]
- Palii, C.G.; Perez-Iratxeta, C.; Yao, Z.; Cao, Y.; Dai, F.; Davison, J.; Atkins, H.; Allan, D.; Dilworth, F.J.; Gentleman, R.; et al. Differential genomic targeting of the transcription factor TAL1 in alternate haematopoietic lineages. EMBO J. 2011, 30, 494–509. [Google Scholar] [CrossRef] [PubMed]
- Basso, K.; Saito, M.; Sumazin, P.; Margolin, A.A.; Wang, K.; Lim, W.K.; Kitagawa, Y.; Schneider, C.; Alvarez, M.J.; Califano, A.; et al. Integrated biochemical and computational approach identifies BCL6 direct target genes controlling multiple pathways in normal germinal center B cells. Blood 2010, 115, 975–984. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Koschmann, J.; Bhar, A.; Stegmaier, P.; Kel, A.E.; Wingender, E. “Upstream Analysis”: An Integrated Promoter-Pathway Analysis Approach to Causal Interpretation of Microarray Data. Microarrays 2015, 4, 270-286. https://doi.org/10.3390/microarrays4020270
Koschmann J, Bhar A, Stegmaier P, Kel AE, Wingender E. “Upstream Analysis”: An Integrated Promoter-Pathway Analysis Approach to Causal Interpretation of Microarray Data. Microarrays. 2015; 4(2):270-286. https://doi.org/10.3390/microarrays4020270
Chicago/Turabian StyleKoschmann, Jeannette, Anirban Bhar, Philip Stegmaier, Alexander E. Kel, and Edgar Wingender. 2015. "“Upstream Analysis”: An Integrated Promoter-Pathway Analysis Approach to Causal Interpretation of Microarray Data" Microarrays 4, no. 2: 270-286. https://doi.org/10.3390/microarrays4020270