Next-Generation Sequencing as a Tool to Detect Vaginal Microbiota Disturbances during Pregnancy

 , , , , and
, , , , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Patients
2.2. Samples
2.3. Library Preparation
2.4. Next-Generation Sequencing
2.5. Bioinformatics Analysis
3. Results
3.1. Characteristics of the Study Population
3.2. Metagenomic Sequencing
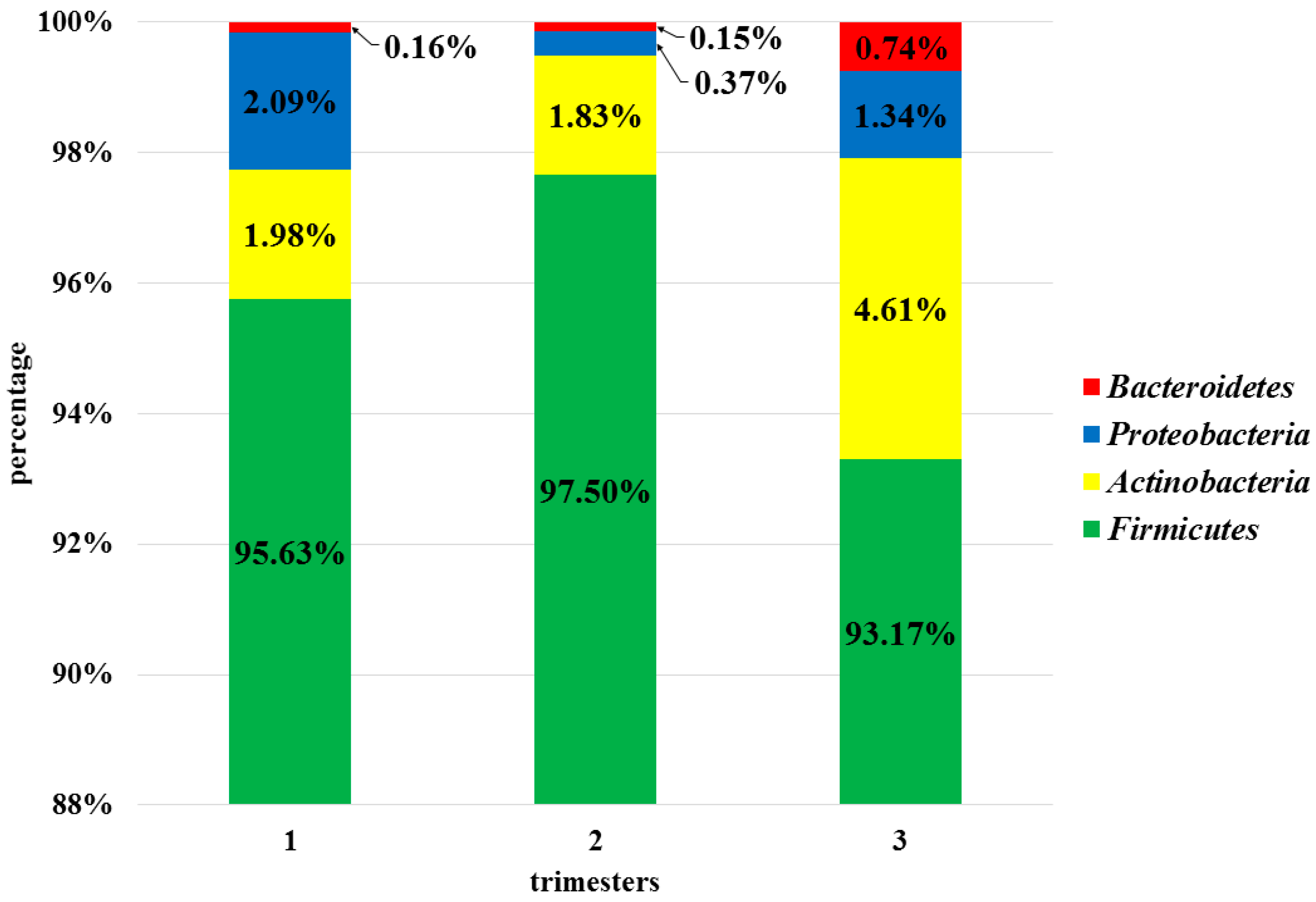
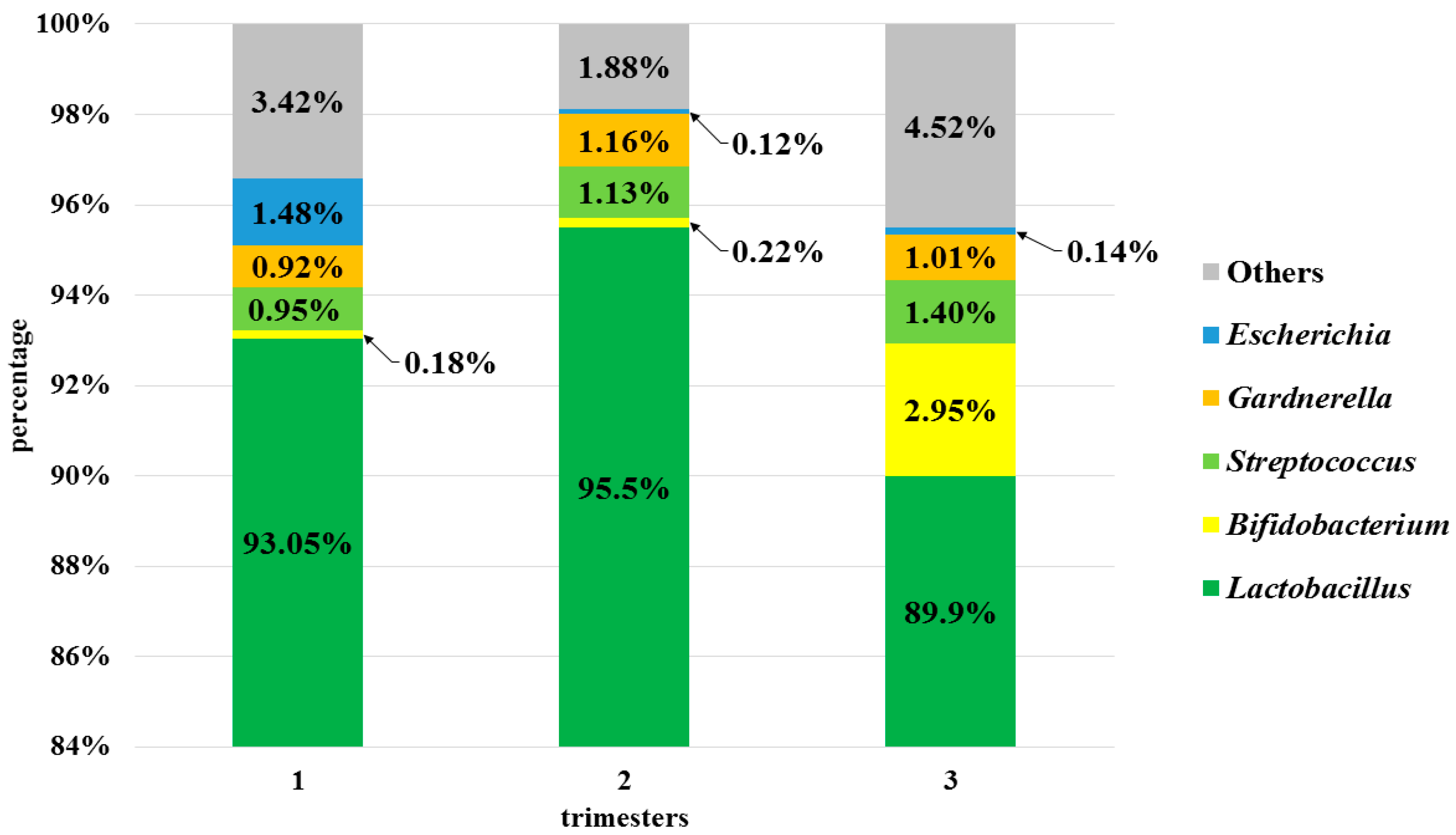
3.3. Analysis of the Vaginal Microbiota in Each Trimester in the Pregnant Women Group
3.4. Semi-Quantitative and Qualitative Composition of the Physiological Microbiota in Individual Patients
3.4.1. Stable Microbiota during Pregnancy
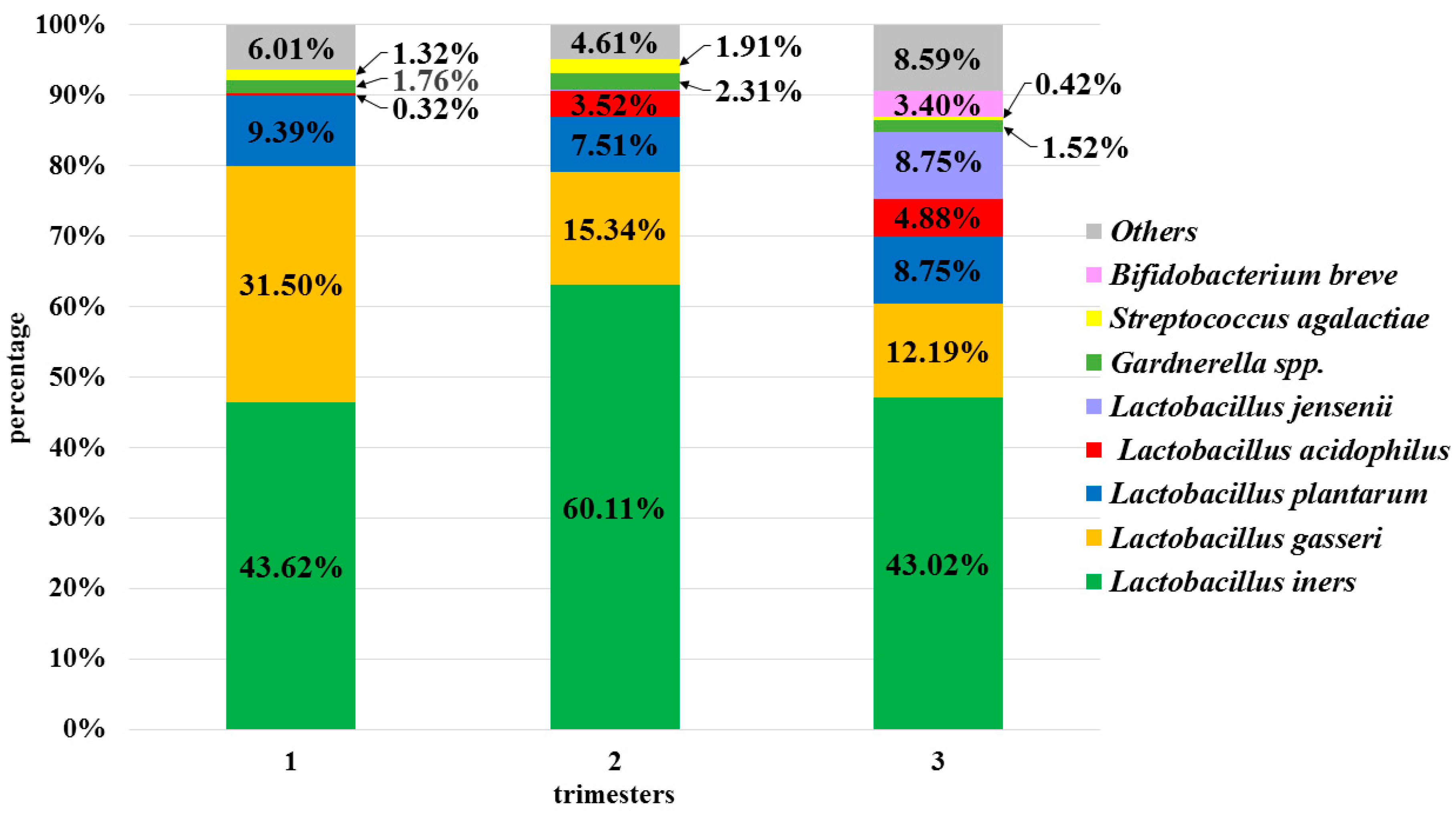
3.4.2. Fluctuations in the Non-Pathogenic Potential Lactobacillus Species Composition of the Vaginal Microbiota
3.4.3. Fluctuations in the Pathogenic Potential Species Composition of the Vaginal Microbiota
4. Discussion
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Hickey, R.J.; Zhou, X.; Pierson, J.D.; Ravel, J.; Forney, L.J. Understanding vaginal microbiome complexity from an ecological perspective. Transl. Res. 2012, 160, 267–282. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, B.; Forney, L.J.; Ravel, J. Vaginal Microbiome: Rethinking Health and Disease. Annu. Rev. Microbiol. 2012, 66, 371–389. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van De Wijgert, J.; Borgdorff, H.; Verhelst, R.; Crucitti, T.; Francis, S.; Verstraelen, H.; Jespers, V. The Vaginal Microbiota: What Have We Learned after a Decade of Molecular Characterization? PLoS ONE 2014, 9, e105998. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nuriel-Ohayon, M.; Neuman, H.; Koren, O. Microbial Changes during Pregnancy, Birth, and Infancy. Front. Microbiol. 2016, 7, 1031. [Google Scholar] [CrossRef] [Green Version]
- Kumar, R.; Cliver, S.P.; Zhi, D.; Szychowski, J.M.; Abramovici, A.; Biggio, J.R.; Lefkowitz, E.J.; Morrow, C.; Edwards, R.K.; Subramaniam, A. Vaginal Microbiota in Pregnancy: Evaluation Based on Vaginal Flora, Birth Outcome, and Race. Am. J. Perinatol. 2016, 33, 401–408. [Google Scholar] [CrossRef] [Green Version]
- Fettweis, J.M.; Serrano, M.G.; Girerd, P.H.; Jefferson, K.K.; Buck, G.A. A New Era of the Vaginal Microbiome: Advances Using Next-Generation Sequencing. Chem. Biodivers. 2012, 9, 965–976. [Google Scholar] [CrossRef]
- Dunlop, A.L.; Mulle, J.G.; Ferranti, E.P.; Edwards, S.; Dunn, A.B.; Corwin, E.J. Maternal Microbiome and Pregnancy Outcomes That Impact Infant Health: A Review. Adv. Neonatal Care 2015, 15, 377–385. [Google Scholar] [CrossRef] [Green Version]
- Ravel, J.; Brotman, R.M. Translating the vaginal microbiome: Gaps and challenges. Genome Med. 2016, 8, 35. [Google Scholar] [CrossRef] [Green Version]
- Prince, A.L.; Chu, D.M.; Seferovic, M.D.; Antony, K.M.; Ma, J.; Aagaard, K.M. The Perinatal Microbiome and Pregnancy: Moving Beyond the Vaginal Microbiome. Cold Spring Harb. Perspect. Med. 2015, 5, a023051. [Google Scholar] [CrossRef]
- MacIntyre, D.A.; Chandiramani, M.; Lee, Y.S.; Kindinger, L.; Smith, A.; Angelopoulos, N.; Lehne, B.; Arulkumaran, S.; Brown, R.; Teoh, T.G.; et al. The vaginal microbiome during pregnancy and the postpartum period in a European population. Sci. Rep. 2015, 5, 8988. [Google Scholar] [CrossRef] [Green Version]
- Brzychczy-Włoch, M.; Ochońska, D.; Bulanda, M. Carriage of Group B Streptococci in Pregnant Women from The Region of Krakow and their Antibiotic Resistance in the Years 2008–2012. Pol. J. Microbiol. 2013, 62, 427–433. [Google Scholar] [CrossRef] [PubMed]
- Eloe-Fadrosh, E.A.; Rasko, D.A. The Human Microbiome: From Symbiosis to Pathogenesis. Annu. Rev. Med. 2013, 64, 145–163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haque, M.M.; Merchant, M.; Kumar, P.N.; Dutta, A.; Mande, S.S. First-trimester vaginal microbiome diversity: A potential indicator of preterm delivery risk. Sci. Rep. 2017, 7, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bagga, R.; Arora, P. Genital micro-organisms in pregnancy. Front. Public Health 2020, 8, 225. [Google Scholar] [CrossRef]
- Brzychczy-Wloch, M.; Pabian, W.; Majewska, E.; Zuk, M.G.; Kielbik, J.; Gosiewski, T.; Bulanda, M.G. Dynamics of colonization with group B streptococci in relation to normal flora in women during subsequent trimesters of pregnancy. New Microbiol. 2014, 37, 307–319. [Google Scholar]
- Nugent, R.P.; Krohn, M.A.; Hillier, S.L. Reliability of diagnosing bacterial vaginosis is improved by a standardized method of gram stain interpretation. J. Clin. Microbiol. 1991, 29, 297–301. [Google Scholar] [CrossRef] [Green Version]
- Gosiewski, T.; Szała, L.; Pietrzyk, A.; Brzychczy-Włoch, M.; Heczko, P.B.; Bulanda, M. Comparison of Methods for Isolation of Bacterial and Fungal DNA from Human Blood. Curr. Microbiol. 2014, 68, 149–155. [Google Scholar] [CrossRef] [Green Version]
- 16S Metagenomic Sequencing Library Preparation; Preparing 16S Ribosomal RNA Gene Amplicons for the Illumina MiSeq System. Available online: https://Support.illumina.com (accessed on 14 October 2020).
- Wang, Q.; Garrity, G.M.; Tiedje, J.M.; Cole, J.R. Native Bayesian Classifier for Rapid Assignment of rRNA Sequences into the New Bacterial Taxonomy. Appl. Environ. Microbiol. 2007, 73, 5261–5267. [Google Scholar] [CrossRef] [Green Version]
- Edgar, R.C.; Haas, B.J.; Clemente, J.C.; Quince, C.; Knight, R. UCHIME improves sensitivity and speed of chimera detection. Bioinformatics 2011, 27, 2194–2200. [Google Scholar] [CrossRef] [Green Version]
- Alishum, A. DADA2 formatted 16S rRNA gene sequences for both bacteria and archaea. Res. Data 2019. [Google Scholar]
- Chong, J.; Liu, P.; Zhou, G.; Xia, J. Using Microbiome Analyst for comprehensive statistical, functional, and meta-analysis of microbiome data. Nat. Protoc. 2020, 15, 799–821. [Google Scholar] [CrossRef] [PubMed]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoav, B.; Hochberg, Y. Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing. J. R. Stat. Soc. Ser. B 1995, 57, 289–300. [Google Scholar]
- JASP; Version 0.14; Computer Software; JASP Team: Amsterdam, The Netherlands, 2020.
- Romero, R.; Hassan, S.S.; Gajer, P.; Tarca, A.L.; Fadrosh, D.W.; Nikita, L.; Galuppi, M.; Lamont, R.F.; Chaemsaithong, P.; Miranda, J.; et al. The composition and stability of the vaginal microbiota of normal pregnant women is different from that of non-pregnant women. Microbiome 2014, 2, 4. [Google Scholar] [CrossRef] [Green Version]
- DiGiulio, D.B.; Callahan, B.J.; McMurdie, P.J.; Costello, E.K.; Lyell, D.J.; Robaczewska, A.; Sun, C.L.; Goltsman, D.S.A.; Wong, R.J.; Shaw, G.; et al. Temporal and spatial variation of the human microbiota during pregnancy. Proc. Natl. Acad. Sci. USA 2015, 112, 11060–11065. [Google Scholar] [CrossRef] [Green Version]
- Hyman, R.W.; Bs, M.F.; Jiang, H.; Fung, E.; Rand, L.; Bs, B.J.; Vo, K.C.; Caughey, A.B.; Hilton, J.F.; Davis, R.W.; et al. Diversity of the vaginal microbiome correlates with preterm birth. Reprod. Sci. 2014, 21, 32–40. [Google Scholar] [CrossRef] [Green Version]
- Aagaard, K.M.; Riehle, K.; Ma, J.; Segata, N.; Mistretta, T.-A.; Coarfa, C.; Raza, S.; Rosenbaum, S.; Veyver, I.V.D.; Milosavljevic, A.; et al. A Metagenomic Approach to Characterization of the Vaginal Microbiome Signature in Pregnancy. PLoS ONE 2012, 7, e36466. [Google Scholar] [CrossRef]
- Walther-António, M.R.S.; Jeraldo, P.; Miller, M.E.B.; Yeoman, C.J.; Nelson, K.E.; Wilson, B.A.; White, B.A.; Chia, N.; Creedon, D.J. Pregnancy’s Stronghold on the Vaginal Microbiome. PLoS ONE 2014, 9, e98514. [Google Scholar] [CrossRef] [Green Version]
- Hernández-Rodríguez, C.; Romero-González, R.; Albani-Campanario, M.; Figueroa-Damián, R.; Meraz-Cruz, N.; Hernández-Guerrero, C. Vaginal microbiota of healthy pregnant mexican women is constituted by four Lactobacillus species and several vaginosis-associated bacteria. Infect. Dis. Obstet. Gynecol. 2011, 2011, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Linhares, I.M.; Kanninen, T.T.; Orfanelli, T.; Jayaram, A.; Doulaveris, G.; Witkin, S.S. The Vaginal Microbiome: New Findings Bring New Opportunities: The Vaginal Microbiome. Drug Dev. Res. 2013, 74, 360–364. [Google Scholar] [CrossRef]
- Jakobsson, T.; Forsum, U. Lactobacillus iners: A marker of changes in the vaginal flora? J. Clin. Microbiol. 2007, 45, 3145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petrova, M.I.; Reid, G.; Vaneechoutte, M.; Lebeer, S. Lactobacillus iners: Friend or foe? Trends Microbiol. 2017, 25, 182–191. [Google Scholar] [CrossRef] [PubMed]
- Verstraelen, H.; Verhelst, R.; Claeys, G.; De Backer, E.; Temmerman, M.; Vaneechoutte, M. Longitudinal analysis of the vaginal microflora in pregnancy suggests that L. crispatus promotes the stability of the normal vaginal microflora and that L. gasseri and/or L. iners are more conducive to the occurrence of abnormal vaginal microflora. BMC Microbiol. 2009, 9, 116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Borgdorff, H.; Van Der Veer, C.; Van Houdt, R.; Alberts, C.J.; De Vries, H.J.; Bruisten, S.M.; Snijder, M.B.; Prins, M.; Geerlings, S.E.; Van Der Loeff, M.F.S.; et al. The association between ethnicity and vaginal microbiota composition in Amsterdam, the Netherlands. PLoS ONE 2017, 12, e0181135. [Google Scholar] [CrossRef] [Green Version]
- Dobrut, A.; Gosiewski, T.; Pabian, W.; Bodaszewska-Lubaś, M.; Ochońska, D.; Bulanda, M.; Brzychczy-Włoch, M. The dynamics of vaginal and rectal Lactobacillus spp. flora in subsequent trimesters of pregnancy in healthy Polish women, assessed using the Sanger sequencing method. BMC Pregnancy Childbirth 2018, 18, 350. [Google Scholar] [CrossRef]
- Atassi, F.; Brassart, D.; Grob, P.; Graf, F.; Servin, A.L. Lactobacillus strains isolated from the vaginal microbiota of healthy women inhibit Prevotella bivia and Gardnerella vaginalis in coculture and cell culture. FEMS Immunol. Med Microbiol. 2006, 48, 424–432. [Google Scholar] [CrossRef] [Green Version]
- Wong, Y.P.; Tan, G.C.; Wong, K.K.; Anushia, S.; Cheah, F.C. Gardnerella vaginalis in perinatology: An overview of the clinicopathological correlation. Malays. J. Pathol. 2018, 40, 267–286. [Google Scholar]
- Janulaiiene, M.; Paliulye, V.; Pleckaityte, M. Prevalence and distribution of Gardnerella vaginalis subgroups in women with and without bacterial vaginosis. BMC Infect. Dis. 2017, 17, 394. [Google Scholar]
- Recommendation of the Polish Society of Gynecologists and Obstetricians Regarding the Use of Antiseptics in Cases Non-Specific Vaginitis. 2020. Available online: http://ptgin.pl/ (accessed on 12 November 2020).
- Nuriel-Ohayon, M.; Neuman, H.; Ziv, O.; Belogolovski, A.; Barsheshet, Y.; Bloch, N.; Uzan, A.; Lahav, R.; Peretz, A.; Frishman, S.; et al. Progesterone increases Bifidobacterium relative abundance during late pregnancy. Cell Rep. 2019, 27, 730–736. [Google Scholar] [CrossRef] [Green Version]
- Ruiz, L.; Delgado, S.; Ruas-Madiedo, P.; Sanchez, B.; Margolles, A. Bifidobacteria and Their Molecular Communication with the Immune System. Front. Microbiol. 2017, 8, 2345. [Google Scholar] [CrossRef] [Green Version]
- Peirotén, A.; Arqués, J.L.; Medina, M.; Rodríguez Mínguez, E. Bifidobacterial strains shared by mother and child as source of probiotics. Benef. Microbes 2018, 9, 231–238. [Google Scholar] [CrossRef] [PubMed]
- Goltsman, D.S.A.; Sun, C.L.; Relman, D.; Proctor, D.M.; DiGiulio, D.B.; Robaczewska, A.; Thomas, B.C.; Shaw, G.M.; Stevenson, D.K.; Holmes, S.P.; et al. Metagenomic analysis with strain-level resolution reveals fine-scale variation in human pregnancy microbiome. Genome Res. 2018, 28, 1467–1480. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mendz, G.; Kaakoush, N.; Quinlivan, J. New techniques to characterise the vaginal microbiome in pregnancy. AIMS Microbiol. 2016, 2, 55–68. [Google Scholar] [CrossRef]
- Virtanen, S.; Kalliala, I.; Nieminen, P.; Salonen, A. Comparative analysis of vaginal microbiota sampling using 16S rRNA gene analysis. PLoS ONE 2017, 12, e0181477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cole, J.R.; Wang, Q.; Fish, J.A.; Chai, B.; McGarrell, D.M.; Sun, Y.; Brown, C.T.; Porras-Alfaro, A.; Kuske, C.R.; Tiedje, J.M. Ribosomal Database Project: Data and tools for high throughput rRNA analysis. Nucleic Acids Res. 2014, 42, 633–642. [Google Scholar] [CrossRef] [Green Version]
- Chaudhary, N.; Sharma, A.K.; Agarwal, P.; Gupta, A.; Sharma, V.K. 16S Classifier: A Tool for Fast and Accurate Taxonomic Classification of 16S rRNA Hypervariable Regions in Metagenomic Datasets. PLoS ONE 2015, 10, e0116106. [Google Scholar] [CrossRef]
- Martínez-Porchas, M.; Villalpando-Canchola, E.; Vargas-Albores, F. Significant loss of sensitivity and specificity in the taxonomic classification occurs when short 16S rRNA gene sequences are used. Heliyon 2016, 2, e00170. [Google Scholar] [CrossRef] [Green Version]
- Quast, C.; Pruesse, E.; Yilmaz, P.; Gerken, J.; Schweer, T.; Yarza, P.; Peplies, J.; Glöckner, F.O. The SILVA ribosomal RNA gene database project: Improved data processing and web-based tools. Nucleic Acids Res. 2012, 41, D590–D596. [Google Scholar] [CrossRef]
- DeSantis, T.Z.; Hugenholtz, P.; Larsen, N.; Rojas, M.; Brodie, E.L.; Keller, K.; Huber, T.; Dalevi, D.; Hu, P.; Andersen, G.L. Greengenes, a Chimera-Checked 16S rRNA Gene Database and Workbench Compatible with ARB. Appl. Environ. Microbiol. 2006, 72, 5069–5072. [Google Scholar] [CrossRef] [Green Version]
- Caporaso, J.G.; Kuczynski, J.; Stombaugh, J.; Bittinger, K.; Bushman, F.D.; Costello, E.K.; Fierer, N.; Peña, A.G.; Goodrich, J.K.; Gordon, J.I.; et al. QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 2010, 7, 335–336. [Google Scholar] [CrossRef] [Green Version]
- Gao, X.; Lin, H.; Revanna, K.; Dong, Q. A Bayesian taxonomic classification method for 16S rRNA gene sequences with improved species-level accuracy. BMC Bioinform. 2017, 18, 247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tuzhikov, A.; Panchin, A.; Shestopalov, V.I. TUIT, a BLAST-based tool for taxonomic classification of nucleotide sequences. BioTechniques 2014, 56, 78–84. [Google Scholar] [CrossRef] [PubMed]
- Lindgreen, S.; Adair, K.L.; Gardner, P.P. An evaluation of the accuracy and speed of metagenome analysis tools. Sci. Rep. 2016, 6, 19233. [Google Scholar] [CrossRef] [PubMed] [Green Version]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Inclusion Criteria | Exclusion Criteria |
|---|---|
| - women in the first trimester of pregnancy, aged 18–40 years - absence of clinical signs of urogenital infection - lack of antibiotic or probiotic use for up to 30 days before getting pregnant and during pregnancy - value of 0–6 at the 10 -point Nugent score in the first trimester as a confirmation of physiological flora of the genitourinary tract- written consent to participate in the study | - pregnant women under the age of 18 and over 40 years old - women with a high-risk pregnancy - rupture of the membranes - gestational diabetes - use of antibiotics for up to 30 days before becoming pregnant or during pregnancy - diagnosis of bacterial vaginosis - result of 7–10 at the 10-point Nugent score in the first trimester - clinical symptoms of urinary tract infection - lack of written consent to participate in the research |
| Final Volume: 25 µL | Thermal Profile | ||||
|---|---|---|---|---|---|
| H2O | 10.5 µL | 95 °C - | 5 min | ||
| Kapa Biosystems (Roche) | 12.5 µL | 95 °C - | 30 s |  | |
| Primer 1 (F) (Genomed) | 0.5 µL | 55 °C - | 30 s | 30 × | |
| Primer 2 (R) (Genomed) | 0.5 µL | 72 °C - | 30 s | ||
| DNA | 1.0 µL | 72 °C - | 5 min | ||
| Taxonomic Level | Percent 1 of Reads in 1st Trimester | Percent 1 of Reads in 2nd Trimester | Percent 1 of Reads in 3rd Trimester |
|---|---|---|---|
| kingdom | 98.82% | 98.17% | 98.32% |
| phylum | 98.72% | 98.07% | 98.19% |
| class | 98.66% | 98.01% | 98.11% |
| order | 98.58% | 97.95% | 98.02% |
| family | 98.47% | 97.85% | 97.89% |
| genus | 98.11% | 97.55% | 97.14% |
| species | 47.58% | 45.22% | 57.09% |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sroka-Oleksiak, A.; Gosiewski, T.; Pabian, W.; Gurgul, A.; Kapusta, P.; Ludwig-Słomczyńska, A.H.; Wołkow, P.P.; Brzychczy-Włoch, M. Next-Generation Sequencing as a Tool to Detect Vaginal Microbiota Disturbances during Pregnancy. Microorganisms 2020, 8, 1813. https://doi.org/10.3390/microorganisms8111813
Sroka-Oleksiak A, Gosiewski T, Pabian W, Gurgul A, Kapusta P, Ludwig-Słomczyńska AH, Wołkow PP, Brzychczy-Włoch M. Next-Generation Sequencing as a Tool to Detect Vaginal Microbiota Disturbances during Pregnancy. Microorganisms. 2020; 8(11):1813. https://doi.org/10.3390/microorganisms8111813
Chicago/Turabian StyleSroka-Oleksiak, Agnieszka, Tomasz Gosiewski, Wojciech Pabian, Artur Gurgul, Przemysław Kapusta, Agnieszka H. Ludwig-Słomczyńska, Paweł P. Wołkow, and Monika Brzychczy-Włoch. 2020. "Next-Generation Sequencing as a Tool to Detect Vaginal Microbiota Disturbances during Pregnancy" Microorganisms 8, no. 11: 1813. https://doi.org/10.3390/microorganisms8111813