Compartmentalized Replication of SARS-Cov-2 in Upper vs. Lower Respiratory Tract Assessed by Whole Genome Quasispecies Analysis

 , ,
, ,  ,
,  , , , and
, , , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Next Generation Sequencing (NGS) of Clinical Samples
2.2. Data Analysis
3. Results
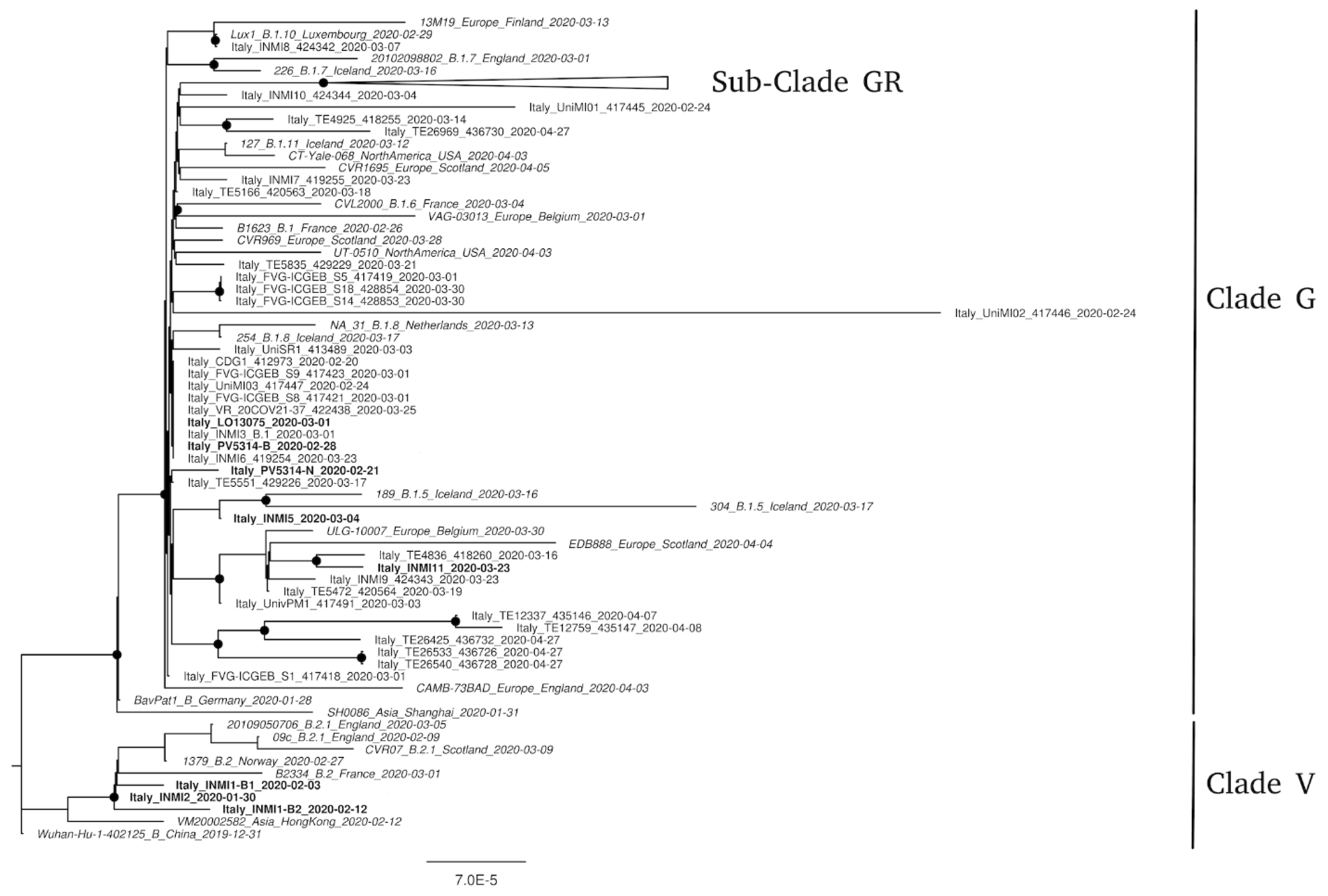
3.1. Phylogenetic Analysis
3.2. Genetic Variability of Whole-Genome Consensus Sequences
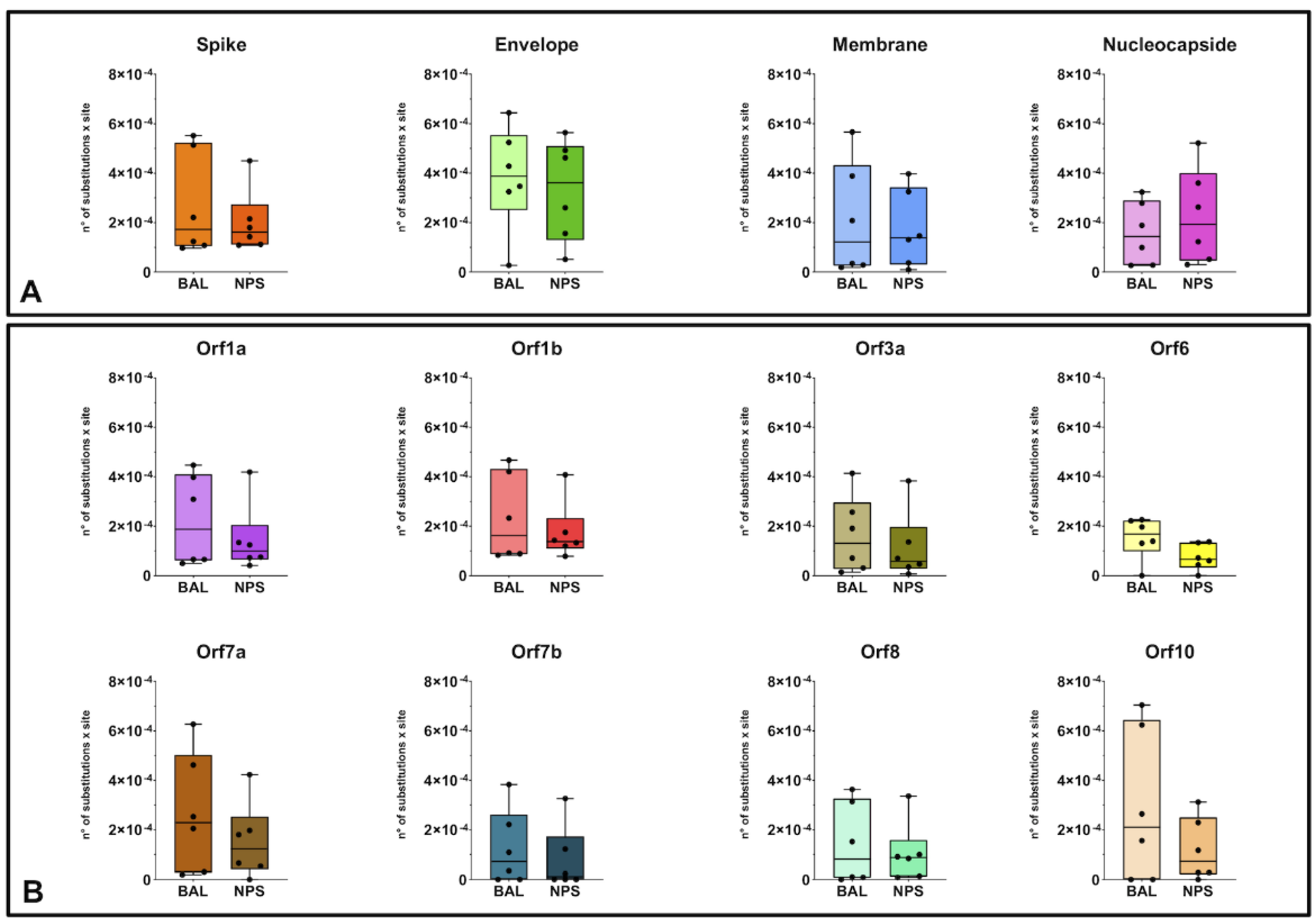
3.3. Diversity and Variation Analysis
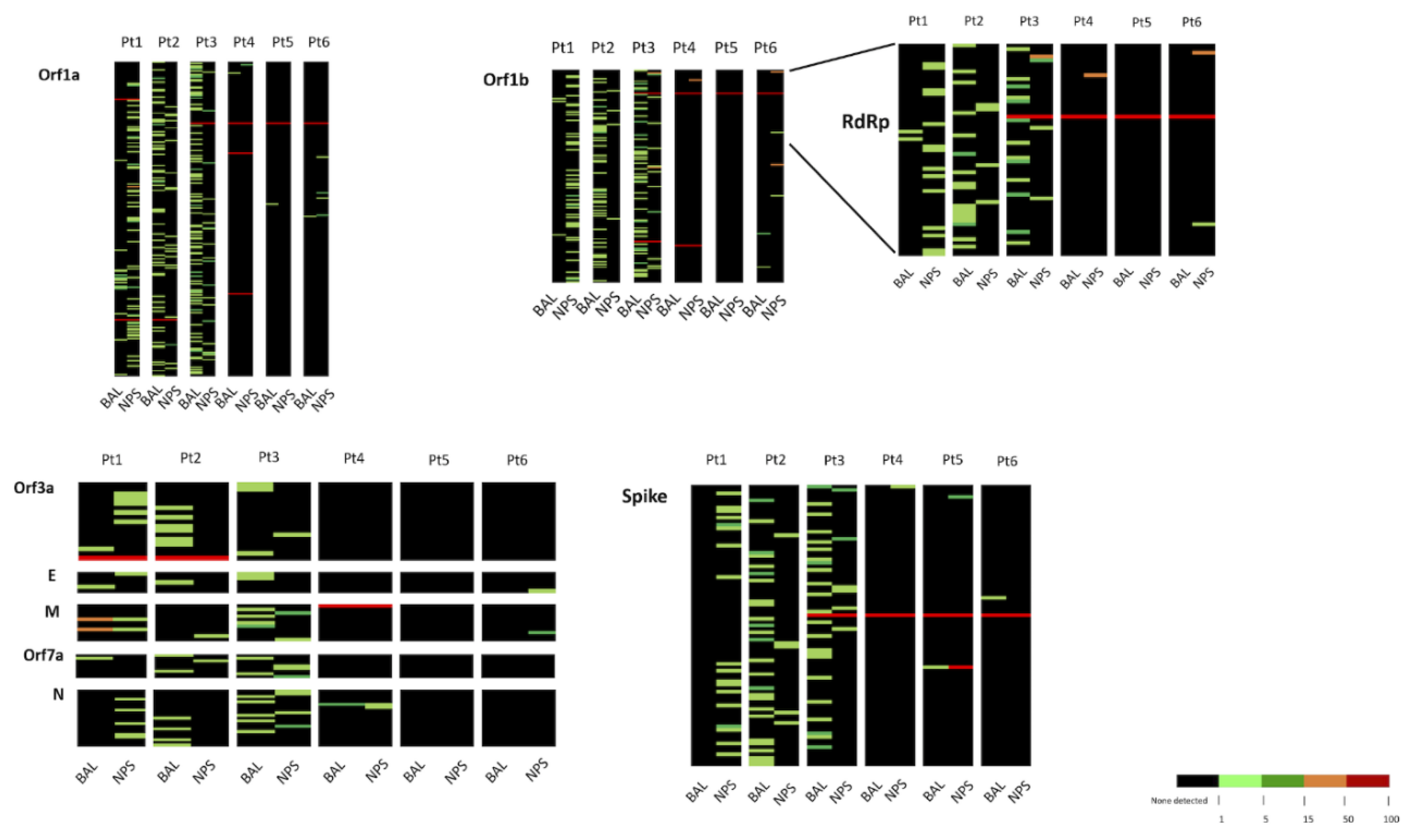
3.4. Analysis of Minority Variants in BAL and NPS
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Disclaimer
| INMI1-B1: EPI_ISL_451298; MT509660 |
| INMI1-B2: EPI_ISL_451299; MT509652 |
| INMI1-N: EPI_ISL_451300; MT509661 |
| INMI2-B: EPI_ISL_451301; MT509653 |
| INMI2-N: EPI_ISL_451302; MT509662 |
| INMI5-B: EPI_ISL_417923; MT509668 |
| INMI5-N: EPI_ISL_451303; MT509654 |
| INMI11-B: EPI_ISL_451304; MT509663 |
| INMI11-N: EPI_ISL_451305; MT509655 |
| PV-5314-B: EPI_ISL_451306; MT509667 |
| PV-5314-N: EPI_ISL_451307; MT509664 |
| LO-13075-B: EPI_ISL_451308; MT509665 |
| LO-13075-N: EPI_ISL_451309; MT509666 |
References
- WHO. Coronavirus Disease COVID-2019; Situation Report 126; World Health Organization: Geneva, Switzerland, 25 May 2020. [Google Scholar]
- Capobianchi, M.R.; Rueca, M.; Messina, F.; Giombini, E.; Carletti, F.; Colavita, F.; Castilletti, C.; Lalle, E.; Bordi, L.; Vairo, F. Molecular characterization of SARS-CoV-2 from the first case of COVID-19 in Italy. Clin. Microbiol. Infect. 2020, 26, 3–5. [Google Scholar] [CrossRef] [PubMed]
- Corman, V.M.; Landt, O.; Kaiser, M.; Molenkamp, R.; Meijer, A.; Chu, D.K.; Bleicker, T.; Brünink, S.; Schneider, J.; Schmidt, M.L.; et al. Detection of 2019 novel coronavirus (2019-nCoV) by real-time RT-PCR. Eurosurveillance 2020, 25, 2000045. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grabherr, M.G.; Haas, B.J.; Yassour, M.; Levin, J.Z.; Thompson, D.A.; Amit, I.; Adiconis, X.; Fan, L.; Raychowdhury, R.; Zeng, Q.; et al. Full-length transcriptome assembly from RNA-seq data without a reference genome. Nat. Biotechnol. 2011, 29, 644–652. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elbe, S.; Buckland-Merrett, G. Data, disease and diplomacy: GISAID′s innovative contribution to global health. Glob. Chall. 2017, 1, 33–46. [Google Scholar] [CrossRef] [Green Version]
- Fu, L.; Niu, B.; Zhu, Z.; Wu, S.; Li, W. CD-HIT: Accelerated for clustering the next-generation sequencing data. Bioinformatics 2012, 28, 3150–3152. [Google Scholar] [CrossRef]
- Rambaut, A.; Holmes, E.C.; Hill, V.; OToole, A.; McCrone, J.; Ruis, C.; du Plessis, L.; Pybus, O. A dynamic nomenclature proposal for SARS-CoV-2 to assist genomic epidemiology. bioRxiv 2020. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [Green Version]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [Green Version]
- Li, H. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv 2013, arXiv:1303.3997. [Google Scholar]
- Bartolini, B.; Rueca, M.; Gruber, C.E.M.; Messina, F.; Carletti, F.; Giombini, E.; Lalle, E.; Bordi, L.; Matusali, G.; Colavita, F.; et al. SARS-CoV-2 phylogenetic analysis in Lazio region, Italy (February–March 2020). Emerg. Infect. Dis. 2020, 26, 1842–1845. [Google Scholar] [CrossRef]
- Jary, A.; Leducq, V.; Malet, I.; Marot, S.; Klement-Frutos, E.; Teyssou, E.; Soulié, C.; Abdi, B.; Wirden, M.; Pourcher, V.; et al. Evolution of viral quasispecies during SARS-CoV-2 infection. Clin. Microbiol. Infect. 2020, in press. [Google Scholar] [CrossRef] [PubMed]
- Chiara, M.; Horner, D.S.; Pesole, G. Comparative genomics suggests limited variability and similar evolutionary patterns between major clades of SARS-Cov-2. bioRxiv 2020. [Google Scholar] [CrossRef]
- Shen, Z.; Xiao, Y.; Kang, L.; Ma, W.; Shi, L.; Zhang, L.; Zhou, Z.; Yang, J.; Zhong, J.; Yang, D.; et al. Genomic diversity of SARS-CoV-2 in Coronavirus Disease 2019 patients. Clin. Infect. Dis. 2020, 15, 1–27. [Google Scholar] [CrossRef] [Green Version]
- Van Dorp, L.; Acman, M.; Richard, D.; Shaw, L.P.; Ford, C.E.; Ormond, L.; Owen, C.J.; Pang, J.; Tan, C.C.; Boshier, F.A.; et al. Emergence of genomic diversity and recurrent mutations in SARS-CoV-2. Infect. Genet. Evol. 2020, 83, 104351. [Google Scholar] [CrossRef] [PubMed]
- Bartolini, B.; Chillemi, G.; Abbate, I.; Bruselles, A.; Rozera, G.; Castrignanò, T.; Paoletti;, D.; Picardi, E.; Desideri, A.; Pesole, G.; et al. Assembly and characterization of pandemic influenza A H1N1 genome in nasopharyngeal swabs using high-throughput pyrosequencing. New Microbiol. 2011, 34, 391–397. [Google Scholar] [PubMed]
- Han, Q.; Jones, J.A.; Nicely, N.I.; Reed, R.K.; Shen, X.; Mansouri, K.; Louder, M.; Trama, A.M.; Alam, S.M.; Edwards, R.J.; et al. Difficult-to-neutralize global HIV-1 isolates are neutralized by antibodies targeting open envelope conformations. Nat. Commun. 2019, 10, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Khera, T.; Behrendt, P.; Bankwitz, D.; Brown, R.J.; Todt, D.; Doepke, M.; Khan, A.G.; Schulze, K.; Law, J.; Logan, M.; et al. Functional and immunogenic characterization of diverse HCV glycoprotein E2 variants. J. Hepatol. 2019, 70, 593–602. [Google Scholar] [CrossRef]
- Wrapp, D.; Wang, N.; Corbett, K.S.; Goldsmith, J.A.; Hsieh, C.L.; Abiona, O.; Graham, B.S.; McLellan, J.S. Cryo-EM structure of the 2019-nCoV spike in the prefusion conformation. Science 2020, 367, 1260–1263. [Google Scholar] [CrossRef] [Green Version]
- Chu, H.; Chan, J.F.W.; Yuen, T.T.T.; Shuai, H.; Yuan, S.; Wang, Y.; Hu, B.; Yip, C.C.Y.; Tsang, J.O.L.; Huang, X.; et al. Comparative tropism, replication kinetics, and cell damage profiling of SARS-CoV-2 and SARS-CoV with implications for clinical manifestations, transmissibility, and laboratory studies of COVID-19: An observational study. Lancet Microb. 2020, 1, e14–e23. [Google Scholar] [CrossRef]
- Chan, J.F.W.; Kok, K.H.; Zhu, Z.; Chu, H.; To, K.K.W.; Yuan, S.; Yuen, K.Y. Genomic characterization of the 2019 novel human-pathogenic coronavirus isolated from a patient with atypical pneumonia after visiting Wuhan. Emerg. Microb. Infect. 2020, 9, 221–236. [Google Scholar] [CrossRef] [Green Version]
- Fung, T.S.; Liu, D.X. Human Coronavirus: Host-Pathogen Interaction. Annu. Rev. Microbiol. 2019, 9, 529–560. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, W.-B. Decoding evolution and transmissions of novel pneumonia coronavirus (SARS-CoV-2) using the whole genomic data. Zool. Res. 2020. [Google Scholar] [CrossRef] [PubMed]
- Korber, B.; Fischer, W.; Gnanakaran, S.G.; Yoon, H.; Theiler, J.; Abfalterer, W.; Foley, B.; Giorgi, E.E.; Bhattacharya, T.; Parker, M.D.; et al. Spike mutation pipeline reveals the emergence of a more transmissible form of SARS-CoV-2. bioRxiv 2020. [Google Scholar] [CrossRef]
- Park, D.; Huh, H.J.; Kim, Y.J.; Son, D.S.; Jeon, H.J.; Im, E.H.; Kim, J.W.; Lee, N.Y.; Kang, E.S.; Kang, C.I.; et al. Analysis of intrapatient heterogeneity uncovers the microevolution of Middle East respiratory syndrome coronavirus. Mol. Case Stud. 2016, 2, 001214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, D.; Zhang, Z.; Wang, F.S. SARS-Associated Coronavirus Quasispecies in Individual Patients. N. Engl. J. Med. 2004, 350, 1366–1367. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Patient | Pt1B T1 | Pt1B T2 | Pt1N | Pt2B | Pt2N | Pt3B | Pt3N | Pt4B | Pt4N | Pt5B | Pt5N | Pt6B | Pt6N |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Type of Sample | BAL T1 | BAL T2 | NPS | BAL | NPS | BAL | NPS | BAL | NPS | BAL | NPS | BAL | NPS |
| Sex | female | male | male | female | male | male | |||||||
| Age | 66 | 67 | 53 | 49 | 75 | 54 | |||||||
| Epidemiological Link, Region (Country) | Wuhan (China) | Wuhan (China) | Lazio (Italy) | Lazio (Italy) | Lombardia (Italy) | Lombardia (Italy) | |||||||
| Date in 2020 | February 4 | February 12 | February 3 | February 3 | January 30 | March 4 | March 4 | March 23 | March 23 | February 21 | February 21 | March 1 | March 1 |
| Ct | 18.0 * | 23.3 * | 21.5 * | 19.9 * | 28.5 * | n.d. | n.d. | 14.1 ** | 16.8 ** | n.d. | 24.0 * | 21.5 * | 20.7 * |
| Pt ID Strain Designation | ||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Region | nt | Ref * | Pt1B T1 INMI1 BAL1 | Pt1B T2 INMI1 AL2 | Pt1N INMI1 NPS | Pt2B INMI2 BAL | Pt2N INMI2NPS | Pt3 INMI5BAL | Pt3N INMI5NPS | Pt4B INMI11BAL | Pt4N INMI11NPS | Pt5B PV-5314 BAL | Pt5N PV-5314 NPS | Pt6B LO-13075 BAL | Pt6N LO-13075 NPS | AA Change |
| 5′ UTR | 241 | C | T | T | T | T | T | T | T | T | - | |||||
| Orf1ab | 2269 | A | T | T | Syn | |||||||||||
| 3037 | C | T | T | T | T | T | T | T | T | Syn | ||||||
| 4255 | G | T | T | Syn | ||||||||||||
| 10150 | T | C | C | Syn | ||||||||||||
| 11083 | G | T | T | T | T | T | L3606F | |||||||||
| 14408 | C | T | T | T | T | T | T | T | T | P4715L | ||||||
| 20268 | A | G | G | Syn | ||||||||||||
| 20355 | A | G | G | Syn | ||||||||||||
| S | 23403 | A | G | G | G | G | G | G | G | G | D614G | |||||
| 24077 | G | T | D839Y | |||||||||||||
| Orf3a | 26144 | G | T | T | T | T | T | G251V | ||||||||
| M | 26530 | A | G | G | D3G | |||||||||||
| 26681 | C | T | Syn | |||||||||||||
| 26754 | G | T | G78C | |||||||||||||
| nt Position | Ref * | Substitution | Pt1N-February 3 (%) | Pt1B T1- February 4 (%) | Pt1B T2- February 12 (%) | AA |
|---|---|---|---|---|---|---|
| 26681 | C | T | 4.63 | 16.85 | 87.91 | Syn |
| 26751 | A | G | 0.00 | 0.00 | 2.30 | T77A |
| 26754 | G | T | 4.20 | 16.69 | 88.51 | G78C |
| 26844 | T | C | 1.20 | 0.00 | 0.00 | S108P |
| 27084 | G | A | 1.50 | 0.00 | 0.00 | A188T |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rueca, M.; Bartolini, B.; Gruber, C.E.M.; Piralla, A.; Baldanti, F.; Giombini, E.; Messina, F.; Marchioni, L.; Ippolito, G.; Di Caro, A.; et al. Compartmentalized Replication of SARS-Cov-2 in Upper vs. Lower Respiratory Tract Assessed by Whole Genome Quasispecies Analysis. Microorganisms 2020, 8, 1302. https://doi.org/10.3390/microorganisms8091302
Rueca M, Bartolini B, Gruber CEM, Piralla A, Baldanti F, Giombini E, Messina F, Marchioni L, Ippolito G, Di Caro A, et al. Compartmentalized Replication of SARS-Cov-2 in Upper vs. Lower Respiratory Tract Assessed by Whole Genome Quasispecies Analysis. Microorganisms. 2020; 8(9):1302. https://doi.org/10.3390/microorganisms8091302
Chicago/Turabian StyleRueca, Martina, Barbara Bartolini, Cesare Ernesto Maria Gruber, Antonio Piralla, Fausto Baldanti, Emanuela Giombini, Francesco Messina, Luisa Marchioni, Giuseppe Ippolito, Antonino Di Caro, and et al. 2020. "Compartmentalized Replication of SARS-Cov-2 in Upper vs. Lower Respiratory Tract Assessed by Whole Genome Quasispecies Analysis" Microorganisms 8, no. 9: 1302. https://doi.org/10.3390/microorganisms8091302
APA StyleRueca, M., Bartolini, B., Gruber, C. E. M., Piralla, A., Baldanti, F., Giombini, E., Messina, F., Marchioni, L., Ippolito, G., Di Caro, A., & Capobianchi, M. R. (2020). Compartmentalized Replication of SARS-Cov-2 in Upper vs. Lower Respiratory Tract Assessed by Whole Genome Quasispecies Analysis. Microorganisms, 8(9), 1302. https://doi.org/10.3390/microorganisms8091302