New Insights to Adenovirus-Directed Innate Immunity in Respiratory Epithelial Cells

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Human Adenoviruses
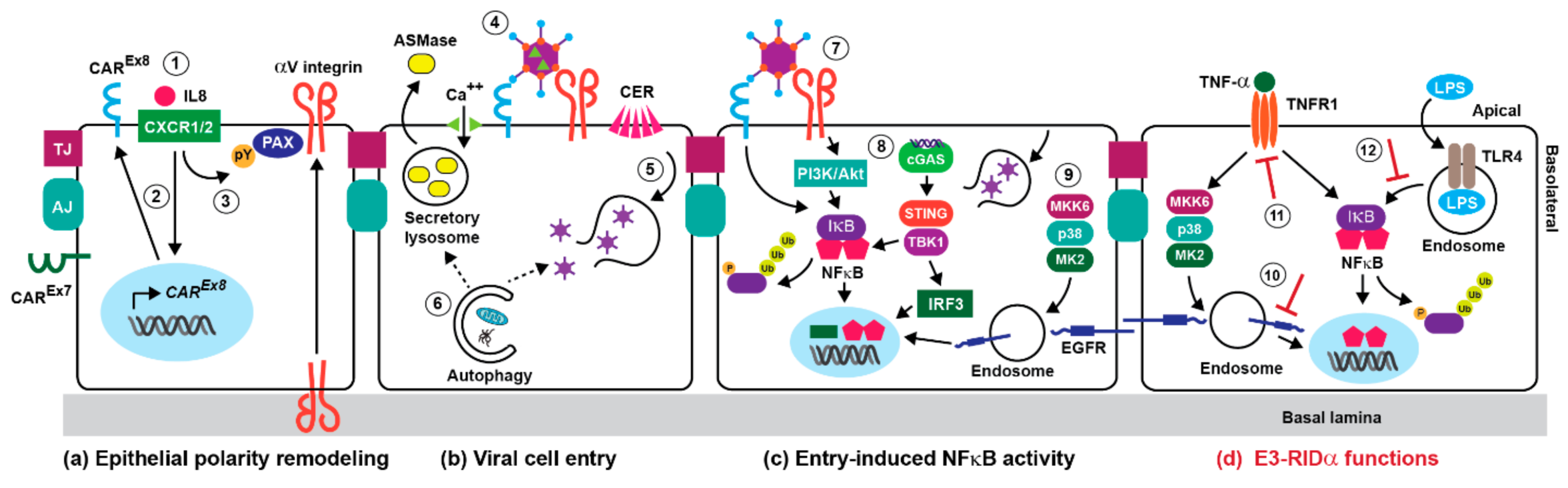
3. Adenoviral Cell Entry and Innate Immunity
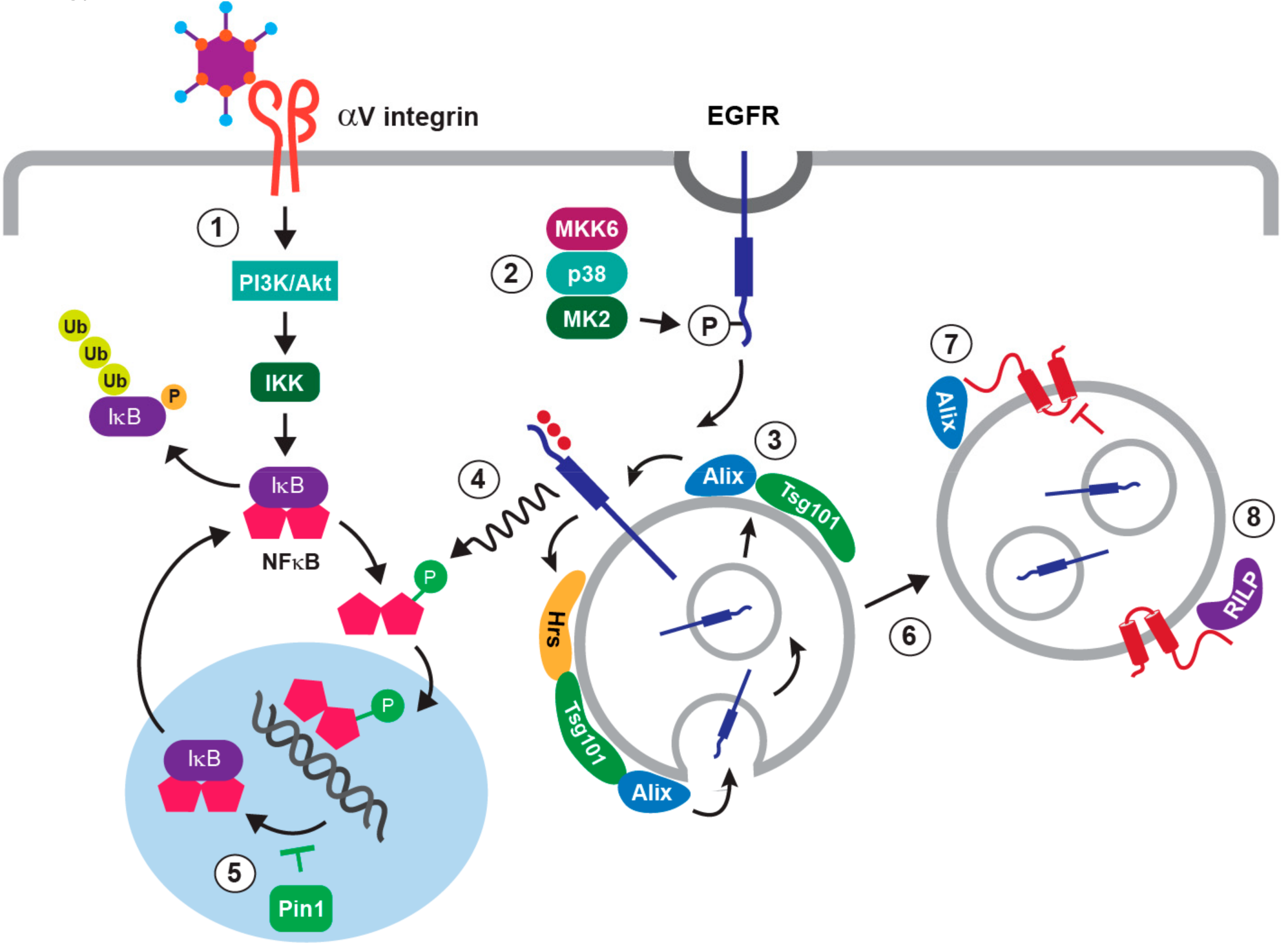
4. The NFκB Pathway in Adenovirus-Directed Innate Immunity
5. Stress-Induced EGFR Responses
6. Innate Immunity and Early Adenoviral-Induced Gene Products
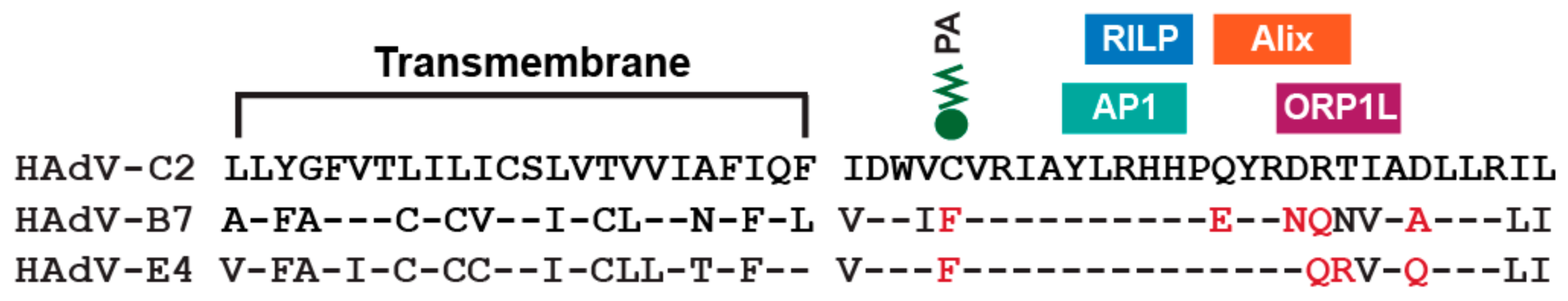
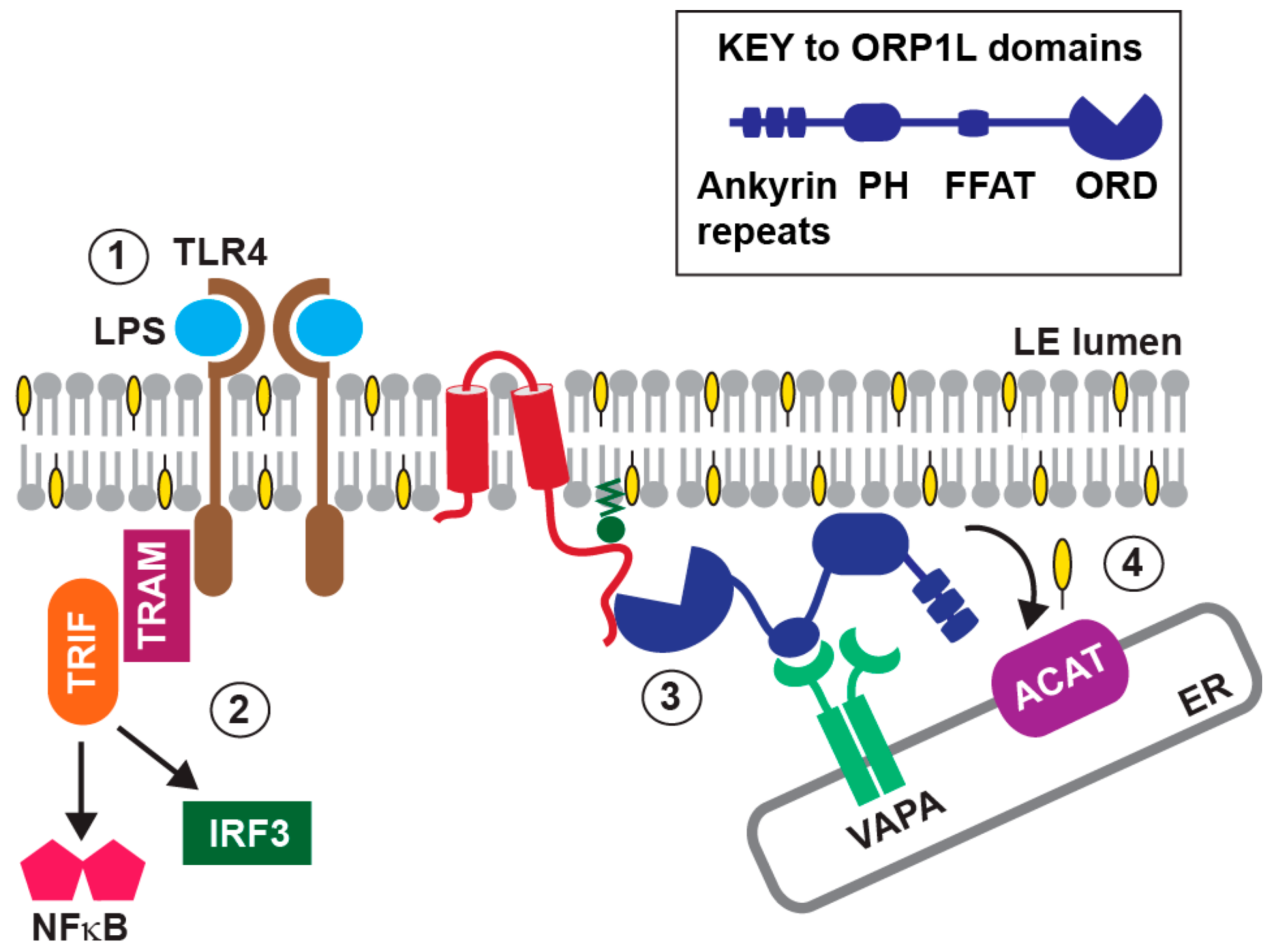
7. Innate Immunity and the E3-RIDα Protein
8. Conclusions and Future Perspectives
Funding
Acknowledgments
Conflicts of Interest
References
- Burgert, H.; Kvist, S. An adenovirus type 2 glycoprotein blocks cell surface expression of human histocompatibility class I antigens. Cell 1985, 41, 987–997. [Google Scholar] [CrossRef]
- Berget, S.M.; Moore, C.; Sharp, P.A. Spliced segments at the 5’ terminus of adenovirus 2 late mRNA. Proc. Natl. Acad. Sci. USA 1977, 74, 3171–3175. [Google Scholar] [CrossRef] [PubMed]
- Yabe, Y.; Samper, L.; Taylor, G.; Trentin, J. Cancer induction in hamsters by human type 12 adenovirus. Effect of route of injection. Proc. Soc. Exp. Biol. Med. 1963, 113, 221–224. [Google Scholar] [CrossRef] [PubMed]
- Chinnadurai, G. Opposing oncogenic activities of small DNA tumor virus transforming proteins. Trends Microbiol. 2011, 19, 174–183. [Google Scholar] [CrossRef] [PubMed]
- Flatt, J.; Butcher, S. Adenovirus flow in host cell networks. Open Biol. 2019, 9, 190012. [Google Scholar] [CrossRef]
- Cuconati, A.; White, E. Viral homologs of BCL-2: Role of apoptosis in the regulation of virus infection. Genes Dev. 2002, 16, 2465–2478. [Google Scholar] [CrossRef]
- Burgert, H.; Blusch, J. Immunomodulatory functions encoded by the E3 transcription unit of adenoviruses. Virus Genes 2000, 21, 13–25. [Google Scholar] [CrossRef]
- Weitzman, M. Functions of the adenovirus E4 proteins and their impact on viral vectors. Front. Biosci. 2005, 10, 1106–1117. [Google Scholar] [CrossRef]
- Windheim, M.; Hilgendorf, A.; Burgert, H. Immune evasion by adenovirus E3 proteins: Exploitation of intracellular trafficking pathways. Curr. Top Microbiol. Immunol. 2004, 273, 229–285. [Google Scholar]
- Horwitz, M.S. Function of adenovirus E3 proteins and their interactions with immunoregulatory cell proteins. J. Gene Med. 2004, 6 (Suppl. 1), S172–S183. [Google Scholar] [CrossRef]
- Cianciola, N.; Carlin, C. Human adenoviruses, cholesterol trafficking, and NF-κB signaling. J. Immunol. Sci. 2018, 2, 9–14. [Google Scholar] [PubMed]
- Muruve, D.A. The innate immune response to adenovirus vectors. Hum. Gene Ther. 2004, 15, 1157–1166. [Google Scholar] [CrossRef] [PubMed]
- Hendrickx, R.; Stichling, N.; Koelen, J.; Kuryk, L.; Lipiec, A.; Greber, U. Innate immunity to adenovirus. Hum. Gene Ther. 2014, 25, 265–284. [Google Scholar] [CrossRef] [PubMed]
- Rowe, W.P.; Huebner, R.J.; Gilmore, L.K.; Parrott, R.H.; Ward, T.G. Isolation of a cytopathogenic agent from human adenoids undergoing spontaneous degeneration in tissue culture. Exp. Biol. Med. 1953, 84, 570–573. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, G.; Yawata, N.; Aoki, K.; Kitaichi, N. Challenges in management of epidemic keratoconjunctivitis with emerging recombinant human adenoviruses. J. Clin. Virol. 2019, 112, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Huang, G.; Yao, W.; Yu, W.; Mao, L.; Sun, H.; Yao, W.; Tian, J.; Wang, L.; Bo, Z.; Zhu, Z.; et al. Outbreak of epidemic keratoconjunctivitis caused by human adenovirus type 56, China, 2012. PLoS ONE 2014, 9, e110781. [Google Scholar] [CrossRef] [PubMed]
- Rebelo-de-Andrade, H.; Pereira, C.; Giria, M.; Prudencio, E.; Brito, M.J.; Cale, E.; Taveira, N. Outbreak of acute respiratory infection among infants in Lisbon, Portugal, caused by human adenovirus serotype 3 and a new 7/3 recombinant strain. J. Clin. Microbiol. 2019, 48, 1391–1396. [Google Scholar] [CrossRef]
- Lynch, J.P.; Fishbein, M.; Echavarria, M. Adenovirus. Semin. Respir. Crit. Care Med. 2011, 32, 494–511. [Google Scholar] [CrossRef]
- Arnold, A.; MacMahon, E. Adenovirus infections. Medicine 2017, 45, 777–780. [Google Scholar] [CrossRef]
- Potter, R.; Cantrell, J.; Mallak, C.; Gaydos, J. Adenovirus-associated deaths in US military during postvaccination period, 1999–2010. Emerg. Infect. Dis. 2012, 18, 507–509. [Google Scholar] [CrossRef]
- Kajon, A.E.; Lamson, D.M.; Bair, C.R.; Lu, X.; Landry, M.L.; Menegus, M.; George, K.S. Adenovirus type 4 respiratory infections among civilian adults, northeastern United States, 2011–2015. Emerg. Infect. Dis. 2018, 24, 201–209. [Google Scholar] [CrossRef]
- Kajon, A.E.; Lamson, D.M.; George, K.S. Emergence and re-emergence of respiratory adenoviruses in the United States. Curr. Opin. Virol. 2019, 34, 63–69. [Google Scholar] [CrossRef] [PubMed]
- Berk, A. Adenoviridae. In Fields Virology, 6th ed.; Knipe, D., Howley, P., Eds.; Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2013; pp. 1704–1731. [Google Scholar]
- Tol, M.J.; Kroes, A.C.; Schinkel, J.; Dinkelaar, W.; Claas, E.C.; der Jol-van Zijde, C.M.; Vossen, J.M. Adenovirus infection in paediatric stem cell transplant recipients: Increased risk in young children with a delayed immune recovery. Bone Marrow Transplant. 2005, 36. [Google Scholar] [CrossRef]
- Hoffman, J.A. Adenovirus infections in solid organ transplant recipients. Curr. Opin. Organ Transplant. 2009, 14, 625–633. [Google Scholar] [CrossRef] [PubMed]
- Chakrabarti, S.; Milligan, D.W.; Moss, P.A.H.; Mautner, V. Adenovirus infections in stem cell transplant recipients: Recent developments in understanding of pathogenesis, diagnosis and management. Leuk. Lymphoma 2004, 45, 873–885. [Google Scholar] [CrossRef] [PubMed]
- Cook, J.; Radke, J. Mechanisms of pathogenesis of emerging adenoviruses. F1000Research 2017, 6, 90. [Google Scholar] [CrossRef] [PubMed]
- Majorant, D.; Qiu, F.; Kalil, A.C.; Wilson, N.; Florescu, D.F. Adenovirus—A deadly disease in the solid organ transplant population: Risk factors and outcomes. Transplant. Proc. 2018, 50, 3769–3774. [Google Scholar] [CrossRef]
- Sandkovsky, U.; Vargas, L.; Florescu, D.F. Adenovirus: Current epidemiology and emerging approaches to prevention and treatment. Curr. Infect. Dis. Rep. 2015, 16, 1–8. [Google Scholar] [CrossRef]
- Lugthart, G.; Oomen, M.A.; der Jol-van Zijde, C.M.; Ball, L.M.; Bresters, D.; Kollen, W.J.W.; Smiers, F.J.; Vermont, C.L.; Bredius, R.G.M.; Schilham, M.W.; et al. The effect of Cidofovir on adenovirus plasma DNA levels in stem cell transplantation recipients without T cell reconstitution. Biol. Blood Marrow Transplant. 2015, 21, 293–299. [Google Scholar] [CrossRef]
- Ljungman, P.; Ribaud, P.; Eyrich, M.; Matthes-Martin, S.; Einsele, H.; Bleakley, M.; Machaczka, M.; Bierings, M.; Bosi, A.; Gratecos, N.; et al. Cidofovir for adenovirus infections after allogeneic hematopoietic stem cell transplantation: A survey by the Infectious Diseases Working Party of the European Group for Blood and Marrow Transplantation. Bone Marrow Transplant. 2003, 31. [Google Scholar] [CrossRef]
- Florescu, D.F.; Keck, M.A. Development of CMX001 (Brincidofovir) for the treatment of serious diseases or conditions caused by dsDNA viruses. Expert Rev. Anti-Infect. Ther. 2014, 12, 1171–1178. [Google Scholar] [CrossRef] [PubMed]
- Hartman, Z.C.; Appledorn, D.M.; Serra, D.; Glass, O.; Mendelson, T.B.; Clay, T.M.; Amalfitano, A. Replication-attenuated Human Adenoviral Type 4 vectors elicit capsid dependent enhanced innate immune responses that are partially dependent upon interactions with the complement system. Virology 2008, 374, 453–467. [Google Scholar] [CrossRef] [PubMed]
- Biggs, H.M.; Lu, X.; Dettinger, L.; Sakthivel, S.; Watson, J.T.; Boktor, S.W. Adenovirus-associated influenza-like illness among college students, Pennsylvania, USA. Emerg. Inf. Dis. 2018, 24, 2117–2119. [Google Scholar] [CrossRef] [PubMed]
- McConnell, M.J.; Imperiale, D.M.J. Biology of adenovirus and its use as a vector for gene therapy. Hum. Gene Ther. 2004, 15, 1022–1033. [Google Scholar] [CrossRef] [PubMed]
- Ghebremedhin, B. Human adenovirus: Viral pathogen with increasing importance. Eur. J. Microbiol. Immunol. 2014, 4, 26–33. [Google Scholar] [CrossRef] [PubMed]
- Meier, O.; Greber, U.F. Adenovirus endocytosis. J. Gene Med. 2004, 6, S152–S163. [Google Scholar] [CrossRef] [PubMed]
- Bluscha, J.H.; Deryckereb, F.; Windheima, M.; Ruzsicsa, Z.; Arnbergc, N.; Adriand, T.; Burgerta, H.-G. The novel early region 3 protein E3/49K is specifically expressed by adenoviruses of subgenus D: Implications for epidemic keratoconjunctivitis and adenovirus evolution. Virology 2002, 296, 94–106. [Google Scholar] [CrossRef]
- Windheim, M.; Southcombe, J.H.; Kremmer, E.; Chaplin, L.; Urlaub, D.; Falk, C.S.; Claus, M.; Mihm, J.; Braithwaite, M.; Dennehy, K.; et al. A unique secreted adenovirus E3 protein binds to the leukocyte common antigen CD45 and modulates leukocyte functions. Proc. Natl. Acad. Sci. USA 2013, 110, E4884–E4893. [Google Scholar] [CrossRef]
- Jiang, H.; White, E.J.; Rios-Vicil, C.I.; Xu, J.; Gomez-Manzano, C.; Fueyo, J. Human adenovirus type 5 induces cell lysis through autophagy and autophagy-triggered caspase activity. J. Virol. 2011, 85, 4720–4729. [Google Scholar] [CrossRef]
- Crystal, R.G. Adenovirus: The first effective in vivo gene delivery vector. Hum. Gene Ther. 2014, 25, 3–11. [Google Scholar] [CrossRef]
- Lee, C.S.; Bishop, E.S.; Zhang, R.; Yu, X.; Farina, E.M.; Yan, S.; Zhao, C.; Zeng, Z.; Shu, Y.; Wu, X.; et al. Adenovirus-mediated gene delivery: Potential applications for gene and cell-based therapies in the new era of personalized medicine. Genes Dis. 2017, 4, 43–63. [Google Scholar] [CrossRef] [PubMed]
- Ginsberg, H.S.; Lundholm-Beauchamp, U.; Horswoord, R.L.; Pernis, B.; Wold, W.S.M.; Chanock, R.M.; Prince, G.A. Role of early region 3 (E3) in pathogenesis of adenovirus disease. Proc. Natl. Acad. Sci. USA 1989, 86, 3823–3827. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Muruve, D.A. Molecular basis of the inflammatory response to adenovirus vectors. Gene Ther. 2003, 10, 935–950. [Google Scholar] [CrossRef] [PubMed]
- Bergelson, J.M.; Cunningham, J.A.; Droguett, G.; Kurt-Jones, E.A.; Krithivas, A.; Hong, J.S.; Horwitz, M.S.; Crowell, R.L.; Finberg, R.W. Isolation of a common receptor for coxsackie B viruses and adenoviruses 2 and 5. Science 1997, 275, 1320–1323. [Google Scholar] [CrossRef] [PubMed]
- Gaggar, A.; Shayakhmetov, D.M.; Lieber, A. CD46 is a cellular receptor for group B adenoviruses. Nat. Med. 2003, 9, 1408–1412. [Google Scholar] [CrossRef]
- Lenman, A.; Liaci, A.M.; Liu, Y.; Ardahl, C.; Rajan, A.; Nilsson, E.; Bradford, W.; Kaeshammer, L.; Jones, M.S.; Frangsmyr, L.; et al. Human adenovirus 52 uses sialic acid-containing glycoproteins and the coxsackie and adenovirus receptor for binding to target cells. PLoS Pathog. 2015, 11, e1004657. [Google Scholar] [CrossRef] [PubMed]
- Arnberg, N.; Pring-Akerblom, P.; Wadell, G. Adenovirus type 37 uses sialic acid as a cellular receptor on Chang C cells. J. Virol. 2002, 76, 8834–8841. [Google Scholar] [CrossRef]
- Wickham, T.J.; Mathias, P.; Cheresh, D.A.; Nemerow, G.R. Integrins αvβ3 and αvβ5 promote adenovirus internalization but not virus attachment. Cell 1993, 73, 309–319. [Google Scholar] [CrossRef]
- Nemerow, G. Cell receptors involved in adenovirus entry. Virology 2000, 274, 1–4. [Google Scholar] [CrossRef]
- Ashbourne, E.K.J.D.; Moninger, T.; Zabner, J. The Coxsackie B Virus and Adenovirus receptor resides in a distinct membrane microdomain. J. Virol. 2003, 77, 2559–2567. [Google Scholar] [CrossRef]
- Kotha, P.L.N.; Sharma, P.; Kolawole, A.O.; Yan, R.; Alghamri, M.S.; Brockman, T.L.; Gomez-Cambronero, J.; Excoffon, K.J.D.A. Adenovirus entry from the apical surface of polarized epithelia is facilitated by the host innate immune response. PLoS Pathog. 2015, 11, e1004696. [Google Scholar] [CrossRef] [PubMed]
- Lütschg, V.; Boucke, K.; Hemmi, S.; Greber, U. Chemotactic antiviral cytokines promote infectious apical entry of human adenovirus into polarized epithelial cells. Nat. Commun. 2011, 2, 391–394. [Google Scholar] [CrossRef] [PubMed]
- Maier, O.; Marvin, S.A.; Wodrich, H.; Campbell, E.M.; Wiethoff, C.M. Spatiotemporal dynamics of adenovirus membrane rupture and endosomal escape. J. Virol. 2012, 86, 10821–10828. [Google Scholar] [CrossRef] [PubMed]
- Gastaldelli, M.; Imelli, N.; Boucke, K.; Amstutz, B.; Meier, O.; Greber, U.F. Infectious adenovirus type 2 transport through early but not late endosomes. Traffic 2008, 9, 2265–2278. [Google Scholar] [CrossRef] [PubMed]
- Wiethoff, C.M.; Wodrich, H.; Gerace, L.; Nemerow, G.R. Adenovirus protein VI mediates membrane disruption following capsid disassembly. J. Virol. 2005, 79, 1992–2000. [Google Scholar] [CrossRef] [PubMed]
- Andrews, N.W.; Almeida, P.E.; Corrotte, M. Damage control: Cellular mechanisms of plasma membrane repair. Trends Cell Biol. 2014, 24, 734–742. [Google Scholar] [CrossRef]
- Luisoni, S.; Suomalainen, M.; Boucke, K.; Tanner Lukas, B.; Wenk Markus, R.; Guan Xue, L.; Grzybek, M.; Coskun, A.; Greber, U.F. Co-option of membrane wounding enables virus penetration into cells. Cell Host Microbe 2016, 18, 75–85. [Google Scholar] [CrossRef] [PubMed]
- Burckhardt, C.J.; Suomalainen, M.; Schoenenberger, P.; Boucke, K.; Hemmi, S.; Greber, U.F. Drifting motions of the adenovirus receptor CAR and immobile integrins initiate virus uncoating and membrane lytic protein exposure. Cell Host Microbe 2011, 10, 105–117. [Google Scholar] [CrossRef]
- Ryter, S.W.; Choi, A.M.K. Autophagy in the lung. Proc. Am. Thorac. Soc. 2010, 7, 13–21. [Google Scholar] [CrossRef]
- Zeng, X.; Carlin, C.R. Host cell autophagy modulates early stages of adenovirus infections in airway epithelial cells. J. Virol. 2013, 87, 2307–2319. [Google Scholar] [CrossRef]
- Kreibich, S.; Emmenlauer, M.; Fredlund, J.; Ramo, P.; Munz, C.; Dehio, C.; Enninga, J.; Hardt, W.-D. Autophagy proteins promote repair of endosomal membranes damaged by the Salmonella type three secretion system 1. Cell Host Microbe 2015, 18, 527–537. [Google Scholar] [CrossRef] [PubMed]
- DeSelm, C.J.; Miller, B.C.; Zou, W.; Beatty, W.L.; van Meel, E.; Takahata, Y.; Klumperman, J.; Tooze, S.A.; Teitelbaum, S.L.; Virgin, H.W. Autophagy proteins regulate the secretory component of osteoclastic bone resorption. Dev. Cell 2011, 21, 966–974. [Google Scholar] [CrossRef] [PubMed]
- Miyazawa, N.; Crystal, R.G.; Leopold, P.L. Adenovirus serotype 7 retention in a late endosomal compartment prior to cytosol escape is modulated by fiber protein. J. Virol. 2001, 75, 1387–1400. [Google Scholar] [CrossRef] [PubMed]
- Pasparakis, M. Regulation of tissue homeostasis by NF-κB signalling: Implications for inflammatory diseases. Nat. Rev. Immunol. 2009, 9, 778–788. [Google Scholar] [CrossRef] [PubMed]
- Tamanini, A.; Nicolis, E.; Bonizzato, A.; Bezzerri, V.; Melotti, P.; Assael, B.M.; Cabrini, G. Interaction of adenovirus type 5 fiber with the Coxsackievirus and Adenovirus Receptor activates inflammatory response in human respiratory cells. J. Virol. 2006, 80, 11241–11254. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; White, L.R.; Clark, S.A.; Heffner, D.J.; Winston, B.W.; Tibbles, L.A.; Muruve, D.A. Akt/protein kinase B activation by adenovirus vectors contributes to NFκB-dependent CXCL10 expression. J. Virol. 2005, 79, 14507–14515. [Google Scholar] [CrossRef] [PubMed]
- Lam, E.; Stein, S.; Falck-Pedersen, E. Adenovirus detection by the cGAS/STING/TBK1 DNA sensing cascade. J. Virol. 2014, 88, 974–981. [Google Scholar] [CrossRef]
- Barber, G.N. STING: Infection, inflammation and cancer. Nat. Rev. Immunol. 2015, 15, 760–770. [Google Scholar] [CrossRef]
- Suomalainen, M.; Nakano, M.Y.; Boucke, K.; Keller, S.; Greber, U.F. Adenovirus-activated PKA and p38/MAPK pathways boost microtubule-mediated nuclear targeting of virus. EMBO J. 2001, 20, 1310–1319. [Google Scholar] [CrossRef]
- Togo, T. Long-term potentiation of wound-induced exocytosis and plasma membrane repair is dependent on cAMP-response element-mediated transcription via a protein kinase C- and p38 MAPK-dependent pathway. J. Biol. Chem. 2004, 279, 44996–45003. [Google Scholar] [CrossRef]
- Tibbles, L.A.; Spurrell, J.C.L.; Bowen, G.P.; Liu, Q.; Lam, M.; Zaiss, A.K.; Robbins, S.M.; Hollenberg, M.D.; Wickham, T.J.; Muruve, D.A. Activation of p38 and ERK signaling during adenovirus vector cell entry lead to expression of the C-X-C chemokine IP-10. J. Virol. 2002, 76, 1559–1568. [Google Scholar] [CrossRef] [PubMed]
- Zeng, X.; Carlin, C. Adenovirus early region 3 RIDa protein limits NFkB signaling through stress-activated EGF receptors. PLoS Pathog. 2019. in review. [Google Scholar]
- Ginsberg, H.; Moldawer, L.; Sehgal, P.; Redington, M.; Kilian, P.; Chanock, R.; Prince, G. A mouse model for investigating the molecular pathogenesis of adenovirus pneumonia. Proc. Natl. Acad. Sci. USA 1991, 88, 1651–1655. [Google Scholar] [CrossRef] [PubMed]
- Haveman, L.M.; De Jager, W.; Van Loon, A.M.; Claas, E.C.J.; Prakken, B.J.; Bierings, M. Different cytokine signatures in children with localized and invasive adenovirus infection after stem cell transplantation. Ped. Transpl. 2010, 14, 520–528. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, S.; Hogg, J.C. Adenovirus infections and lung disease. Curr. Opin. Pharmacol. 2007, 7, 237–243. [Google Scholar] [CrossRef] [PubMed]
- Muruve, D.A.; Barnes, M.J.; Stillman, I.E.; Libermann, T.A. Adenoviral gene therapy leads to rapid induction of multiple chemokines and acute neutrophil-dependent hepatic injury in vivo. Hum. Gene Ther. 1999, 10, 965–976. [Google Scholar] [CrossRef]
- Wang, V.Y.-F.; Huang, W.; Asagiri, M.; Spann, N.; Hoffmann, A.; Glass, C.; Ghosh, G. The transcriptional specificity of NF-κB dimers is coded within the κB DNA response elements. Cell Rep. 2014, 2, 824–839. [Google Scholar] [CrossRef]
- Napetschnig, J.; Wu, H. Molecular basis of NF-κB signaling. Annu. Rev. Biophys. 2013, 42, 443–468. [Google Scholar] [CrossRef]
- Hacker, H.; Karin, M. Regulation and function of IKK and IKK-related kinases. Sci. STKE 2006, 2006, re13. [Google Scholar] [CrossRef]
- Durand, J.K.; Zhang, Q.; Baldwin, A.S. Roles for the IKK-related kinases TBK1 and IKKε in cancer. Cells 2018, 7, 139–146. [Google Scholar] [CrossRef]
- Romashkova, J.A.; Makarov, S.S. NF-κB is a target of AKT in anti-apoptotic PDGF signalling. Nature 1999, 401, 86–90. [Google Scholar] [CrossRef] [PubMed]
- Oeckinghaus, A.; Ghosh, S. The NF-κB family of transcription factors and its regulation. CSH Perspect. Biol. 2009, 1, a000034. [Google Scholar] [CrossRef]
- Lu, K.P.; Zhou, X.Z. The prolyl isomerase PIN1: A pivotal new twist in phosphorylation signalling and disease. Nat. Rev. Mol. Cell Biol. 2007, 8, 904. [Google Scholar] [CrossRef] [PubMed]
- Xing, D.; Gong, K.; Feng, W.; Nozell, S.E.; Chen, Y.-F.; Chatham, J.C.; Oparil, S. O-GlcNAc modification of NFkB p65 inhibits TNF-a-induced inflammatory mediator expression in rat aortic smooth muscle cells. PLoS ONE 2011, 6, e24021. [Google Scholar] [CrossRef]
- Ryo, A.; Suizu, F.; Yoshida, Y.; Perrem, K.; Liou, Y.-C.; Wulf, G.; Rottapel, R.; Yamaoka, S.; Lu, K.P. Regulation of NF-kB signaling by Pin1-dependent prolyl isomerization and ubiquitin-mediated proteolysis of p65/RelA. Mol. Cell 2003, 12, 1413–1426. [Google Scholar] [CrossRef]
- Butterfield, D.A.; Abdul, H.M.; Opii, W.; Newman, S.F.; Joshi, G.; Ansari, M.A.; Sultana, R. Review: Pin1 in Alzheimer’s disease. J. Neurochem. 2006, 98, 1697–1706. [Google Scholar] [CrossRef] [PubMed]
- Yeh, E.S.; Means, A.R. PIN1, the cell cycle and cancer. Nat. Rev. Cancer 2007, 7, 381–386. [Google Scholar] [CrossRef] [PubMed]
- Nath, P.R.; Isakov, N. Insights into peptidyl-prolyl cis-trans isomerase structure and function in immunocytes. Immunol. Lett. 2015, 163, 120–131. [Google Scholar] [CrossRef]
- Goutagny, N.; Severa, M.; Fitzgerald, K.A. Pin-ning down immune responses to RNA viruses. Nat. Immunol. 2006, 7, 555–557. [Google Scholar] [CrossRef] [PubMed]
- Kojima, Y.; Ryo, A. Pinning down viral proteins: A new prototype for virus-host cell interaction. Front. Microbiol. 2010, 1, 107–112. [Google Scholar] [CrossRef]
- Higginbotham, J.N.; Seth, P.; Blaese, R.M.; Ramsey, W.J. The release of inflammatory cytokines from human peripheral blood mononuclear cells in vitro following exposure to adenovirus variants and capsid. Hum. Gene Ther. 2002, 13, 129–141. [Google Scholar] [CrossRef] [PubMed]
- Zsengellar, Z.; Otake, K.; Hossain, S.-A.; Berclaz, P.-Y.; Trapnell, B.C. Internalization of adenovirus by alveolar macrophages initiates early proinflammatory signaling during acute respiratory tract infection. J. Virol. 2000, 74, 9655–9667. [Google Scholar] [CrossRef] [PubMed]
- Carey, B.; Staudt, M.K.; Bonaminio, D.; van der Loo, J.C.M.; Trapnell, B.C. PU.1 redirects adenovirus to lysosomes in alveolar macrophages, uncoupling internalization from infection. J. Immunol. 2007, 178, 2440–2447. [Google Scholar] [CrossRef] [PubMed]
- Lee, B.H.; Kushwah, R.; Wu, J.; Ng, P.; Palaniyar, N.; Grinstein, S.; Philpott, D.J.; Hu, J. Adenoviral vectors stimulate innate immune responses in macrophages through cross-talk with epithelial cells. Immunol. Lett. 2010, 134, 93–102. [Google Scholar] [CrossRef] [PubMed]
- Schlessinger, J. Cell signaling by receptor tyrosine kinases. Cell 2000, 103, 211–225. [Google Scholar] [CrossRef]
- Von Zastrow, M.; Sorkin, A. Signaling on the endocytic pathway. Curr. Opin. Cell Biol. 2007, 19, 436–445. [Google Scholar] [CrossRef]
- Sorkin, A.; von Zastrow, M. Endocytosis and signalling: Intertwining molecular networks. Nat. Rev. Mol. Cell Biol. 2009, 10, 609–622. [Google Scholar] [CrossRef]
- Raiborg, C.; Malerod, L.; Pedersen, N.M.; Stenmark, H. Differential functions of Hrs and ESCRT proteins in endocytic membrane trafficking. Exp. Cell Res. 2008, 314, 801–813. [Google Scholar] [CrossRef]
- Hurley, J.H.; Emr, S.D. The ESCRT complexes: Structure and mechanism of a membrane-trafficking network. Annu. Rev. Biophys. Biomol. Struct. 2006, 35, 277–298. [Google Scholar] [CrossRef]
- Wollert, T.; Hurley, J.H. Molecular mechanism of multivesicular body biogenesis by ESCRT complexes. Nature 2010, 464, 864–873. [Google Scholar] [CrossRef]
- Wollert, T.; Wunder, C.; Lippincott-Schwartz, J.; Hurley, J.H. Membrane scission by the ESCRT-III complex. Nature 2009, 458, 172–177. [Google Scholar] [CrossRef] [PubMed]
- Tomas, A.; Futter, C.E.; Eden, E.R. EGF receptor trafficking: Consequences for signaling and cancer. Trends Cell Biol. 2014, 24, 26–34. [Google Scholar] [CrossRef] [PubMed]
- Tan, X.; Lambert, P.F.; Rapraeger, A.C.; Anderson, R.A. Stress-induced EGFR trafficking: Mechanisms, functions, and therapeutic implications. Trends Cell Biol. 2016, 26, 352–366. [Google Scholar] [CrossRef] [PubMed]
- Zwang, Y.; Yarden, Y. p38 MAP kinase mediates stress-induced internalization of EGFR: Implications for cancer chemotherapy. EMBO J. 2006, 25, 4195–4206. [Google Scholar] [CrossRef] [PubMed]
- Tomas, A.; Vaughan, S.O.; Burgoyne, T.; Sorkin, A.; Hartley, J.A.; Hochhauser, D.; Futter, C.E. WASH and Tsg101/ALIX-dependent diversion of stress-internalized EGFR from the canonical endocytic pathway. Nat. Commun. 2015, 6, 7324. [Google Scholar] [CrossRef] [PubMed]
- Bissig, C.; Gruenberg, J. ALIX and the multivesicular endosome: ALIX in Wonderland. Trends Cell Biol. 2013, 24, 19–25. [Google Scholar] [CrossRef] [PubMed]
- Dores, M.R.; Chen, B.; Lin, H.; Soh, U.J.K.; Paing, M.M.; Montagne, W.A.; Meerloo, T.; Trejo, J. ALIX binds a YPX3L motif of the GPCR PAR1 and mediates ubiquitin-independent ESCRT-III/MVB sorting. J. Cell Biol. 2012, 197, 407–419. [Google Scholar] [CrossRef] [PubMed]
- Amano, Y.; Yamashita, Y.; Kojima, K.; Yoshino, K.; Tanaka, N.; Sugamura, K.; Takeshita, T. Hrs recognizes a hydrophobic amino acid cluster in cytokine receptors during ubiquitin-independent endosomal sorting. J. Biol. Chem. 2011, 286, 15458–15472. [Google Scholar] [CrossRef]
- Luyet, P.-P.; Falguières, T.; Pons, V.; Pattnaik, A.K.; Gruenberg, J. The ESCRT-I subunit TSG101 controls endosome-to-cytosol release of viral RNA. Traffic 2008, 9, 2279–2290. [Google Scholar] [CrossRef]
- Shostak, K.; Chariot, A. EGFR and NF-κB: Partners in cancer. Trends Mol. Med. 2015, 21, 385–393. [Google Scholar] [CrossRef]
- Grandal, M.V.; Grøvdal, L.M.; Henriksen, L.; Andersen, M.H.; Holst, M.R.; Madshus, I.H.; van Deurs, B. Differential roles of Grb2 and AP-2 in p38MAPK- and EGF-Induced EGFR internalization. Traffic 2012, 13, 576–585. [Google Scholar] [CrossRef] [PubMed]
- Hoffman, P.H.; Rajakumar, P.; Hoffman, B.; Heuertz, R.; Wold, W.S.M.; Carlin, C.R. Evidence for intracellular down-regulation of the epidermal growth factor receptor during adenovirus infection by an EGF-independent mechanism. J. Virol. 1992, 66, 197–203. [Google Scholar] [PubMed]
- Hoffman, P.; Carlin, C. Adenovirus E3 protein causes constitutively internalized EGF receptors to accumulate in a prelysosomal compartment, resulting in enhanced degradation. Mol. Cell Biol. 1994, 14, 3695–3706. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Tsacoumangos, A.; Kil, S.J.; Ma, L.; Sonnichsen, F.D.; Carlin, C. A novel dileucine lysosomal-sorting-signal mediates intracellular EGF-receptor retention independently of protein ubiquitylation. J. Cell Sci. 2005, 118, 3959–3971. [Google Scholar] [CrossRef] [PubMed]
- Barrow-McGee, R.; Kermorgant, S. Met endosomal signalling: In the right place, at the right time. Int. J. Biochem. Cell Biol. 2014, 49, 69–74. [Google Scholar] [CrossRef]
- Kostenko, O.; Tsacoumangos, A.; Crooks, D.M.; Kil, S.J.; Carlin, C.R. Gab1 signaling is regulated by EGF receptor sorting in early endosomes. Oncogene 2006, 25, 6604–6617. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Cunnick, J.M.; Mei, L.; Doupnik, C.A.; Wu, J. Phosphotyrosines 627 and 659 of Gab1 constitute a bisphosphoryl tyrosine-based activation motif (BTAM) conferring binding and activation of SHP2. J. Biol. Chem. 2001, 276, 24380–24387. [Google Scholar] [CrossRef]
- Zheng, K.; Kitazato, K.; Wang, Y. Viruses exploit the function of epidermal growth factor receptor. Rev. Med. Virol. 2014, 24, 274–286. [Google Scholar] [CrossRef]
- Jindal, S.; Malkovsky, M. Stress responses to viral infection. Trends Microbiol. 1994, 2, 89–91. [Google Scholar] [CrossRef]
- Tsutsumi-Ishii, Y.; Nagaoka, I. Modulation of human b-defensin-2 transcription in pulmonary epithelial cells by lipopolysaccharide-stimulated mononuclear phagocytes via proinflammatory cytokine production. J. Immunol. 2003, 170, 4226–4236. [Google Scholar] [CrossRef]
- Hayashi, S. Latent adenovirus infection in COPD. Chest 2002, 121, 183S–187S. [Google Scholar] [CrossRef] [PubMed]
- Keicho, N.; Elliott, W.M.; Hogg, J.C.; Hayashi, S. Adenovirus E1A upregulates interleukin-8 expression induced by endotoxin in pulmonary epithelial cells. Am. J. Physiol. Lung Cell Mol. Physiol. 1997, 272, L1046–L1052. [Google Scholar] [CrossRef] [PubMed]
- Thorne, P.S.; Paul, B. McCray, J.; Howe, T.S.; O’Neill, M.A. Early-onset inflammatory responses in vivo to adenoviral vectors in the presence or absence of lipopolysaccharide-induced inflammation. Am. J. Respir. Cell Mol. Biol. 1999, 20, 1155–1164. [Google Scholar] [CrossRef] [PubMed]
- Harrod, K.S.; Mounday, A.D.; Whitsett, J.A. Adenoviral E3-14.7K protein in LPS-induced lung inflammation. Am. J. Physiol. Lung Cell Mol. Physiol. 2000, 278, L631–L639. [Google Scholar] [CrossRef] [PubMed]
- Burgert, H.G.; Maryanski, J.L.; Kvist, S. “E3/19K” protein of adenovirus type 2 inhibits lysis of cytolytic T lymphocytes by blocking cell surface expression of histocompability class I antigens. Proc. Natl. Acad. Sci. USA 1987, 84, 1356–1360. [Google Scholar] [CrossRef] [PubMed]
- Flomenberg, P.; Gutierrez, E.; Hogan, K. Identification of class I MHC regions which bind to the adenovirus E3-19k protein. Mol. Immunol. 1994, 31, 1277–1284. [Google Scholar] [CrossRef]
- Feuerbach, D.; Etteldorf, S.; Ebenau-Jehle, C.; Abastado, J.P.; Madden, D.; Burgert, H.G. Identification of amino acids within the MHC molecule important for the interaction with the adenovirus protein E3/19K. J. Immunol. 1994, 153, 1626–1636. [Google Scholar] [PubMed]
- Pahl, H.L.; Baeuerle, P.A. The ER-overload response: Activation of NF-κB. Trends Biochem. Sci. 1997, 22, 63–67. [Google Scholar] [CrossRef]
- Choi, S.; Park, Y.S.; Koga, T.; Treloar, A.; Kim, K.C. TNF-α is a key regulator of MUC1, an anti-inflammatory molecule, during airway Pseudomonas aeruginosa infection. Am. J. Respir. Cell Mol. Biol. 2011, 44, 255–260. [Google Scholar] [CrossRef]
- Pobezinskaya, Y.L.; Liu, Z. The role of TRADD in death receptor signaling. Cell Cycle 2012, 11, 871–876. [Google Scholar] [CrossRef]
- Carmody, R.J.; Maguschak, K.; Chen, Y.H. A novel mechanism of nuclear factor-kappaB regulation by adenoviral protein 14.7K. Immunology 2006, 117, 188–195. [Google Scholar] [CrossRef] [PubMed]
- Ye, J.; Xie, X.; Tarassishin, L.; Horwitz, M.S. Regulation of the NF-κB activation pathway by isolated domains of FIP3/IKKγ, a component of the IkB-α kinase complex. J. Biol. Chem. 2000, 275, 9882–9889. [Google Scholar] [CrossRef] [PubMed]
- Machitani, M.; Yamaguchi, T.; Shimizu, K.; Sakurai, F.; Katayama, K.; Kawabata, K.; Mizuguchi, H. Adenovirus vector-derived VA-RNA-mediated innate immune responses. Pharmaceutics 2011, 3, 338–353. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, T.; Kawabata, K.; Kouyama, E.; Ishii, K.J.; Katayama, K.; Suzuki, T.; Kurachi, S.; Sakurai, F.; Akira, S.; Mizuguchi, H. Induction of type I interferon by adenovirus-encoded small RNAs. Proc. Natl. Acad. Sci. USA 2010, 107, 17286–17291. [Google Scholar] [CrossRef] [PubMed]
- Hoffman, P.; Yaffe, M.B.; Hoffman, B.L.; Yei, S.; Wold, W.S.M.; Carlin, C. Characterization of the adenovirus E3 protein that down-regulates the epidermal growth factor receptor. J. Biol. Chem. 1992, 267, 13480–13487. [Google Scholar] [PubMed]
- Crooks, D.; Kil, S.J.; McCaffery, J.M.; Carlin, C. E3-13.7 integral membrane proteins encoded by human adenoviruses alter epidermal growth factor receptor trafficking by interacting directly with receptors in early endosomes. Mol. Biol. Cell 2000, 11, 3559–3572. [Google Scholar] [CrossRef] [PubMed]
- Cianciola, N.L.; Greene, D.J.; Morton, R.E.; Carlin, C.R. Adenovirus RIDa uncovers a novel pathway requiring ORP1L for lipid droplet formation independent of NPC1. Mol. Biol. Cell 2013, 24, 3309–3325. [Google Scholar] [CrossRef] [PubMed]
- Cianciola, N.L.; Crooks, D.; Shah, A.H.; Carlin, C. A tyrosine-based signal plays a critical role in the targeting and function of adenovirus RID {alpha} protein. J. Virol. 2007, 81, 10437–10450. [Google Scholar] [CrossRef]
- Van der Kant, R.; Fish, A.; Janssen, L.; Janssen, H.; Krom, S.; Ho, N.; Brummelkamp, T.; Carette, J.; Rocha, N.; Neefjes, J. Late endosomal transport and tethering are coupled processes controlled by RILP and the cholesterol sensor ORP1L. J. Cell Sci. 2013, 126, 3462–3474. [Google Scholar] [CrossRef]
- Johansson, M.; Rocha, N.; Zwart, W.; Jordens, I.; Janssen, L.; Kuijl, C.; Olkkonen, V.; Neefjes, J. Activation of endosomal dynein motors by stepwise assembly of Rab7-RILP-p150Glued, ORP1L, and the receptor {beta}lll spectrin. J. Cell Biol. 2007, 12, 459–471. [Google Scholar] [CrossRef]
- Zhao, K.; Ridgway, N.D. Oxysterol-binding protein-related protein 1L regulates cholesterol egress from the endo-lysosomal system. Cell Rep. 2017, 19, 1807–1818. [Google Scholar] [CrossRef] [PubMed]
- Cianciola, N.L.; Chung, S.; Manor, D.; Carlin, C.R. Adenovirus modulates Toll-like receptor 4 signaling by reprogramming ORP1L-VAP protein contacts for cholesterol transport from endosomes to the endoplasmic reticulum. J. Virol. 2017, 91, e01904-16. [Google Scholar] [CrossRef] [PubMed]
- Vinogradova, O.; Carlin, C.R.; Sonnichsen, F.D.; Sanders, C.R. A membrane setting for the sorting motifs present in the adenovirus E3-13.7 protein which down-regulates the epidermal growth factor receptor. J. Biol. Chem. 1998, 273, 17343–17350. [Google Scholar] [CrossRef] [PubMed]
- Cianciola, N.L.; Carlin, C.R. Adenovirus RID-α activates an autonomous cholesterol regulatory mechanism that rescues defects linked to Niemann-Pick disease type C. J. Cell Biol. 2009, 187, 537–552. [Google Scholar] [CrossRef] [PubMed]
- Guan, X.; Fierke, C.A. Understanding protein palmitoylation: Biological significance and enzymology. Sci. China Chem. 2014, 54, 1888–1897. [Google Scholar] [CrossRef] [PubMed]
- Shah, A.H.; Cianciola, N.L.; Mills, J.L.; Sonnichsen, F.D.; Carlin, C. Adenovirus RIDα regulates endosome maturation by mimicking GTP-Rab7. J. Cell Biol. 2007, 179, 965–980. [Google Scholar] [CrossRef] [PubMed]
- Guerra, F.; Bucci, C. Multiple roles of the small GTPase Rab7. Cells 2016, 5, 34–40. [Google Scholar] [CrossRef]
- Lin, X.; Zhang, J.; Chen, L.; Chen, Y.; Xu, X.; Hong, W.; Wang, T. Tyrosine phosphorylation of Rab7 by Src kinase. Cell Signal. 2017, 35, 84–94. [Google Scholar] [CrossRef]
- McNees, A.L.; Garnett, C.T.; Gooding, L.R. The adenovirus E3 RID complex protects some cultured human T and B lymphocytes from Fas-induced apoptosis. J. Virol. 2002, 76, 9716–9723. [Google Scholar] [CrossRef]
- Hilgendorf, A.; Lindberg, J.; Ruzsics, Z.; Honing, S.; Elsing, A.; Lofqvist, M.; Engelmann, H.; Burgert, H.-G. Two distinct transport motifs in the adenovirus E3 proteins act in concert to down-modulate apoptosis receptors and the epidermal growth factor receptor. J. Biol. Chem. 2003, 278, 51872–51884. [Google Scholar] [CrossRef]
- Chin, Y.R.; Horwitz, M.S. Mechanism for removal of tumor necrosis factor receptor 1 from the cell surface by the adenovirus RID{alpha}/{beta} complex. J. Virol. 2005, 79, 13606–13617. [Google Scholar] [CrossRef] [PubMed]
- Hoffman, B.L.; Ullrich, A.; Wold, W.; Carlin, C. Retrovirus-mediated transfer of an adenovirus gene encoding an integral membrane protein is sufficient to down regulate the receptor for epidermal growth factor. Mol. Cell Biol. 1990, 10, 5521–5524. [Google Scholar] [CrossRef] [PubMed]
- Blom, T.; Somerharju, P.; Ikonen, E. Synthesis and biosynthetic trafficking of membrane lipids. CSH Perspect. Biol. 2011, 3, a004713. [Google Scholar] [CrossRef] [PubMed]
- Van Meer, G.; Voelker, D.; Feigenson, G. Membrane lipids: Where they are and how they behave. Nat. Rev. Mol. Cell Biol. 2008, 9, 112–124. [Google Scholar] [CrossRef] [PubMed]
- D’Angelo, G.; Vicinanza, M.; MA, D.M. Lipid-transfer proteins in biosynthetic pathways. Curr. Opin. Cell Biol. 2008, 20, 360–370. [Google Scholar] [CrossRef] [PubMed]
- Lev, S. Non-vesicular lipid transport by lipid-transfer proteins and beyond. Nat. Rev. Mol. Cell Biol. 2010, 11, 739–750. [Google Scholar] [CrossRef] [PubMed]
- Olkkonen, V.M.; Beaslas, O.; Nissila, E. Oxysterols and their cellular effectors. Biomolecules 2012, 2, 76–103. [Google Scholar] [CrossRef]
- Johansson, M.; Lehto, M.; Tanhuanpaa, K.; Cover, T.L.; Olkkonen, V.M. The oxysterol-binding protein homologue ORP1L interacts with Rab7 and alters functional properties of late endocytic compartments. Mol. Biol. Cell 2005, 16, 5480–5492. [Google Scholar] [CrossRef]
- Rocha, N.; Kuijl, C.; van der Kant, R.; Janssen, L.; Houben, D.; Janssen, H.; Zwart, W.; Neefjes, J. Cholesterol sensor ORP1L contacts the ER protein VAP to control Rab7-RILP-p150Glued and late endosome positioning. J. Cell Biol. 2009, 185, 1209–1225. [Google Scholar] [CrossRef]
- Chang, T.-Y.; Reid, P.C.; Sugii, S.; Ohgami, N.; Cruz, J.C.; Chang, C.C.Y. Niemann-Pick type C disease and intracellular cholesterol trafficking. J. Biol. Chem. 2005, 280, 20917–20920. [Google Scholar] [CrossRef]
- Suzuki, M.; Sugimoto, Y.; Ohsaki, Y.; Ueno, M.; Kato, S.; Kitamura, Y.; Hosokawa, H.; Davies, J.; Ioannou, Y.; Vanier, M.; et al. Endosomal accumulation of Toll-like receptor 4 causes constitutive secretion of cytokines and activation of signal transducers and activators of transcription in Niemann-Pick disease type C (NPC) fibroblasts: A potential basis for glial cell activation in the NPC brain. J. Neurosci. 2007, 27, 1879–1891. [Google Scholar] [PubMed]
- Kagan, J.C.; Su, T.; Horng, T.; Chow, A.; Akira, S.; Medzhitov, R. TRAM couples endocytosis of Toll-like receptor 4 to the induction of interferon-β. Nat. Immunol. 2008, 9, 361. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, M.; Sato, S.; Hemmi, H.; Hoshino, K.; Kaisho, T.; Sanjo, H.; Takeuchi, O.; Sugiyama, M.; Okabe, M.; Takeda, K.; et al. Role of adaptor TRIF in the MyD88-independent Toll-Like receptor signaling pathway. Science 2003, 301, 640–643. [Google Scholar] [CrossRef] [PubMed]
- Triantafilou, M.; Miyake, K.; Golenbock, D.T.; Triantafilou, K. Mediators of innate immune recognition of bacteria concentrate in lipid rafts and facilitate lipopolysaccharide-induced cell activation. J. Cell Sci. 2002, 115, 2603–2611. [Google Scholar] [PubMed]
- Olsson, S.; Sundler, R. The role of lipid rafts in LPS-induced signaling in a macrophage cell line. Mol. Immunol. 2006, 43, 607–612. [Google Scholar] [CrossRef]
- Lusa, S.; Blom, T.S.; Eskelinen, E.L.; Kuismanen, E.; Mansson, J.E.; Simons, K.; Ikonen, E. Depletion of rafts in late endocytic membranes is controlled by NPC1-dependent recycling of cholesterol to the plasma membrane. J. Cell Sci. 2001, 114, 1893–1900. [Google Scholar] [PubMed]
- Hiscott, J.; Kwon, H.; Genin, P. Hostile takeovers: Viral appropriation of the NF-κB pathway. J. Clin. Investig. 2001, 107, 143–151. [Google Scholar] [CrossRef]
- Shike, H.; Shimizu, C.; Kanegaye, J.; Foley, J.L.; Burns, J.C. Quantitation of adenovirus genome during acute infection in normal children. Pediatr. Infect. Dis. J. 2005, 24, 29–33. [Google Scholar] [CrossRef]
- Flint, S.J.; Racaniello, V.R.; Enquist, L.W.; Skalka, A.M. Principles of Virology, 3rd ed.; Flint, S.J., Enquist, L.W., Racaniello, V.R., Eds.; ASM Press: Washington, DC, USA, 2009. [Google Scholar]
- Gueret, V.; Negrete-Virgen, J.A.; Lyddiatt, A.; Al-Rubeai, M. Rapid titration of adenoviral infectivity by flow cytometry in batch culture of infected HEK293 cells. Cytotechnology 2002, 38, 87–97. [Google Scholar] [CrossRef]
- Moore, J.D.; Potter, A. Pin1 inhibitors: Pitfalls, progress and cellular pharmacology. Bioorganic Med. Chem. Lett. 2013, 23, 4283–4291. [Google Scholar] [CrossRef]
- Nagy, P.D.; Strating, J.R.P.M.; van Kuppeveld, F.J.M. Building viral replication organelles: Close encounters of the membrane types. PLoS Pathog. 2016, 12, e1005912. [Google Scholar] [CrossRef] [PubMed]
- Oishi, Y.; Spann, N.J.; Link, V.M.; Muse, E.D.; Strid, T.; Edillor, C.; Kolar, M.J.; Matsuzaka, T.; Hayakawa, S.; Tao, J.; et al. SREBP1 contributes to resolution of pro-inflammatory TLR4 signaling by reprogramming fatty acid metabolism. Cell Metab. 2017, 25, 412–427. [Google Scholar] [CrossRef] [PubMed]
- Killerby, M.E.; Rozwadowski, F.; Lu, X.; Caulcrick-Grimes, M.; McHugh, L.; Haldeman, A.M.; Fulton, T.; Schneider, E.; Sakthivel, S.K.; Bhatnagar, J.; et al. Respiratory illness associated with emergent human adenovirus genome type 7d, New Jersey, 2016–2017. Open Forum Infect. Dis. 2019, 6, ofz017. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Fu, J.; Bouvier, M. Allele- and locus-specific recognition of class I MHC molecules by the immunomodulatory E3-19K protein from adenovirus. J. Immunol. 2007, 178, 4567–4575. [Google Scholar] [CrossRef] [PubMed]
- Fu, J.; Li, L.; Bouvier, M. Adenovirus E3-19K proteins of different serotypes and subgroups have similar, yet distinct, immunomodulatory functions toward major histocompatibility class I molecules. J. Biol. Chem. 2011, 286, 17631–17639. [Google Scholar] [CrossRef]
- Garnett, C.T.; Erdman, D.; Xu, W.; Gooding, L.R. Prevalence and quantitation of species C adenovirus DNA in human mucosal lymphocytes. J. Virol. 2002, 76, 10608–10616. [Google Scholar] [CrossRef]
- Lion, T.; Kosulin, K.; Landlinger, C.; Rauch, M.; Preuner, S.; Jugovic, D.; Potschger, U.; Lawitschka, A.; Peters, C.; Fritsch, G.; et al. Monitoring of adenovirus load in stool by real-time PCR permits early detection of impending invasive infection in patients after allogeneic stem cell transplantation. Leukemia 2010, 24, 706. [Google Scholar] [CrossRef]
- Al Qurashi, Y.M.A.; Guiver, M.; Cooper, R.J. Sequence typing of adenovirus from samples from hematological stem cell transplant recipients. J. Med. Virol. 2011, 83, 1951–1958. [Google Scholar] [CrossRef]
- Dhingra, A.; Hage, E.; Ganzenmueller, T.; Bottcher, S.; Hofmann, J.; Hamprecht, K.; Obermeier, P.; Rath, B.; Hausmann, F.; Dobner, T.; et al. Molecular evolution of human adenovirus (HAdV) species C. Sci. Rep. 2019, 9, 1039. [Google Scholar] [CrossRef]
- Shisler, J.; Yang, C.; Walter, B.; Ware, C.; Gooding, L. The adenovirus E3-10.4K/14.5K complex mediates loss of cell surface Fas (CD95) and resistance to Fas-induced apoptosis. J. Virol. 1997, 71, 8299–8306. [Google Scholar]
- Gooding, L.R.; Ranheim, T.S.; Tollefson, A.E.; Aquino, L.; Duerksen-Hughes, P.; Horton, T.M.; Wold, W.S. The 10,400- and 14,500-dalton proteins encoded by region E3 of adenovirus function together to protect many but not all mouse cell lines against lysis by tumor necrosis factor. J. Virol. 1991, 65, 4114–4123. [Google Scholar] [PubMed]
- Lion, T. Adenovirus infections in immunocompetent and immunocompromised patients. Clin. Microbiol. Rev. 2017, 27, 441–462. [Google Scholar] [CrossRef] [PubMed]
- Heise, C.; Hermiston, T.; Johnson, L.; Brooks, G.; Sampson-Johannes, A.; Williams, A.; Hawkins, L.; Kirn, D. An adenovirus E1A mutant that demonstrates potent and selective systemic anti-tumoral efficacy. Nat. Med. 2000, 6, 1134. [Google Scholar] [CrossRef]
- Weigert, M.; Binks, A.; Dowson, S.; Leung, E.Y.L.; Athineos, D.; Yu, X.; Mullin, M.; Walton, J.B.; Orange, C.; Ennis, D.; et al. RIPK3 promotes adenovirus type 5 activity. Cell Death Dis. 2017, 8, 3206. [Google Scholar] [CrossRef] [PubMed]
- Tabas, I. Consequences of cellular cholesterol accumulation: Basic concepts and physiological implications. J. Clin. Investig. 2002, 110, 905–911. [Google Scholar] [CrossRef] [PubMed]
- Tall, A.R.; Yvan-Charvet, L. Cholesterol, inflammation and innate immunity. Nat. Rev. Immunol. 2015, 15, 104. [Google Scholar] [CrossRef] [PubMed]




© 2019 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Carlin, C.R. New Insights to Adenovirus-Directed Innate Immunity in Respiratory Epithelial Cells. Microorganisms 2019, 7, 216. https://doi.org/10.3390/microorganisms7080216
Carlin CR. New Insights to Adenovirus-Directed Innate Immunity in Respiratory Epithelial Cells. Microorganisms. 2019; 7(8):216. https://doi.org/10.3390/microorganisms7080216
Chicago/Turabian StyleCarlin, Cathleen R. 2019. "New Insights to Adenovirus-Directed Innate Immunity in Respiratory Epithelial Cells" Microorganisms 7, no. 8: 216. https://doi.org/10.3390/microorganisms7080216
APA StyleCarlin, C. R. (2019). New Insights to Adenovirus-Directed Innate Immunity in Respiratory Epithelial Cells. Microorganisms, 7(8), 216. https://doi.org/10.3390/microorganisms7080216