Comparative Analysis of CNV Calling Algorithms: Literature Survey and a Case Study Using Bovine High-Density SNP Data



Abstract
:1. Introduction
2. CNV Detection Using SNP Arrays
3. Algorithms for CNV Detection
4. Comparing the CNV Detection Algorithms Using Human Data

{kind=link}
| Authors | Year | Algorithm | Data | Platform | Vendor | Conclusion | Comment | |
|---|---|---|---|---|---|---|---|---|
| Lai [66] | 2005 | CGHseq, Quantreg, CLAC, GLAD, CBS, HMM, Wavelet, Lowess, ChARM, GA and ACE | Simulation and empirical samples for Glioblastoma | array CGH | Custom cDNA array | Several general characteristics of future program development were suggested. | Earlier programs for array CGH. | |
| Baross [67] | 2007 | CNAG, dChip, CNAT, GLAD | Simulation and empirical mental retardation 100K Affymetrix SNP array | SNP array | Affymetrix | Multiple programs were needed to find all real aberrations. | False positive deletions was substantial, but could be greatly reduced by using the SNP genotype information to confirm loss of heterozygosity. | |
| Winchester [35] | 2009 | Birdsuite, CNAT, GADA, PennCNV, QuantiSNP | NA12156, NA15510 | SNP array | Affymetrix, Illumina | Multiple predictions from different software. | Use software designed for the platform. | |
| Dellinger [68] | 2010 | CBS, cnvFinder, cnvPartition, GALD, Nexus, PennCNV and QuantiSNP | Simulation and empirical samples from Singapore cohort study of the risk factors for Myopia | SNP array | Illumina | QuantiSNP outperformed other methods based on ROC curve residuals over most datasets. Nexus Rank and SNPRank have low specificity and high power. Nexus Rank calls oversized CNVs. PennCNV detects one of the fewest numbers of CNVs. | The normalized singleton ratio (NSR) is proposed as a metric for parameter optimization. | |
| Tsuang [69] | 2010 | PennCNV, QuantiSNP, HMMSeg, and cnvPartition | 48 Schizophrenia samples | SNP array | Illumina | Both guidelines for the identification of CNVs inferred from high-density arrays and the establishment of a gold standard for validation of CNVs are needed. | Given the variety of methods used, there will be many false positives and false negatives. | |
| Zhang [70] | 2011 | Birdsuite, Partek Genomics Suite, HelixTree, and PennCNV-affy | ~1,000 Bipolar + 270 HapMap samples | SNP array | Affymetrix | Birdsuite and Partek had higher positive predictive values. | Poor overlap between 2 gold standards (Kidd et al. and Conrad et al.). | |
| Marenne [71] | 2011 | cnvPartition, PennCNV, and QuantiSNP | 96 pair samples from Spanish Bladder Cancer/EPICURO study | SNP array | Illumina | PennCNV was the most reliable algorithm when assessing the number of copies. | Current calling algorithms should be improved for high performance CNV analysis in genome-wide scans. | |
| Pinto [49] | 2011 | Birdsuite, cnvFinder, cnvPartition, dCHIP, ADM-2 (DNA Analytics), Genotyping Console (GTC), iPattern, Nexus Copy Number, Partek Genomics Suite, PennCNV, QuantiSNP | 6 samples in triplicate on 11 array platforms | array CGH, SNP array, and BAC array | Agilent, NimbleGen, Affymetrix, and Illumina | Different analytic tools applied to the same raw data typically yield CNV calls with <50% concordance. Moreover, reproducibility in replicate experiments is <70% for most platforms. | The CNV resource presented here allows independent data evaluation and provides a means to benchmark new algorithms. CNV calls are disproportionally affected by genome complexity as they tend to overlap SDs and a single CNV is detected as multiple smaller variants. | |
| Koike [48] | 2011 | Birdsuite, Birdseye, PennCNV, CGHseg, DNAcopy | HapMap samples | SNP array | Affymetrix | Hidden Markov model-based programs PennCNV and Birdseye (part of Birdsuite), or Birdsuite show better detection performance. | Segmental duplications and interspersed repeats (LINEs) are involved in CNVs. | |
| Eckel-Passow [72] | 2011 | Affymetrix Power Tools (APT), Aroma.Affymetrix, PennCNV and CRLMM | 1,418 GENOA (Genetic Epidemiology Network of Atherosclerosis)/FBPP (Family Blood Pressure Program) samples | SNP array | Affymetrix | Recommended trying multiple algorithms, evaluating concordance/discordance and subsequently consider the union of regions for downstream association tests. | Advocated that software developers need to provide guidance with respect to evaluating and choosing optimal settings in order to obtain optimal results for an individual dataset. | |
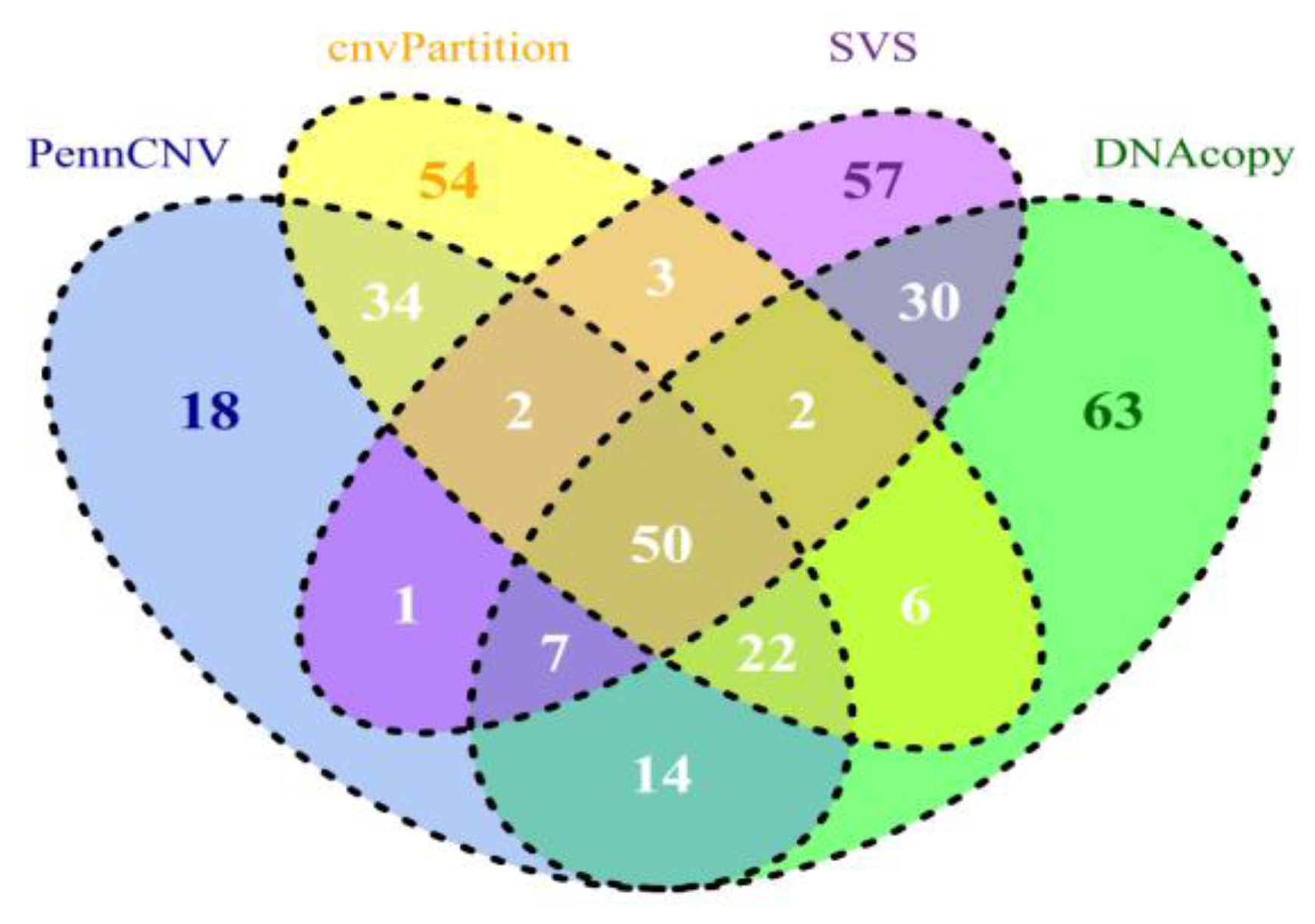
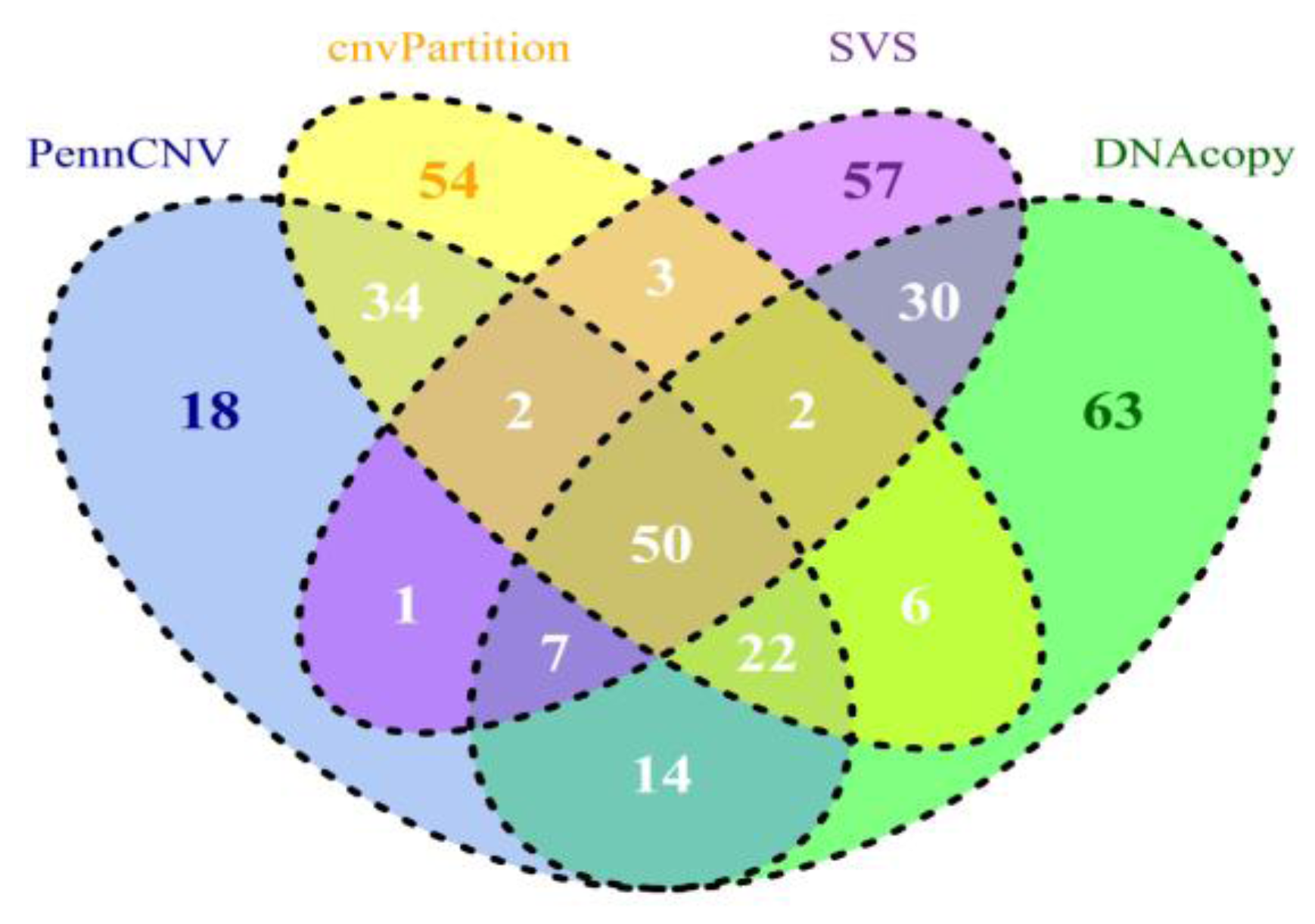
5. Comparing CNV Detection Algorithms Using Bovine High-Density SNP Data
| Tool | Event | Count | Gain | Loss | Average Length |
|---|---|---|---|---|---|
| PennCNV | CNV | 46,751 (74.2) | 17,796 (28.2) | 28,955 (46.0) | 2,334,244,479 (49,929) |
| CNVR | 3,364 a | 1,382 b | 2,376 c | 147,476,461 (43,840) | |
| cnvPartition | CNV | 16,566 (26.3) | 5,021 (8.0) | 11,545 (18.3) | 2,191,528,246 (132,291) |
| CNVR | 1,298 a | 541 b | 916 c | 172,378,730 (132,803) | |
| SVS | CNV | 92,463 (146.8) | 205 (0.3) | 92,258 (146.4) | 2,234,601,290 (24,168) |
| CNVR | 7,099 a | 78 b | 7,056 c | 151,471,634 (21,337) | |
| DNAcopy | CNV | 41,858 (66.4) | 4,469 (7.1) | 37,389 (59.3) | 1,863,930,368 (44,530) |
| CNVR | 5,961 a | 1,457 b | 5,284 c | 194,287,154 (32,593) |
| Count | Length (base pair) | ||||||
|---|---|---|---|---|---|---|---|
| Tool1 | Tool2 | Intersection a | Union a | Percentage | Intersection b | Union b | Percentage |
| PennCNV | cnvPartition | 1,420 | 3,242 | 43.80% | 107,775,740 | 212,079,451 | 50.82% |
| PennCNV | DNAcopy | 2,355 | 6,970 | 33.79% | 93,149,061 | 248,614,554 | 37.47% |
| PennCNV | SVS | 1,264 | 9,199 | 13.74% | 59,557,597 | 239,390,498 | 24.88% |
| cnvPartition | DNAcopy | 1,284 | 5,975 | 21.49% | 79,825,624 | 286,840,260 | 27.83% |
| cnvPartition | SVS | 981 | 7,416 | 13.23% | 56,569,347 | 267,281,017 | 21.16% |
| DNAcopy | SVS | 2,332 | 10,728 | 21.74% | 88,864,805 | 256,893,983 | 34.59% |
6. Conclusions
Acknowledgments
Conflict of Interest
References
- Sebat, J.; Lakshmi, B.; Troge, J.; Alexander, J.; Young, J.; Lundin, P.; Maner, S.; Massa, H.; Walker, M.; Chi, M.; et al. Large-scale copy number polymorphism in the human genome. Science 2004, 305, 525–528. [Google Scholar] [CrossRef]
- Redon, R.; Ishikawa, S.; Fitch, K.R.; Feuk, L.; Perry, G.H.; Andrews, T.D.; Fiegler, H.; Shapero, M.H.; Carson, A.R.; Chen, W.; et al. Global variation in copy number in the human genome. Nature 2006, 444, 444–454. [Google Scholar] [CrossRef]
- Conrad, D.F.; Pinto, D.; Redon, R.; Feuk, L.; Gokcumen, O.; Zhang, Y.; Aerts, J.; Andrews, T.D.; Barnes, C.; Campbell, P.; et al. Origins and functional impact of copy number variation in the human genome. Nature 2009, 464, 704–712. [Google Scholar]
- Altshuler, D.M.; Gibbs, R.A.; Peltonen, L.; Dermitzakis, E.; Schaffner, S.F.; Yu, F.L.; Bonnen, P.E.; de Bakker, P.I.W.; Deloukas, P.; Gabriel, S.B.; et al. Integrating common and rare genetic variation in diverse human populations. Nature 2010, 467, 52–58. [Google Scholar] [CrossRef]
- Mills, R.E.; Walter, K.; Stewart, C.; Handsaker, R.E.; Chen, K.; Alkan, C.; Abyzov, A.; Yoon, S.C.; Ye, K.; Cheetham, R.K.; et al. Mapping copy number variation by population-scale genome sequencing. Nature 2011, 470, 59–65. [Google Scholar] [CrossRef]
- Graubert, T.A.; Cahan, P.; Edwin, D.; Selzer, R.R.; Richmond, T.A.; Eis, P.S.; Shannon, W.D.; Li, X.; McLeod, H.L.; Cheverud, J.M.; et al. A high-resolution map of segmental DNA copy number variation in the mouse genome. PLoS. Genet. 2007, 3, e3. [Google Scholar] [CrossRef]
- Guryev, V.; Saar, K.; Adamovic, T.; Verheul, M.; van Heesch, S.A.; Cook, S.; Pravenec, M.; Aitman, T.; Jacob, H.; Shull, J.D.; et al. Distribution and functional impact of DNA copy number variation in the rat. Nat. Genet. 2008, 40, 538–545. [Google Scholar] [CrossRef]
- She, X.; Cheng, Z.; Zollner, S.; Church, D.M.; Eichler, E.E. Mouse segmental duplication and copy number variation. Nat. Genet. 2008, 40, 909–914. [Google Scholar] [CrossRef]
- Yalcin, B.; Wong, K.; Agam, A.; Goodson, M.; Keane, T.M.; Gan, X.C.; Nellaker, C.; Goodstadt, L.; Nicod, J.; Bhomra, A.; et al. Sequence-based characterization of structural variation in the mouse genome. Nature 2011, 477, 326–329. [Google Scholar] [CrossRef]
- Chen, W.K.; Swartz, J.D.; Rush, L.J.; Alvarez, C.E. Mapping DNA structural variation in dogs. Genome Res. 2009, 19, 500–509. [Google Scholar]
- Nicholas, T.J.; Cheng, Z.; Ventura, M.; Mealey, K.; Eichler, E.E.; Akey, J.M. The genomic architecture of segmental duplications and associated copy number variants in dogs. Genome Res. 2009, 19, 491–499. [Google Scholar]
- Nicholas, T.J.; Baker, C.; Eichler, E.E.; Akey, J.M. A high-resolution integrated map of copy number polymorphisms within and between breeds of the modern domesticated dog. BMC Genomics 2011, 12, 414. [Google Scholar] [CrossRef]
- Liu, G.E.; van Tassell, C.P.; Sonstegard, T.S.; Li, R.W.; Alexander, L.J.; Keele, J.W.; Matukumalli, L.K.; Smith, T.P.; Gasbarre, L.C. Detection of germline and somatic copy number variations in cattle. Dev. Biol. 2008, 132, 231–237. [Google Scholar]
- Liu, G.E.; Hou, Y.; Zhu, B.; Cardone, M.F.; Jiang, L.; Cellamare, A.; Mitra, A.; Alexander, L.J.; Coutinho, L.L.; Dell’aquila, M.E.; et al. Analysis of copy number variations among diverse cattle breeds. Genome Res. 2010, 20, 693–703. [Google Scholar] [CrossRef]
- Volker, M.; Backstrom, N.; Skinner, B.M.; Langley, E.J.; Bunzey, S.K.; Ellegren, H.; Griffin, D.K. Copy number variation, chromosome rearrangement, and their association with recombination during avian evolution. Genome Res. 2010, 20, 503–511. [Google Scholar] [CrossRef]
- Wang, X.F.; Nahashon, S.; Feaster, T.K.; Bohannon-Stewart, A.; Adefope, N. An initial map of chromosomal segmental copy number variations in the chicken. BMC Genomics 2010, 11, 351. [Google Scholar] [CrossRef]
- Fadista, J.; Nygaard, M.; Holm, L.E.; Thomsen, B.; Bendixen, C. A snapshot of CNVs in the pig genome. PLoS ONE 2008, 3, e3916. [Google Scholar] [CrossRef]
- Ramayo-Caldas, Y.; Castelló, A.; Pena, R.N.; Alves, E.; Mercadé, A.; Souza, C.A.; Fernández, A.I.; Perez-Enciso, M.; Folch, J.M. Copy number variation in the porcine genome inferred from a 60 k SNP BeadChip. BMC Genomics 2010, 11, 593. [Google Scholar] [CrossRef]
- Fontanesi, L.; Beretti, F.; Martelli, P.L.; Colombo, M.; Dall’olio, S.; Occidente, M.; Portolano, B.; Casadio, R.; Matassino, D.; Russo, V. A first comparative map of copy number variations in the sheep genome. Genomics 2011, 97, 158–165. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Zhang, L.; Xu, L.; Ren, H.; Lu, J.; Zhang, X.; Zhang, S.; Zhou, X.; Wei, C.; Zhao, F.; et al. Analysis of copy number variations in the sheep genome using 50 k SNP BeadChip array. BMC Genomics 2013, 14, 229. [Google Scholar] [CrossRef]
- Fontanesi, L.; Martelli, P.L.; Beretti, F.; Riggio, V.; Dall’olio, S.; Colombo, M.; Casadio, R.; Russo, V.; Portolano, B. An initial comparative map of copy number variations in the goat (Capra hircus) genome. BMC Genomics 2010, 11, 639. [Google Scholar] [CrossRef] [Green Version]
- Hou, Y.; Liu, G.E.; Bickhart, D.M.; Cardone, M.F.; Wang, K.; Kim, E.S.; Matukumalli, L.K.; Ventura, M.; Song, J.; Vanradan, P.M.; et al. Genomic characteristics of cattle copy number variations. BMC Genomics 2011, 12, 127. [Google Scholar] [CrossRef]
- Bae, J.S.; Cheong, H.S.; Kim, L.H.; NamGung, S.; Park, T.J.; Chun, J.Y.; Kim, J.Y.; Pasaje, C.F.; Lee, J.S.; Shin, H.D. Identification of copy number variations and common deletion polymorphisms in cattle. BMC Genomics 2010, 11, 232. [Google Scholar] [CrossRef]
- Fadista, J.; Thomsen, B.; Holm, L.E.; Bendixen, C. Copy number variation in the bovine genome. BMC Genomics 2010, 11, 284. [Google Scholar] [CrossRef]
- Seroussi, E.; Glick, G.; Shirak, A.; Yakobson, E.; Weller, J.I.; Ezra, E.; Zeron, Y. Analysis of copy loss and gain variations in Holstein cattle autosomes using BeadChip SNPs. BMC Genomics 2010, 11, 673. [Google Scholar] [CrossRef]
- Pinto, D.; Pagnamenta, A.T.; Klei, L.; Anney, R.; Merico, D.; Regan, R.; Conroy, J.; Magalhaes, T.R.; Correia, C.; Abrahams, B.S.; et al. Functional impact of global rare copy number variation in autism spectrum disorders. Nature 2010, 466, 368–372. [Google Scholar] [CrossRef] [Green Version]
- Cook, E.H., Jr.; Scherer, S.W. Copy-number variations associated with neuropsychiatric conditions. Nature 2008, 455, 919–923. [Google Scholar] [CrossRef]
- Sebat, J.; Lakshmi, B.; Malhotra, D.; Troge, J.; Lese-Martin, C.; Walsh, T.; Yamrom, B.; Yoon, S.; Krasnitz, A.; Kendall, J.; et al. Strong association of de novo copy number mutations with autism. Science 2007, 316, 445–449. [Google Scholar] [CrossRef]
- Aitman, T.J.; Dong, R.; Vyse, T.J.; Norsworthy, P.J.; Johnson, M.D.; Smith, J.; Mangion, J.; Roberton-Lowe, C.; Marshall, A.J.; Petretto, E.; et al. Copy number polymorphism in Fcgr3 predisposes to glomerulonephritis in rats and humans. Nature 2006, 439, 851–855. [Google Scholar] [CrossRef]
- Liu, G.E.; Brown, T.; Hebert, D.A.; Cardone, M.F.; Hou, Y.L.; Choudhary, R.K.; Shaffer, J.; Amazu, C.; Connor, E.E.; Ventura, M.; et al. Initial analysis of copy number variations in cattle selected for resistance or susceptibility to intestinal nematodes. Mamm. Genome 2011, 22, 111–121. [Google Scholar] [CrossRef]
- Hou, Y.; Liu, G.E.; Bickhart, D.M.; Matukumalli, L.K.; Li, C.; Song, J.; Gasberre, L.C.; van Tassell, C.P.; Sonstegard, T.S. Genomic regions showing copy number variations associate with resistance or susceptibility to gastrointestinal nematodes in Angus cattle. Funct. Integr. Genomics 2011, 12, 81–92. [Google Scholar]
- Hou, Y.; Bickhart, D.M.; Chung, H.; Hutchison, J.L.; Norman, H.D.; Connor, E.E.; Liu, G.E. Analysis of copy number variations in Holstein cows identify potential mechanisms contributing to differences in residual feed intake. Funct. Integr. Genomics 2012, 12, 717–723. [Google Scholar] [CrossRef]
- LaFramboise, T. Single nucleotide polymorphism arrays: A decade of biological, computational and technological advances. Nucleic Acids Res. 2009, 37, 4181–4193. [Google Scholar] [CrossRef]
- Rincon, G.; Weber, K.L.; van Eenennaam, A.L.; Golden, B.L.; Medrano, J.F. Hot topic: Performance of bovine high-density genotyping platforms in Holsteins and Jerseys. J. Dairy Sci. 2011, 94, 6116–6121. [Google Scholar] [CrossRef]
- Winchester, L.; Yau, C.; Ragoussis, J. Comparing CNV detection methods for SNP arrays. Brief. Funct. Genomic Proteomic 2009, 8, 353–366. [Google Scholar] [CrossRef]
- Sharp, A.J.; Locke, D.P.; McGrath, S.D.; Cheng, Z.; Bailey, J.A.; Vallente, R.U.; Pertz, L.M.; Clark, R.A.; Schwartz, S.; Segraves, R.; et al. Segmental duplications and copy-number variation in the human genome. Am. J. Hum. Genet. 2005, 77, 78–88. [Google Scholar] [CrossRef]
- Marques-Bonet, T.; Girirajan, S.; Eichler, E.E. The origins and impact of primate segmental duplications. Trends Genet. 2009, 25, 443–454. [Google Scholar] [CrossRef]
- Alkan, C.; Kidd, J.M.; Marques-Bonet, T.; Aksay, G.; Antonacci, F.; Hormozdiari, F.; Kitzman, J.O.; Baker, C.; Malig, M.; Mutlu, O.; et al. Personalized copy number and segmental duplication maps using next-generation sequencing. Nat. Genet. 2009, 41, 1061–1067. [Google Scholar] [CrossRef]
- McCarroll, S.A.; Kuruvilla, F.G.; Korn, J.M.; Cawley, S.; Nemesh, J.; Wysoker, A.; Shapero, M.H.; de Bakker, P.I.; Maller, J.B.; Kirby, A.; et al. Integrated detection and population-genetic analysis of SNPs and copy number variation. Nat. Genet. 2008, 40, 1166–1174. [Google Scholar] [CrossRef]
- Estivill, X.; Armengol, L. Copy number variants and common disorders: Filling the gaps and exploring complexity in genome-wide association studies. PLoS Genet. 2007, 3, 1787–1799. [Google Scholar]
- Locke, D.P.; Sharp, A.J.; McCarroll, S.A.; McGrath, S.D.; Newman, T.L.; Cheng, Z.; Schwartz, S.; Albertson, D.G.; Pinkel, D.; Altshuler, D.M.; et al. Linkage disequilibrium and heritability of copy-number polymorphisms within duplicated regions of the human genome. Am. J. Hum. Genet. 2006, 79, 275–290. [Google Scholar] [CrossRef]
- Campbell, C.D.; Sampas, N.; Tsalenko, A.; Sudmant, P.H.; Kidd, J.M.; Malig, M.; Vu, T.H.; Vives, L.; Tsang, P.; Bruhn, L.; et al. Population-genetic properties of differentiated human copy-number polymorphisms. Am. J. Human Genet. 2011, 88, 317–332. [Google Scholar] [CrossRef]
- Illumina—Sequencing and Array-Based Solutions for Genetic Research. Available online: http://www.illumina.com (accessed on 6 June 2013).
- Korn, J.M.; Kuruvilla, F.G.; McCarroll, S.A.; Wysoker, A.; Nemesh, J.; Cawley, S.; Hubbell, E.; Veitch, J.; Collins, P.J.; Darvishi, K.; et al. Integrated genotype calling and association analysis of SNPs, common copy number polymorphisms and rare CNVs. Nat. Genet. 2008, 40, 1253–1260. [Google Scholar] [CrossRef]
- Wang, K.; Li, M.; Hadley, D.; Liu, R.; Glessner, J.; Grant, S.F.; Hakonarson, H.; Bucan, M. PennCNV: An integrated hidden Markov model designed for high-resolution copy number variation detection in whole-genome SNP genotyping data. Genome Res. 2007, 17, 1665–1674. [Google Scholar] [CrossRef]
- Pique-Regi, R.; Monso-Varona, J.; Ortega, A.; Seeger, R.C.; Triche, T.J.; Asgharzadeh, S. Sparse representation and Bayesian detection of genome copy number alterations from microarray data. Bioinformatics 2008, 24, 309–318. [Google Scholar] [CrossRef]
- Yavas, G.; Koyuturk, M.; Ozsoyoglu, M.; Gould, M.P.; LaFramboise, T. An optimization framework for unsupervised identification of rare copy number variation from SNP array data. Genome Biol. 2009, 10, R119. [Google Scholar] [CrossRef]
- Koike, A.; Nishida, N.; Yamashita, D.; Tokunaga, K. Comparative analysis of copy number variation detection methods and database construction. BMC Genet. 2011, 12, 29. [Google Scholar] [CrossRef]
- Pinto, D.; Darvishi, K.; Shi, X.H.; Rajan, D.; Rigler, D.; Fitzgerald, T.; Lionel, A.C.; Thiruvahindrapuram, B.; MacDonald, J.R.; Mills, R.; et al. Comprehensive assessment of array-based platforms and calling algorithms for detection of copy number variants. Nat. Biotechnol. 2011, 29, 512–520. [Google Scholar] [CrossRef]
- Birdsuite FAQ. Broad Institute of MIT and Harvard. Available online: http://www.broadinstitute.org/science/programs/medical-and-population-genetics/birdsuite/birdsuite-faq (accessed on 6 June 2013).
- Colella, S.; Yau, C.; Taylor, J.M.; Mirza, G.; Butler, H.; Clouston, P.; Bassett, A.S.; Seller, A.; Holmes, C.C.; Ragoussis, J. QuantiSNP: An objective bayes hidden-Markov model to detect and accurately map copy number variation using SNP genotyping data. Nucleic Acids Res. 2007, 35, 2013–2025. [Google Scholar] [CrossRef]
- Marioni, J.C.; Thorne, N.P.; Valsesia, A.; Fitzgerald, T.; Redon, R.; Fiegler, H.; Andrews, T.D.; Stranger, B.E.; Lynch, A.G.; Dermitzakis, E.T.; et al. Breaking the waves: Improved detection of copy number variation from microarray-based comparative genomic hybridization. Genome Biol. 2007, 8, R228. [Google Scholar] [CrossRef]
- Diskin, S.J.; Li, M.; Hou, C.; Yang, S.; Glessner, J.; Hakonarson, H.; Bucan, M.; Maris, J.M.; Wang, K. Adjustment of genomic waves in signal intensities from whole-genome SNP genotyping platforms. Nucleic Acids Res. 2008, 36, e126. [Google Scholar] [CrossRef]
- QuantiSNP. Available online: http://sites.google.com/site/quantisnp/ (accessed on 6 June 2013).
- Olshen, A.B.; Venkatraman, E.S.; Lucito, R.; Wigler, M. Circular binary segmentation for the analysis of array-based DNA copy number data. Biostatistics 2004, 5, 557–572. [Google Scholar] [CrossRef]
- Genetic Association Software, Genome-Wide Association (GWAS) Software for SNP, CNV, and NGS. Available online: http://www.goldenhelix.com/SNP_Variation/ (accessed on 6 June 2013).
- Breheny, P.; Chalise, P.; Batzler, A.; Wang, L.; Fridley, B.L. Genetic association studies of copy-number variation: Should assignment of copy number states precede testing? PLoS ONE 2012, 7, e34262. [Google Scholar] [CrossRef]
- Storey, J.D.; Tibshirani, R. Statistical significance for genomewide studies. Proc. Natl. Acad. Sci. USA 2003, 100, 9440–9445. [Google Scholar] [CrossRef]
- Benjamini, Y.; Yekutieli, D. The control of the false discovery rate in multiple testing under dependency. Ann. Stat. 2001, 29, 1165–1188. [Google Scholar] [CrossRef]
- Li, B.; Leal, S.M. Methods for detecting associations with rare variants for common diseases: Application to analysis of sequence data. Am. J. Hum. Genet. 2008, 83, 311–321. [Google Scholar] [CrossRef]
- Yang, H.C.; Hsieh, H.Y.; Fann, C.S. Kernel-based association test. Genetics 2008, 179, 1057–1068. [Google Scholar] [CrossRef]
- Baladandayuthapani, V.; Ji, Y.; Talluri, R.; Nieto-Barajas, L.E.; Morris, J.S. Bayesian random segmentation models to identify shared copy number aberrations for array CGH data. J. Am. Stat. Assoc. 2010, 105, 1358–1375. [Google Scholar] [CrossRef]
- Nowak, G.; Hastie, T.; Pollack, J.R.; Tibshirani, R. A fused lasso latent feature model for analyzing multi-sample aCGH data. Biostatistics 2011, 12, 776–791. [Google Scholar] [CrossRef]
- Glessner, J.T.; Li, J.; Hakonarson, H. ParseCNV integrative copy number variation association software with quality tracking. Nucleic Acids Res. 2013, 41, e64. [Google Scholar] [CrossRef]
- Scherer, S.W.; Lee, C.; Birney, E.; Altshuler, D.M.; Eichler, E.E.; Carter, N.P.; Hurles, M.E.; Feuk, L. Challenges and standards in integrating surveys of structural variation. Nat. Genet. 2007, 39, S7–S15. [Google Scholar] [CrossRef]
- Lai, W.R.; Johnson, M.D.; Kucherlapati, R.; Park, P.J. Comparative analysis of algorithms for identifying amplifications and deletions in array CGH data. Bioinformatics 2005, 21, 3763–3770. [Google Scholar] [CrossRef]
- Baross, A.; Delaney, A.D.; Li, H.I.; Nayar, T.; Flibotte, S.; Qian, H.; Chan, S.Y.; Asano, J.; Ally, A.; Cao, M.; et al. Assessment of algorithms for high throughput detection of genomic copy number variation in oligonucleotide microarray data. BMC Bioinformatics 2007, 8, 368. [Google Scholar] [CrossRef]
- Dellinger, A.E.; Saw, S.M.; Goh, L.K.; Seielstad, M.; Young, T.L.; Li, Y.J. Comparative analyses of seven algorithms for copy number variant identification from single nucleotide polymorphism arrays. Nucleic Acids Res. 2010, 38, e105. [Google Scholar] [CrossRef]
- Tsuang, D.W.; Millard, S.P.; Ely, B.; Chi, P.; Wang, K.; Raskind, W.H.; Kim, S.; Brkanac, Z.; Yu, C.E. The effect of algorithms on copy number variant detection. PLoS ONE 2010, 5, e14456. [Google Scholar] [CrossRef]
- Zhang, D.; Qian, Y.; Akula, N.; Alliey-Rodriguez, N.; Tang, J.; Gershon, E.S.; Liu, C. Accuracy of CNV detection from GWAS data. PLoS ONE 2011, 6, e14511. [Google Scholar] [CrossRef]
- Marenne, G.; Rodriguez-Santiago, B.; Closas, M.G.; Perez-Jurado, L.; Rothman, N.; Rico, D.; Pita, G.; Pisano, D.G.; Kogevinas, M.; Silverman, D.T.; et al. Assessment of copy number variation using the Illumina Infinium 1M SNP-array: A comparison of methodological approaches in the Spanish Bladder Cancer/EPICURO study. Hum. Mutat. 2011, 32, 240–248. [Google Scholar] [CrossRef]
- Eckel-Passow, J.E.; Atkinson, E.J.; Maharjan, S.; Kardia, S.L.; de Andrade, M. Software comparison for evaluating genomic copy number variation for Affymetrix 6.0 SNP array platform. BMC Bioinformatics 2011, 12, 220. [Google Scholar] [CrossRef]
- Hou, Y.; Bickhart, D.M.; Hvinden, M.L.; Li, C.; Song, J.; Boichard, D.A.; Fritz, S.; Eggen, A.; Denise, S.; Wiggans, G.R.; et al. Fine mapping of copy number variations on two cattle genome assemblies using high density SNP array. BMC Genomics 2012, 13, 376. [Google Scholar] [CrossRef]
- Matsunami, N.; Hadley, D.; Hensel, C.H.; Christensen, G.B.; Kim, C.; Frackelton, E.; Thomas, K.; da Silva, R.P.; Stevens, J.; Baird, L.; et al. Identification of rare recurrent copy number variants in high-risk autism families and their prevalence in a large ASD population. PLoS ONE 2013, 8, e52239. [Google Scholar] [CrossRef]
- Carter, N.P. Methods and strategies for analyzing copy number variation using DNA microarrays. Nat. Genet. 2007, 39, S16–S21. [Google Scholar] [CrossRef]
- Bickhart, D.M.; Hou, Y.; Schroeder, S.G.; Alkan, C.; Cardone, M.F.; Matukumalli, L.K.; Song, J.; Schnabel, R.D.; Ventura, M.; Taylor, J.F.; et al. Copy number variation of individual cattle genomes using next-generation sequencing. Genome Res. 2012, 22, 778–790. [Google Scholar] [CrossRef]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Xu, L.; Hou, Y.; Bickhart, D.M.; Song, J.; Liu, G.E. Comparative Analysis of CNV Calling Algorithms: Literature Survey and a Case Study Using Bovine High-Density SNP Data. Microarrays 2013, 2, 171-185. https://doi.org/10.3390/microarrays2030171
Xu L, Hou Y, Bickhart DM, Song J, Liu GE. Comparative Analysis of CNV Calling Algorithms: Literature Survey and a Case Study Using Bovine High-Density SNP Data. Microarrays. 2013; 2(3):171-185. https://doi.org/10.3390/microarrays2030171
Chicago/Turabian StyleXu, Lingyang, Yali Hou, Derek M. Bickhart, Jiuzhou Song, and George E. Liu. 2013. "Comparative Analysis of CNV Calling Algorithms: Literature Survey and a Case Study Using Bovine High-Density SNP Data" Microarrays 2, no. 3: 171-185. https://doi.org/10.3390/microarrays2030171