
Membrane Association and Catabolite Repression of the Sulfolobus solfataricus α-Amylase

Abstract
:1. Introduction
2. Materials and Methods
2.1. Archaeal Strains and Cultivation
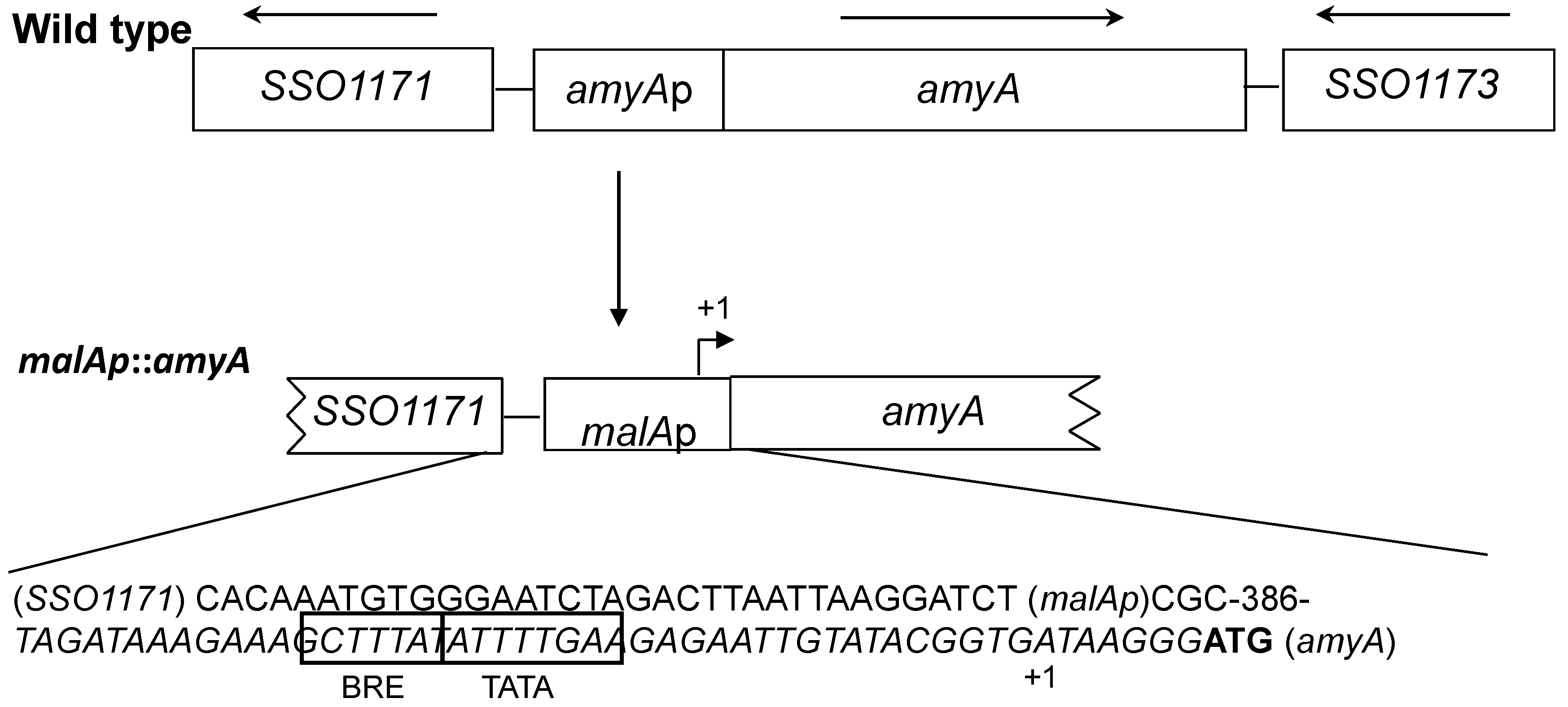
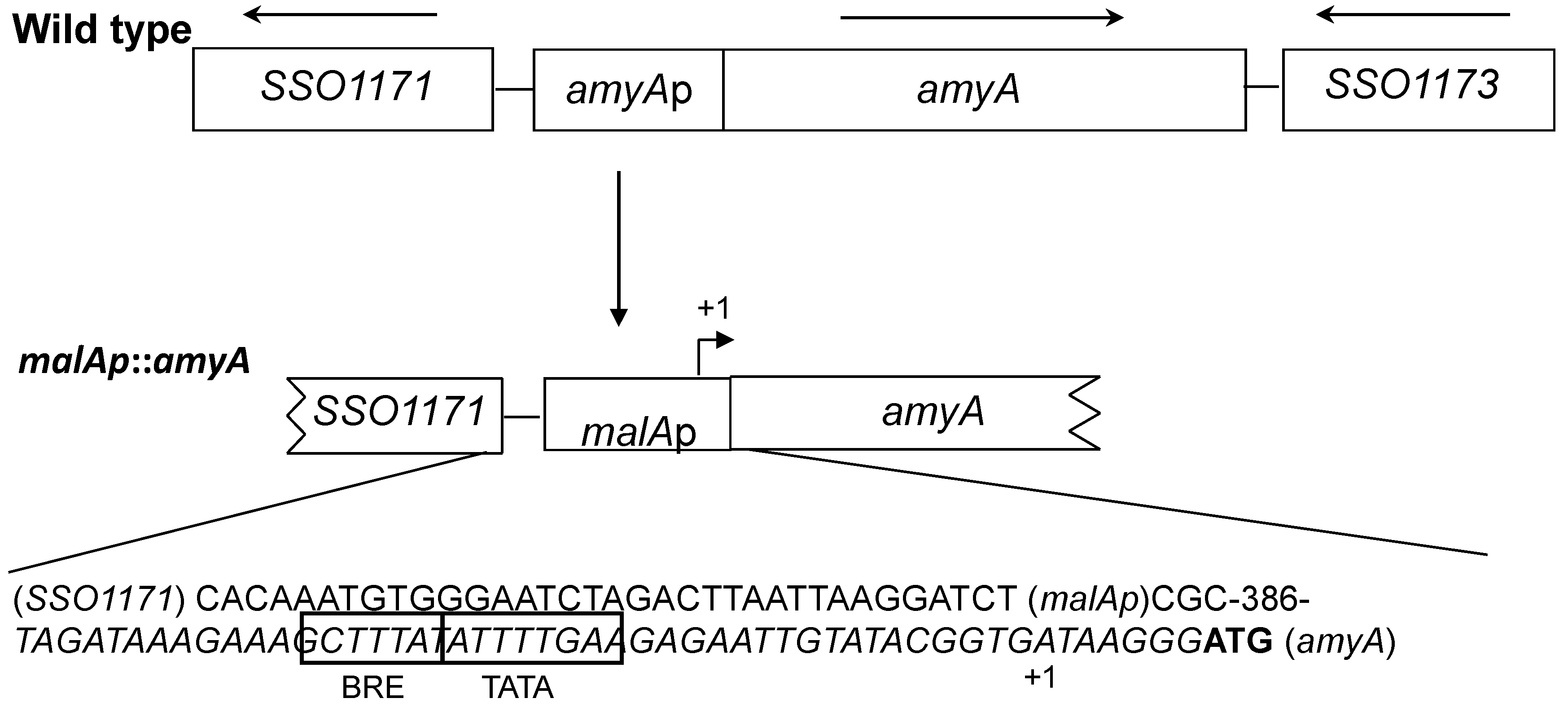
2.2. Molecular Biology Methods and Strain Constructions

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Genotype | Source Or Derivation | |
|---|---|---|
| Strains | ||
| PBL2004 | amyA::lacS | PBL2002 [19] |
| PBL2025 | amyA + Δ(SSO3004-3050) | PBL2000 [35,36] |
| PBL2058 | malAp::amyA | PBL2025 by markerless exchange (ME) |
| PBL2059 | malAp::amyA (G877Stop) | PBL2058 by ME |
| PBL2064 | malAp::amyA Δ(I2-C31) | PBL2058 by ME |
| PBL2065 | malAp::amyA (K898Stop) | PBL2058 by ME |
| Plasmids | ||
| pUC19 | bla | New England BioLabs |
| pPB1035 | lacS-KpnI | pUC19 [34] |
| pBN1081 | 1171::malAp::amyA (fusion at M9) | pUC19 (this work) |
| pBN1062 | malAp::amyA | pPB1035 (this work) |
| pBN1063 | amyA (G877Stop) | pPB1035 (this work) |
| pBN1064 | amyA Δ(I2-C31) | pPB1035 (this work) |
| pBN1065 | amyA (K898Stop) | pPB1035 (this work) |
2.3. Protein Purification
2.4. Α-Amylase Activity Assay
2.5. Starch Plates Diffusion Assay
2.6. Sub-Cellular Fractionation
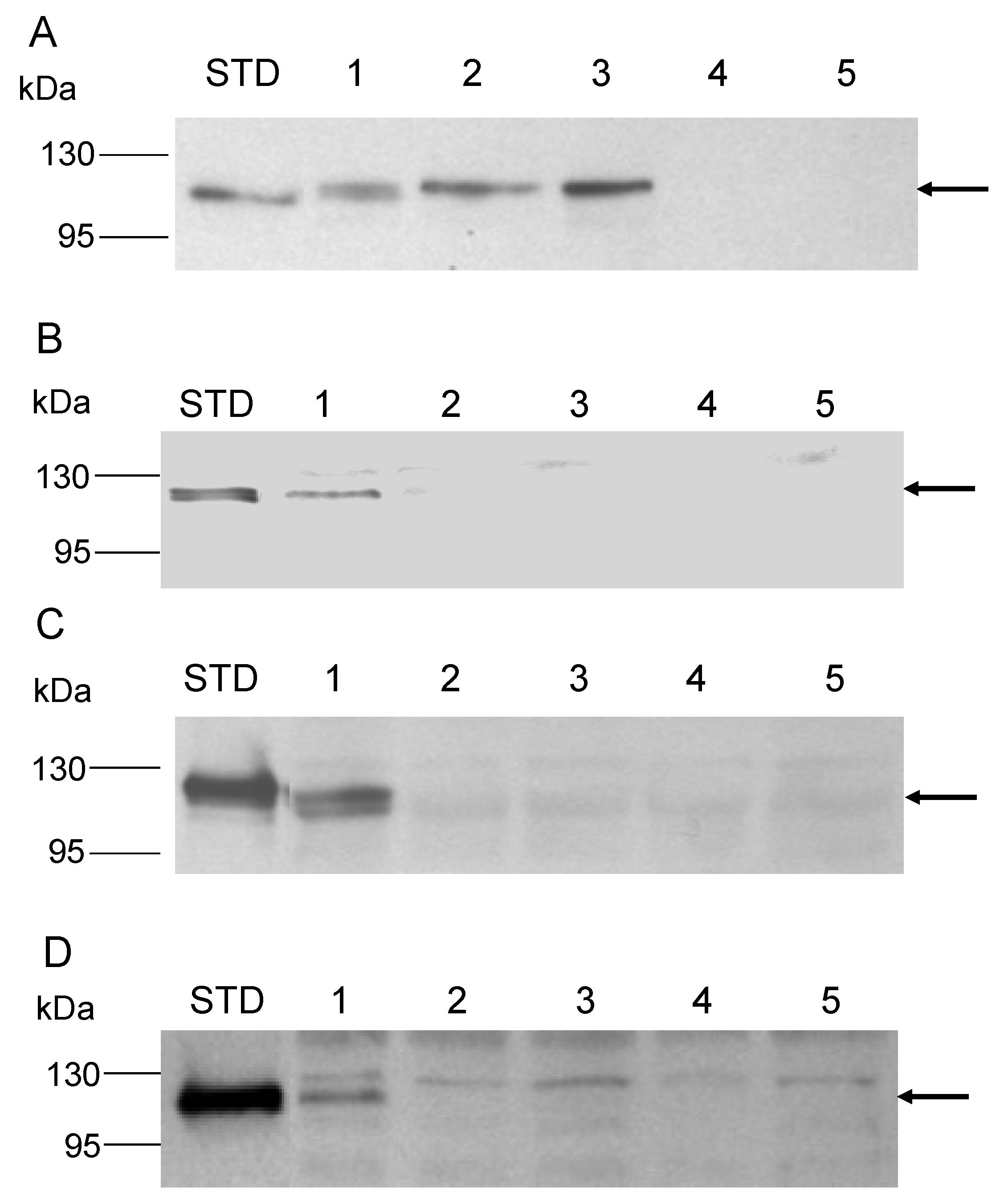
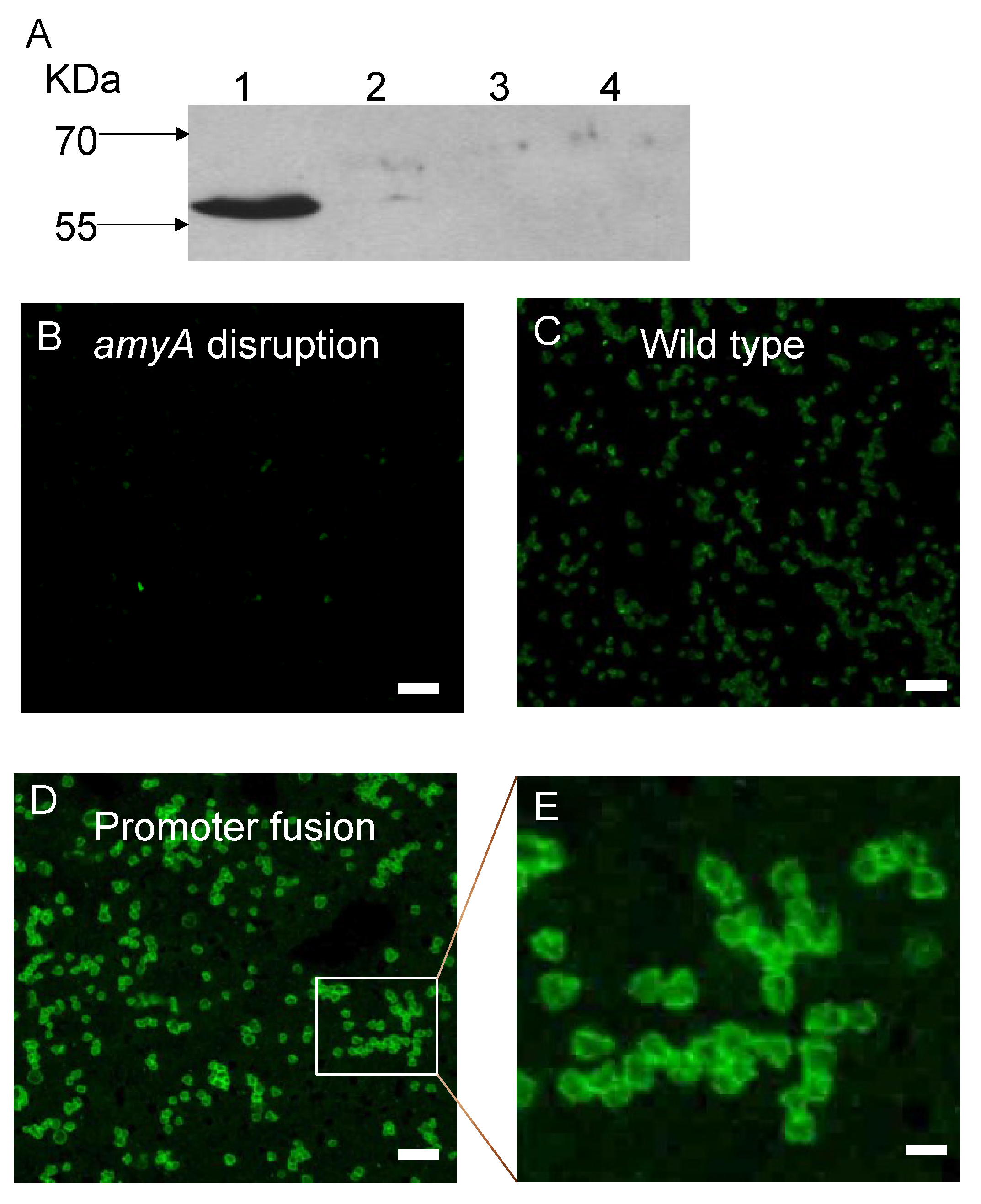
2.7. Protein Electrophoresis and Western Blot Analysis
2.8. Bioinformatic Analysis
3. Results
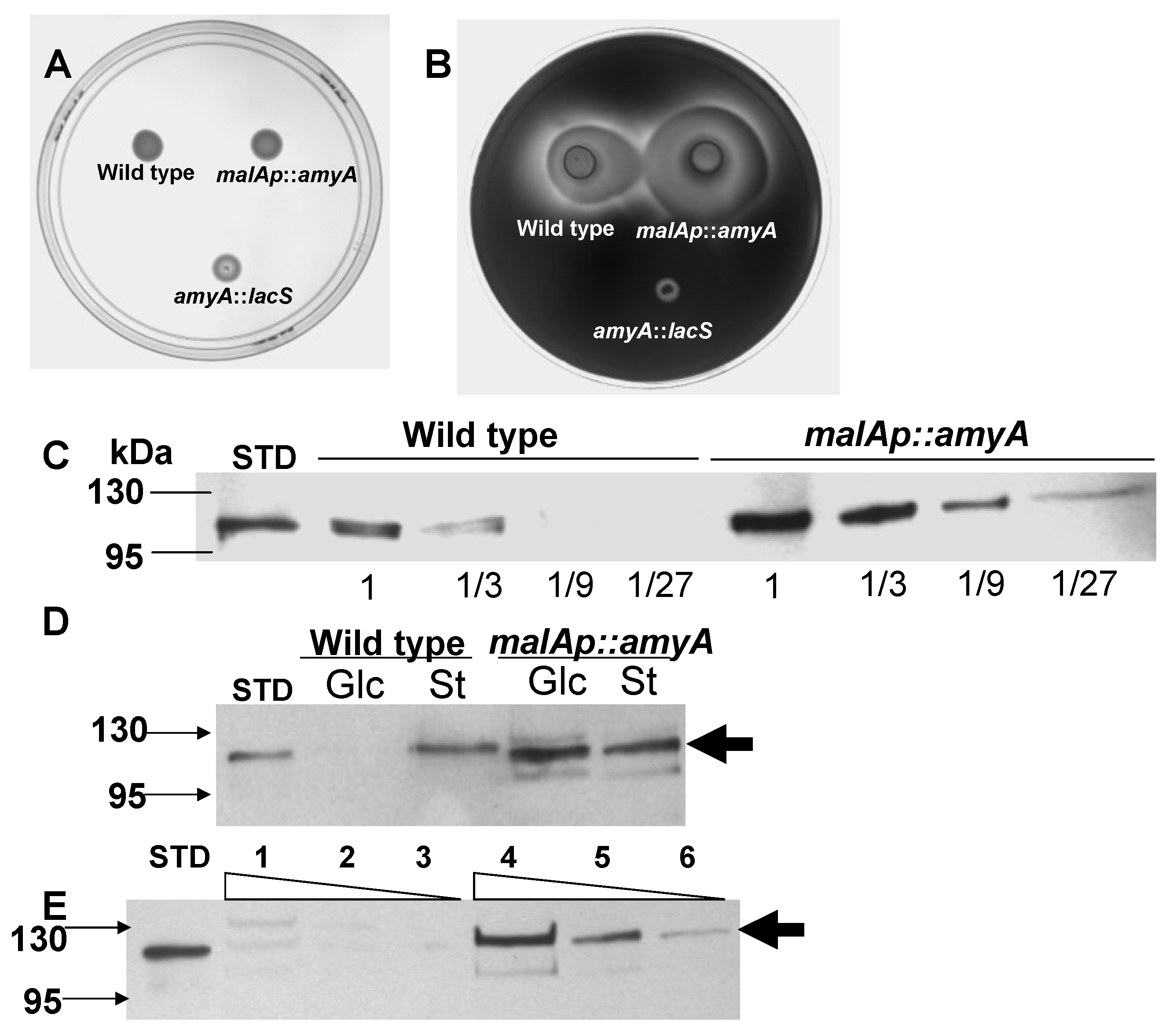
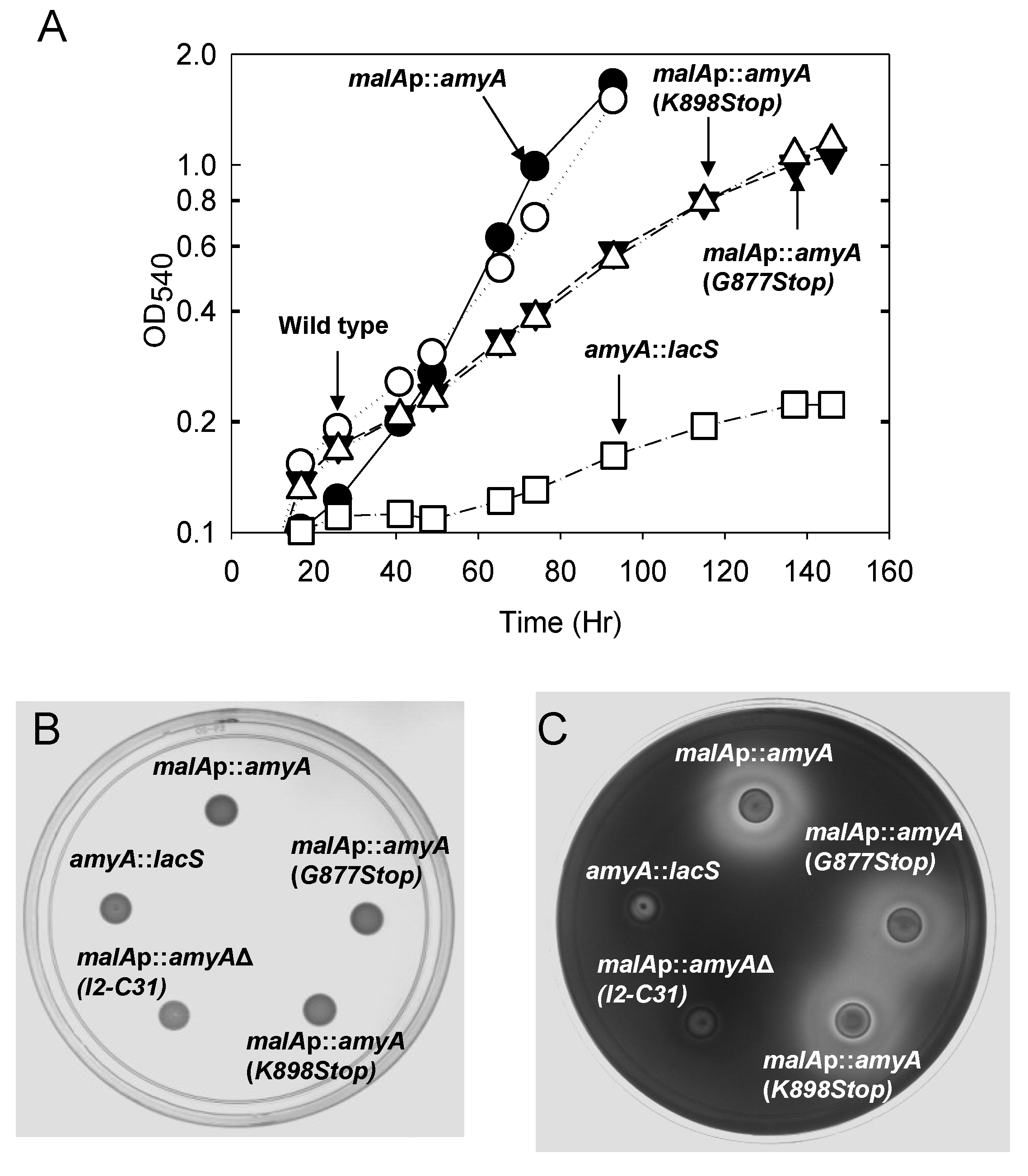
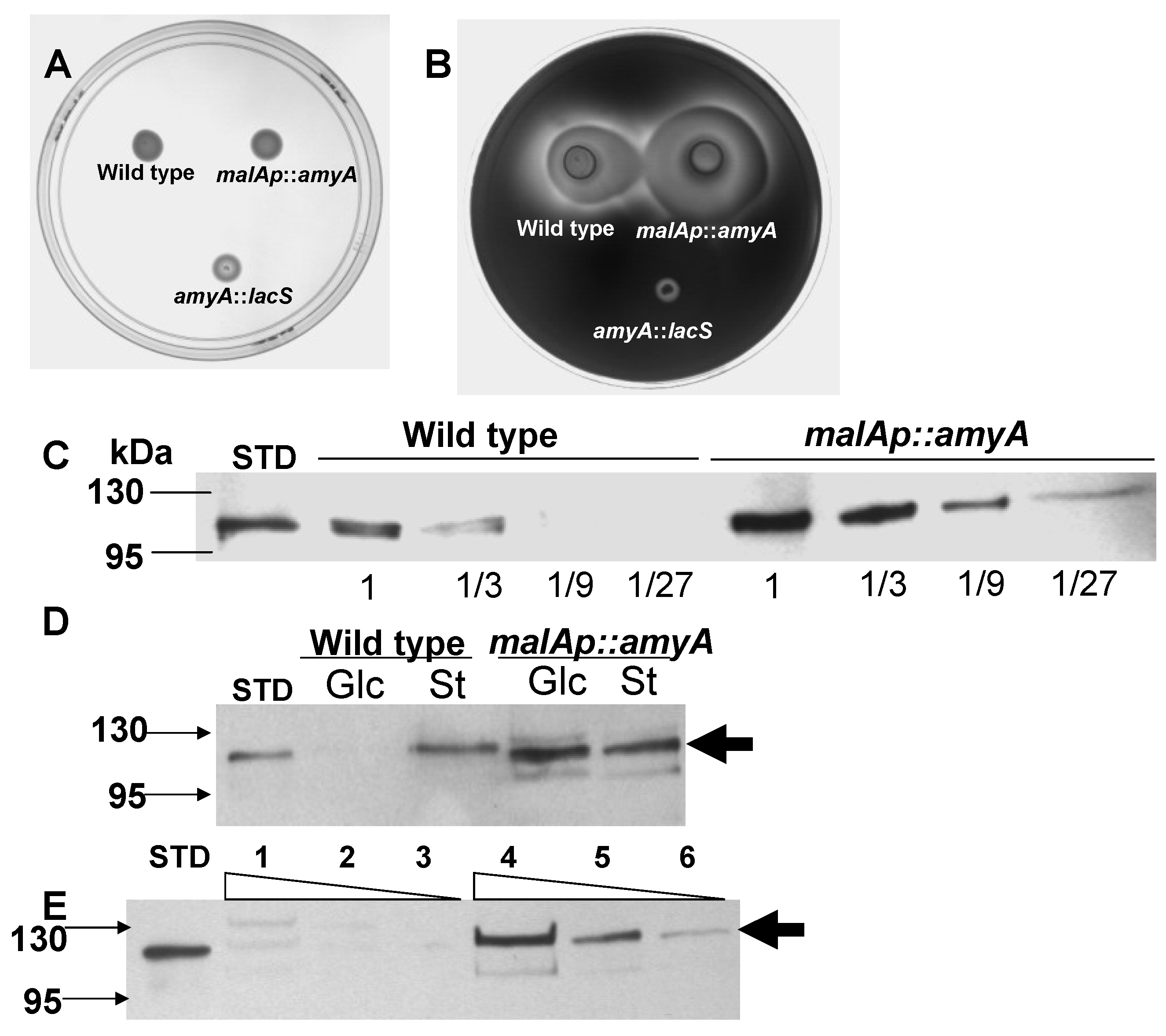
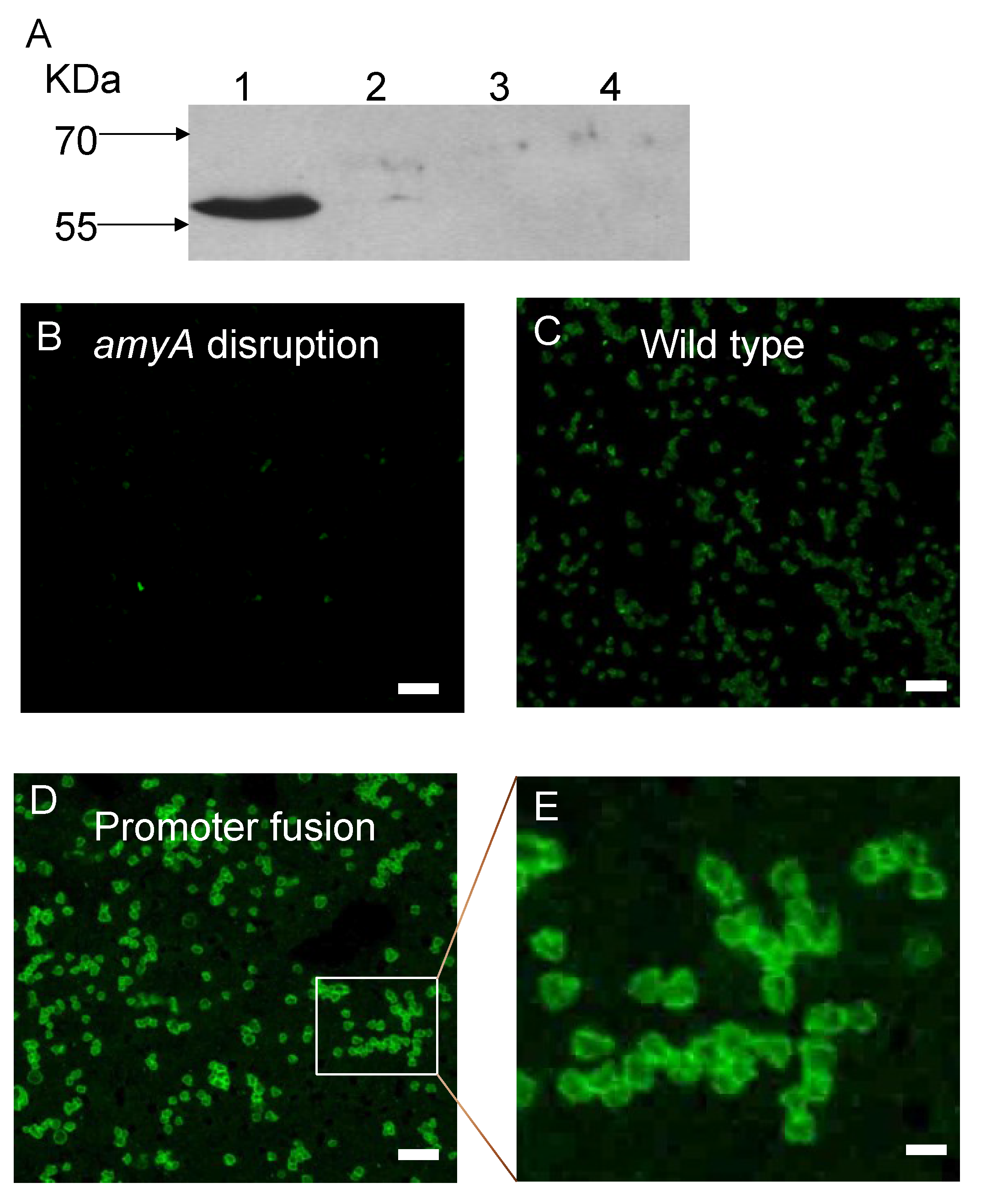
3.1. Promoter Substitution and Catabolite Repression
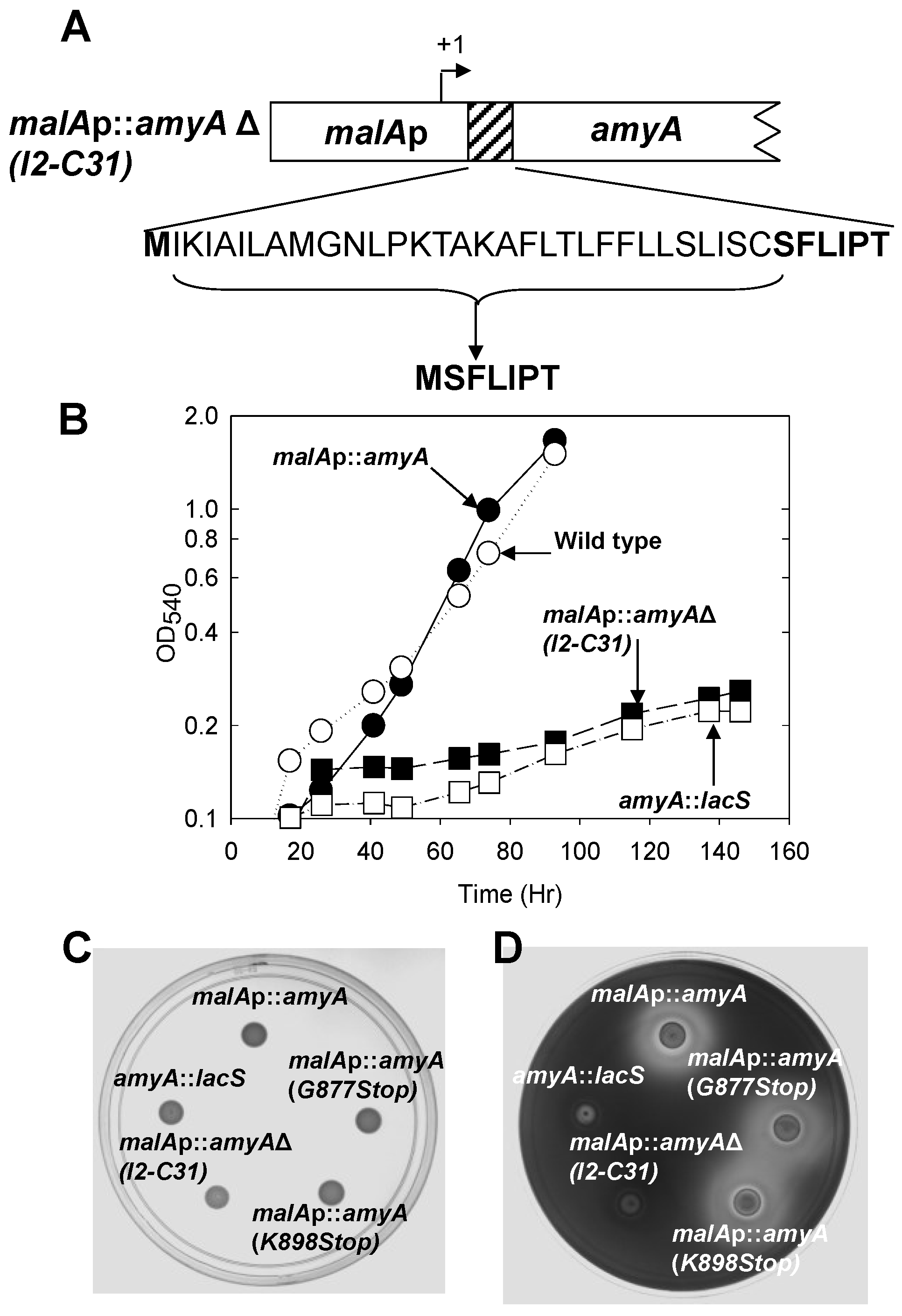
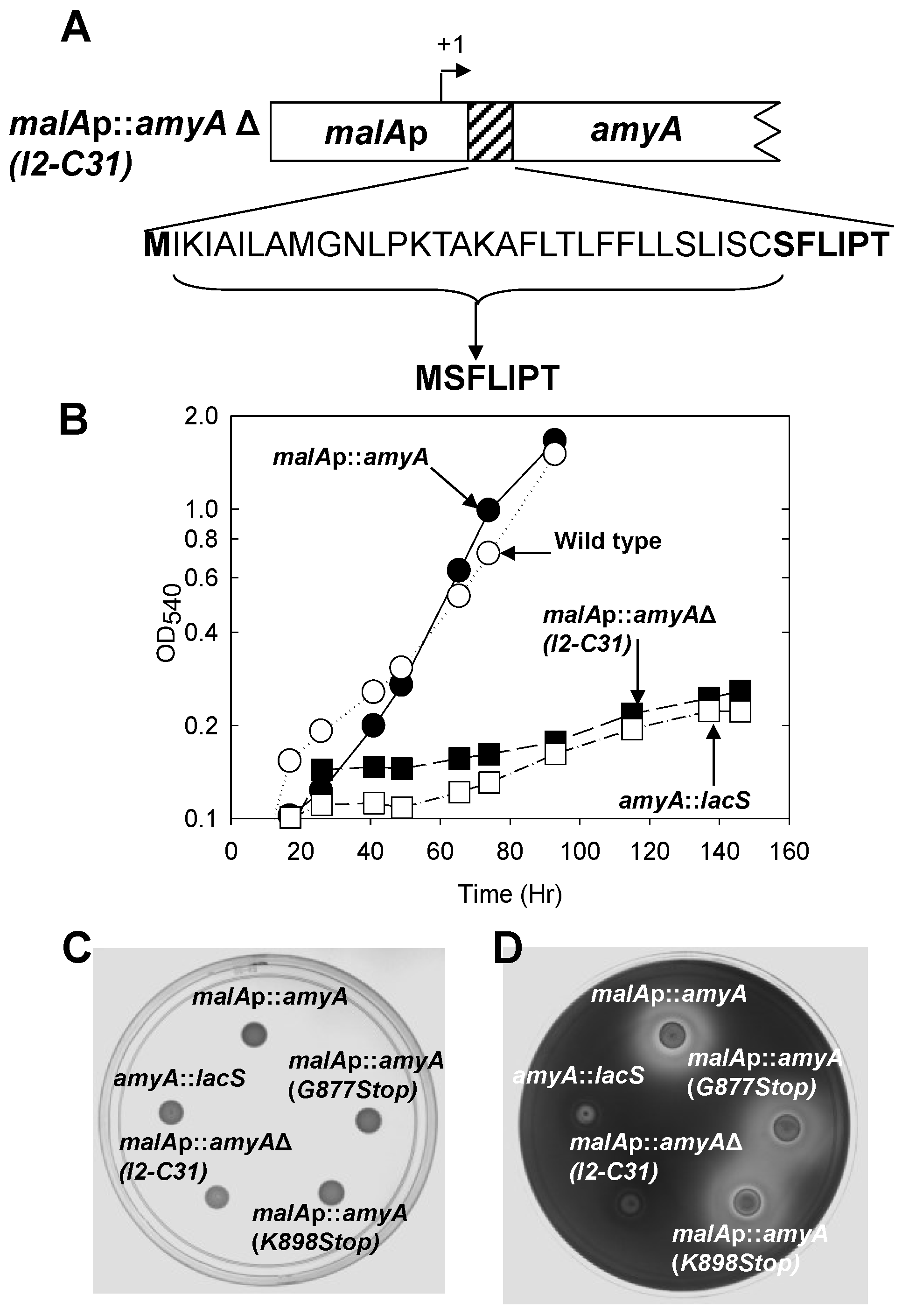
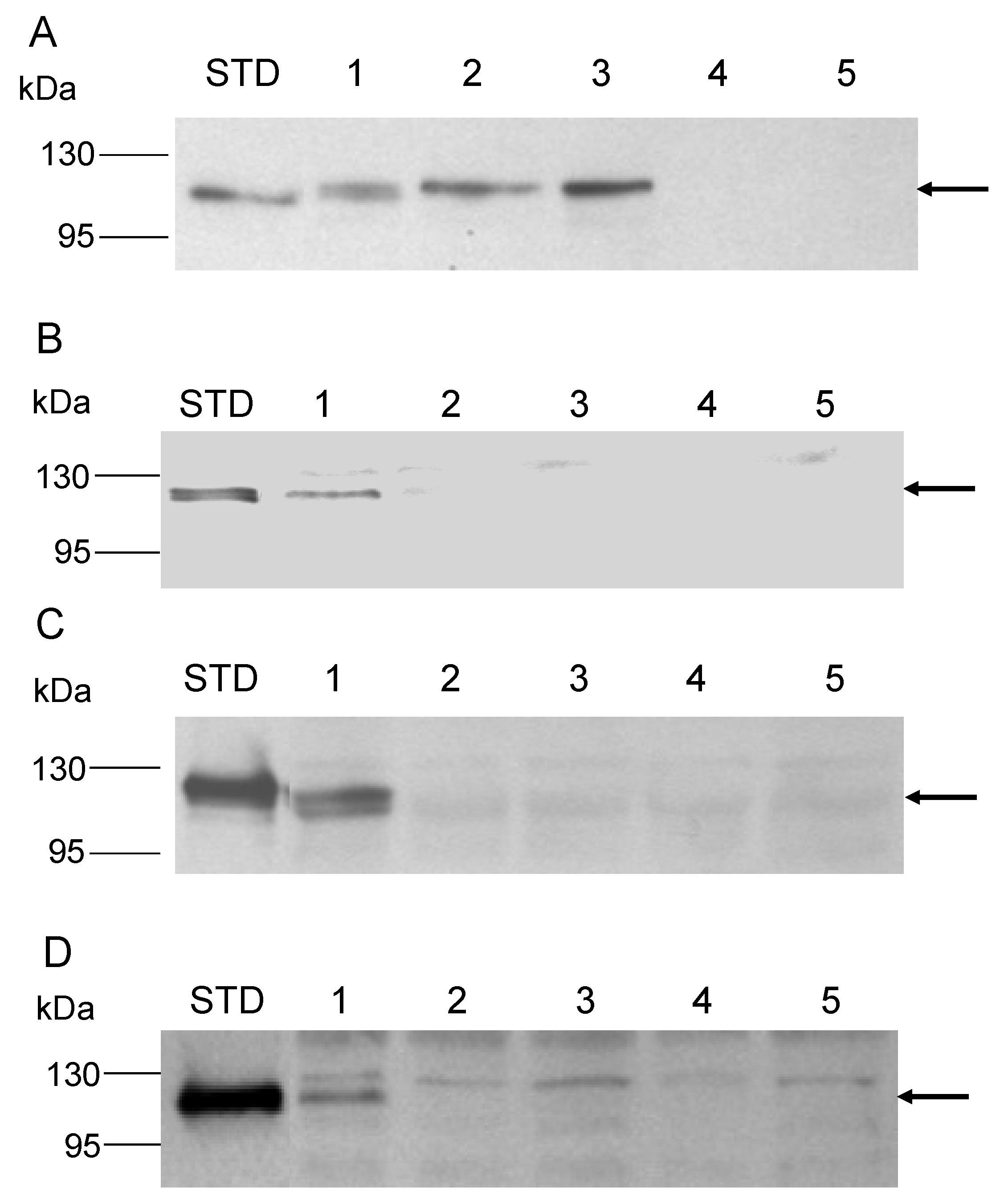
3.2. Role of the Class II Amya Leader Sequence
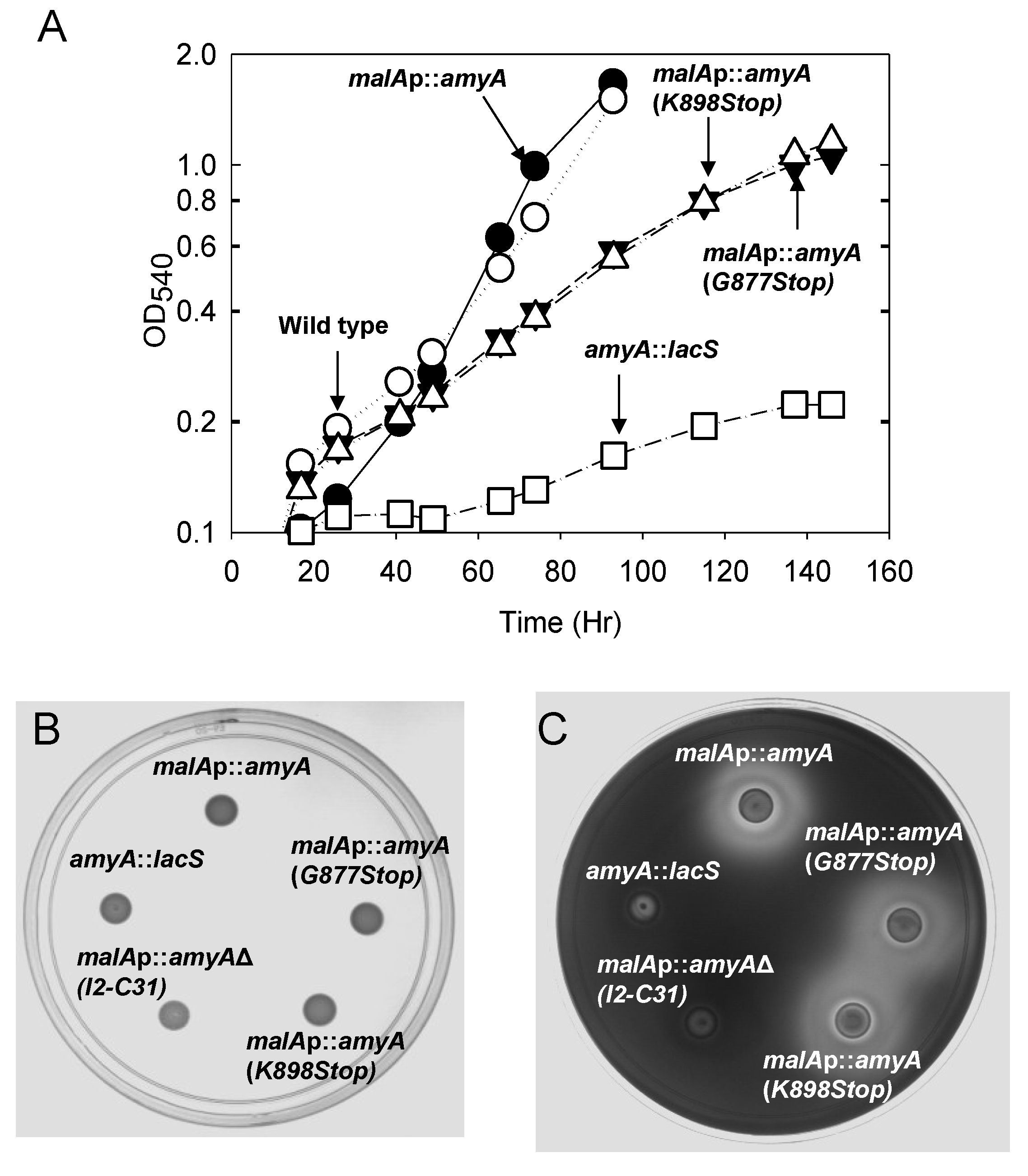
| Strain | α-Amylase Activity * (U/mL) | |
|---|---|---|
| Supernatant | Cell Extracts | |
| AmyA+ (PBL2025) | 0.23 ± 0.01 | 0.13 ± 0.02 |
| malAp::amyA (PBL2058) | 0.16 ± 0.01 | 0.18 ± 0.01 |
| malAp::amyA G877 to STOP (PBL2059) | 0.17 ± 0.01 | 0.03 ± 0.01 |
| malAp::amyA K898 to STOP (PBL2065) | 0.15 ± 0.01 | 0.02 ± 0.01 |
| malAp::amyA Δ I2 to C31 (PBL2064) | <0.01 | 0.01 |
| AmyA− (PBL2004) | 0.02 ± 0.03 | 0.02 ± 0.06 |
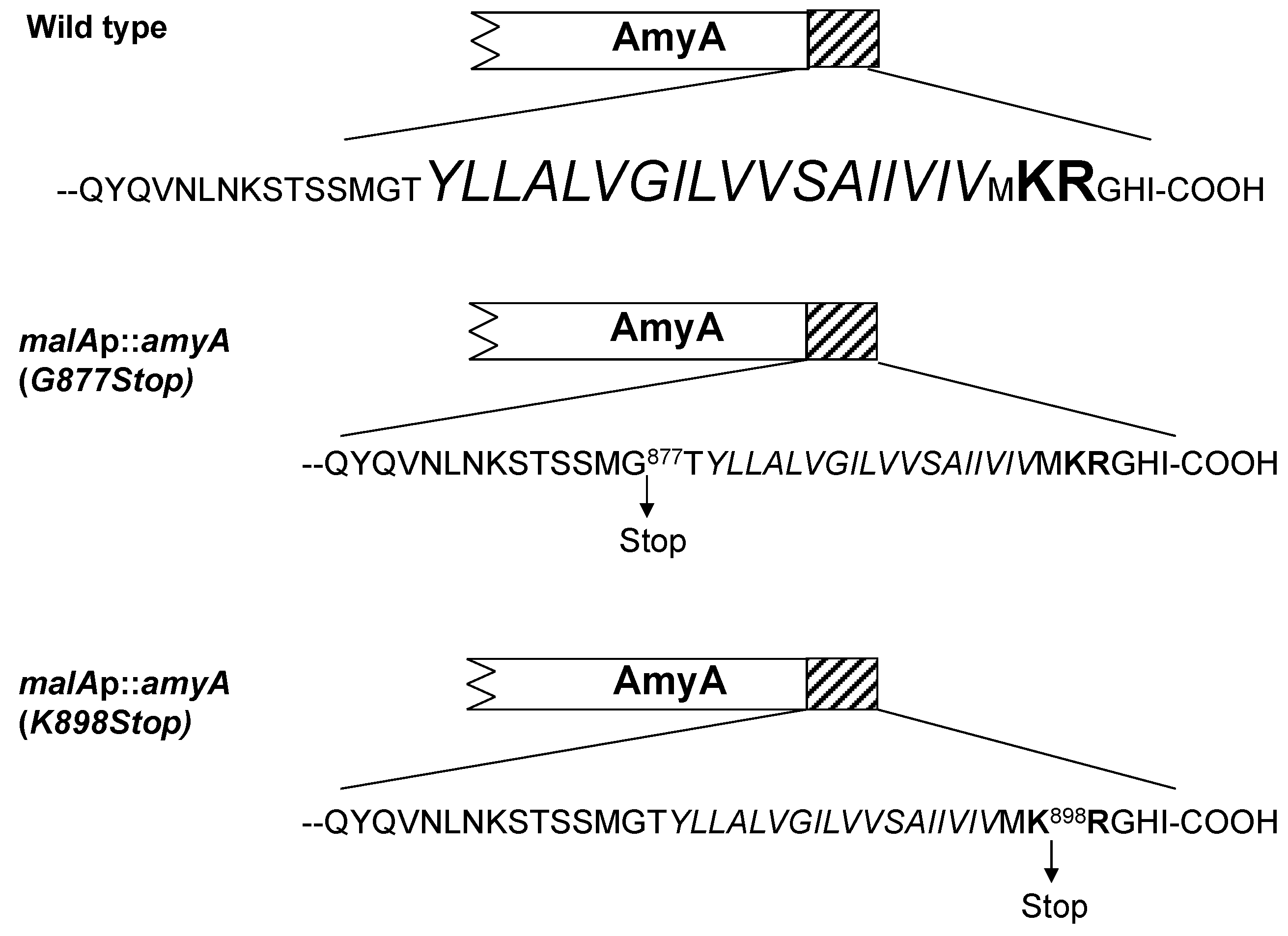
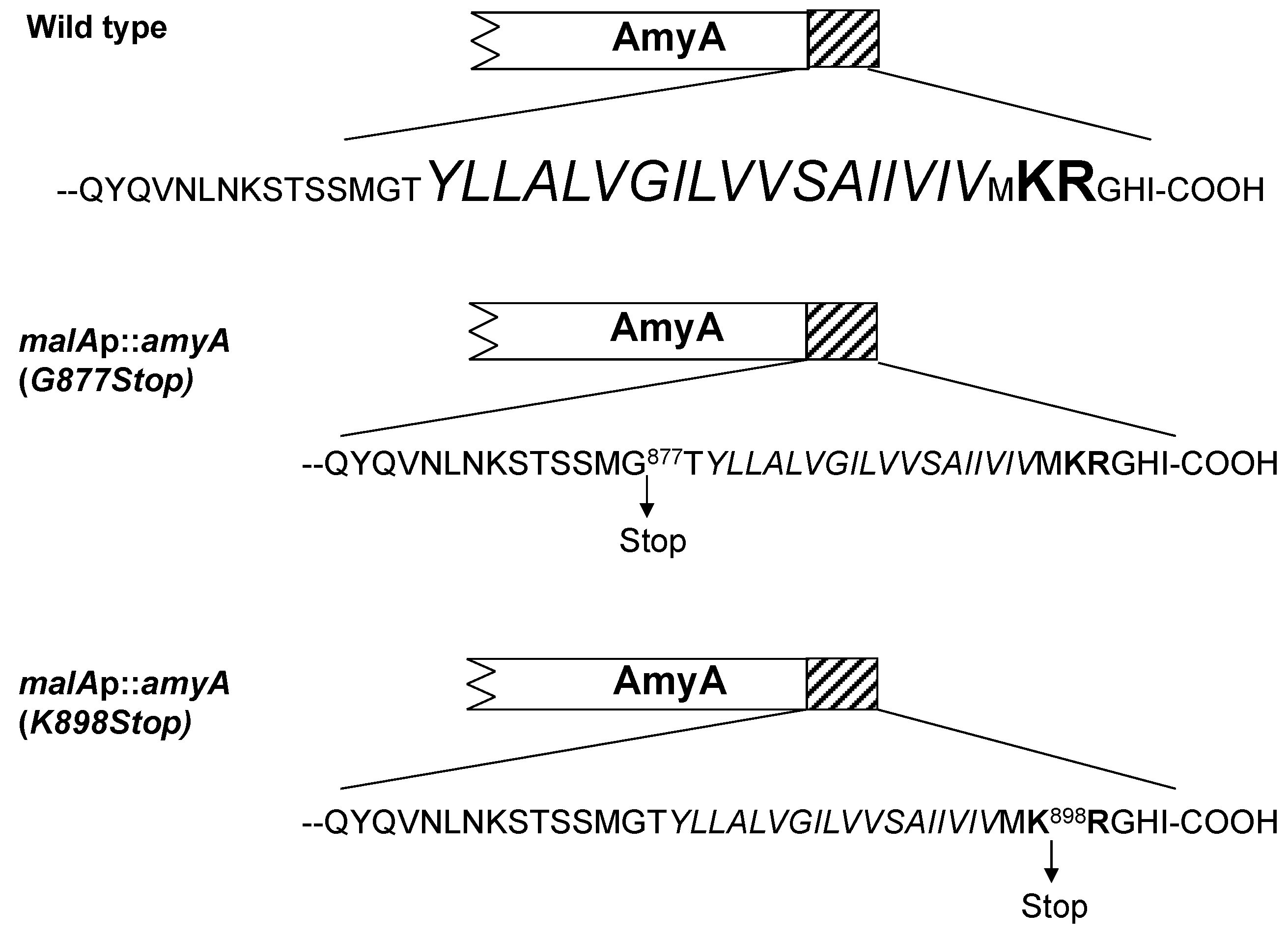
3.3. Role of the Amya C-Terminal Motif
4. Discussion
Supplementary Files
Supplementary File 1Acknowledgements
Author Contributions
Conflicts of Interest
References
- Albers, S.V.; Szabo, Z.; Driessen, A.J. Protein secretion in the archaea: Multiple paths towards a unique cell surface. Nat. Rev. Microbiol. 2006, 4, 537–547. [Google Scholar] [CrossRef] [PubMed]
- Pohlschroder, M.; Hartmann, E.; Hand, N.J.; Dilks, K.; Haddad, A. Diversity and evolution of protein translocation. Annu. Rev. Microbiol. 2005, 59, 91–111. [Google Scholar] [CrossRef] [PubMed]
- Ring, G.; Eichler, J. Extreme secretion: Protein translocation across the archael plasma membrane. J. Bioenerg. Biomembr. 2004, 36, 35–45. [Google Scholar] [CrossRef] [PubMed]
- Bardy, S.L.; Eichler, J.; Jarrell, K.F. Archaeal signal peptides—A comparative survey at the genome level. Protein Sci. 2003, 12, 1833–1843. [Google Scholar] [CrossRef] [PubMed]
- Albers, S.V.; Driessen, A.M. Signal peptides of secreted proteins of the archaeon Sulfolobus solfataricus: A genomic survey. Arch. Microbiol. 2002, 177, 209–216. [Google Scholar] [CrossRef] [PubMed]
- Ng, S.Y.; Chaban, B.; van Dyke, D.J.; Jarrell, K.F. Archaeal signal peptidases. Microbiology 2007, 153, 305–314. [Google Scholar] [CrossRef] [PubMed]
- Zolghadr, B.; Weber, S.; Szabo, Z.; Driessen, A.J.; Albers, S.V. Identification of a system required for the functional surface localization of sugar binding proteins with class III signal peptides in Sulfolobus solfataricus. Mol. Microbiol. 2007, 64, 795–806. [Google Scholar] [CrossRef] [PubMed]
- Szabo, Z.; Stahl, A.O.; Albers, S.V.; Kissinger, J.C.; Driessen, A.J.; Pohlschroder, M. Identification of diverse archaeal proteins with class III signal peptides cleaved by distinct archaeal prepilin peptidases. J. Bacteriol. 2007, 189, 772–778. [Google Scholar] [CrossRef] [PubMed]
- Szabo, Z.; Pohlschroder, M. Diversity and subcellular distribution of archaeal secreted proteins. Front. Microbiol. 2012, 3. [Google Scholar] [CrossRef] [PubMed]
- Pohlschroder, M.; Dilks, K.; Hand, N.J.; Wesley Rose, R. Translocation of proteins across archaeal cytoplasmic membranes. FEMS Microbiol. Rev. 2004, 28, 3–24. [Google Scholar] [CrossRef] [PubMed]
- Gold, V.A.; Duong, F.; Collinson, I. Structure and function of the bacterial Sec translocon. Mol. Membr. Biol. 2007, 24, 387–394. [Google Scholar] [CrossRef] [PubMed]
- Dilks, K.; Rose, R.W.; Hartmann, E.; Pohlschroder, M. Prokaryotic utilization of the twin-arginine translocation pathway: A genomic survey. J. Bacteriol. 2003, 185, 1478–1483. [Google Scholar] [CrossRef] [PubMed]
- Rose, R.W.; Bruser, T.; Kissinger, J.C.; Pohlschroder, M. Adaptation of protein secretion to extremely high-salt conditions by extensive use of the twin-arginine translocation pathway. Mol. Microbiol. 2002, 45, 943–950. [Google Scholar] [CrossRef] [PubMed]
- Haseltine, C.; Rolfsmeier, M.; Blum, P. The glucose effect and regulation of alpha-amylase synthesis in the hyperthermophilic archaeon Sulfolobus solfataricus. J. Bacteriol. 1996, 178, 945–950. [Google Scholar] [PubMed]
- Chong, P.K.; Wright, P.C. Identification and characterization of the Sulfolobus solfataricus P2 proteome. J. Proteome. Res. 2005, 4, 1789–1798. [Google Scholar] [CrossRef] [PubMed]
- Szabo, Z.; Sani, M.; Groeneveld, M.; Zolghadr, B.; Schelert, J.; Albers, S.V.; Blum, P.; Boekema, E.J.; Driessen, A.J. Flagellar motility and structure in the hyperthermoacidophilic archaeon Sulfolobus solfataricus. J. Bacteriol. 2007, 189, 4305–4309. [Google Scholar] [CrossRef] [PubMed]
- Ellen, A.; Albers, S.-V.; Driessen, A.M. Comparative study of the extracellular proteome of Sulfolobus species reveals limited secretion. Extremophiles 2010, 14, 87–98. [Google Scholar] [CrossRef] [PubMed]
- Miller, P.S.; Blum, P.H. Extremophile-inspired strategies for enzymatic biomass saccharification. Environ. Sci. 2010, 31, 1005–1015. [Google Scholar] [CrossRef] [PubMed]
- Worthington, P.; Hoang, V.; Perez-Pomares, F.; Blum, P. Targeted disruption of the alpha-amylase gene in the hyperthermophilic archaeon Sulfolobus solfataricus. J. Bacteriol. 2003, 185, 482–488. [Google Scholar] [CrossRef] [PubMed]
- Imamura, H.; Fushinobu, S.; Jeon, B.S.; Wakagi, T.; Matsuzawa, H. Identification of the catalytic residue of Thermococcus litoralis 4-alpha-glucanotransferase through mechanism-based labeling. Biochemistry 2001, 40, 12400–12406. [Google Scholar] [CrossRef] [PubMed]
- Kang, S.; Vieille, C.; Zeikus, J.G. Identification of Pyrococcus furiosus amylopullulanase catalytic residues. Appl. Microbiol. Biotechnol. 2005, 66, 408–413. [Google Scholar] [CrossRef] [PubMed]
- Zona, R.; Chang-Pi-Hin, F.; O’Donohue, M.J.; Janecek, S. Bioinformatics of the glycoside hydrolase family 57 and identification of catalytic residues in amylopullulanase from Thermococcus hydrothermalis. Eur. J. Biochem. 2004, 271, 2863–2872. [Google Scholar] [CrossRef] [PubMed]
- Lalithambika, S.; Peterson, L.; Dana, K.; Blum, P. Carbohydrate hydrolysis and transport in the extreme thermoacidophile Sulfolobus solfataricus. Appl. Environ. Microbiol. 2012, 78, 7931–7938. [Google Scholar] [CrossRef] [PubMed]
- Canganella, F.; Andrade, M.; Antranakian, G. Characterization of amylolytic and pullulytic enzymes from thermophilic archaea and from a new Fervidobacterium species. Appl. Microbiol. Biotechnol. 1994, 42, 239–245. [Google Scholar] [CrossRef]
- Rudiger, A.; Jorgensen, P.L.; Antranikian, G. Isolation and characterization of a heat-stable pullulanase from the hyperthermophilic archaeon Pyrococcus woesei after cloning and expression of its gene in Escherichia coli. Appl. Environ. Microbiol. 1995, 61, 567–575. [Google Scholar] [PubMed]
- Worthington, P.; Blum, P.; Perez-Pomares, F.; Elthon, T. Large-scale cultivation of acidophilic hyperthermophiles for recovery of secreted proteins. Appl. Environ. Microbiol. 2003, 69, 252–257. [Google Scholar] [CrossRef] [PubMed]
- Worthington, P.L. The isolation, identification, characterization and regulation of alpha-amylase In Sulfolobus Solfataricus. Available online: http://digitalcommons.unl.edu/dissertations/AAI3142109 (accessed on 14 September 2015).
- Haseltine, C.; Montalvo-Rodriguez, R.; Bini, E.; Carl, A.; Blum, P. Coordinate transcriptional control in the hyperthermophilic archaeon Sulfolobus solfataricus. J. Bacteriol. 1999, 181, 3920–3927. [Google Scholar] [PubMed]
- Allen, M.B. Studies with Cyanidium caldarium, an anomalously pigmented chlorophyte. Arch. Mikrobiol. 1959, 32, 270–277. [Google Scholar] [CrossRef] [PubMed]
- Brock, T.D.; Brock, K.M.; Belly, R.T.; Weiss, R.L. Sulfolobus: A new genus of sulfur-oxidizing bacteria living at low pH and high temperature. Arch. Mikrobiol. 1972, 84, 54–68. [Google Scholar] [CrossRef] [PubMed]
- Rolfsmeier, M.; Haseltine, C.; Bini, E.; Clark, A.; Blum, P. Molecular characterization of the alpha-glucosidase gene (malA) from the hyperthermophilic archaeon Sulfolobus solfataricus. J. Bacteriol. 1998, 180, 1287–1295. [Google Scholar] [PubMed]
- Rockabrand, D.; Livers, K.; Austin, T.; Kaiser, R.; Jensen, D.; Burgess, R.; Blum, P. Roles of DnaK and RpoS in starvation-induced thermotolerance of Escherichia coli. J. Bacteriol. 1998, 180, 846–854. [Google Scholar] [PubMed]
- Higuchi, R.; Krummel, B.; Saiki, R.K. A general method of in vitro preparation and specific mutagenesis of DNA fragments: Study of protein and DNA interactions. Nucleic. Acids. Res. 1988, 16, 7351–7367. [Google Scholar] [CrossRef] [PubMed]
- Schelert, J.; Drozda, M.; Dixit, V.; Dillman, A.; Blum, P. Regulation of mercury resistance in the crenarchaeote Sulfolobus solfataricus. J. Bacteriol. 2006, 188, 7141–7150. [Google Scholar] [CrossRef] [PubMed]
- Manning, G.B.; Campbell, L.L. Thermostable alpha-amylase of Bacillus stearothermophilus. I. Crystallization and some general properties. J. Biol. Chem. 1961, 236, 2952–2957. [Google Scholar] [PubMed]
- Grogan, D.W. Organization and interactions of cell envelope proteins of the extreme thermoacidophile Sulfolobus acidocaldarius. Can. J. Microbiol. 1996, 42, 1163–1171. [Google Scholar] [CrossRef]
- Rybicki, E.P.; von Wechmar, M.B.; Burger, J.T. Monospecific antibody preparation for use in the detection of viruses. In World Perspectives on Barley Yellow Dwarf; Burnett, P.A., Ed.; CIMMYT: El Batan, Mexico, 1990; pp. 149–153. [Google Scholar]
- Hoang, V.; Bini, E.; Dixit, V.; Drozda, M.; Blum, P. The role of cis-acting sequences governing catabolite repression control of lacS expression in the archaeon Sulfolobus solfataricus. Genetics 2004, 167, 1563–1572. [Google Scholar] [CrossRef] [PubMed]
- Lubelska, J.M.; Jonuscheit, M.; Schleper, C.; Albers, S.V.; Driessen, A.J. Regulation of expression of the arabinose and glucose transporter genes in the thermophilic archaeon Sulfolobus solfataricus. Extremophiles 2006, 10, 383–391. [Google Scholar] [CrossRef] [PubMed]
- Szabo, Z.; Albers, S.V.; Driessen, A.J. Active-site residues in the type IV prepilin peptidase homologue pibD from the archaeon Sulfolobus solfataricus. J. Bacteriol. 2006, 188, 1437–1443. [Google Scholar] [CrossRef] [PubMed]
- Elferink, M.G.; Albers, S.V.; Konings, W.N.; Driessen, A.J. Sugar transport in Sulfolobus solfataricus is mediated by two families of binding protein-dependent abc transporters. Mol. Microbiol. 2001, 39, 1494–1503. [Google Scholar] [CrossRef] [PubMed]
- Erra-Pujada, M.; Debeire, P.; Duchiron, F.; O’Donohue, M.J. The type II pullulanase of Thermococcus hydrothermalis: Molecular characterization of the gene and expression of the catalytic domain. J. Bacteriol. 1999, 181, 3284–3287. [Google Scholar] [PubMed]
- Chami, M.; Bayan, N.; Peyret, J.L.; Gulik-Krzywicki, T.; Leblon, G.; Shechter, E. The S-layer protein of Corynebacterium glutamicum is anchored to the cell wall by its C-terminal hydrophobic domain. Mol. Microbiol. 1997, 23, 483–492. [Google Scholar] [CrossRef] [PubMed]
- Linstedt, A.D.; Foguet, M.; Renz, M.; Seelig, H.P.; Glick, B.S.; Hauri, H.P. A C-terminally-anchored golgi protein is inserted into the endoplasmic reticulum and then transported to the golgi apparatus. Proc. Natl. Acad. Sci. USA 1995, 92, 5102–5105. [Google Scholar] [CrossRef] [PubMed]
- Schneewind, O.; Mihaylova-Petkov, D.; Model, P. Cell wall sorting signals in surface proteins of Gram-positive bacteria. Embo. J. 1993, 12, 4803–4811. [Google Scholar] [PubMed]
- Alami, M.; Luke, I.; Deitermann, S.; Eisner, G.; Koch, H.G.; Brunner, J.; Muller, M. Differential interactions between a twin-arginine signal peptide and its translocase in Escherichia coli. Mol. Cell 2003, 12, 937–946. [Google Scholar] [CrossRef]
- Berks, B.C.; Palmer, T.; Sargent, F. Protein targeting by the bacterial twin-arginine translocation (Tat) pathway. Curr. Opin. Microbiol. 2005, 8, 174–181. [Google Scholar] [CrossRef] [PubMed]
- Paterson, G.K.; Mitchell, T.J. The biology of Gram-positive sortase enzymes. Trends Microbiol. 2004, 12, 89–95. [Google Scholar] [CrossRef] [PubMed]
- Fischetti, V.A.; Pancholi, V.; Schneewind, O. Conservation of a hexapeptide sequence in the anchor region of surface proteins from Gram-positive cocci. Mol. Microbiol. 1990, 4, 1603–1605. [Google Scholar] [CrossRef] [PubMed]
- Schneewind, O.; Model, P.; Fischetti, V.A. Sorting of protein A to the staphylococcal cell wall. Cell 1992, 70, 267–281. [Google Scholar] [CrossRef]
- Navarre, W.W.; Schneewind, O. Proteolytic cleavage and cell wall anchoring at the LPXTG motif of surface proteins in Gram-positive bacteria. Mol. Microbiol. 1994, 14, 115–121. [Google Scholar] [CrossRef] [PubMed]
- Perry, A.M.; Ton-That, H.; Mazmanian, S.K.; Schneewind, O. Anchoring of surface proteins to the cell wall of Staphylococcus aureus. III. Lipid II is an in vivo peptidoglycan substrate for sortase-catalyzed surface protein anchoring. J Biol Chem 2002, 277, 16241–16248. [Google Scholar] [CrossRef] [PubMed]
- Desvaux, M.; Dumas, E.; Chafsey, I.; Hebraud, M. Protein cell surface display in Gram-positive bacteria: From single protein to macromolecular protein structure. FEMS Microbiol. Lett. 2006, 256, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Haft, D.H.; Payne, S.H.; Selengut, J. Archaeosortases and exosortases are widely distributed systems linking membrane transit with posttranslational modification. J. Bacteriol. 2012, 194, 36–48. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, K.; Fujita, Y.; Itoh, Y.; Yamane, K. Modification of length, hydrophobic properties and electric charge of Bacillus subtilis alpha-amylase signal peptide and their different effects on the production of secretory proteins in B. Subtilis and Escherichia coli cells. Mol. Gen. Genet. 1989, 216, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.H.; Huang, L.F.; Li, H.M.; Chen, Y.R.; Yu, S.M. Signal peptide-dependent targeting of a rice alpha-amylase and cargo proteins to plastids and extracellular compartments of plant cells. Plant. Physiol. 2004, 135, 1367–1377. [Google Scholar] [CrossRef] [PubMed]
- Ohmura, K.; Nakamura, K.; Yamazaki, H.; Shiroza, T.; Yamane, K.; Jigami, Y.; Tanaka, H.; Yoda, K.; Yamasaki, M.; Tamura, G. Length and structural effect of signal peptides derived from Bacillus subtilis alpha-amylase on secretion of Escherichia coli beta-lactamase in B. Subtilis cells. Nucleic. Acids. Res. 1984, 12, 5307–5319. [Google Scholar] [CrossRef] [PubMed]
- Cusano, A.M.; Parrilli, E.; Duilio, A.; Sannia, G.; Marino, G.; Tutino, M.L. Secretion of psychrophilic alpha-amylase deletion mutants in Pseudoalteromonas haloplanktis TAC125. FEMS Microbiol. Lett. 2006, 258, 67–71. [Google Scholar] [CrossRef] [PubMed]
- Snyder, A.; Vasil, A.I.; Zajdowicz, S.L.; Wilson, Z.R.; Vasil, M.L. Role of the Pseudomonas aeruginosa PlcH Tat signal peptide in protein secretion, transcription, and cross-species Tat secretion system compatibility. J. Bacteriol. 2006, 188, 1762–1774. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Soo, E.; Rudrappa, D.; Blum, P. Membrane Association and Catabolite Repression of the Sulfolobus solfataricus α-Amylase. Microorganisms 2015, 3, 567-587. https://doi.org/10.3390/microorganisms3030567
Soo E, Rudrappa D, Blum P. Membrane Association and Catabolite Repression of the Sulfolobus solfataricus α-Amylase. Microorganisms. 2015; 3(3):567-587. https://doi.org/10.3390/microorganisms3030567
Chicago/Turabian StyleSoo, Edith, Deepak Rudrappa, and Paul Blum. 2015. "Membrane Association and Catabolite Repression of the Sulfolobus solfataricus α-Amylase" Microorganisms 3, no. 3: 567-587. https://doi.org/10.3390/microorganisms3030567