
Complete Genome Sequence, Molecular Characterization and Phylogenetic Relationships of a Temminck’s Stint Calicivirus: Evidence for a New Genus within Caliciviridae Family

 , and
, and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Sampling
2.2. Sample Preparation and Sequencing
2.3. Assembly and Genome Annotation
2.4. Comparative Analysis
2.5. Phylogenetic Analysis
2.6. Species Demarcation
2.7. Protein 3D Structure Prediction
3. Results
3.1. Annotation of TsCV Genome and Comparative Analysis
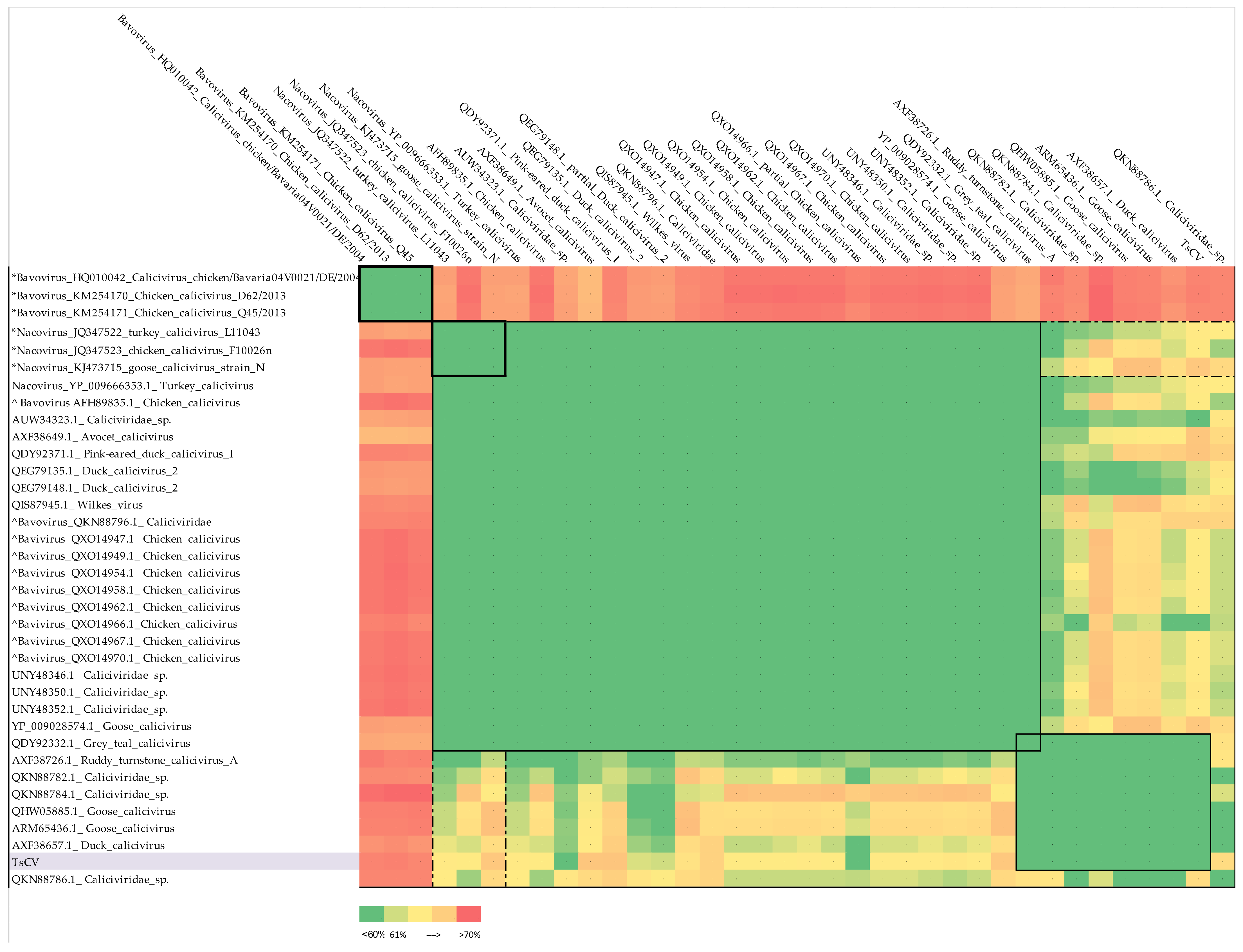
3.2. Taxonomic Classification of TsCV by ICTV Criteria
- 1.
- Goose calicivirus, (ARM65436.1 and QHW05885.1), duck calicivirus (AXF38657.1) and Caliciviridae sp. with accession numbers QKN88782.1, QKN88786.1 and QKN88784.1, as well as TsCV, cannot be assigned to the Nacovirus genus and can be combined into one separate genus with a member of the proposed Sanovirus genus included (the spread of divergence values within the proposed genus is 49.8 ± 7.8%, the divergence with members of the Nacovirus genus is 62.7 ± 1.7%).
- 2.
- Ruddy turnstone calicivirus A (AXF38726.1) has borderline divergence values from representatives of the genus Nacovirus (60.3 ± 0.8%) but is much closer to the proposed genus Sanovirus (values of pairwise divergence with goose calicivirus 53.3% and with putative members 56.1 ± 2.5%).
- 3.
- The inclusion of ruddy turnstone calicivirus A (AXF38726.1) virus in the proposed genus Sanovirus does not violate the demarcation criterion (the spread of divergence values within the proposed genus is 52.1 ± 7.2% and the divergence with members of the Nacovirus genus is 62.3 ± 1.8%).
- 4.
- Grey teal calicivirus (QDY92332.1) cannot be classified according to the accepted criterion, since the divergence values of its VP1 amino acid sequence are less than 60%, both in comparison with representative sequences of Nacovirus and with putative members of the proposed Sanovirus genus.
- 5.
- Chicken caliciviruses accession numbers QXO14947, QXO14949, QXO14954, QXO14958, QXO14962, QXO14966, QXO14967, QXO14970, AFH89835.1 and Caliciviridae sp. QKN88796 appear to be misclassified to the genus Bavovirus and should be moved to the genus Nacovirus.
3.3. Phylogenetic Analysis
3.4. Species Demarcation
- 1.
- Chicken calicivirus Q45/2013 (KM254171) and chicken calicivirus D62/2013 (KM254170) belonging the genus Bavovirus;
- 2.
- Bovine enteric calicivirus NB (AY082891) and Newbury-1 virus (DQ013304) belonging the genus Nebovirus;
- 3.
- Chiba virus/GVIII (AJ844470) and Yuzawa virus GVIII (KJ196291) belonging the genus Norovirus;
- 4.
- Calicivirus pig/AB104/CAN (FJ355930), calicivirus_pig/AB90/CAN (FJ355928) and calicivirus_pig/F15-10/CAN (FJ355929) belonging the genus Valovirus;
- 5.
- Sapovirus Angelholm virus SW278 (DQ125333), Ehime_virus (DQ058829) and Houston virus 7-1181 (AF435814) belonging the genus Sapovirus.
- 6.
- NongKhai-24 virus (AY646856) and Arg39 virus (AY289803) belonging the genus Sapovirus;
- 7.
- Sapovirus MT-2010/1982 (HM002617), sapovirus U65427 and Manchester virus (X86560) belonging the genus Sapovirus;
- 8.
- London_virus/29845 (U95645) and Bristol_virus (AJ249939) belonging the genus Sapovirus;
- 9.
- Rabbit_calicivirus-1 (X96868) and rabbit hemorrhagic disease virus-FRG (M67473) belonging the genus Lagovirus;
- 10.
- Unclassified duck calicivirus 2 MN175552 and MN175556;
- 11.
- Unclassified goose calicivirus KY399947 and MN068022;
- 12.
- Chicken calicivirus F10026n (JQ347523, Nacovirus according to the ICTV Caliciviridae report [4]), unclassified Caliciviridae_sp. OM469263 and OM469262 and six chicken caliciviruses strains (MW684845, MW684838, MW684835, MW684844, MW684834 and MW684840) presumably misclassified to the genus Bavovirus.
- 1.
- Goose calicivirus strain N (KJ473715) belonging the genus Nacovirus according to the ICTV Caliciviridae report [4] and ucclassified goose calicivirus (NC_024078);
- 2.
- Caliciviridae sp. OM469263.1 and OM469260.
3.5. Protein 3D-Structure Prediction
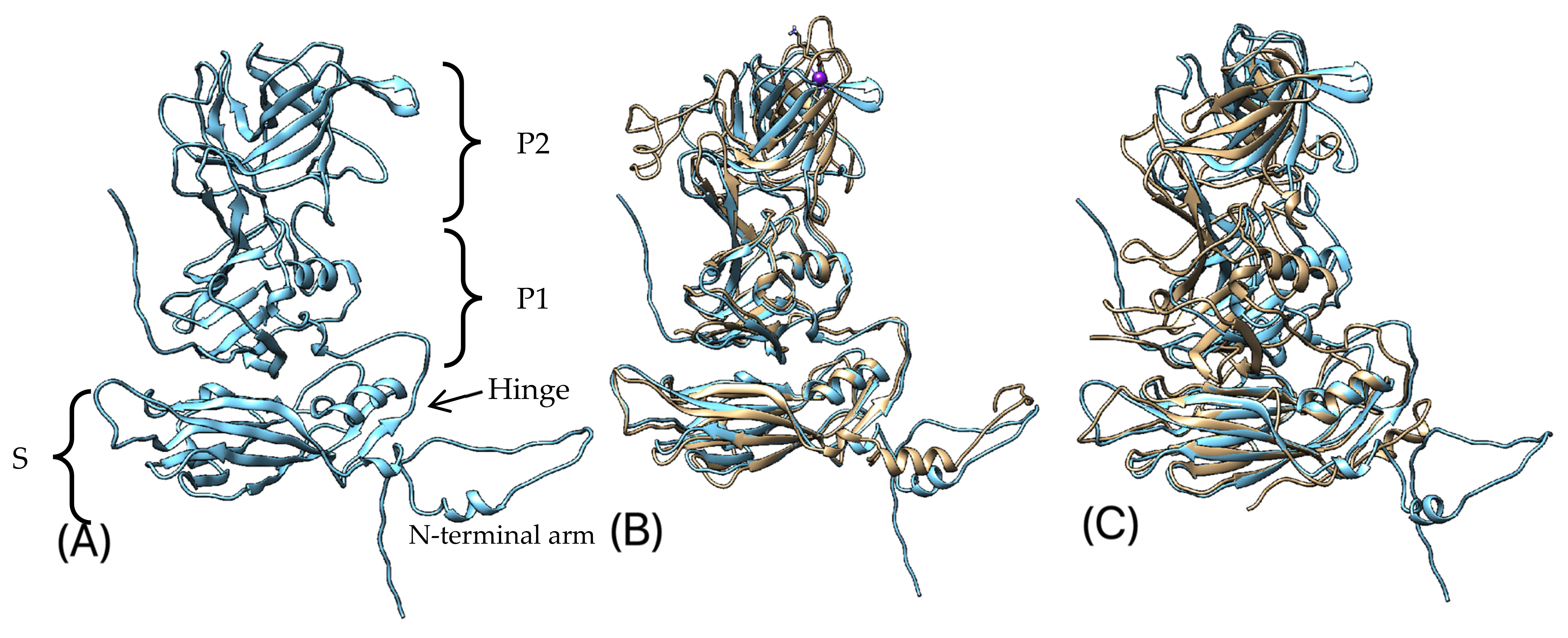

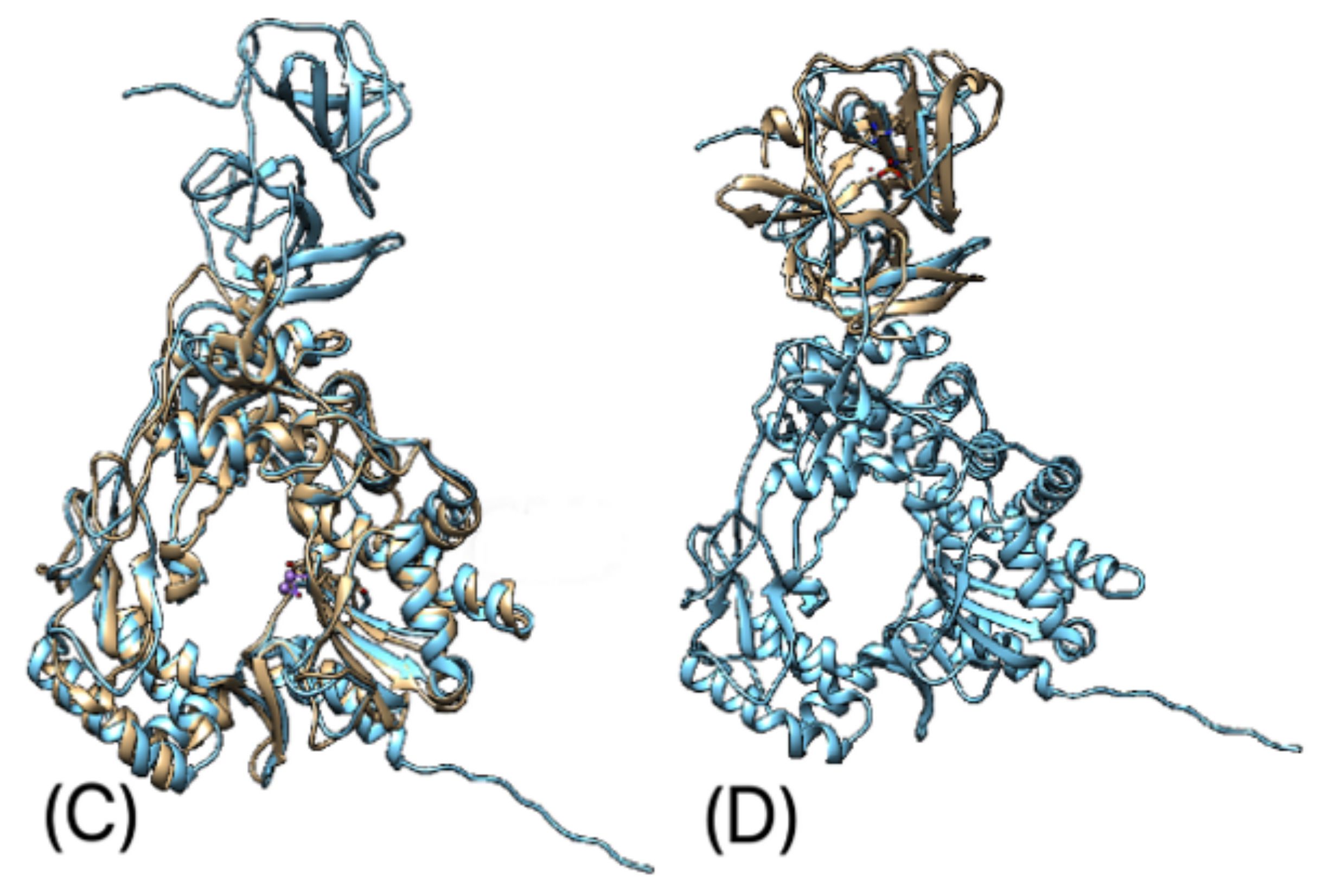
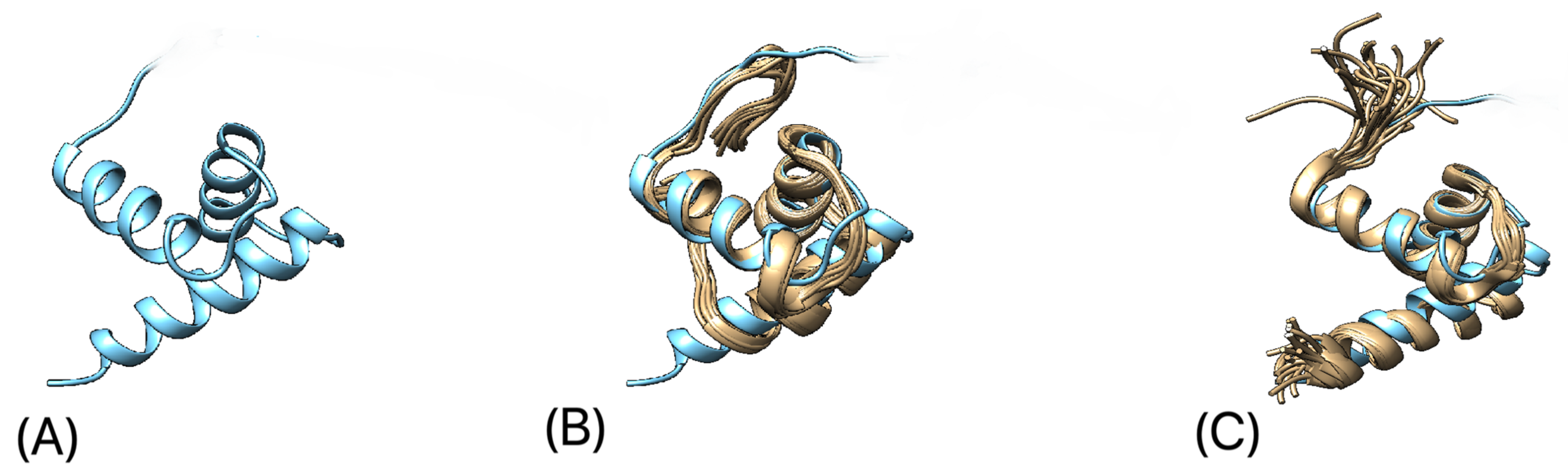
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Taxa | Number of Contigs | Total Length of Contigs | Group |
|---|---|---|---|
| Scolopacidae/Calidris | 21 | 19,912 | Bird |
| Turdidae/Erithacus | 2 | 2730 | |
| Sylviidae/Curruca | 3 | 2430 | |
| Amphipleuraceae/Halamphora | 3 | 3460 | Algae |
| Bacillariaceae/Cylindrotheca | 3 | 2757 | |
| Stauroneidaceae/Stauroneis | 3 | 2574 | |
| Danionidae/Danio | 6 | 7526 | Fish |
| Serranidae/Epinephelus | 4 | 3906 | |
| Batrachoididae/Thalassophryne | 2 | 2494 | |
| Apogonidae/Sphaeramia | 3 | 2442 | |
| Bovichtidae/Cottoperca | 2 | 2420 | |
| Echeneidae/Echeneis | 2 | 2005 | |
| Acanthosomatidae/Elasmucha | 5 | 5581 | Insect |
| Chironomidae/Chironomus | 4 | 4822 | |
| Apidae/Apis | 2 | 2064 | |
| Ostreidae/Crassostrea | 5 | 6755 | Mollusc |
| Eimeriidae/Eimeria | 7 | 7273 | Parasite |
| Rhabditidae/Caenorhabditis | 2 | 3541 | |
| Cornaceae/Cornus | 8 | 7911 | Plant |
| Malvaceae/Gossypium | 4 | 4437 | |
| Solanaceae/Solanum | 4 | 4272 | |
| Poaceae/Panicum | 2 | 2716 | |
| Brassicaceae/Brassica | 3 | 2585 | |
| Fabaceae/Cercis | 1 | 2406 | |
| Fabaceae/Cicer | 1 | 2348 | |
| Hominidae/Homo | 4 | 4027 | Human |
| Cercopithecidae/Macaca | 1 | 2868 | Primate |
| Amoebidiaceae/Amoebidium | 4 | 3011 | Protozoa |
| Protaspidae/Cryothecomonas | 1 | 2136 | |
| Octodontidae/Octodon | 1 | 2964 | Rodent |
| Sciuridae/Sciurus | 2 | 2732 | |
| Vespertilionidae/Eptesicus | 1 | 2138 | Bat |
| Genus | Sequence Name | GenBank Accession |
|---|---|---|
| Bavovirus | calicivirus chicken/Bavaria04V0021/DE/2004 | HQ010042 |
| Bavovirus | chicken calicivirus D62/2013 | KM254170 |
| Bavovirus | chicken calicivirus Q45/2013 | KM254171 |
| Lagovirus | rabbit hemorrhagic disease virus isolate Sr12 2 | KC741409 |
| Lagovirus | rabbit hemorrhagic disease virus-FRG | M67473 |
| Lagovirus | rabbit calicivirus-1 | X96868 |
| Lagovirus | European brown hare syndrome virus | Z69620 |
| Minovirus | fathead minnow calicivirus-USA/MN/2012 | KX371097 |
| Nacovirus | turkey calicivirus L11043 | JQ347522 |
| Nacovirus | chicken calicivirus F10026n | JQ347523 |
| Nacovirus | goose calicivirus strain N | KJ473715 |
| Nebovirus | Newbury-1 virus | DQ013304 |
| Nebovirus | bovine enteric calicivirus NB | AY082891 |
| Nebovirus | bovine calicivirus Kirklareli | KT119483 |
| Norovirus | lion norovirus GIV.2/Pistoia/387/06/ITA | EF450827 |
| Norovirus | swine calicivirus Sw918 | AB074893 |
| Norovirus | bovine norovirus Newbury2 | AF097917 |
| Norovirus | Hu/NLV/Alphatron/98-2/1998/NET | AF195847 |
| Norovirus | bovine calicivirus Jena | AJ011099 |
| Norovirus | Chiba virus/GVIII | AJ844470 |
| Norovirus | murine norovirus 1 | AY228235 |
| Norovirus | sheep norovirus Norsewood | EU193658 |
| Norovirus | dog norovirus GVI1/HKU Ca026F/2007/HKG | FJ692500 |
| Norovirus | dog norovirus GVI.1/Bari/91/2007/ITA | FJ875027 |
| Norovirus | dog norovirus Viseu | GQ443611 |
| Norovirus | Rn/GV/HKU CT2/HKG/2011 | JX486101 |
| Norovirus | Yuzawa virus GVIII | KJ196291 |
| Norovirus | bat norovirus-YN2010 | KJ790198 |
| Norovirus | Southampton virus | L07418 |
| Norovirus | Norwalk virus | M87661 |
| Norovirus | California sea lion norovirus strain Csl/NoV2/PF080916-2 | MG572715 |
| Norovirus | Hawaii calicivirus | U07611 |
| Norovirus | SapporoHK299 virus GIX.1 | KJ196290 |
| Norovirus | Lordsdale virus | X86557 |
| Recovirus | Tulane virus | EU391643 |
| Recovirus | human recovirus Bangladesh | JQ745645 |
| Recovirus | human recovirus Venezuela | MG571787 |
| Recovirus | WUHARV Calicivirus 1 | JX627575 |
| Recovirus | Tulane virus FT205 | KC662363 |
| Salovirus | Atlantic salmon calicivirus Nordland/2011 | KJ577139 |
| Salovirus | Atlantic salmon calicivirus AL V901 | KJ577140 |
| Sapovirus | porcine enteric calicivirus Cowden | AF182760 |
| Sapovirus | Mex340 virus | AF435812 |
| Sapovirus | Houston virus 7-1181 | AF435814 |
| Sapovirus | Arg39 virus | AY289803 |
| Sapovirus | NongKhai-24 virus | AY646856 |
| Sapovirus | porcine sapovirus JJ681 | AY974192 |
| Sapovirus | Ehime virus | DQ058829 |
| Sapovirus | Angelholm virus SW278 | DQ125333 |
| Sapovirus | porcine sapovirus 2053P4 | DQ359100 |
| Sapovirus | porcine sapovirus 43 | EU221477 |
| Sapovirus | porcine sapovirus sav1 | FJ387164 |
| Sapovirus | porcine sapovirus F19-10 | FJ498786 |
| Sapovirus | Sapovirus MT-2010/1982 | HM002617 |
| Sapovirus | Sapporo virus | U65427 |
| Sapovirus | Houston virus/90 | U95644 |
| Sapovirus | Manchester virus | X86560 |
| Sapovirus | Bristol virus | AJ249939 |
| Sapovirus | London virus/29845 | U95645 |
| Valovirus | calicivirus pig/AB90/CAN | FJ355928 |
| Valovirus | calicivirus pig/F15-10/CAN | FJ355929 |
| Valovirus | calicivirus pig/AB104/CAN | FJ355930 |
| Vesivirus | canine calicivirus-no48 | AF053720 |
| Vesivirus | Pan-1 virus | AF091736 |
| Vesivirus | vesicular exanthema of swine virus strain A48 | AF181082 |
| Vesivirus | walrus calicivirus | AF321298 |
| Vesivirus | calicivirus 2117 | AY343325 |
| Vesivirus | canine vesivirus Bari/212/07/ITA | JN204722 |
| Vesivirus | Feline calicivirus-9 | M86379 |
| Vesivirus | San Miguel sea lion virus-1 | M87481 |
| Vesivirus | San Miguel sea lion virus-4 | M87482 |
| Vesivirus | feline calicivirus CFI/68 | U13992 |
| Vesivirus | San Miguel sea lion virus-17 | U52005 |
| GenBank Accession | Sequence Name |
|---|---|
| QKN88784.1 | Caliciviridae sp. |
| QHW05885.1 | Goose calicivirus |
| AXF38657.1 | Duck calicivirus |
| ARM65436.1 | Goose calicivirus |
| QKN88782.1 | Caliciviridae sp. |
| AXF38726.1 | Ruddy turnstone calicivirus A |
| AUW34323.1 | Caliciviridae sp. |
| QKN88786.1 | Caliciviridae sp. |
| QDY92332.1 | Grey teal calicivirus |
| QKN88796.1 | Caliciviridae sp. |
| YP_9666353.1 | Turkey calicivirus |
| QIS87945.1 | Wilkes virus |
| AFH89835.1 | Chicken calicivirus |
| QEG79135.1 | Duck calicivirus 2 |
| QEG79148.1 | Duck calicivirus 2 |
| QDY92371.1 | Pink-eared duck calicivirus I |
| QXO14949.1 | Chicken calicivirus |
| QXO14947.1 | Chicken calicivirus |
| QXO14962.1 | Chicken calicivirus |
| QXO14958.1 | Chicken calicivirus |
| UNY48352.1 | Caliciviridae sp. |
| UNY48346.1 | Caliciviridae sp. |
| QXO14954.1 | Chicken calicivirus |
| UNY48350.1 | Caliciviridae sp. |
| QXO14967.1 | Chicken calicivirus |
| YP_9028574.1 | Goose calicivirus |
| AXF38649.1 | Avocet calicivirus |
| QXO14966.1 | Chicken calicivirus |
| QXO14970.1 | Chicken calicivirus |
| Virus | ASAP | bPTP ML | bPTP Bayesian | GMYC (Single Threshold) |
|---|---|---|---|---|
| Minovirus_KX371097_fathead_minnow_calicivirus-USA/MN/2012 | 1 | 1 | 1 | outgroup |
| Salovirus_KJ577139_Atlantic_salmon_calicivirus_Nordland/2011 | 1 | 1 | 1 | outgroup |
| Salovirus_KJ577140_Atlantic_salmon_calicivirus_AL_V901 | 1 | 1 | 1 | outgroup |
| Bavovirus_HQ010042_Calicivirus_chicken/Bavaria04V0021/DE/2004 | 3 | 3 | 3 | 2 |
| Bavovirus_KM254171_Chicken_calicivirus_Q45/2013 | ||||
| Bavovirus_KM254170_Chicken_calicivirus_D62/2013 | 1 | |||
| Temminck’s stint calicivirus | 1 | 1 | 1 | 1 |
| Unclassified_MT138020_Caliciviridae_sp. | 1 | 1 | 1 | 1 |
| Nebovirus_KT119483_bovine_calicivirus_Kirklareli | 1 | 1 | 1 | 1 |
| Nebovirus_AY082891_Bovine_enteric_calicivirus_NB | 2 | 2 | 2 | 2 |
| Nebovirus_DQ013304_Newbury-1_virus | ||||
| ^Bavovirus_MW684845_Chicken_calicivirus | 9 | 9 | 9 | 9 |
| ^Bavovirus_MW684838_Chicken_calicivirus | ||||
| ^Bavovirus_MW684835_Chicken_calicivirus | ||||
| ^Bavovirus_MW684844_partial_Chicken_calicivirus | ||||
| ^Bavovirus_MW684834_Chicken_calicivirus | ||||
| ^Bavovirus_MW684840_Chicken_calicivirus | ||||
| Nacovirus_JQ347523_chicken_calicivirus_F10026n | ||||
| Unclassified_OM469263_Caliciviridae_sp. | ||||
| Unclassified_OM469262_Caliciviridae_sp. | ||||
| Unclassified_MK204392_Grey_teal_calicivirus | 1 | 1 | 1 | 1 |
| Unclassified_MH453861_Ruddy_turnstone_calicivirus_A | 1 | 1 | 1 | 1 |
| Valovirus_FJ355930_Calicivirus_pig/AB104/CAN | 3 | 3 | 3 | 3 |
| Valovirus_FJ355928_Calicivirus_pig/AB90/CAN | ||||
| Valovirus_FJ355929_Calicivirus_pig/F15-10/CAN | ||||
| Unclassified_MH453804_Avocet_calicivirus | 1 | 1 | 1 | 1 |
| Unclassified_MT138018_Caliciviridae_sp. | 1 | 1 | 1 | 1 |
| Unclassified_MK204416_Pink-eared_duck_calicivirus_I | 1 | 1 | 1 | 1 |
| Nacovirus_KJ473715_goose_calicivirus_strain_N | 1 | 1 | 1 | 1 |
| Sapovirus_DQ125333_Angelholm_virus_SW278 | 3 | 3 | 3 | 3 |
| Sapovirus_DQ058829_Ehime_virus | ||||
| Sapovirus_AF435814_Houston_virus_7-1181 | ||||
| Norovirus_KJ790198_bat_norovirus-YN2010 | 1 | 1 | 1 | 1 |
| Norovirus_FJ692500_dog_norovirus_GVI1/HKU_Ca026F/2007/HKG | 1 | 1 | 1 | 1 |
| Unclassified_KY312552_Caliciviridae_sp. | 1 | 1 | 1 | 1 |
| Nacovirus_JQ347522_turkey_calicivirus_L11043 | 1 | 1 | 1 | 1 |
| Unclassified_MH453811_Duck_calicivirus | 1 | 1 | 1 | 1 |
| Unclassified_MT138017_Caliciviridae_sp. | 1 | 1 | 1 | 1 |
| Unclassified_KY399947_Goose_calicivirus | 2 | 2 | 2 | 2 |
| Unclassified_MN068022_Goose_calicivirus | ||||
| Norovirus_MG572715_California_sea_lion_norovirus_strain_Csl/NoV2/PF080916-2 | 1 | 1 | 1 | 1 |
| Norovirus_AJ844470_Chiba_virus/GVIII | 2 | 2 | 2 | 2 |
| Norovirus_KJ196291_Yuzawa_virus_GVIII | ||||
| Sapovirus_DQ359100_porcine_sapovirus_2053P4 | 1 | 1 | 1 | 1 |
| Sapovirus_AY974192_Porcine_sapovirus_JJ681 | 1 | 1 | 1 | 1 |
| Unclassified_MT138028_Caliciviridae | 1 | 1 | 1 | 1 |
| Unclassified_MT025075_Wilkes_virus | 1 | 1 | 1 | 1 |
| Unclassified_MN175552_Duck_calicivirus_2 | 2 | 2 | 2 | 2 |
| Unclassified_MN175556_partial_Duck_calicivirus_2 | ||||
| Sapovirus_AY646856_NongKhai-24_virus | 2 | 2 | 2 | 2 |
| Sapovirus_AY289803_Arg39_virus | ||||
| Norovirus_KJ196290_SapporoHK299_virus_GIX.1 | 1 | 1 | 1 | 1 |
| Norovirus_FJ875027_dog_norovirus_GVI.1/Bari/91/2007/ITA | 1 | 1 | 1 | 1 |
| Norovirus_GQ443611_dog_norovirus_Viseu | 1 | 1 | 1 | 1 |
| Norovirus_U07611_Hawaii_calicivirus | 1 | 1 | 1 | 1 |
| Norovirus_JX486101_Rn/GV/HKU_CT2/HKG/2011 | 1 | 1 | 1 | 1 |
| Norovirus_AY228235_murine_norovirus_1 | 1 | 1 | 1 | 1 |
| Norovirus_AB074893_Swine_calicivirus_Sw918 | 1 | 1 | 1 | 1 |
| Norovirus_X86557_Lordsdale_virus | 1 | 1 | 1 | 1 |
| Norovirus_AF195847_Hu/NLV/Alphatron/98-2/1998/NET | 1 | 1 | 1 | 1 |
| Norovirus_EF450827_lion_norovirus_GIV.2/Pistoia/387/06/ITA | 1 | 1 | 1 | 1 |
| Norovirus_L07418_Southampton_virus | 1 | 1 | 1 | 1 |
| Norovirus_M87661_Norwalk_virus | 1 | 1 | 1 | 1 |
| Vesivirus_AF053720_Canine_calicivirus-no48 | 1 | 1 | 1 | 1 |
| Recovirus_MG571787_Human_Recovirus_Venezuela | 1 | 1 | 1 | 1 |
| Recovirus_JQ745645_human_recovirus_Bangladesh | 1 | 1 | 1 | 1 |
| Norovirus_AF097917_bovine_norovirus_Newbury2 | 1 | 1 | 1 | 1 |
| Norovirus_AJ011099_bovine_calicivirus_Jena | 1 | 1 | 1 | 1 |
| Norovirus_EU193658_sheep_norovirus_Norsewood | 1 | 1 | 1 | 1 |
| Sapovirus_U95644_Houston_virus/90 | 1 | 1 | 1 | 1 |
| Sapovirus_HM002617_Sapovirus_MT-2010/1982 | 2 | 2 | 2 | 2 |
| Sapovirus_X86560_Manchester_virus | ||||
| Vesivirus_AF091736_Pan-1_virus | 1 | 1 | 4 | 1 |
| Vesivirus_U52005_San_Miguel_sea_lion_virus-17 | 1 | 1 | 1 | |
| Vesivirus_M87482_San_Miguel_sea_lion_virus-4 | 2 | 1 | 1 | |
| Vesivirus_AF181082_Vesicular_exanthema_of_swine_virus_strain_A48 | 1 | 1 | ||
| Lagovirus_Z69620_European_brown_hare_syndrome_virus | 1 | 1 | 1 | 1 |
| Lagovirus_KC741409_rabbit_hemorrhagic_disease_virus_isolate_Sr12_2 | 3 | 3 | 3 | 1 |
| Lagovirus_X96868_rabbit_calicivirus-1 | 2 | |||
| Lagovirus_M67473_rabbit_hemorrhagic_disease_virus-FRG | ||||
| Sapovirus_FJ498786_porcine_sapovirus_F19-10 | 1 | 1 | 1 | 1 |
| Sapovirus_EU221477_porcine_sapovirus_43 | 1 | 1 | 1 | 1 |
| Recovirus_JX627575_WUHARV_Calicivirus_1 | 1 | 1 | 1 | 1 |
| Recovirus_EU391643_Tulane_virus | 2 | 2 | 2 | 1 |
| Recovirus_KC662363_Tulane_Virus_FT205 | 1 | |||
| Vesivirus_JN204722_canine_vesivirus_Bari/212/07/ITA | 2 | 1 | 1 | 1 |
| Vesivirus_AY343325_calicivirus_2117 | 1 | 1 | 1 | |
| Vesivirus_AF321298_walrus_calicivirus | 1 | 1 | 1 | 1 |
| Vesivirus_M87481_San_Miguel_sea_lion_virus-1 | 1 | 1 | 1 | 1 |
| Sapovirus_AF435812_Mex340_virus | 1 | 1 | 1 | 1 |
| Sapovirus_U95645_London_virus/29845 | 2 | 2 | 2 | 2 |
| Sapovirus_AJ249939_Bristol_virus | ||||
| Sapovirus_FJ387164_porcine_sapovirus_sav1 | 2 | 1 | 2 | 1 |
| Sapovirus_AF182760_porcine_enteric_calicivirus_Cowden | 1 | 1 | ||
| Vesivirus_M86379_Feline_calicivirus-9 | 2 | 1 | 2 | 1 |
| Vesivirus_U13992_Feline_calicivirus_CFI/68 | 1 | 1 | ||
| TOTAL: | 68 | 72 | 67 | 72 |
References
- Green, K.Y.; Ando, T.; Balayan, M.S.; Berke, T.; Clarke, I.N.; Estes, M.K.; Matson, D.O.; Nakata, S.; Neill, J.D.; Studdert, M.J.; et al. Taxonomy of the Caliciviruses. J. Infect. Dis. 2000, 181, S322–S330. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- L’Homme, Y.; Sansregret, R.; Plante-Fortier, É.; Lamontagne, A.-M.; Ouardani, M.; Lacroix, G.; Simard, C. Genomic Characterization of Swine Caliciviruses Representing a New Genus of Caliciviridae. Virus Genes 2009, 39, 66–75. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, S.; Malik, Y.S.; Kobayashi, N. Animal Caliciviruses. In Animal-Origin Viral Zoonoses; Livestock Diseases and Management; Malik, Y.S., Singh, R.K., Dhama, K., Eds.; Springer: Singapore, 2020; pp. 81–109. ISBN 9789811526503. [Google Scholar]
- Vinjé, J.; Estes, M.K.; Esteves, P.; Green, K.Y.; Katayama, K.; Knowles, N.J.; L’Homme, Y.; Martella, V.; Vennema, H.; White, P.A.; et al. ICTV Virus Taxonomy Profile: Caliciviridae. J. Gen. Virol. 2019, 100, 1469–1470. [Google Scholar] [CrossRef] [PubMed]
- Desselberger, U. Caliciviridae Other Than Noroviruses. Viruses 2019, 11, 286. [Google Scholar] [CrossRef] [Green Version]
- Wang, F.; Wang, M.; Dong, Y.; Zhang, B.; Zhang, D. Genetic Characterization of a Novel Calicivirus from a Goose. Arch. Virol. 2017, 162, 2115–2118. [Google Scholar] [CrossRef] [PubMed]
- Ng, T.F.F.; Marine, R.; Wang, C.; Simmonds, P.; Kapusinszky, B.; Bodhidatta, L.; Oderinde, B.S.; Wommack, K.E.; Delwart, E. High Variety of Known and New RNA and DNA Viruses of Diverse Origins in Untreated Sewage. J. Virol. 2012, 86, 12161–12175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Royall, E.; Locker, N. Translational Control during Calicivirus Infection. Viruses 2016, 8, 104. [Google Scholar] [CrossRef] [Green Version]
- Smertina, E.; Hall, R.N.; Urakova, N.; Strive, T.; Frese, M. Calicivirus Non-Structural Proteins: Potential Functions in Replication and Host Cell Manipulation. Front. Microbiol. 2021, 12, 712710. [Google Scholar] [CrossRef]
- Li, T.-F.; Hosmillo, M.; Schwanke, H.; Shu, T.; Wang, Z.; Yin, L.; Curry, S.; Goodfellow, I.G.; Zhou, X. Human Norovirus NS3 Has RNA Helicase and Chaperoning Activities. J. Virol. 2018, 92, e01606-17. [Google Scholar] [CrossRef] [Green Version]
- Cotton, B.T.; Hyde, J.L.; Sarvestani, S.T.; Sosnovtsev, S.V.; Green, K.Y.; White, P.A.; Mackenzie, J.M. The Norovirus NS3 Protein Is a Dynamic Lipid- and Microtubule-Associated Protein Involved in Viral RNA Replication. J. Virol. 2017, 91, e02138-16. [Google Scholar] [CrossRef] [Green Version]
- Goodfellow, I. The Genome-Linked Protein VPg of Vertebrate Viruses—A Multifaceted Protein. Curr. Opin. Virol. 2011, 1, 355–362. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boniotti, B.; Wirblich, C.; Sibilia, M.; Meyers, G.; Thiel, H.J.; Rossi, C. Identification and Characterization of a 3C-like Protease from Rabbit Hemorrhagic Disease Virus, a Calicivirus. J. Virol. 1994, 68, 6487–6495. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mahar, J.E.; Bok, K.; Green, K.Y.; Kirkwood, C.D. The Importance of Intergenic Recombination in Norovirus GII.3 Evolution. J. Virol. 2013, 87, 3687–3698. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thackray, L.B.; Wobus, C.E.; Chachu, K.A.; Liu, B.; Alegre, E.R.; Henderson, K.S.; Kelley, S.T.; Virgin, H.W. Murine Noroviruses Comprising a Single Genogroup Exhibit Biological Diversity despite Limited Sequence Divergence. J. Virol. 2007, 81, 10460–10473. [Google Scholar] [CrossRef] [Green Version]
- Schaffer, F.L.; Bachrach, H.L.; Brown, F.; Gillespie, J.H.; Burroughs, N.; Madin, S.H.; Madeley, R.; Povey, C.; Scott, F.; Smith, A.W.; et al. Caliciviridae. Intervirology 1980, 14, 1–6. [Google Scholar] [CrossRef]
- Conley, M.J.; McElwee, M.; Azmi, L.; Gabrielsen, M.; Byron, O.; Goodfellow, I.G.; Bhella, D. Calicivirus VP2 Forms a Portal-like Assembly Following Receptor Engagement. Nature 2019, 565, 377–381. [Google Scholar] [CrossRef] [Green Version]
- De Souza, W.M.; Fumagalli, M.J.; de Araujo, J.; Ometto, T.; Modha, S.; Thomazelli, L.M.; Durigon, E.L.; Murcia, P.R.; Figueiredo, L.T.M. Discovery of Novel Astrovirus and Calicivirus Identified in Ruddy Turnstones in Brazil. Sci. Rep. 2019, 9, 5556. [Google Scholar] [CrossRef] [Green Version]
- Wolf, S.; Reetz, J.; Hoffmann, K.; Gründel, A.; Schwarz, B.-A.; Hänel, I.; Otto, P.H. Discovery and Genetic Characterization of Novel Caliciviruses in German and Dutch Poultry. Arch. Virol. 2012, 157, 1499–1507. [Google Scholar] [CrossRef] [PubMed]
- Liao, Q.; Wang, X.; Wang, D.; Zhang, D. Complete Genome Sequence of a Novel Calicivirus from a Goose. Arch. Virol. 2014, 159, 2529–2531. [Google Scholar] [CrossRef]
- Ahlers, L.R.H.; Goodman, A.G. The Immune Responses of the Animal Hosts of West Nile Virus: A Comparison of Insects, Birds, and Mammals. Front. Cell. Infect. Microbiol. 2018, 8, 96. [Google Scholar] [CrossRef] [Green Version]
- Luo, H.; Wang, T. Recent Advances in Understanding West Nile Virus Host Immunity and Viral Pathogenesis. F1000Research 2018, 7, 338. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wheeler, S.S.; Taff, C.C.; Reisen, W.K.; Townsend, A.K. Mosquito Blood-Feeding Patterns and Nesting Behavior of American Crows, an Amplifying Host of West Nile Virus. Parasit. Vectors 2021, 14, 331. [Google Scholar] [CrossRef] [PubMed]
- Mulvey, P.; Duong, V.; Boyer, S.; Burgess, G.; Williams, D.T.; Dussart, P.; Horwood, P.F. The Ecology and Evolution of Japanese Encephalitis Virus. Pathogens 2021, 10, 1534. [Google Scholar] [CrossRef]
- Horman, W.S.J.; Nguyen, T.H.O.; Kedzierska, K.; Bean, A.G.D.; Layton, D.S. The Drivers of Pathology in Zoonotic Avian Influenza: The Interplay Between Host and Pathogen. Front. Immunol. 2018, 9, 1812. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blagodatski, A.; Trutneva, K.; Glazova, O.; Mityaeva, O.; Shevkova, L.; Kegeles, E.; Onyanov, N.; Fede, K.; Maznina, A.; Khavina, E.; et al. Avian Influenza in Wild Birds and Poultry: Dissemination Pathways, Monitoring Methods, and Virus Ecology. Pathogens 2021, 10, 630. [Google Scholar] [CrossRef]
- Liu, W.J.; Wu, Y.; Bi, Y.; Shi, W.; Wang, D.; Shi, Y.; Gao, G.F. Emerging HxNy Influenza A Viruses. Cold Spring Harb. Perspect. Med. 2022, 12, a038406. [Google Scholar] [CrossRef]
- Kocher, J.F.; Lindesmith, L.C.; Debbink, K.; Beall, A.; Mallory, M.L.; Yount, B.L.; Graham, R.L.; Huynh, J.; Gates, J.E.; Donaldson, E.F.; et al. Bat Caliciviruses and Human Noroviruses Are Antigenically Similar and Have Overlapping Histo-Blood Group Antigen Binding Profiles. mBio 2018, 9, e00869-18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berry, E.S.; Skilling, D.E.; Barlough, J.E.; Vedros, N.A.; Gage, L.J.; Smith, A.W. New Marine Calicivirus Serotype Infective for Swine. Am. J. Vet. Res. 1990, 51, 1184–1187. [Google Scholar]
- Ayginin, A.A.; Pimkina, E.V.; Matsvay, A.D.; Speranskaya, A.S.; Safonova, M.V.; Blinova, E.A.; Artyushin, I.V.; Dedkov, V.G.; Shipulin, G.A.; Khafizov, K. The Study of Viral RNA Diversity in Bird Samples Using De Novo Designed Multiplex Genus-Specific Primer Panels. Adv. Virol. 2018, 2018, 3248285. [Google Scholar] [CrossRef]
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; Prjibelski, A.D.; et al. SPAdes: A New Genome Assembly Algorithm and Its Applications to Single-Cell Sequencing. J. Comput. Biol. 2012, 19, 455–477. [Google Scholar] [CrossRef] [Green Version]
- McGinnis, S.; Madden, T.L. BLAST: At the Core of a Powerful and Diverse Set of Sequence Analysis Tools. Nucleic Acids Res. 2004, 32, W20–W25. [Google Scholar] [CrossRef] [PubMed]
- Federhen, S. The NCBI Taxonomy Database. Nucleic Acids Res. 2012, 40, D136–D143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.; Durbin, R. Fast and Accurate Short Read Alignment with Burrows-Wheeler Transform. Bioinforma. Oxf. Engl. 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. 1000 Genome Project Data Processing Subgroup The Sequence Alignment/Map Format and SAMtools. Bioinform. Oxf. Engl. 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Milne, I.; Stephen, G.; Bayer, M.; Cock, P.J.A.; Pritchard, L.; Cardle, L.; Shaw, P.D.; Marshall, D. Using Tablet for Visual Exploration of Second-Generation Sequencing Data. Brief. Bioinform. 2013, 14, 193–202. [Google Scholar] [CrossRef] [PubMed]
- NCBI Software Tool to Search for Open Reading Frames (ORFs) in the DNA Sequence Open Reading Frame Finder (RRID:SCR_016643). Available online: https://www.ncbi.nlm.nih.gov/orffinder (accessed on 1 May 2022).
- Boratyn, G.M.; Schäffer, A.A.; Agarwala, R.; Altschul, S.F.; Lipman, D.J.; Madden, T.L. Domain Enhanced Lookup Time Accelerated BLAST. Biol. Direct 2012, 7, 12. [Google Scholar] [CrossRef] [Green Version]
- SnapGen Viewer Software by Dotmatics (from Insightful Science; Available at Snapgene.Com); Insightful Science: San Diego, CA, USA, 2022.
- Clark, K.; Karsch-Mizrachi, I.; Lipman, D.J.; Ostell, J.; Sayers, E.W. GenBank. Nucleic Acids Res. 2016, 44, D67–D72. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katoh, K. MAFFT: A Novel Method for Rapid Multiple Sequence Alignment Based on Fast Fourier Transform. Nucleic Acids Res. 2002, 30, 3059–3066. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cock, P.J.A.; Antao, T.; Chang, J.T.; Chapman, B.A.; Cox, C.J.; Dalke, A.; Friedberg, I.; Hamelryck, T.; Kauff, F.; Wilczynski, B.; et al. Biopython: Freely Available Python Tools for Computational Molecular Biology and Bioinformatics. Bioinform. Oxf. Engl. 2009, 25, 1422–1423. [Google Scholar] [CrossRef]
- Tamura, K.; Stecher, G.; Kumar, S. MEGA11: Molecular Evolutionary Genetics Analysis Version 11. Mol. Biol. Evol. 2021, 38, 3022–3027. [Google Scholar] [CrossRef]
- Lanfear, R.; Frandsen, P.B.; Wright, A.M.; Senfeld, T.; Calcott, B. PartitionFinder 2: New Methods for Selecting Partitioned Models of Evolution for Molecular and Morphological Phylogenetic Analyses. Mol. Biol. Evol. 2017, 34, 772–773. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ducasse, J.; Ung, V.; Lecointre, G.; Miralles, A. LIMES: A Tool for Comparing Species Partition. Bioinformatics 2020, 36, 2282–2283. [Google Scholar] [CrossRef] [PubMed]
- Castresana, J. Selection of Conserved Blocks from Multiple Alignments for Their Use in Phylogenetic Analysis. Mol. Biol. Evol. 2000, 17, 540–552. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kosakovsky Pond, S.L.; Poon, A.F.Y.; Velazquez, R.; Weaver, S.; Hepler, N.L.; Murrell, B.; Shank, S.D.; Magalis, B.R.; Bouvier, D.; Nekrutenko, A.; et al. HyPhy 2.5—A Customizable Platform for Evolutionary Hypothesis Testing Using Phylogenies. Mol. Biol. Evol. 2020, 37, 295–299. [Google Scholar] [CrossRef] [PubMed]
- Kozlov, A.M.; Darriba, D.; Flouri, T.; Morel, B.; Stamatakis, A. RAxML-NG: A Fast, Scalable and User-Friendly Tool for Maximum Likelihood Phylogenetic Inference. Bioinformatics 2019, 35, 4453–4455. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abadi, S.; Azouri, D.; Pupko, T.; Mayrose, I. Model Selection May Not Be a Mandatory Step for Phylogeny Reconstruction. Nat. Commun. 2019, 10, 934. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spielman, S.J. Relative Model Fit Does Not Predict Topological Accuracy in Single-Gene Protein Phylogenetics. Mol. Biol. Evol. 2020, 37, 2110–2123. [Google Scholar] [CrossRef] [PubMed]
- Letunic, I.; Bork, P. Interactive Tree Of Life (ITOL) v5: An Online Tool for Phylogenetic Tree Display and Annotation. Nucleic Acids Res. 2021, 49, W293–W296. [Google Scholar] [CrossRef]
- Matsvay, A.; Dyachkova, M.; Mikhaylov, I.; Kiselev, D.; Say, A.; Burskaia, V.; Artyushin, I.; Khafizov, K.; Shipulin, G. Complete Genome Sequence, Molecular Characterization and Phylogenetic Relationships of a Novel Tern Atadenovirus. Microorganisms 2021, 10, 31. [Google Scholar] [CrossRef]
- Fujisawa, T.; Barraclough, T.G. Delimiting Species Using Single-Locus Data and the Generalized Mixed Yule Coalescent Approach: A Revised Method and Evaluation on Simulated Data Sets. Syst. Biol. 2013, 62, 707–724. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Kapli, P.; Pavlidis, P.; Stamatakis, A. A General Species Delimitation Method with Applications to Phylogenetic Placements. Bioinformatics 2013, 29, 2869–2876. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Puillandre, N.; Brouillet, S.; Achaz, G. ASAP: Assemble Species by Automatic Partitioning. Mol. Ecol. Resour. 2021, 21, 609–620. [Google Scholar] [CrossRef] [PubMed]
- Tamura, K.; Tao, Q.; Kumar, S. Theoretical Foundation of the RelTime Method for Estimating Divergence Times from Variable Evolutionary Rates. Mol. Biol. Evol. 2018, 35, 1770–1782. [Google Scholar] [CrossRef] [PubMed]
- Tamura, K.; Battistuzzi, F.U.; Billing-Ross, P.; Murillo, O.; Filipski, A.; Kumar, S. Estimating Divergence Times in Large Molecular Phylogenies. Proc. Natl. Acad. Sci. USA 2012, 109, 19333–19338. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chan, J.F.-W.; To, K.K.-W.; Chen, H.; Yuen, K.-Y. Cross-Species Transmission and Emergence of Novel Viruses from Birds. Curr. Opin. Virol. 2015, 10, 63–69. [Google Scholar] [CrossRef]
- Paradis, E.; Claude, J.; Strimmer, K. APE: Analyses of Phylogenetics and Evolution in R Language. Bioinformatics 2004, 20, 289–290. [Google Scholar] [CrossRef] [Green Version]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly Accurate Protein Structure Prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef]
- Suzek, B.E.; Wang, Y.; Huang, H.; McGarvey, P.B.; Wu, C.H.; UniProt Consortium. UniRef Clusters: A Comprehensive and Scalable Alternative for Improving Sequence Similarity Searches. Bioinformatics 2015, 31, 926–932. [Google Scholar] [CrossRef] [Green Version]
- Mitchell, A.L.; Almeida, A.; Beracochea, M.; Boland, M.; Burgin, J.; Cochrane, G.; Crusoe, M.R.; Kale, V.; Potter, S.C.; Richardson, L.J.; et al. MGnify: The Microbiome Analysis Resource in 2020. Nucleic Acids Res. 2020, 48, D570–D578. [Google Scholar] [CrossRef]
- Steinegger, M.; Söding, J. Clustering Huge Protein Sequence Sets in Linear Time. Nat. Commun. 2018, 9, 2542. [Google Scholar] [CrossRef] [Green Version]
- Mirdita, M.; von den Driesch, L.; Galiez, C.; Martin, M.J.; Söding, J.; Steinegger, M. Uniclust Databases of Clustered and Deeply Annotated Protein Sequences and Alignments. Nucleic Acids Res. 2017, 45, D170–D176. [Google Scholar] [CrossRef] [PubMed]
- Berman, H.M. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera? A Visualization System for Exploratory Research and Analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wille, M.; Eden, J.; Shi, M.; Klaassen, M.; Hurt, A.C.; Holmes, E.C. Virus–Virus Interactions and Host Ecology Are Associated with RNA Virome Structure in Wild Birds. Mol. Ecol. 2018, 27, 5263–5278. [Google Scholar] [CrossRef] [PubMed]
- Wille, M.; Shi, M.; Klaassen, M.; Hurt, A.C.; Holmes, E.C. Virome Heterogeneity and Connectivity in Waterfowl and Shorebird Communities. ISME J. 2019, 13, 2603–2616. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sosnovtsev, S.V.; Belliot, G.; Chang, K.-O.; Prikhodko, V.G.; Thackray, L.B.; Wobus, C.E.; Karst, S.M.; Virgin, H.W.; Green, K.Y. Cleavage Map and Proteolytic Processing of the Murine Norovirus Nonstructural Polyprotein in Infected Cells. J. Virol. 2006, 80, 7816–7831. [Google Scholar] [CrossRef] [Green Version]
- Prasad, B.V.V.; Hardy, M.E.; Dokland, T.; Bella, J.; Rossmann, M.G.; Estes, M.K. X-ray Crystallographic Structure of the Norwalk Virus Capsid. Science 1999, 286, 287–290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zeitler, C.E.; Estes, M.K.; Venkataram Prasad, B.V. X-ray Crystallographic Structure of the Norwalk Virus Protease at 1.5-Å Resolution. J. Virol. 2006, 80, 5050–5058. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ng, K.K.S.; Cherney, M.M.; Vázquez, A.L.; Machın, Á.; Alonso, J.M.M.; Parra, F.; James, M.N.G. Crystal Structures of Active and Inactive Conformations of a Caliciviral RNA-Dependent RNA Polymerase. J. Biol. Chem. 2002, 277, 1381–1387. [Google Scholar] [CrossRef] [Green Version]
- Ng, K.K.-S.; Pendás-Franco, N.; Rojo, J.; Boga, J.A.; Machín, À.; Alonso, J.M.M.; Parra, F. Crystal Structure of Norwalk Virus Polymerase Reveals the Carboxyl Terminus in the Active Site Cleft. J. Biol. Chem. 2004, 279, 16638–16645. [Google Scholar] [CrossRef] [Green Version]
- Leen, E.N.; Kwok, K.Y.R.; Birtley, J.R.; Simpson, P.J.; Subba-Reddy, C.V.; Chaudhry, Y.; Sosnovtsev, S.V.; Green, K.Y.; Prater, S.N.; Tong, M.; et al. Structures of the Compact Helical Core Domains of Feline Calicivirus and Murine Norovirus VPg Proteins. J. Virol. 2013, 87, 5318–5330. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hwang, H.-J.; Min, H.J.; Yun, H.; Pelton, J.G.; Wemmer, D.E.; Cho, K.-O.; Kim, J.-S.; Lee, C.W. Solution Structure of the Porcine Sapovirus VPg Core Reveals a Stable Three-Helical Bundle with a Conserved Surface Patch. Biochem. Biophys. Res. Commun. 2015, 459, 610–616. [Google Scholar] [CrossRef] [PubMed]
- Lambden, P.R.; Clarke, I.N. Genome Organization in the Caliciviridae. Trends Microbiol. 1995, 3, 261–265. [Google Scholar] [CrossRef]
- Powell, M.L.; Brown, T.D.K.; Brierley, I. Translational Termination–Re-Initiation in Viral Systems. Biochem. Soc. Trans. 2008, 36, 717–722. [Google Scholar] [CrossRef] [Green Version]
- Napthine, S.; Lever, R.A.; Powell, M.L.; Jackson, R.J.; Brown, T.D.K.; Brierley, I. Expression of the VP2 Protein of Murine Norovirus by a Translation Termination-Reinitiation Strategy. PLoS ONE 2009, 4, e8390. [Google Scholar] [CrossRef] [Green Version]
- Chen, R.; Neill, J.D.; Estes, M.K.; Prasad, B.V.V. X-ray Structure of a Native Calicivirus: Structural Insights into Antigenic Diversity and Host Specificity. Proc. Natl. Acad. Sci. USA 2006, 103, 8048–8053. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simmonds, P.; Adams, M.J.; Benkő, M.; Breitbart, M.; Brister, J.R.; Carstens, E.B.; Davison, A.J.; Delwart, E.; Gorbalenya, A.E.; Harrach, B.; et al. Virus Taxonomy in the Age of Metagenomics. Nat. Rev. Microbiol. 2017, 15, 161–168. [Google Scholar] [CrossRef]
- de Vries, J.J.C.; Brown, J.R.; Couto, N.; Beer, M.; Le Mercier, P.; Sidorov, I.; Papa, A.; Fischer, N.; Oude Munnink, B.B.; Rodriquez, C.; et al. Recommendations for the Introduction of Metagenomic Next-Generation Sequencing in Clinical Virology, Part II: Bioinformatic Analysis and Reporting. J. Clin. Virol. 2021, 138, 104812. [Google Scholar] [CrossRef]
- van Regenmortel, M.H.V.; Bishop, D.H.L.; Fauquet, C.M.; Mayo, M.A.; Maniloff, J.; Calisher, C.H. Guidelines to the Demarcation of Virus Species. Arch. Virol. 1997, 142, 1505–1518. [Google Scholar] [CrossRef]
- Konstantinidis, K.T.; Tiedje, J.M. Genomic Insights That Advance the Species Definition for Prokaryotes. Proc. Natl. Acad. Sci. USA 2005, 102, 2567–2572. [Google Scholar] [CrossRef] [Green Version]
- Bodewes, R. Novel Viruses in Birds: Flying through the Roof or Is a Cage Needed? Vet. J. 2018, 233, 55–62. [Google Scholar] [CrossRef] [PubMed]
- Zockler, C. Patterns in Biodiversity in Arctic Birds. WCMC Biodiversity Bulletin; WCMC: Cambridge, UK, 1998; Volume 3, pp. 1–16. [Google Scholar]
- Stepanian, L.S.; Pavlov, D.S. Conspectus of the Ornithological Fauna of Russia and Adjacent Territories (within the Borders of the USSR as a Historic Region); IKTS Akademkniga: Moscow, Russia, 2003; ISBN 978-5-94628-093-8. [Google Scholar]


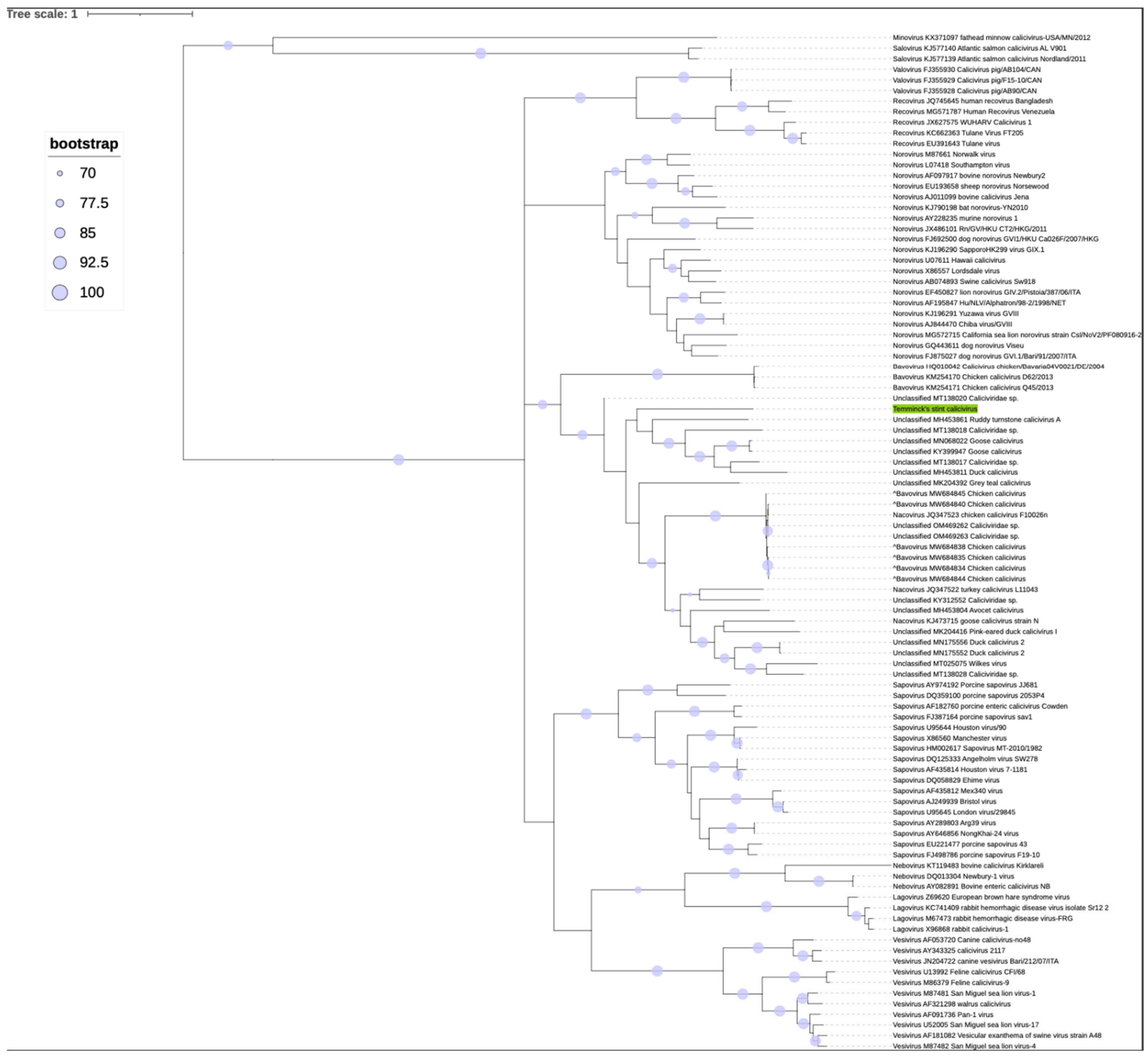
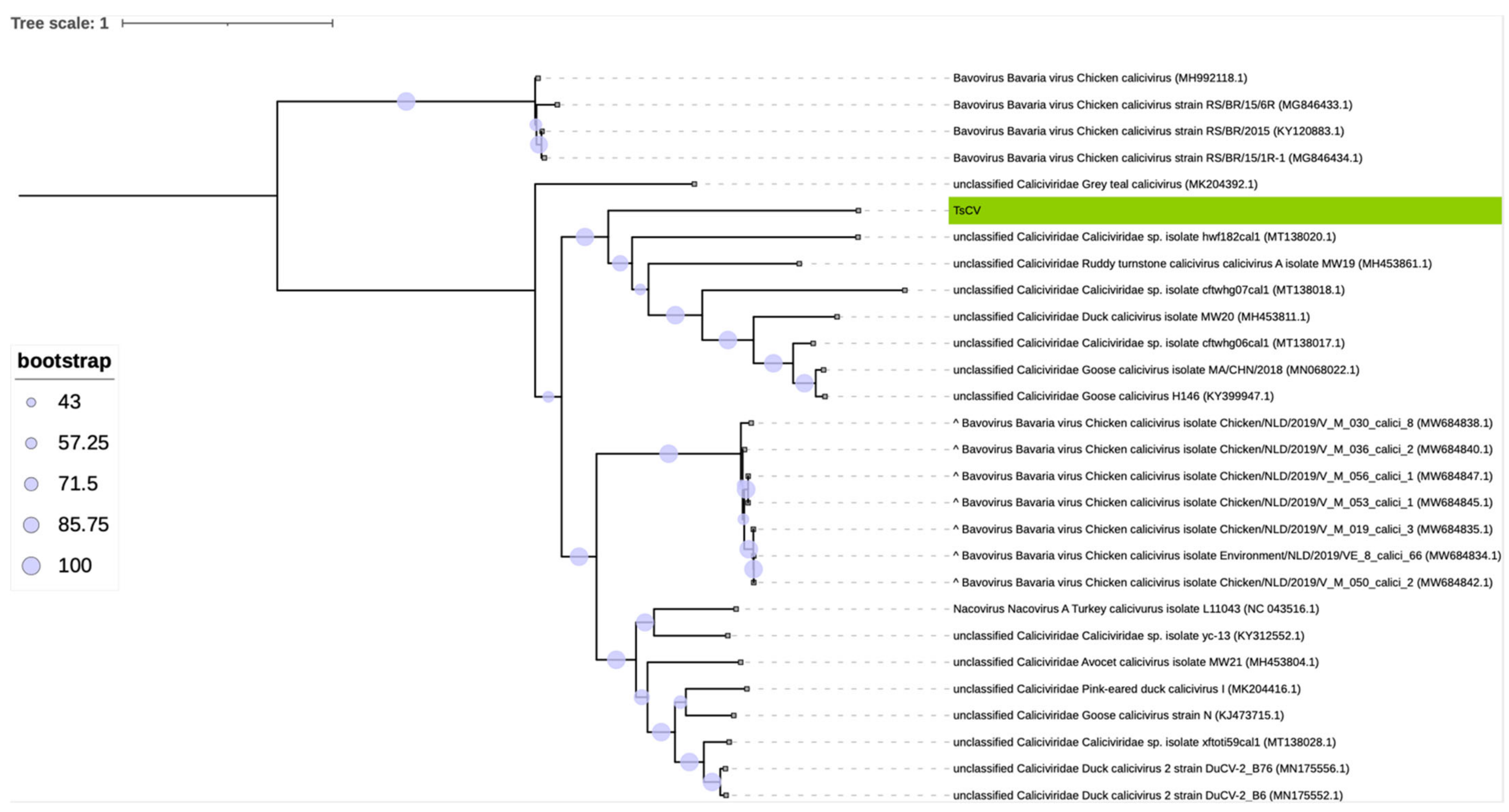
| TsCV | Ruddy turnstone calicivirus A [67] (MH453861.1) | Duck Calicivirus [67] (MH453811.1) | Goose Calicivirus (MN068022.1) | Goose Calicivirus [6] (KY399947.1) | Caliciviridae sp. (MT138017.1) | Caliciviridae sp. (MT138020.1) | |
|---|---|---|---|---|---|---|---|
| %GC | 51.73% | 51.77% | 50.71% | 48.66% | 49.13% | 47.93% | 50.78% |
| Genome nucleotide identity to TsCV | 43.68% | 40.33% | 42.39% | 41.82% | 42.64% | 42.37% | |
| VP1 protein divergence with TsCV | 59.70% | 58.42% | 57.44% | 57.44% | 59.69% | 59.18% | |
| 3′ UTR length | 816 nt | 521 nt | 901 nt | 15 nt | 18 nt | 225 nt | 731 nt |
| ORF1 length | 6849 nt | 7221 nt | 7827 nt | 7254 nt | 7254 nt | 7827 nt | 7173 nt |
| Distance between ORF1 and ORF2 | 1 nt | −17 nt | −74 nt | −8 nt | −8 nt | −8 nt | −10 nt |
| ORF2 length | 711 nt | 621 nt | 765 nt | 855 nt | 855 nt | 852 nt | 957 nt |
| 5′ URT length | 198 nt | 452 nt | 289 nt | 323 nt | 330 nt | 99 nt | 92 nt |
| Genome length | 8575 nt | 8798 nt | 9780 nt | 8439 nt | 8449 nt | 8995 nt | 8943 nt |
| Bavovirus | Lagovirus | Minovirus | Nacovirus | Nebovirus | Norovirus | Recovirus | Salovirus | Sapovirus | Valovirus | Vesivirus | |
|---|---|---|---|---|---|---|---|---|---|---|---|
| TsCV | 70.8 ± 0.3% | 78.4 ± 0.3% | 87.7 | 63.1 ± 1.7% | 78.1 ± 0.3% | 82.0 ± 1.4% | 83.7 ± 0.2% | 87.4 ± 0.9% | 71.2 ± 1.2% | 82.7 ± 0.3% | 75.1 ± 1.1% |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Matsvay, A.; Dyachkova, M.; Sai, A.; Burskaia, V.; Artyushin, I.; Shipulin, G. Complete Genome Sequence, Molecular Characterization and Phylogenetic Relationships of a Temminck’s Stint Calicivirus: Evidence for a New Genus within Caliciviridae Family. Microorganisms 2022, 10, 1540. https://doi.org/10.3390/microorganisms10081540
Matsvay A, Dyachkova M, Sai A, Burskaia V, Artyushin I, Shipulin G. Complete Genome Sequence, Molecular Characterization and Phylogenetic Relationships of a Temminck’s Stint Calicivirus: Evidence for a New Genus within Caliciviridae Family. Microorganisms. 2022; 10(8):1540. https://doi.org/10.3390/microorganisms10081540
Chicago/Turabian StyleMatsvay, Alina, Marina Dyachkova, Anna Sai, Valentina Burskaia, Ilya Artyushin, and German Shipulin. 2022. "Complete Genome Sequence, Molecular Characterization and Phylogenetic Relationships of a Temminck’s Stint Calicivirus: Evidence for a New Genus within Caliciviridae Family" Microorganisms 10, no. 8: 1540. https://doi.org/10.3390/microorganisms10081540